

Abstract
Monolysocardiolipin (MLCL) is a three-tailed variant of cardiolipin (CL), the signature lipid of mitochondria. MLCL is not normally found in healthy tissue but accumulates in mitochondria of people with Barth syndrome (BTHS), with an overall increase in the MLCL:CL ratio. The reason for MLCL accumulation remains to be fully understood. The effect of MLCL build-up and decreased CL content in causing the characteristics of BTHS are also unclear. In both cases, an understanding of the nature of MLCL interaction with mitochondrial proteins will be key. Recent work has shown that MLCL associates less tightly than CL with proteins in the mitochondrial inner membrane, suggesting that MLCL accumulation is a result of CL degradation, and that the lack of MLCL–protein interactions compromises the stability of the protein-dense mitochondrial inner membrane, leading to a decrease in optimal respiration. There is some data on MLCL–protein interactions for proteins involved in the respiratory chain and in apoptosis, but there remains much to be understood regarding the nature of MLCL–protein interactions. Recent developments in structural, analytical and computational approaches mean that these investigations are now possible. Such an understanding will be key to further insights into how MLCL accumulation impacts mitochondrial membranes. In turn, these insights will help to support the development of therapies for people with BTHS and give a broader understanding of other diseases involving defective CL content.
Keywords: cardiolipin, mitochondria, monolysocardiolipin, protein–lipid interactions
Introduction
Monolysocardiolipin (MLCL) is a three-tailed glycerol-phospholipid and an intermediate product in the biosynthesis and degradation of cardiolipin (CL) [1]. In eukaryotes, CL is found uniquely in mitochondrial membranes, comprising 10–20% of the lipid content of the inner mitochondrial membrane [2]. CL is also found in bacteria, reflecting the common evolutionary origins of mitochondria and bacteria [3]. MLCL accumulates in the mitochondrial membranes of people with Barth syndrome (BTHS) [4], a genetic disease caused by mutations in a transacylase enzyme, tafazzin, involved in CL remodelling.
CL is involved in many mitochondrial processes: CL has a crucial role in mitochondrial energy production, interacting with and enhancing the activity of all the major respiratory chain proteins [5] as well as roles in cristae morphology [6], apoptosis [7], mitophagy [8], and mitochondrial fusion and fission [9,10]. It is, therefore, not surprising that the deficiency of tafazzin, resulting in the accumulation of MLCL and other CL variants, and a decrease in total CL content, might cause problems [11]. However, there remains much to be understood regarding the molecular mechanisms underlying the effect of the change in lipid content and its impact in disease [11–13].
Here, the reason for MLCL accumulation, the effect of this build-up in mitochondrial membranes, and what we know about the protein–MLCL interactions involved are reviewed.
The structure and physical properties of MLCL
The chemical structure of MLCL and the mature (remodelled) form of CL is shown in Figure 1. The distinctive features of CL, as compared with other lipids found in eukaryotic cells, are its geometric and electrostatic properties: CL has a conical shape due to its small headgroup and four acyl chains, and its two phosphate groups confer a double negative charge. These properties help to inform an understanding of the physical properties of MLCL.
Figure 1. Chemical structure of MLCL (A) and mature, tetralinoleoyl-CL (B).

MLCL, CL and membrane curvature
Due to its geometry, CL has been shown in vitro to be sorted to membranes with negative curvature [14]. Molecular dynamics simulations of CL-containing membranes also show localisation of CL to curved regions of the membrane [15–17]. MLCL, on the other hand, has one less acyl chain, therefore, would be expected to have less of a propensity to localise to curved membrane regions, indeed MLCL-containing membranes have been shown to have a greater preference for a lamellar phase than CL [18]. Molecular dynamics simulations have also shown that MLCL does not localise specifically to curved regions of the bilayer [19] and that MLCL bilayers take on less negative curvature than bilayers containing CL [20].
MLCL and CL headgroup charge
There has been some controversy over the charge of CL at physiological pH. CL has been shown to contain only one negative charge in bulk bilayers at physiological pH, rationalised by assuming that the headgroup ‘traps’ one proton on one of the phosphate moieties by forming a tight bicyclic H-bonding structure with the oxygen from the connecting glycerol moiety [21]. The finding led to the proposal that CL acts as a proton shuttle [22]. However, subsequent work has since shown that CL exists with a double negative charge at physiological pH [23–25]. Furthermore, the putative bicyclic H-bonding network has a particular headgroup geometry; however, this is not observed in crystal structures of protein–CL complexes [26]. It has been demonstrated in silico that MLCL has a double negative charge at physiological pH [24].
