

Climate change causes significant alterations in precipitation and temperature regimes that are predicted to become more extreme throughout the next century. Microorganisms are important members within ecosystems, and how they respond to these changing abiotic stressors has large implications for the functioning of ecosystems, the recycling of nutrients, and the health of the aboveground plant community. Drought stress negatively impacts microbial activity, but the magnitude of this stress response may be dependent on above- and belowground interactions. This study demonstrates that beneficial associations between plants and microbes can enhance tolerance to abiotic stress.
KEYWORDS: bacteria, drought, fungi, Populus
ABSTRACT
Drought stress negatively impacts microbial activity, but the magnitude of stress responses is likely dependent on a diversity of belowground interactions. Populus trichocarpa individuals and no-plant bulk soils were exposed to extended drought (∼0.03% gravimetric water content [GWC] after 12 days), rewet, and a 12-day “recovery” period to determine the effects of plant presence in mediating soil microbiome stability to water stress. Plant metabolomic analyses indicated that drought exposure increased host investment in C and N metabolic pathways (amino acids, fatty acids, phenolic glycosides) regardless of recovery. Several metabolites positively correlated with root-associated microbial alpha-diversity, but not those of soil communities. Soil bacterial community composition shifted with P. trichocarpa presence and with drought relative to irrigated controls, whereas soil fungal composition shifted only with plant presence. However, root fungal communities strongly shifted with drought, whereas root bacterial communities changed to a lesser degree. The proportion of bacterial water-stress opportunistic operational taxonomic units (OTUs) (enriched counts in drought) was high (∼11%) at the end of drying phases and maintained after rewet and recovery phases in bulk soils, but it declined over time in soils with plants present. For root fungi, opportunistic OTUs were high at the end of recovery in drought treatments (∼17% abundance), although relatively not responsive in soils, particularly planted soils (<0.5% abundance for sensitive or opportunistic). These data indicate that plants modulate soil and root-associated microbial drought responses via tight plant-microbe linkages during extreme drought scenarios, but trajectories after extreme drought vary with plant habitat and microbial functional groups.
IMPORTANCE Climate change causes significant alterations in precipitation and temperature regimes that are predicted to become more extreme throughout the next century. Microorganisms are important members within ecosystems, and how they respond to these changing abiotic stressors has large implications for the functioning of ecosystems, the recycling of nutrients, and the health of the aboveground plant community. Drought stress negatively impacts microbial activity, but the magnitude of this stress response may be dependent on above- and belowground interactions. This study demonstrates that beneficial associations between plants and microbes can enhance tolerance to abiotic stress.
INTRODUCTION
Global climate change is causing significant changes to precipitation and temperature regimes that are predicted to become more extreme throughout the next century and occur across larger regions worldwide (1). Heightened drought stress induced by climate change has negative consequences for plant survival, with photosynthetic CO2 fixation suppression and metabolic impairment leading to substantial declines in growth and often eventual plant mortality (2). Due to this abiotic stress, future predictions show that both agricultural (3) and bioenergy crops will have reduced productivity in low-latitude regions due to water deficits (4).
Plants co-occur and associate with diverse archaeal, bacterial, and fungal communities. Plant health and fitness are linked to the identity and function of their microbial partners; plant hosts influence microbiome selection from surrounding soils likely to influence their mutualistic benefits (5, 6). Due to these relationships, environmental stressors that influence plant hosts or their microbiome are likely to influence the other partner. Drought stress represents an abiotic disturbance that can concomitantly influence plant host productivity, growth, and physiology, while also shifting both exogenous soil and host-associated microbiomes. Extended water stress may select for known drought-tolerant microbes within rhizospheres or roots, such as Actinobacteria, Chloroflexi, and Firmicutes (6–9), and restructure and destabilize microbial networks, particularly bacteria (10). Furthermore, switching of mycorrhizal groups from ectomycorrhizae (EcMs) to arbuscular mycorrhizae fungi (AMF) (11) or dominance of specific drought-tolerant EcM species, such as Cenococcum geophilum, may also occur and influence plant resistance to reduced water availability (12, 13). However, experimental drought manipulations often target one large drought event or multiple drying/rewet events without consideration of recovery dynamics (i.e., a drought-selected outcome once water availability is continuously unlimited) for both plant host metabolism and the microbiome.
Populus is a woody, perennial plant species used as a model organism for plant-microbe interactions research and in applications for bioenergy crops (14, 15). Prior work demonstrates the selection of microbiomes among Populus genotypes and species (16–19), across plant tissues and compartments (18, 20), and in response to abiotic stress (21). Timm et al. (21) found that the Populus deltoides (Eastern Cottonwood) rhizosphere’s bacterial alpha-diversity is reduced under drought stress, yet root endosphere (microbial communities within root tissue) diversity increased. Furthermore, several “core” operational taxonomic units (OTUs) correlated with plant metabolites within roots (i.e., amino acids and aromatics), suggesting a link between plant physiological state and belowground microbial community ecologies during water stress events. Such responses are noteworthy for this specific plant host. Populus species and genotypes do express drought tolerance variability and may in fact exhibit drought sensitivity. This sensitivity is indicated by a reduction in biomass due to high water demands for metabolic maintenance and is particularly prevalent for productive genotypes used for industrial purposes (22). However, it has been shown in other host-microbiome research that low drought tolerance within various plant hosts may be ameliorated by beneficial microbial symbioses and associations, thereby enhancing host tolerance to abiotic stresses (23, 24).
In this study, we tested how an extreme experimental drought, and subsequent rewet and recovery periods (i.e., continuous irrigation), influenced host physiology of a model perennial tree species, Black Cottonwood (Populus trichocarpa) via metabolomic profiling of root tissues in unison with targeted analyses of belowground bacterial/archaeal and fungal communities. We further examined microbial community taxon responses to drought, rewet, and recovery within root tissues of potted P. trichocarpa individuals (endophytes), compared to bulk soil communities alone without plants. This study’s goals are to understand (i) how P. trichocarpa trees physiologically respond and concomitantly interact and modulate bacterial/archaeal and fungal belowground microbiomes during water stress and (ii) how root endophyte and soil microbiomes differentially respond and recover after an extreme drought event (i.e., low water potential causing leaf die-off) when in association with P. trichocarpa. We hypothesized that (i) P. trichocarpa would invest in amino acid production belowground to reduce cellular damage during water deficits as well as metabolites responsive to environmental stress (e.g., proline derivatives [25]) and would reduce microbial sensitivity (i.e., greater microbial resistance) to water stress (comparative to bulk soils with no plants), and this effect would be stronger for fungi than bacteria/archaea, and that (ii) the root-associated microbiomes directly associated with P. trichocarpa would shift in response to specific metabolic changes in root secondary metabolism (i.e., amino acid production) more so than rhizospheres due to the greater linkages between root-associated microbiomes comparative to those in surrounding soils.
RESULTS
Root metabolite profiling.
Several metabolite groups differed in concentration between drought treatments: phenolic glycosides (F1,23 = 5.75, Fig. 1C), amino acids (F1,23 = 29.3, Fig. 1D), fatty acids (F1,23 = 8.98, Fig. 1E), and sugar alcohols (F1,23 = 4.66, Fig. 1F) had greater production in plant roots which underwent drought (P < 0.05). Amino acids had a significant drought treatment-by-time interaction (F2,22 = 4.27, P = 0.03): at the end of drying (day 12), amino acids were produced, on average, in ∼110% greater concentrations in drought; at rewet (day 13), they were similar among controls and droughts; and at recovery (day 25), they had greater production in drought relative to control roots (Fig. 1D). Twenty-two out of 39 metabolites identified through the SIMPER analysis differed across time, and 18 metabolites had a significant interaction between the main effects of treatment (drought, control) and time (drying, rewet, recovery; see Table S2 in the supplemental material). Notably, the amino acids alanine, gamma-aminobutyric acid (GABA), glycine, serine, leucine, and threonine (P < 0.05) and the carbohydrates fructose, glucose, and raffinose (P < 0.01) were enriched in drought-treatment roots after drying, rewet, and recovery, or across all three periods, compared to controls (Table S2). Citric acid (P ≤ 0.01) was depleted in drought treatments relative to controls (Table S2). The organic acids maleic acid and succinic acid were enriched under drought treatments, but only after the drying period (P ≤ 0.01, Table S2).
FIG 1.
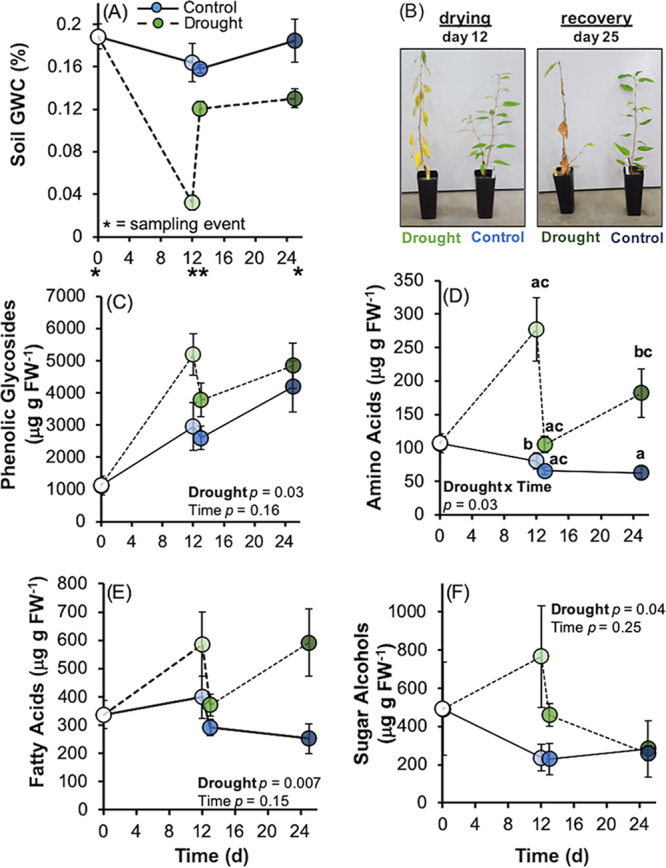
Mean ± standard error (SE) soil gravimetric water content (GWC) of irrigated control and drought treatments (A) which underwent a drought period resulting in significant leaf wilt, a rewet, and recovery period with irrigation maintained at rewet (images provided in panel B). Plant metabolomic profiling of phenolic glycosides (C), amino acids (D), fatty acids (E), and sugar alcohols (F) indicates plants underwent significant metabolic changes after drought. Letters denote pairwise differences for a drought-by-time significant interaction for amino acids only. Other metabolites differed only by drought treatment and not by time. Blue points indicate control treatments, green points indicate drought treatments, and shading indicates increases from drying through rewet to recovery periods.
Microbial alpha- and beta-diversity.
Soil bacterial/archaeal phylogenetic diversity (PD) significantly differed with plant presence (P < 0.001), between drought treatments (P = 0.02), and across time (drying, rewet, recovery; P < 0.001; Table S3). Soil bacterial/archaeal taxonomic diversity (H′) differed only between drought treatments (P = 0.005). PD was, on average, ∼8% greater in bulk soils relative to soils with plants, and both PD and H′ were, on average, ∼5% and 2% lower in drought treatments compared to controls, respectively. Soil fungal H′ differed with plant presence (P < 0.001) only and was, on average, ∼10% greater in bulk soils relative to planted soils (Table S3). In contrast to soils, drought elicited 21% and 12% increases in bacterial/archaeal PD and H′ in roots compared to controls (P = 0.03 and 0.003, respectively), and bacterial/archaeal root PD was greater at the end of recovery compared to rewet (P = 0.04). Fungal diversity in roots did not change between drought treatments or across time (P ≥ 0.08; Table S3).
Bacterial/archaeal richness and diversity indices including PD, H′, and fungal H′ did not correlate with metabolite functional categories in planted soils (full regression model: P > 0.10). However, within roots (PD full model: adjusted [Adj.] R2 = 0.30, F5,19 = 3.02, P = 0.04; H′ full model: Adj. R2 = 20, F2,22 = 4.05, P = 0.03), bacterial/archaeal PD (T = −2.97, P = 0.007), and H′ (T = −2.01, P = 0.057) negatively correlated with carbohydrates, whereas PD also positively correlated with fatty acids (T = 2.71, P = 0.01), and H′ correlated with amino acids (T = 2.82, P = 0.01). Root fungal H′ also positively correlated with amino acids (T = 2.16, P = 0.04) and negatively with sugar alcohols (T = −2.32, P = 0.03).
For bacteria/archaea in soils, plant presence (bulk versus planted soils) explained the most variation (10%), and time (8%) and drought treatments explained a lower proportion of variation (∼5%) in community composition. All two-way interactions between main effects were significant (Table 1). The main effect of drought versus control in planted soils marginally influenced bacterial/archaeal community composition (plant presence × treatment interaction P = 0.01; false-discovery rate [FDR]-corrected P = 0.07). Drought versus control bulk soil communities did significantly differ in composition (Fig. 2A). However, communities did not significantly shift between drought and controls until the end of the recovery period (treatment × time interaction: FDR-corrected P < 0.05, Table 1). For soil fungi, plant presence explained almost 2× the variation (∼17%) compared to bacteria/archaea. Drought treatments did not significantly influence soil fungal community composition, whereas time (drying, rewet, recovery) accounted for ∼4% of community composition variation (Table 1; Fig. 2B). No two- or three-way interactions were significant for soil fungal communities (P > 0.10). Root-associated bacterial/archaeal communities did not respond to main effect of drought treatment (P = 0.72) but did have a marginally significant interaction between treatment and time (Table 1; Fig. 2C). However, post hoc FDR tests indicated no significant differences were found in composition with treatment × time (FDR-corrected P > 0.20). Fungal communities in roots responded to both the drought treatment and time (Table 1, Fig. 2D, and Fig. S2D) and also had a marginally significant interaction. Unlike bacteria/archaea in roots, fungal communities at day 12 (drying) and day 25 (recovery) differed in drought compared to controls, but at rewet, fungal communities were similar in community composition (Table 1).
TABLE 1.
Permutational multivariate ANOVA results for bacteria/archaea and fungi using Euclidean distance matrices after a centered log-ratio transformation was applied to raw OTU countsa
| Microbial group | Model factor | F value | R2 | P value |
|---|---|---|---|---|
| Bacteria/archaea—soils | Plant presence | 7.34 | 0.10 | 0.001 |
| Treatment | 2.09 | 0.03 | 0.002 | |
| Time | 2.70 | 0.08 | 0.001 | |
| PP × Trt | 1.68 | 0.02 | 0.014 | |
| PP × time | 1.67 | 0.05 | 0.002 | |
| Trt × time | 1.40 | 0.04 | 0.009 | |
| PP × Trt × time | 1.19 | 0.03 | 0.10 | |
| Residuals | 0.65 | |||
| Bacteria/archaea—root | Treatment | 0.96 | 0.04 | 0.72 |
| Time | 1.12 | 0.09 | 0.025 | |
| Trt × time | 1.07 | 0.08 | 0.08 | |
| Residuals | 0.77 | |||
| Fungi—soils | Plant presence | 12.87 | 0.17 | 0.001 |
| Treatment | 1.11 | 0.01 | 0.23 | |
| Time | 1.68 | 0.04 | 0.01 | |
| PP × Trt | 1.26 | 0.02 | 0.11 | |
| PP × time | 1.25 | 0.03 | 0.11 | |
| Trt × time | 0.98 | 0.03 | 0.44 | |
| PP × Trt × time | 1.17 | 0.03 | 0.15 | |
| Residuals | 0.67 | |||
| Fungi—root | Treatment | 1.90 | 0.06 | 0.001 |
| Time | 1.29 | 0.08 | 0.01 | |
| Trt × time | 1.13 | 0.07 | 0.08 | |
| Residuals | 0.78 | |||
Main effects include plant presence (PP; present or not present), treatment (Trt; drought versus control), and time of sampling (at drought, after rewet, and recovery). Nine hundred ninety-nine permutations were implemented to calculate significance values. Significant main effects are boldfaced.
FIG 2.
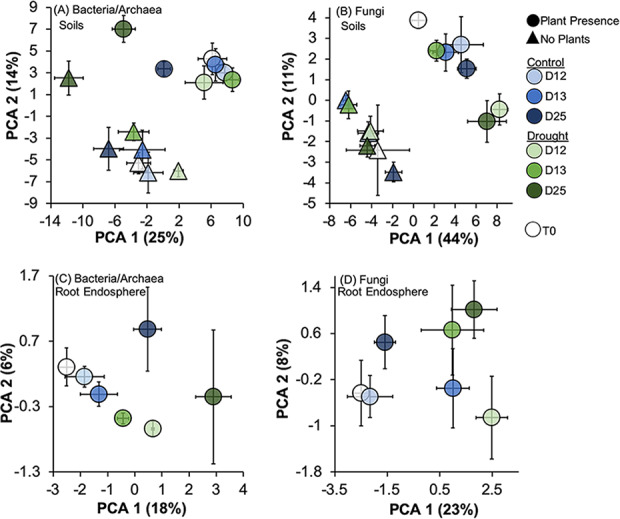
Centroids (mean ordination scores) with standard error (SE) bars of principal-component analysis (PCA) for bacteria/archaea (A and C) and fungal communities (B and D) in soils (A and B) and roots (C and D). Raw OTU counts were centered log-ratio transformed, and Euclidean distances were calculated and input in PCAs. Symbols represent plant presence or no presence for bulk soils (circle = plant presence, triangle = no plants), colors denote treatment (blue = control, green = drought), and shading from light to dark refers to time of sampling.