MLCL vs. CL headgroup geometry
All-atom molecular dynamics simulations comparing MLCL and CL behaviour in bilayers suggest that the hydroxyl group on the lyso side of the MLCL headgroup (Figure 1A) is prone to orient itself more towards the interfacial polar region than the hydrophobic core of the membrane, causing the lyso side of MLCL to be ‘pulled’ towards the solvent phase and leading to headgroup tilt, whereas the headgroup of CL displayed no such tilting [19]. The tilting of the MLCL headgroup means that the phosphate on the lyso side of MLCL is also positioned slightly further from the bilayer hydrophobic core than either phosphate moiety in the CL headgroup. This has possible implications for MLCL–protein interactions as compared with CL–protein interactions, given that the two phosphates on CL often form ionic interactions with positively charged protein side-chains [27]. Molecular dynamics simulations also showed that the acyl chain on the lyso side of MLCL was more ordered than other acyl chains [19].
MLCL and CL acyl chain content
The acyl chain content of MLCL in BTHS cells has a low degree of unsaturation [28,29] whereas remodelled, mature CL has a higher level of unsaturation, although this does vary between tissue types [30]: (18:2)4 acyl chain content is particularly tightly controlled in mammalian cardiac tissue [31], while brain CL is more diverse [32–34].
How does MLCL accumulate?
MLCL is produced from CL by a lipase removing a single acyl chain from CL, which can occur as part of CL remodelling or degradation. To understand the accumulation of MLCL, it is first useful to briefly review CL biosynthesis and remodelling. The biosynthesis of nascent CL occurs in the mitochondrial inner membrane from phosphatidylglycerol (PG) and CDP-diacylglycerol [35]. The acyl chain composition of nascent CL is remodelled, that is, acyl chains are removed and replaced, to form the mature form of CL, which typically has higher unsaturated content. In yeast, CL deacylation is performed by a CL-specific phospholipase Cld1, which has a specificity for saturated tails [36,37], whereas in mammals, phospholipase A2 (PLA2) is responsible [38–40], most likely the calcium-independent iPLA2 gamma in vivo [41]. Tafazzin can then reacylate MLCL [42] (Figure 2). Tafazzin uses acyl chains from other phospholipids to reacylate MLCL, or to act as a CL-transacylase. The specificity of tafazzin remains controversial: it has been demonstrated that tafazzin may have some acyl specificity [43]; however, the work from Schlame et al. [44] shows that the specificity of tafazzin is dependent on the physical properties of the bilayer, particularly the ability of the bilayer to form non-bilayer phases. There are other remodelling enzymes: CL can also be reacylated by acyl-CoA:lysocardolipin actyltransferase 1 (or ALCAT1) in mitochondrial-associated membranes [45] and in mitochondrial membranes by monolysocardiolipin acyltransferase 1 (or MLCLAT1) [46]. There is also evidence that the trifunctional protein alpha subunit, which is similar to MLCLAT1, is involved in CL remodelling [47–49].
Figure 2. Schematic of CL remodelling.
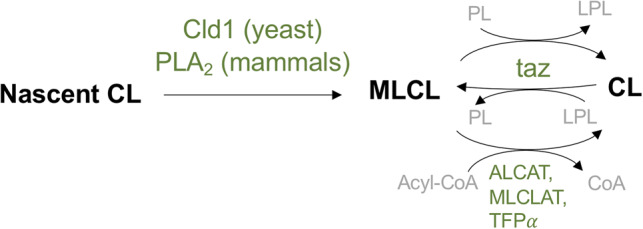
Nascent CL is deacylated by Cld1 in yeast; phospholipase A2 (PLA2) in mammals. Tafazzin transfers an acyl chain from another phospholipid (PL) to MLCL, forming CL and a lysophospholipid (LPL). After one or more cycles, this will result in the mature form of CL. CL acyltransferase enzymes, which take MLCL and acyl-CoA as substrate, are also shown: acyl-CoA:lysocardolipin actyltransferase 1 (ALCAT), monolysocardiolipin acyltransferase 1 (MLCLAT), and trifunctional protein alpha subunit (TFPα). Enzyme names are coloured green and lipid names are in black.