Within soils, Actinobacteria, Planctomycetes, Bacteroidetes, and Gammaproteobacteria differed with plant presence (Table 2): Actinobacteria, Bacteroidetes, and Gammaproteobacteria were enriched in planted soils relative to bulk soil whereas Planctomycetes and the fungal Basidiomycota and Mortierellomycota were enriched in bulk soils relative to planted (Fig. 3). Actinobacteria and Firmicutes also shifted between drought treatments and had a plant presence × drought treatment interaction (P = 0.002; Table 2). In bulk soils, drought elicited ∼40% and 24% increases in Actinobacteria and Firmicutes, respectively, whereas drought had no effect on their abundances when plants were present (Fig. 3A and B). On average, Acidobacteria and Chloroflexi increased by ∼1 to 2% in abundance in drought soils relative to controls regardless of whether plants were present or not (Table 2). The fungal phylum Ascomycota increased by an average of ∼8% when exposed to drought. Deltaproteobacteria declined ∼11% in drought compared to controls regardless of plant presence or time (Table 2). Alpha- and Betaproteobacteria had significant interactions with drought and time. Specifically, at drying and rewet, their relative abundance did not vary between drought and control (Tukey’s honestly significant difference [HSD], P > 0.10), yet drought elicited an increase in their mean relative abundance after 12 days of high water availability (end of recovery period) regardless of plant presence. In roots, drought elicited an average ∼22% and 51% increase in Alphaproteobacteria and Verrucomicrobia relative to controls, respectively (P = 0.06, Fig. 3C and Table S4), but none had a drought × time interaction (Table S4). Although not abundant, Glomeromycota had lower relative abundances in drought (mean 0.4% relative abundance) relative to controls (P < 0.001) (1.2%; Table S4).
TABLE 2.
Dominant bacterial and fungal phylum three-way ANOVA model results with plant presence, treatment (drought, control), and time (end of drought, at rewet, and recovery) as fixed, main effectsa
| Phylumb | Rel. abund.c | Plant presence |
Treatment |
Time |
|||
|---|---|---|---|---|---|---|---|
| F statistic | P value | F statistic | P value | F statistic | P value | ||
| Bacteria | |||||||
| Alphaproteobacteria* | 17.5 | 3.57 | 0.07 | 6.82 | 0.01 | 2.41 | 0.10 |
| Actinobacteria** | 16.2 | 8.88 | 0.005 | 29.31 | <0.001 | 0.87 | 0.42 |
| Acidobacteria | 15.9 | 2.32 | 0.13 | 10.20 | 0.003 | 3.71 | 0.03 |
| Betaproteobacteria*** | 10.4 | 0.15 | 0.70 | 18.66 | <0.001 | 4.50 | 0.02 |
| Planctomycetes | 6.1 | 88.18 | <0.001 | 0.63 | 0.43 | 29.62 | <0.001 |
| Deltaproteobacteria*** | 5.8 | 2.00 | 0.17 | 5.49 | 0.02 | 0.63 | 0.54 |
| Verrucomicrobia | 5.4 | 0.83 | 0.37 | 2.70 | 0.11 | 2.12 | 0.13 |
| Bacteroidetes | 4.3 | 4.65 | 0.04 | 0.70 | 0.41 | 0.63 | 0.54 |
| Gammaproteobacteria | 4.2 | 49.82 | <0.001 | 0.0 | 0.99 | 0.49 | 0.61 |
| Chloroflexi | 4.1 | 0.09 | 0.77 | 6.18 | 0.02 | 8.39 | <0.001 |
| Firmicutes** | 2.4 | 0.79 | 0.38 | 3.42 | 0.07 | 4.49 | 0.02 |
| Gemmatimonadetes | 2.3 | 0.10 | 0.76 | 11.62 | 0.001 | 0.55 | 0.58 |
| Fungi | |||||||
| Ascomycota | 41.3 | 0.08 | 0.78 | 8.00 | 0.007 | 3.20 | 0.05 |
| Basidiomycota | 41.1 | 41.85 | <0.001 | 1.25 | 0.27 | 1.87 | 0.16 |
| Mortierellomycota | 5.7 | 15.39 | <0.001 | 0.001 | 0.97 | 1.58 | 0.22 |
| Chytridiomycota | 1.4 | 0.27 | 0.60 | 0.03 | 0.86 | 0.71 | 0.50 |
Model results are shown for soils only. Phyla which significantly varied with at least one main effect are boldfaced. Results provided used centered log-ratio-transformed data based on raw, absolute phylum-level sequence counts.
Significance is indicated as follows: *, significant two-way interaction of treatment and time; **, significant two-way interaction of plant presence and treatment; ***, significant three-way interactions of plant presence, treatment, and time.
Rel. abund., relative abundance.
FIG 3.

Relative abundance of dominant (≥1.0%) bacterial/archaeal phyla (and class for Proteobacteria) (A to C) and fungal phyla (D to F) across drying, rewet, and recovery periods within bulk soils (A and D), planted soils (B and E), and root endospheres (C and F).
Differentially abundant OTUs across drying/rewet/recovery periods.
Bulk soils and planted soils had sensitive (water stress depleted, total = 304) and opportunistic (water stress enriched, total = 231) bacterial/archaeal OTUs at dry-down, rewet, and recovery (Fig. 4, Table S5, and Fig. S3A and B) whereas fungi had lower numbers of both sensitive (total = 10) and opportunistic (total = 4; Table S5; Fig. S3) OTUs. In roots, there was only one opportunistic OTU for bacteria, but for fungi, there were 4 sensitive and 19 opportunistic OTUs over time (Table S5; Fig. S4).
FIG 4.

The absolute log2 fold value change of DESeq-detected differential bacterial/archaeal OTUs that were opportunistic (enriched in drought versus controls) or sensitive (depleted in drought versus controls) over time at end of drying (A and D), rewet (B and E), and recovery (C and F) for bulk soils (A to C) and planted soils (D to E). Each dot represented a significantly differentially abundant OTU. Only one bacterial OTU over time was differentially abundant in roots. Asterisks denote if strategy (sensitivity or opportunism) fold change significantly differed at each time for bulk soils and planted soil separately.
For both drying and rewet, bacterial/archaeal sensitive OTUs were more responsive than opportunistic OTUs (i.e., greater log2 fold change; t test, P ≤ 0.06; Fig. 4A and B). However, the greatest proportion of water-stress sensitivity (summed sequence abundance for differentially depleted OTUs) occurred for bulk soils at recovery (∼7% depleted relative to 0.2% for planted soils; Fig. 4C and Fig. S2A). Recovery-sensitive OTUs in soils included a large range of phyla including Crenarchaeota, Alpha-, Beta-, Delta-, and Gammaproteobacteria, Cyanobacteria, and Planctomycetes, among others (Fig. S3A).
Fungal sensitivity, in general, was low but was greatest for bulk soils at recovery (3.9%), and sensitive fungi were classified primarily to the Entomophthoromycota order Basidiobolales, mycorrhizal Hebeloma sacchariolens, and unclassified Rozellomycota (Fig. S3B). Notably, fungal drying sensitivity was also greater for bulk soils (0.9%) relative to planted soils (no sensitive OTUs) and included unclassified Rozellomycota (Fig. S3B). Four fungal OTUs were sensitive in roots and made up ∼0.8% and 1.4% sequence proportions (Fig. S4A): 2 at drying (primarily Hebeloma sacchariolens and to a lesser degree an unclassified Ascomycota, and 2 at recovery (primarily Paxillus involutus). Root fungal sensitive OTUs were, on average, more responsive than opportunistic OTUs (log2 fold abundance) at recovery (Fig. 5C).
FIG 5.

The absolute log2 fold value change of DESeq-detected differential fungal OTUs that were opportunistic (enriched in drought versus controls) or sensitive (depleted in drought versus controls) over time at end of drying (A), rewet (B), and recovery (C) for roots. Each dot represents a significant differentially abundant OTU. No sensitive OTUs were detected for fungal communities at rewet (B). Asterisks denote if strategy (sensitivity or opportunism) fold change significantly differed at each time.
Bacterial/archaeal water stress opportunism was more responsive than sensitivity (greater log2 fold change) for planted soils at recovery (t test: P = 0.03; Fig. 4F), but the abundance of opportunistic OTUs was greatest for both planted soils (12.4%) and bulk soils (11.5%) at the end of drying (Fig. 4B; Fig. S3B). However, drying opportunistic OTUs in planted soils were more enriched by Acidobacteria, whereas bulk soils were more enriched by Alphaproteobacteria, Actinobacteria, and Gemmatimonadetes (Fig. S3B). Interestingly, the proportion of opportunistic bacterial/archaeal OTUs declined after drying (day 12) for planted soils (∼2% at rewet and recovery) but remained relatively high for bulk soils overtime (∼10% enriched at rewet, 8.8% enriched at recovery; Fig. S3). Opportunistic OTUs in bulk soils at rewet were represented by Betaproteobacteria, Actinobacteria, and Firmicutes whereas at recovery, Beta- and Alphaproteobacteria comprised the majority of opportunistic strategists (Fig. S3). Opportunistic OTUs in planted soils at rewet were primarily Acidobacteria, whereas those at recovery were primarily Alphaproteobacteria (Fig. S3B).
Opportunistic fungal OTUs were most abundant in bulk soils at recovery (1.4%) and were comprised of saprotrophic Minimedusa polyspora (Fig. S3D). At drying and rewet, there were no opportunistic fungal OTUs in bulk soils whereas in planted soils at drying (0.8%), rewet (0.3%), and recovery (0.08%), opportunists were low but present and classified to Hebeloma sacchariolens (Fig. S3D). Only one root bacterial OTU was opportunistic and classified to Cyanobacteria order YS2 at recovery. There were 4 fungal OTUs in roots during both drying and rewet, and 11 by recovery (Fig. 5). Opportunistic fungal OTUs at the end of drying were more responsive than sensitive OTUs (t test, P = 0.03; Fig. 5A). There were 13 differential root OTUs at recovery, primarily opportunistic. Several were not abundant and were classified to putative pathogenic taxa, such as Alternaria, Trichoderma, and Didymella glomerata. The abundant opportunistic OTUs at recovery were primarily classified to Sordariomycetes, Diaporthales, and Xylariales (Fig. S4B).
DISCUSSION
In this study, we found that an extreme drought caused plant physiology to shift (greater amino acids, organic acids, and other metabolite groups shunted belowground) even after a high-water-availability recovery period and that these changes in specific plant metabolites predicted variation in microbial diversity, particularly in roots. In planted soils, drought conditions led to shifts in the bacterial/archaeal microbiome but not fungal, whereas in root-associated microbiomes, the opposite was found, although bacteria/archaea were weakly affected by drought in roots. Root fungal communities were more responsive to drought than root-associated bacteria/archaea, but this effect was switched in surrounding soils. This supports the hypothesis that soil fungi are drought tolerant (26–28) and that their communities remain more stable under drought stress (10), and instead, fungi tend to be more responsive to plant inputs or physiological host state. Our data show that fungi may be more responsive to plants undergoing water stress and less so to physical desiccation by itself (abiotic effect only). These trends highlight differences in the importance of abiotic (water availability) and biotic (plant-microbe) interactions for bacterial versus fungal communities belowground during environmental stress.
Plant host metabolic stress and the root microbiome.
After 12 days of no irrigation and subsequent rewet, P. trichocarpa had heavy leaf abscission (although not total leaf loss for most individuals) and increased allocation of amino acids, fatty acids, sugar alcohols, and phenolic glycosides belowground (Fig. 1), likely for increased investment in root growth due to reduction or absence of photosynthetic capacity (29, 30). Furthermore, metabolites indicative of water stress, such as 5-oxo-proline (25) and fructose (31), were upregulated at the end of drying, and proline remained so at the end of recovery (see Table S2 in the supplemental material). Although plants did survive and “recover” after drying, the drought severity was high enough to prolong the stress response belowground by concentrating sugars and amino acids which may enhance water uptake (32). Furthermore, both bacterial/archaeal and fungal taxonomic diversity positively correlated with amino acid accumulation within the roots, suggesting that drought stress may influence belowground colonization dynamics. Root secondary metabolites, and likely other root exudates (i.e., primary metabolites), varied substantially over time in response to drought, which may promote host resiliency by microbially induced responses (33). Specifically, root metabolite accumulation of organic or amino acids, which was greater in plants undergoing drought, may serve to attract microbial endophytes (34, 35) and confer plant growth-promoting traits (36). Beneficial microorganisms can promote specific exudate secretion, osmolyte production (37), or peroxidase production which removes reactive oxygen species prevalent during water stress. Rhizosphere microbiomes (communities in soils surrounding roots) also often respond, if not more strongly, to changes in root function relative to endophytes (5, 19) due to greater nutrient availability within surrounding soils (38, 39). In support of our hypothesis, we found no evidence of a correlation between rhizosphere microbial diversity and secondary metabolite production; therefore, it is reasonable to suggest that nutrient supply within rhizospheres may shift plant host-microbiome interactions during water stress.
Although soil fungi were more responsive to plant presence than drought in bulk soils (Table 1), root fungal communities may be responsive to shifts in plant host stress responses. It cannot be ruled out that drought and the subsequent recovery period may in fact allow for greater pathogen colonization as several opportunistic OTUs at recovery included putative plant pathogens (e.g., Alternaria and Didymella glomerata). Although these putative plant pathogens were in low abundance at recovery (mean relative abundance in drought treatments at recovery: Alternaria, 0.1%; Didymella glomerata, 0.13%), extreme drought may suppress plant immunity and allow greater initial pathogen colonization (40–42).
Microbial recovery from drought: bacterial and fungal responses.
At the end of soil drying, soil bacterial and archaeal taxa were generally resistant to water stress long term, as relative abundances of sensitive and opportunistic taxa did not exceed ∼13%, and this became more pronounced by the end of recovery (Fig. S3A and B). At recovery, planted soils had approximately ∼2% opportunistic taxa and no sensitive taxa, whereas bulk soils had a substantially greater proportion for both, respectively (∼6 to 8% of relative abundances). Bacterial taxa in soil aggregates may readily respond to water fluctuations due to cellular sensitivity to desiccation via their semipermeable membranes (28, 43). Several bacterial phyla—Acidobacteria, Chloroflexi, Actinobacteria, Firmicutes—were enriched under drought conditions, either among bulk soils or in P. trichocarpa planted soils (Table 2). Likewise, a large proportion of bacterial OTUs belonging to these phyla were opportunistic at the end of drought and maintained heightened abundance, even by the end of recovery when plants were not present (Fig. S3), suggesting that numerous taxa, even across broad phylogenetic groups (44), are resilient to water stress in soils. One extreme pulse disturbance, as demonstrated in this study, can have dramatic effects on soil bacterial dynamics, as shown by alpha- and betaproteobacterial dominance in drought treatments and during recovery (Table 2). These more copiotrophic (45, 46) bacterial groups likely carry a competitive advantage in being able to respond more rapidly to changing conditions (i.e., dormancy states via sporulation) and become more dominant over time. Structure-function linkages strongly apply to microbial communities which drive biogeochemical cycling (47); therefore, such trends may have broader importance as drought events become more prevalent globally (1). Furthermore, P. trichocarpa presence resulted in bacterial compositional change and heightened soil bacterial resistance to extreme drought once water stress was alleviated. Plant roots can recruit beneficial microbial partners (24) when present in soils, which may modify abiotic stress responses; Root exudates may in fact “save” some microbial sensitive taxa via enhanced carbon supply and water availability relative to bulk soils (6).
Interestingly, our data demonstrate that differential abundance of root endosphere microorganisms, particularly bacterial and archaeal OTUs, was quite low (Fig. S3 and 4) during drought, rewet, and recovery, while a large effect was seen in rhizosphere microbiomes. This is counter to other studies which indicate that roots are often the “habitat” which holds the strongest selective pressure in host microbiomes relative to other surrounding environments belowground (8, 9, 48). Our results indicate that the root microbiome of poplar may be more resistant to change due to short-term perturbations. Additionally, unlike other plant hosts, poplars are capable of complex interactions with bacteria and fungi in roots and engage with both arbuscular mycorrhizae and ectomycorrhizae, which may confound comparison with other studies targeting different plant hosts. Further, other poplar microbiome studies have shown high variability in root endosphere microbiomes (18), and we lost replicates due to poor 16S rRNA amplification; therefore, our statistical power to detect alpha and beta-diversity changes, along with differential abundance, was limited.
Partially in support of our hypothesis, soil fungal communities differed with the presence of P. trichocarpa but were relatively unaffected by extreme drought in soils. Specific fungal life histories and traits, such as low growth rates, cell wall thickness, and significant osmolyte production (49), likely allow for greater “community-level” resistance to short-term water stress events in soils. However, root communities shifted considerably with drought, both at the end of drying and after irrigation returned. In fact, ectomycorrhizal taxa—the basidiomycete Paxillus involutus—made up a large portion of water-stress-sensitive taxa in roots, and Glomeromycota were also lower in drought treatments relative to controls that were continually irrigated. This serves as preliminary evidence that an extreme drought may cause substantial functional turnover in fungal root-associated communities with declines in mycorrhizal species (likely from turnover in root biomass). However, a significant proportion of responsive fungal taxa in roots (∼17%) were opportunistic by recovery (Fig. S3) and were classified to diverse groups of fungi. Together, these trends suggest that drought conditions may directly modify plant-fungal interactions so that fungi potentially beneficial for plant growth proliferate. This is, in part, supported by the observation that P. trichocarpa individuals were monitored for several weeks after the end of the experiment (data not shown) and had substantial aboveground growth.
This study highlights the complex interactions between a Black Cottonwood (P. trichocarpa) genotype and soil microbiota after extended drought conditions and demonstrates the differential responses of bacteria/archaea and fungi to short-term drought and recovery dynamics in unison with plant metabolic stress responses. Root-associated fungal communities do not follow similar trends as soil fungi and may mitigate abiotic stress response within hosts belowground (34). Likewise, P. trichocarpa moderates microbial responses to environmental stress, allowing for greater stability (communities unchanging to disturbance) in surrounding soils. Additional work should seek to understand how depletion or enrichment of specific microbial taxa within bulk and plant-associated soils may result in functional changes at the ecosystem scale.
MATERIALS AND METHODS
Plant and soil material collections and growth conditions.
During March 2018, 80 cuttings of Populus trichocarpa genotype CBI-9947 were collected from a field-grown common garden population that has been maintained for over a decade in Corvallis, OR. Bulk soils were collected from a replicate common garden located in a nutrient-rich floodplain located approximately 175 km north on the Columbia River in Clatskanie, OR (19). Cuttings and soils were kept on ice, shipped overnight, and maintained at 4°C until rooting took place under climate-controlled greenhouse conditions. In July 2018, cuttings were rinsed with deionized (DI) water and a 1% ZeroTol 2.0 solution and the cutting base had rooting powder added (0.1% indole-3-butyric acid) to elicit root growth. Cuttings were placed in a standard and sterilized potting soil mixture (Fafard 52 mix; Sungro Horticulture, MA, USA) for ∼4 months prior to the experiment.