MLCL accumulation occurs in people with BTHS leading to an increase in the MLCL:CL ratio, and otherwise, MLCL is not present in normal tissue, except for testis [50]. The cause of an increase in the MLCL:CL ratio in BTHS is yet to be fully understood [1]. Initially, MLCL accumulation was thought to be due to tafazzin being part of a deacylation–reacylation cycle, with tafazzin deficiency causing the remodelling process to stall, meaning that MLCL was not being reacylated. However, more recently, it has been shown that MLCL accumulation may occur due to MLCL being an intermediate in CL degradation [51]. This hypothesis is based on the finding that tafazzin deficiency in yeast increases CL turnover, while knock-down of Cld1 decreases CL turnover. The rationale is that due to less favourable interaction of MLCL and saturated CL within complexes of mitochondrial membrane proteins, saturated CL is exposed to degradation to MLCL, which creates a cycle culminating in the accumulation of MLCL.
MLCL accumulation in disease
MLCL accumulates in people with BTHS [4,52,53], leading to the characteristic increased MLCL:CL ratio. MLCL has also been shown to build-up after cardiac arrest in rats [54], and the lack of CL remodelling has also recently been associated with cardiac arrhythmia [47]. However, despite defective or decreased CL content being implicated in many other disease states (e.g. cancer, neurodegenerative disease, and diabetes), the accumulation of MLCL is not widely found in other CL-linked diseases [11]. As introduced above, BTHS is known to be caused by a mutation in the tafazzin gene [55].
People with BTHS present with myopathy, heart failure, muscle weakness, growth retardation and neutropenia [56]. On a cellular and molecular level, cells from people with BTHS have a reduced mitochondrial membrane potential [57], lower electron transport chain protein activity [58,59], decreased respiratory coupling index [60] and increased proton leak [29] (leading to possible further mitochondrial damage via the increased production of reactive oxygen species [29,60]), destabilised respiratory supercomplexes [61], and abnormal cristae structures [62]. Mutations in taffazin also cause altered ATP synthase organisation in Drosophila melanogaster [63].
As well as a lower concentration of CL and altered CL composition (i.e. a higher proportion of CL with saturated tails), an increase in the MLCL:CL ratio is a key marker for BTHS [64,65]: although dependent on cell type, the MLCL:CL ratio typically increases from a range of 0.0–0.2 in control cells to the significantly different range of 0.4–100 in cells from BTHS patients [28,64,66].
The reasons for altered lipid content causing BTHS symptoms, and the functional role of tafazzin remains an open question, although the existence of BTHS underlines its importance [1,11,13,35]. Recently, it has been suggested that the main role of tafazzin is to remodel CL such that it lowers the energetic cost of protein crowding in the protein-dense inner mitochondrial membrane: Xu et al. [51] showed that MLCL associates less tightly with mitochondrial proteins and that the association of CL in respiratory supercomplexes protects CL from degradation. The authors also showed that CL unsaturation promotes protein association, which in turn is thought to protect CL from degradation. In further work, Xu et al. [37] showed that the expression of complexes of the respiratory chain triggers remodelling; knock-down of other mitochondrial proteins, even including others that inhibit the respiratory function, did not alter CL composition. This lead to the suggestion that tafazzin remodels CL to have unsaturated tails since these will lower the energetic cost of the membrane packing around respiratory complexes in the inner mitochondrial membrane. It was argued that as a result of this, respiratory complexes can be incorporated into the mitochondrial membrane with higher density, without loss of membrane stability. This might explain why membranes with high energy requirements are most vulnerable to tafazzin deficiency [56].