Experimental design.
After the initial growth period, 80 cuttings were transferred to 1-liter pots with a 2:1 sand-soil mixture of the floodplain soils described above. In addition, 10 1-liter pots with no plants (referred to as “bulk soils” here) were also placed adjacent to potted P. trichocarpa cuttings in the greenhouse. The potted plants and bulk soils were allowed to acclimate in the sand-soil mixture for ∼4 weeks prior to the drought experiment. During this time, pots were irrigated twice a day (8 a.m., 4 p.m.) for 10 min (approximately 25-ml/min irrigation rate). In August 2018, 5 replicate pots were destructively harvested to represent conditions at the initiation of the experiment (time zero). A representative subsample of the fine rooting system (<1-mm diameter) (∼5 g) was rinsed in DI water, placed in a 15-ml sterile Falcon tube, frozen on liquid nitrogen, and stored at −80°C for plant metabolomic profiling. An additional subset of plant roots and attached soils were placed in sterile 50-ml Falcon tubes, kept on ice until transfer from the greenhouse to the laboratory, and subsequently stored at −80°C until soil-root separation and DNA extraction took place. Lastly, 5 pots of bulk soils were subsampled with a soil corer (2.5-cm diameter) and stored at −80°C until DNA extractions. After soils were subsampled and plants were destructively harvested, irrigation lines were removed from half of potted plants (n = 40 overall) and half (n = 5) of bulk soils to begin the drying period (Fig. 1) and thus referred to as the “drought” treatment (no irrigation) and subsequent treatment phases. The other half of the potted plants and bulk soils had their irrigation lines retained (n = 40 plants, n = 5 bulk soils) and their irrigation regime maintained. These are referred to as “control” plants and bulk soils. At each subsequent phase, replicate plants were destructively harvested from both the drought and control groups, as described above. Plants were deemed alive by noticeable leaf growth and/or maintenance (budding) prior to destructive harvests so that no dead plants were included in this study. No irrigation (drying) was allowed to take place in drought pots until plants were under noticeable water stress (retained some green leaves but had substantial leaf wilting; ∼12 days) (Fig. 1A). After 12 days of drying, replicate drought (n = 10) and control (n = 10) plants and bulk soils (n = 10) were harvested as described above. Subsequently, drought plants and bulk soils were watered with approximately ∼300 ml DI water, and irrigation lines were replaced to elicit a “rewet” phase (Fig. 1A). Twenty-four hours after rewet, drought (n = 5) and control (n = 5) plants and bulk soils (n = 5) were destructively harvested and represented the day 13 rewet event period. Lastly, to understand recovery dynamics after an extreme drought and rewet, after 12 days of irrigated conditions (same amount of time as drying period), remaining drought (n = 5) and control (n = 5) plants and bulk soils (n = 5) were destructively harvested (Fig. 1A).
Plant metabolomic profiling, genomic DNA extractions, and Illumina MiSeq sequencing.
Root tissue metabolomic profiles were analyzed via gas chromatography (GC)-mass spectrometry (MS) as previously described (50). Metabolites were identified using the Wiley Registry, 10th edition, within the NIST 2014 mass spectral database as well as a large, user-created database of trimethylsilyl-derivatized metabolites (50). Sixty-six metabolites were quantified (see Table S1 in the supplemental material) and then grouped into one of eight functional categories: amino acids, phosphorylated metabolites, lipid/fatty acid related, organic acids, phenolics, phenolic glycosides, carbohydrates, and sugar alcohols.
The 66 plant root secondary metabolites detected in this study and SIMPER statistical output for each metabolite. Download Table S1, DOCX file, 0.01 MB (15.4KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Individual metabolites identified through SIMPER which differed between treatment (drought versus control), time (drought, rewet, and recovery), and their interaction produced in P. trichocarpa roots. The direction of change based on two-way ANOVA model output is also given. Metabolites which had a significant treatment and time (T*T) interaction are boldfaced. Download Table S2, DOCX file, 0.02 MB (16.1KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Three-way ANOVA model results for alpha-diversity estimates for bacteria/archaea and fungi. Response variables which significantly varied by a fixed term (presence of plants, drought treatment, and time of drying, rewet, and recovery) are boldfaced. An asterisk denotes a response variable which had a significant treatment-by-time interaction. There were no significant three-way interactions. Download Table S3, DOCX file, 0.01 MB (14.2KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Dominant bacterial and fungal phylum 2-way ANOVA model results with treatment (drought, control) and time (end of drought, rewet, and recovery) as fixed, main effects and their interaction (T*T). Model results are for root endosphere samples only. Phyla which significantly varied with a main effect are boldfaced. Results provided used centered log-ratio-transformed data based on raw, absolute phylum-level sequence counts. Download Table S4, DOCX file, 0.01 MB (15.7KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DESeq2 results for bacteria/archaea and fungi among compartments and time. The number and proportion of OTUs which were differentially abundant within drought versus control treatments are provided. Sensitive refers to OTUs which were depleted under drought, and opportunistic refers to those which were enriched compared to controls. Download Table S5, DOCX file, 0.02 MB (16KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Plant roots and attached soils were rinsed in sterile DI water as previously described (18) to separate soils from root tissue. Fine roots (<1-mm diameter) were separated, frozen in liquid nitrogen, and bead beaten for 1 min with a sterile, 5-mm steel bead prior to extraction. Samples were then placed in liquid nitrogen for 1 min between bead-beating steps (total of 3 bead-beating/freezing cycles). Pulverized tissue had genomic DNA (gDNA) extracted using the Qiagen PowerPlant Pro DNA kit. Separated soils had gDNA extracted using standard protocols for the Qiagen PowerSoil DNA kit (Qiagen, Venlo, the Netherlands). Bulk soil samples also used this same process for gDNA extractions. Lastly, gDNA for roots was purified and concentrated using a Zymo-5 DNA Clean and Concentrator kit (Zymo Research Corporation, Irvine, CA, USA). All root and soil DNA samples were quantified on a Nanodrop 1000 spectrophotometer prior to PCR amplification.
A two-step PCR approach was used with frameshifting nucleotide primers (51) and PCR conditions described elsewhere (18). All samples were pooled based on band intensity (agarose gel, 1.2% [wt/vol]) and purified with Agencourt AMPure XP beads at an 0.7:1 ratio. Both ITS2 and the V4 region of 16S rRNA gene libraries were sequenced using Illumina MiSeq (v.2, 2 × 250 cycles) with 9 pM amplicon concentrations and a 15% PhiX spike.
Bioinformatics.
Paired-end fastq sequence files had primers and adaptors removed individually using the cutadapt program using default parameters with maximum error rate of 0.1, no insertions or deletions allowed, and 3 bp overlap required between read and adaptors. Trimmed fastq files were then joined using QIIME1 and imported into the QIIME2 environment (52). Joined sequences were demultiplexed, and median Phred quality scores were visualized. Due to poor quality on the 3′ end for 16S data, they were trimmed at 250 bp. ITS2 data were not trimmed due to taxon-specific variation in read length for this gene region. Both gene regions were denoised using the DADA2 algorithm in QIIME2 (53), and operational taxonomic units (OTUs) were clustered via the vsearch open-reference workflow at a 97% sequence similarity threshold. Representative sequences were then assigned a taxonomic classification using Naïve Bayes classifier through the sklearn python package. ITS2 representative sequences were assigned a taxonomic classification in QIIME1 using BLAST and the UNITE reference database (54). Contaminants (unassigned reads, mitochondria, and chloroplasts for 16S; Protista, Chromista, Animalia, and Plantae reads for ITS2) were removed. Due to the inclusion of peptide nucleic acid blockers (PNAs) in PCRs (18), these contaminants were a fairly low percentage of total reads for bacteria (∼4%) and fungi (∼23%). However, due to some suspected sequence contamination in 16S libraries, particularly in root endosphere communities, some replicate samples were removed from the final data set. Specifically, 4 instead of 5 replicates are included for bacterial drought time-2 “rewet” samples and both drought and control time-1 “drought” and time-2 samples for root endospheres. Lastly, only 2 out of 5 time zero root endosphere bacterial communities were retained due to a low yield of high-quality sequences. For alpha-diversity estimates, sequence data were rarefied separately for soil (30,000 sequences for bacteria, 5,800 for fungi) and roots (1,000 for bacteria, 5,800 for fungi). For bacteria/archaea, Faith’s phylogenetic diversity (Faith’s PD) was calculated, and for both bacteria/archaea and fungi, the Shannon index (H′) was calculated.
Statistical analysis.
Individual metabolites were grouped into functional categories, and the concentration of each metabolite within each category was summed. A two-way analysis of variance (ANOVA) model was then used to determine if the total concentration of amino acids, phosphorylated metabolites, fatty acids, organic acids, phenolics, phenolic glycosides, and sugar alcohols differed among treatments (drought and control) and with time point representative of drought (day 12), rewet (day 13), and end of recovery (day 25). Furthermore, to determine how each metabolite differed with treatments and time point, all 66 metabolites were input into a multivariate similarity percentage (SIMPER) analysis (55). The SIMPER analysis detected which metabolites significantly contributed to overall secondary metabolite changes in drought versus control treatments and reduced the original data set to 39 metabolites (Table S1). Similar to functional categories of metabolites, individual two-way ANOVA models were used for each of 39 plant metabolites with treatment and time as the fixed independent variables. Data normality was assessed using Q-Q plots, and if nonnormal, data were log10 transformed prior to ANOVA models. The statistical output for time as a main effect is reported, but only to interpret significant interactions among treatment and time.
Alpha-diversity estimates (Faith’s phylogenetic diversity [PD] and Shannon index [H′] for bacteria; only H′ for fungi) were calculated after rarefaction of soils and root samples separately and input as response variables in 3-way ANOVA models. Explanatory variables in models included plant presence (bulk soils, planted soils), drought treatment (drought, irrigated control), and time representative of drought (day 12), rewet (day 13), and recovery (day 25; Fig. 1A). Due to the compositional nature of sequencing data (56), raw OTU counts were centered log-ratio (clr) transformed after a pseudocount of 1 was added via the chemometrics R package (57). Euclidean distance was then calculated and used as input for a principal-component analysis (PCA) to visualize community compositional differences among soils with and without P. trichocarpa and across time. In addition, a pseudocount was added to clr-transformed OTU and sample matrix and Bray-Curtis distance matrices were calculated and similarly input to PCA to compare to Euclidean-based PCAs (Fig. S1 and S2). A permutational multivariate ANOVA (adonis function in the vegan package) (58) model (perMANOVA) (59) was used to determine the proportion of variation attributed to plant presence, drought treatment, time of sampling, and their two- and three-way interactions for both bacteria/archaea and fungi. If deemed statistically significant, post hoc FDR corrections were calculated for comparisons among group levels (pairwise.perm.manova [60]). Raw counts of dominant bacterial and fungal phyla and families were similarly clr transformed, and 3-way ANOVA models were used to understand their responses to plant presence, drought treatments, and time. In one instance, the phylum Euryarchaeota did not meet normality assumptions in the root data set and therefore was log10 transformed prior to analysis. Finally, a multiple regression model with a stepwise selection procedure and Akaike’s information criterion (AIC) minimization was calculated to determine if host-metabolite functional categories correlate with microbial alpha-diversity estimates for planted soils and root samples separately. Regression models were performed using the stepAIC function in the MASS package (61). We delineate type 1 error rates as 0.10, and therefore we report significant results with P values less than 0.10.
Raw PCA scores from Bray-Curtis-based distances for bacteria/archaea and fungi in in soils (planted, bulk) and in roots. Symbols represent plant presence or no presence for bulk soils (circle = plant presence, triangle = no plants), colors denote treatment (blue = control, green = drought), and shading from light to dark refers to time of sampling. Download FIG S1, TIF file, 0.1 MB (114.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Raw PCA scores from Euclidean-based distances for bacteria/archaea and fungi in soils (planted, bulk) and in roots. Symbols represent plant presence or no presence for bulk soils (circle = plant presence, triangle = no plants), colors denote treatment (blue = control, green = drought), and shading from light to dark refers to time of sampling. Download FIG S2, TIF file, 0.3 MB (284.5KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The proportion of sequences belonging to significant differentially sensitive (depleted in drought) and opportunistic (enriched in drought) OTUs across bacterial and archaeal phyla (A and B) and fungal taxonomies (C and D) over time (day 12 = drying period, day 13 = rewet period, day 25 = recovery period). The absence of bars indicates that zero OTUs were differentially abundant for that time except for panel A, where only very rare OTUs (<0.01% relative abundance) were identified. Percent proportion of sensitivity refers to all sequences in a taxonomic group within control samples, and percent proportion of opportunism refers to all sequences in a taxonomic group with drought samples. Download FIG S3, TIF file, 0.4 MB (459.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The proportion of sequences belonging to significant differentially sensitive (depleted in drought) and opportunistic (enriched in drought) OTUs across fungal taxonomies over time (day 12 = drying period, day 13 = rewet period, day 25 = recovery period) within root endospheres. The absence of indicates that zero OTUs were differentially abundant for that time except for panel A, where only very rare OTUs (<0.2% relative abundance) were identified. Percent proportion of sensitivity refers to all sequences in a taxonomic group within control samples, and percent proportion of opportunism refers to all sequences in a taxonomic group with drought samples. Download FIG S4, TIF file, 0.2 MB (208.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
One of the explicit study goals was to assess the relative amount of water-stress-depleted (sensitive) and enriched (opportunistic) OTUs among soils with and without plants and with root-associated OTUs to elucidate the tolerance of microbial communities after a pulse disturbance manipulated through an extreme drying and rewet event. To address this effect, raw read counts of OTUs were log2 transformed and modeled using a negative binomial distribution for generalized linear regression models using the DESeq2 package in R (62). Singleton and doubleton OTUs were removed prior to this analysis for each data set, and DESeq filtering of OTUs which were sparse (independent.filtering=TRUE) was carried out prior to a Wald test with Benjamini-Hochberg post hoc false-discovery rate-corrected P values. OTUs deemed differential by this analysis were either significantly lower or greater in drought versus control. Here, we refer to OTUs that are depleted in drought treatments relative to controls as water-stress sensitive (i.e., drying, rewet, recovery sensitive) and OTUs that are enriched in drought treatments relative to controls as water-stress opportunists. Differentially abundant OTUs were considered for bulk soil, soils with P. trichocarpa, and roots separately as well as each sampling period separately (day 12, day 13, day 25). Lastly, Wald’s 2-sample t test was used to determine if the mean absolute log2 fold change differed among opportunistic and sensitive OTUs within a sampling period and for bulk soils, planted soils, and roots separately. If few differential (<5) OTUs were detected for a category, this test was not performed (root samples for 16S rRNA, bulk soil and planted soils for ITS data).
Data availability.
Metabolomics data have been deposited to the EMBL-EBI MetaboLights database (https://doi.org/10.1093/nar/gkz1019) with the identifier MTBLS1463. The complete data set can be accessed at https://www.ebi.ac.uk/metabolights/MTBLS1463. Both 16S and ITS data have been deposited through NCBI’s Sequence Read Archive (SRA) under BioProject accession no. PRJNA554609.
ACKNOWLEDGMENTS
We thank Mindy Clark and Steven LeBreux for assistance in the greenhouse. We also thank Dawn Klingman for Illumina MiSeq sequencing preparation.
This research was funded by Center for Bioenergy Innovation, a U.S. Department of Energy Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science and by the Genomic Science Program, U.S. Department of Energy, Office of Science, Biological and Environmental Research, as part of the Plant Microbe Interfaces Scientific Focus Area at ORNL (http://pmi.ornl.gov). This research was also supported in part by an appointment to the ORNL Laboratory Technology Associate Program, sponsored by the U.S. Department of Energy and administered by the Oak Ridge Institute for Science and Education. Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the U.S. Department of Energy under contract DEAC05-00OR22725.
We declare no competing interests related to this work.