The nature of MLCL interactions with mitochondrial proteins
The result that MLCL associates less tightly than CL with mitochondrial membrane proteins [51] forms a key part of understanding how the MLCL:CL ratio increases in people with BTHS and why MLCL accumulation might affect CL remodelling. It also provides a rationale for the destabilised respiratory supercomplexes and thereby reduced membrane potential and lower electron transport chain protein activity [58,59] found in people with BTHS. The weaker association of MLCL vs. CL was demonstrated using 31P-NMR spectroscopy, which showed that MLCL was solubilised by digitonin, a relatively weak detergent, whereas CL and MLCL were solubilised by SDS (sodium dodecyl sulphate), a stronger detergent [51]. The nature of MLCL–protein interactions remains unclear but is a vital part of the picture.
MLCL interaction with proteins involved in oxidative phosphorylation
CL is known to interact with, and affect the activity of, all of the key protein complexes of oxidative phosphorylation, namely: Complex I [67,68], cytochrome bc1 (Complex III) [69], cytochrome c oxidase (Complex IV) [70] and ATP synthase [71–73] as well as the ADP/ATP carrier [74], the mitochondrial transporter responsible for shuttling newly synthesised ATP out of the mitochondrial matrix towards the rest of the cell.
In contrast, binding of MLCL to cytochrome c oxidase occurs but with lower affinity than CL. MLCL retained 60% of the activity of cytochrome c oxidase compared with CL, and when CL is depleted altogether, the enzyme has only 30–50% of the original activity [75]. This agrees with the findings of Xu et al. [51] that MLCL interacts less tightly than CL, and further suggests that the loss of the activity of cytochrome c oxidase reconstituted with MLCL rather than CL is related to lower MLCL affinity. The partial retention of activity with MLCL indicates that MLCL is in some ways able to mimic CL interaction.
The ADP/ATP carrier was also reported to retain only 12% of activity compared with CL on reconstitution with MLCL [76]. In this case, it is not necessarily clear whether the loss of activity with MLCL compared with CL is due to the loss of association or whether the MLCL could be as tightly bound, but with a lesser impact on activity; compounding the difficulty in teasing these apart is the lack of knowledge of how exactly CL optimises the activity of this carrier.
MLCL and proteins involved in apoptosis
In cultured lymphoblast cells from people with BTHS, apoptosis was found not to be increased [4]. However, MLCL interactions with many proteins involved in apoptosis have been investigated demonstrating that MLCL interacts with tBid more tightly than CL [77,78], caspase-8 [29] less tightly than CL, and also modifies the release of cytochrome c [78,79].
Bid is a proapoptotic protein and member of the Bcl-2 family. Bid is cleaved by caspase-8 to form tBid, which then associates at the mitochondrial outer membrane, leading to oligomerisation of other Bcl-2 family proteins, Bax and Bak, and the release of cytochrome c [80]. CL had been suggested to target tBid to the OMM during apoptosis [81]. Using electron spray mass spectroscopy, MLCL was shown to enhance Bid and tBid mitochondrial membrane association, relative to CL [78]. MLCL also appeared to enhance the release of cytochrome c [78] and affect the oxidation state of cytochrome c [79]. Furthermore, MLCL was shown to be capable of inducing tBid dissociation from nBid (the other Bid-cleavage product) [77]. Although there exists structural information regarding tBid [82–84], the nature of the interaction with CL and MLCL is not clear [77]. In contrast with Bid, MLCL binds less tightly to active caspase-8 than CL [29]. Thus, whether apoptosis is be enhanced or otherwise by MLCL accumulation remains a somewhat confounding picture.
The enhanced interaction of Bid with MLCL vs. CL is intriguing, as it contrasts with the findings showing that MLCL associates less tightly with mitochondrial proteins [51]. However, Bid is a membrane-associated rather than a membrane-embedded protein.
What does this tell us about MLCL–protein interactions?
It is not clear why the loss of an acyl chain in MLCL causes the loss of interaction with proteins and the loss of activity compared with CL. A computational study of MLCL properties [19], discussed above, showed that due to the loss of an acyl chain, the lyso side of the MLCL headgroup was tilted away from the hydrophobic core, and the acyl chain on the lyso side of MLCL was more ordered. Both of these differences may cause a weakening of the association with cytochrome c oxidase and possibly the ADP/ATP carrier, while retaining some of the characters of the CL interaction. The ordering of the lyso acyl chain in MLCL may also contribute to the preference for MLCL not to be sequestered in respiratory supercomplexes [37,51]. The fact that non-embedded membrane protein Bid interacted more tightly with MLCL than CL may also be due to a headgroup tilt that exposes the lyso phosphate oxygen to the solvent phase.