REFERENCES
- 1.Lobell DB, Schlenker W, Costa-Roberts J. 2011. Climate trends and global crop production since 1980. Science 333:616–620. doi: 10.1126/science.1204531. [DOI] [PubMed] [Google Scholar]
- 2.Oliver RJ, Finch JW, Taylor G. 2009. Second generation bioenergy crops and climate change: a review of the effects of elevated atmospheric CO2 and drought on water use and the implications for yield. Glob Change Biol Bioenergy 1:97–114. doi: 10.1111/j.1757-1707.2009.01011.x. [DOI] [Google Scholar]
- 3.Lesk C, Rowhani P, Ramankutty N. 2016. Influence of extreme weather disasters on global crop production. Nature 529:84–87. doi: 10.1038/nature16467. [DOI] [PubMed] [Google Scholar]
- 4.Tuck G, Glendining MJ, Smith P, House JI, Wattenbach M. 2006. The potential distribution of bioenergy crops in Europe under present and future climate. Biomass Bioenergy 30:183–197. doi: 10.1016/j.biombioe.2005.11.019. [DOI] [Google Scholar]
- 5.Berendsen RL, Pieterse CMJ, Bakker P. 2012. The rhizosphere microbiome and plant health. Trends Plant Sci 17:478–486. doi: 10.1016/j.tplants.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Naylor D, Coleman-Derr D. 2018. Drought stress and root-associated bacterial communities. Front Plant Sci 8:2223. doi: 10.3389/fpls.2017.02223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pérez Castro S, Cleland EE, Wagner R, Sawad RA, Lipson DA. 2019. Soil microbial responses to drought and exotic plants shift carbon metabolism. ISME J 13:1776–1787. doi: 10.1038/s41396-019-0389-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu L, Naylor D, Dong Z, Simmons T, Pierroz G, Hixson KK, Kim Y-M, Zink EM, Engbrecht KM, Wang Y, Gao C, DeGraaf S, Madera MA, Sievert JA, Hollingsworth J, Birdseye D, Scheller HV, Hutmacher R, Dahlberg J, Jansson C, Taylor JW, Lemaux PG, Coleman-Derr D. 2018. Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc Natl Acad Sci U S A 115:E4284–E4293. doi: 10.1073/pnas.1717308115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santos-Medellín C, Edwards J, Liechty Z, Nguyen B, Sundaresan V. 2017. Drought stress results in a compartment-specific restructuring of the rice root-associated microbiomes. mBio 8:e00764-17. doi: 10.1128/mBio.00764-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Vries FT, Griffiths RI, Bailey M, Craig H, Girlanda M, Gweon HS, Hallin S, Kaisermann A, Keith AM, Kretzschmar M, Lemanceau P, Lumini E, Mason KE, Oliver A, Ostle N, Prosser JI, Thion C, Thomson B, Bardgett RD. 2018. Soil bacterial networks are less stable under drought than fungal networks. Nat Commun 9:3033. doi: 10.1038/s41467-018-05516-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Querejeta JI, Egerton-Warburton LM, Allen MF. 2009. Topographic position modulates the mycorrhizal response of oak trees to interannual rainfall variability. Ecology 90:649–662. doi: 10.1890/07-1696.1. [DOI] [PubMed] [Google Scholar]
- 12.Pigott CD. 1982. Survival of mycorrhiza formed by Cenococcum geophilum FR. in dry soils. New Phytol 92:513–517. doi: 10.1111/j.1469-8137.1982.tb03409.x. [DOI] [Google Scholar]
- 13.Courty P-E, Buée M, Diedhiou AG, Frey-Klett P, Le Tacon F, Rineau F, Turpault M-P, Uroz S, Garbaye J. 2010. The role of ectomycorrhizal communities in forest ecosystem processes: new perspectives and emerging concepts. Soil Biol Biochem 42:679–698. doi: 10.1016/j.soilbio.2009.12.006. [DOI] [Google Scholar]
- 14.Rubin EM. 2008. Genomics of cellulosic biofuels. Nature 454:841–845. doi: 10.1038/nature07190. [DOI] [PubMed] [Google Scholar]
- 15.Sannigrahi P, Ragauskas AJ, Tuskan GA. 2010. Poplar as a feedstock for biofuels: a review of compositional characteristics. Biofuel Bioprod Biorefin 4:209–226. doi: 10.1002/bbb.206. [DOI] [Google Scholar]
- 16.Shakya M, Gottel N, Castro H, Yang ZK, Gunter L, Labbé J, Muchero W, Bonito G, Vilgalys R, Tuskan G, Podar M, Schadt CW. 2013. A multifactor analysis of fungal and bacterial community structure in the root microbiome of mature Populus deltoides trees. PLoS One 8:e76382. doi: 10.1371/journal.pone.0076382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonito G, Reynolds H, Robeson MS, Nelson J, Hodkinson BP, Tuskan G, Schadt CW, Vilgalys R. 2014. Plant host and soil origin influence fungal and bacterial assemblages in the roots of woody plants. Mol Ecol 23:3356–3370. doi: 10.1111/mec.12821. [DOI] [PubMed] [Google Scholar]
- 18.Cregger MA, Veach AM, Yang ZK, Crouch MJ, Vilgalys R, Tuskan GA, Schadt CW. 2018. The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome 6:31. doi: 10.1186/s40168-018-0413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veach AM, Morris R, Yip DZ, Yang ZK, Engle NL, Cregger MA, Tschaplinski TJ, Schadt CW. 2019. Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome 7:76. doi: 10.1186/s40168-019-0668-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hacquard S, Schadt CW. 2015. Towards a holistic understanding of the beneficial interactions across the Populus microbiome. New Phytol 205:1424–1430. doi: 10.1111/nph.13133. [DOI] [PubMed] [Google Scholar]
- 21.Timm CM, Carter KR, Carrell AA, Jun S-R, Jawdy SS, Vélez JM, Gunter LE, Yang Z, Nookaew I, Engle NL, Lu T-Y, Schadt CW, Tschaplinski TJ, Doktycz MJ, Tuskan GA, Pelletier DA, Weston DJ. 2018. Abiotic stresses shift belowground Populus-associated bacteria toward a core stress microbiome. mSystems 3:e00070-17. doi: 10.1128/mSystems.00070-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monclus R, Dreyer E, Villar M, Delmotte FM, Delay D, Petit J-M, Barbaroux C, Le Thiec D, Brechet C, Brignolas F. 2006. Impact of drought on productivity and water use efficiency in 29 genotypes of Populus deltoides x Populus nigra. New Phytol 169:765–777. doi: 10.1111/j.1469-8137.2005.01630.x. [DOI] [PubMed] [Google Scholar]
- 23.Compant S, Van Der Heijden MGA, Sessitsch A. 2010. Climate change effects on beneficial plant-microorganism interactions. FEMS Microbiol Ecol 73:197–214. doi: 10.1111/j.1574-6941.2010.00900.x. [DOI] [PubMed] [Google Scholar]
- 24.De-la-Peña C, Loyola-Vargas VM. 2014. Biotic interactions in the rhizosphere: a diverse cooperative enterprise for plant productivity. Plant Physiol 166:701–719. doi: 10.1104/pp.114.241810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verbruggen N, Hermans C. 2008. Proline accumulation in plants: a review. Amino Acids 35:753–759. doi: 10.1007/s00726-008-0061-6. [DOI] [PubMed] [Google Scholar]
- 26.Yuste JC, Peñuelas J, Estiarte M, Garcia-Mas J, Mattana S, Ogaya R, Pujol M, Sardans J. 2011. Drought-resistant fungi control soil organic matter decomposition and its response to temperature: fungal diversity control over soil C balance. Glob Chang Biol 17:1475–1486. doi: 10.1111/j.1365-2486.2010.02300.x. [DOI] [Google Scholar]
- 27.Barnard RL, Osborne CA, Firestone MK. 2013. Responses of soil bacterial and fungal communities to extreme desiccation and rewetting. ISME J 7:2229–2241. doi: 10.1038/ismej.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schimel J, Balser TC, Wallenstein M. 2007. Microbial stress-response physiology and its implications for ecosystem function. Ecology 88:1386–1394. doi: 10.1890/06-0219. [DOI] [PubMed] [Google Scholar]
- 29.Gargallo-Garriga A, Sardans J, Pérez-Trujillo M, Oravec M, Urban O, Jentsch A, Kreyling J, Beierkuhnlein C, Parella T, Peñuelas J. 2015. Warming differentially influences the effects of drought on stoichiometry and metabolomics in shoots and roots. New Phytol 207:591–603. doi: 10.1111/nph.13377. [DOI] [PubMed] [Google Scholar]
- 30.Gargallo-Garriga A, Sardans J, Pérez-Trujillo M, Rivas-Ubach A, Oravec M, Vecerova K, Urban O, Jentsch A, Kreyling J, Beierkuhnlein C, Parella T, Peñuelas J. 2014. Opposite metabolic responses of shoots and roots to drought. Sci Rep 4:6829. doi: 10.1038/srep06829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zang U, Goisser M, Häberle K-H, Matyssek R, Matzner E, Borken W. 2014. Effects of drought stress on photosynthesis, rhizosphere respiration, and fine-root characteristics of beech saplings: a rhizotron field study. J Plant Nutr Soil Sci 177:168–177. doi: 10.1002/jpln.201300196. [DOI] [Google Scholar]
- 32.Brunner I, Herzog C, Dawes MA, Arend M, Sperisen C. 2015. How tree roots respond to drought. Front Plant Sci 6:547. doi: 10.3389/fpls.2015.00547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams A, Vries FT. 2020. Plant root exudation under drought: implications for ecosystem functioning. New Phytol 225:1899–1905. doi: 10.1111/nph.16223. [DOI] [PubMed] [Google Scholar]
- 34.Meena KK, Sorty AM, Bitla UM, Choudhary K, Gupta P, Pareek A, Singh DP, Prabha R, Sahu PK, Gupta VK, Singh HB, Krishanani KK, Minhas PS. 2017. Abiotic stress responses and microbe-mediated mitigation in plants: the omics strategies. Front Plant Sci 8:172. doi: 10.3389/fpls.2017.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andreo-Jimenez B, Vandenkoornhuyse P, Lê Van A, Heutinck A, Duhamel M, Kadam N, Jagadish K, Ruyter-Spira C, Bouwmeester H. 2019. Plant host and drought shape the root associated fungal microbiota in rice. PeerJ 7:e7463. doi: 10.7717/peerj.7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rolli E, Marasco R, Vigani G, Ettoumi B, Mapelli F, Deangelis ML, Gandolfi C, Casati E, Previtali F, Gerbino R, Pierotti Cei F, Borin S, Sorlini C, Zocchi G, Daffonchio D. 2015. Improved plant resistance to drought is promoted by the root-associated microbiome as a water stress-dependent trait: root bacteria protect plants from drought. Environ Microbiol 17:316–331. doi: 10.1111/1462-2920.12439. [DOI] [PubMed] [Google Scholar]
- 37.Allard-Massicotte R, Tessier L, Lécuyer F, Lakshmanan V, Lucier J-F, Garneau D, Caudwell L, Vlamakis H, Bais HP, Beauregard PB. 2016. Bacillus subtilis early colonization of Arabidopsis thaliana roots involves multiple chemotaxis receptors. mBio 7:e01664-16. doi: 10.1128/mBio.01664-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker TS, Bais HP, Grotewold E, Vivanco JM. 2003. Root exudation and rhizosphere biology. Plant Physiol 132:44–51. doi: 10.1104/pp.102.019661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartmann A, Schmid M, van Tuinen D, Berg G. 2009. Plant-driven selection of microbes. Plant Soil 321:235–257. doi: 10.1007/s11104-008-9814-y. [DOI] [Google Scholar]
- 40.Lebeis SL, Paredes SH, Lundberg DS, Breakfield N, Gehring J, McDonald M, Malfatti S, Glavina del Rio T, Jones CD, Tringe SG, Dangl JL. 2015. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349:860–864. doi: 10.1126/science.aaa8764. [DOI] [PubMed] [Google Scholar]
- 41.Fitzpatrick CR, Copeland J, Wang PW, Guttman DS, Kotanen PM, Johnson M. 2018. Assembly and ecological function of the root microbiome across angiosperm plant species. Proc Natl Acad Sci U S A 115:E1157–E1165. doi: 10.1073/pnas.1717617115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desprez-Loustau M-L, Marçais B, Nageleisen L-M, Piou D, Vannini A. 2006. Interactive effects of drought and pathogens in forest trees. Ann For Sci 63:597–612. doi: 10.1051/forest:2006040. [DOI] [Google Scholar]
- 43.Evans SE, Wallenstein MD. 2014. Climate change alters ecological strategies of soil bacteria. Ecol Lett 17:155–164. doi: 10.1111/ele.12206. [DOI] [PubMed] [Google Scholar]
- 44.Lennon JT, Aanderud ZT, Lehmkuhl BK, Schoolmaster DR. 2012. Mapping the niche space of soil microorganisms using taxonomy and traits. Ecology 93:1867–1879. doi: 10.1890/11-1745.1. [DOI] [PubMed] [Google Scholar]
- 45.Ho A, Lonardo DPD, Bodelier P. 2017. Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol Ecol 93:fix006. doi: 10.1093/femsec/fix006. [DOI] [PubMed] [Google Scholar]
- 46.Fierer N, Bradford MA, Jackson RB. 2007. Toward an ecological classification of soil bacteria. Ecology 88:1354–1364. doi: 10.1890/05-1839. [DOI] [PubMed] [Google Scholar]
- 47.Treseder KK, Berlemont R, Allison SD, Martiny AC. 2018. Drought increases the frequencies of fungal functional genes related to carbon and nitrogen acquisition. PLoS One 13:e0206441. doi: 10.1371/journal.pone.0206441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naylor D, DeGraaf S, Purdom E, Coleman-Derr D. 2017. Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J 11:2691–2704. doi: 10.1038/ismej.2017.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crowther TW, Maynard DS, Crowther TR, Peccia J, Smith JR, Bradford MA. 2014. Untangling the fungal niche: the trait-based approach. Front Microbiol 5:579. doi: 10.3389/fmicb.2014.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Veach AM, Yip D, Engle NL, Yang ZK, Bible A, Morrell-Falvey J, Tschaplinski TJ, Kalluri UC, Schadt CW. 2018. Modification of plant cell wall chemistry impacts metabolome and microbiome composition in Populus PdKOR1 RNAi plants. Plant Soil 429:349–361. doi: 10.1007/s11104-018-3692-8. [DOI] [Google Scholar]
- 51.Lundberg DS, Yourstone S, Mieczkowski P, Jones CD, Dangl JL. 2013. Practical innovations for high-throughput amplicon sequencing. Nat Methods 10:999–1002. doi: 10.1038/nmeth.2634. [DOI] [PubMed] [Google Scholar]
- 52.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope E, Da Silva R, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley G, Janssen S, Jarmusch AK, Jiang L, Kaehler B, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MG, Lee J, Ley R, Liu Y-X, Loftfield E, Lozupone C, Maher M, Marotz C, Martin B, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton J, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS II, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CH, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Knight R, Caporaso JG. 2019. Reproducible, interactive, scalable, and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abarenkov K, Henrik Nilsson R, Larsson K-H, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AFS, Tedersoo L, Ursing BM, Vrålstad T, Liimatainen K, Peintner U, Kõljalg U. 2010. The UNITE database for molecular identification of fungi—recent updates and future perspectives: letters. New Phytol 186:281–285. doi: 10.1111/j.1469-8137.2009.03160.x. [DOI] [PubMed] [Google Scholar]
- 55.Clarke KR. 1993. Non-parametric multivariate analyses of changes in community structure. Austral Ecol 18:117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 56.Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. 2017. Microbiome datasets are compositional: and this is not optional. Front Microbiol 8:2224. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wehrens R. 2011. Chemometrics with R: multivariate data analysis in the natural sciences and life sciences. Springer, Heidelberg, Germany. [Google Scholar]
- 58.Oksanen J, Blanchet J, Friendly M, Kindt R, Legendre P, McGlinn D. 2018. Vegan: community ecology package. Ordination methods, diversity analysis, and other functions for community and vegetation analysis. R package.
- 59.Anderson MJ. 2001. Permutation tests for univariate or multivariate analysis of variance and regression. Can J Fish Aquat Sci 58:626–639. doi: 10.1139/f01-004. [DOI] [Google Scholar]
- 60.Herve M. 2019. RVAideMemoire: testing and plotting procedures for biostatistics. R package.
- 61.Venables W, Ripley B. 2002. Modern applied statistics with S, 4th ed. Springer, New York, NY. [Google Scholar]
- 62.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The 66 plant root secondary metabolites detected in this study and SIMPER statistical output for each metabolite. Download Table S1, DOCX file, 0.01 MB (15.4KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Individual metabolites identified through SIMPER which differed between treatment (drought versus control), time (drought, rewet, and recovery), and their interaction produced in P. trichocarpa roots. The direction of change based on two-way ANOVA model output is also given. Metabolites which had a significant treatment and time (T*T) interaction are boldfaced. Download Table S2, DOCX file, 0.02 MB (16.1KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Three-way ANOVA model results for alpha-diversity estimates for bacteria/archaea and fungi. Response variables which significantly varied by a fixed term (presence of plants, drought treatment, and time of drying, rewet, and recovery) are boldfaced. An asterisk denotes a response variable which had a significant treatment-by-time interaction. There were no significant three-way interactions. Download Table S3, DOCX file, 0.01 MB (14.2KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Dominant bacterial and fungal phylum 2-way ANOVA model results with treatment (drought, control) and time (end of drought, rewet, and recovery) as fixed, main effects and their interaction (T*T). Model results are for root endosphere samples only. Phyla which significantly varied with a main effect are boldfaced. Results provided used centered log-ratio-transformed data based on raw, absolute phylum-level sequence counts. Download Table S4, DOCX file, 0.01 MB (15.7KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DESeq2 results for bacteria/archaea and fungi among compartments and time. The number and proportion of OTUs which were differentially abundant within drought versus control treatments are provided. Sensitive refers to OTUs which were depleted under drought, and opportunistic refers to those which were enriched compared to controls. Download Table S5, DOCX file, 0.02 MB (16KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Raw PCA scores from Bray-Curtis-based distances for bacteria/archaea and fungi in in soils (planted, bulk) and in roots. Symbols represent plant presence or no presence for bulk soils (circle = plant presence, triangle = no plants), colors denote treatment (blue = control, green = drought), and shading from light to dark refers to time of sampling. Download FIG S1, TIF file, 0.1 MB (114.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Raw PCA scores from Euclidean-based distances for bacteria/archaea and fungi in soils (planted, bulk) and in roots. Symbols represent plant presence or no presence for bulk soils (circle = plant presence, triangle = no plants), colors denote treatment (blue = control, green = drought), and shading from light to dark refers to time of sampling. Download FIG S2, TIF file, 0.3 MB (284.5KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The proportion of sequences belonging to significant differentially sensitive (depleted in drought) and opportunistic (enriched in drought) OTUs across bacterial and archaeal phyla (A and B) and fungal taxonomies (C and D) over time (day 12 = drying period, day 13 = rewet period, day 25 = recovery period). The absence of bars indicates that zero OTUs were differentially abundant for that time except for panel A, where only very rare OTUs (<0.01% relative abundance) were identified. Percent proportion of sensitivity refers to all sequences in a taxonomic group within control samples, and percent proportion of opportunism refers to all sequences in a taxonomic group with drought samples. Download FIG S3, TIF file, 0.4 MB (459.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The proportion of sequences belonging to significant differentially sensitive (depleted in drought) and opportunistic (enriched in drought) OTUs across fungal taxonomies over time (day 12 = drying period, day 13 = rewet period, day 25 = recovery period) within root endospheres. The absence of indicates that zero OTUs were differentially abundant for that time except for panel A, where only very rare OTUs (<0.2% relative abundance) were identified. Percent proportion of sensitivity refers to all sequences in a taxonomic group within control samples, and percent proportion of opportunism refers to all sequences in a taxonomic group with drought samples. Download FIG S4, TIF file, 0.2 MB (208.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data Availability Statement
Metabolomics data have been deposited to the EMBL-EBI MetaboLights database (https://doi.org/10.1093/nar/gkz1019) with the identifier MTBLS1463. The complete data set can be accessed at https://www.ebi.ac.uk/metabolights/MTBLS1463. Both 16S and ITS data have been deposited through NCBI’s Sequence Read Archive (SRA) under BioProject accession no. PRJNA554609.