EPR studies of the Na+/K+ ATPase show that CL and MLCL have a similar affinity for interaction with the protein [85], which could indicate that for any difference to be observed, there would have to be a tightly interacting CL binding site; since there is no evidence for the existence of a mitochondrial Na+/K+ ATPase [86], it might be assumed that the protein has not evolved to have optimal CL binding. It may be in this case, therefore, that the rearrangements in the MLCL headgroup are too subtle to make a difference.
However, without further studies, it is not certain that the MLCL headgroup rearrangements observed in bulk in bilayers [19] will translate to alterations in protein–lipid interactions.
Confounding any interpretation of MLCL–protein interactions is the problem that the molecular mechanism through which CL optimises protein activity is itself unclear. However, this does mean that a greater understanding of MLCL–protein interactions may also help to shed light on the role of CL.
Conclusions and outlook
A key aspect of understanding the reasons for MLCL accumulation and the effect of MLCL accumulation in mitochondrial membranes lies in understanding MLCL interaction with mitochondrial proteins. Recent work has shown that MLCL interacts less tightly with mitochondrial membrane proteins [51] and that global impairment of respiratory chain complexes triggers MLCL accumulation [37], leading to the hypothesis that an increase in the MLCL:CL ratio impacts the stability of mitochondrial membranes, in particular impairing the tight-packing of the respiratory supercomplexes. To test this hypothesis, it will be important to understand more about the nature of MLCL–protein interactions. Existing data indicate that the loss of an acyl chain in MLCL diminishes membrane-embedded protein association at high-affinity CL interaction sites [75,76], possibly due to slight rearrangement in the MLCL headgroup as compared with CL [19]. MLCL association with peripheral membrane proteins does not necessarily follow the trend [78]. However, it remains unclear how MLCL interacts with mitochondrial membrane proteins and why it does so less tightly than CL with mitochondrial membrane-embedded proteins.
Other possible effects of MLCL accumulation
Altered mitochondrial morphology, i.e. deformed cristae, is a classic feature of BTHS. In a fly BTHS model, ATP synthase organisation is altered, which may influence cristae structure, since it has been shown that rows of ATP synthase dimers form on cristae edges, which drives membrane curvature [87–90]. Given that CL interacts directly with ATP synthase, albeit transiently [91], and that CL is known to localise to curved regions, whereas MLCL does not [19], it is conceivable that differences in MLCL interaction with ATP synthase may contribute to altered mitochondrial morphology in BTHS. However, it could also be that other CL-associated cristae-organising machinery [92] are also affected by abnormal CL composition, which then impacts on ATP synthase organisation. MLCL interaction with ATP synthase dimers and even the localisation of MLCL within cristae structures in vivo is unclear.
More generally, it is also possible that the accumulation of MLCL may disrupt mitochondrial membrane properties, leading to further effects on the mitochondrial protein function. MLCL-containing bilayers, as mentioned above, are more likely to be flat [18–20] and are less susceptible to shape deformation under pressure than CL-containing bilayers [19]. Since the mitochondrial inner membrane is highly curved, there may be many proteins, localised to cristae tips, as discussed above, or at cristae junctions, or which perform dynamic functions during mitochondrial fusion and fission, that therefore could be affected by an increase in the MLCL:CL ratio. MLCL-containing bilayers are also less likely to have small ‘defects’, where the hydrophobic core of the bilayer is exposed [19]. This may mean that, for peripheral proteins where partial interaction of the protein with the hydrophobic core of the membrane is the mode of binding, MLCL-containing bilayers form less favourable interactions.
Advances in investigating protein–lipid interactions
Although there remains much to be understood about the role of MLCL and its interaction with mitochondrial proteins, we are currently at an exciting time for research into protein–lipid interactions, since advances in both experimental and computational methods promise (and already are starting to achieve) significant advances.
While there have been some notable cases of CL being resolved directly in crystal structures (e.g. the ADP/ATP carrier [74,93], cytochrome bc1 [94], and cytochrome c oxidase [95]), the cryo-electron microscopy ‘resolution revolution’ [96] means that there are now many more membrane protein structures with CL resolved, such as the yeast respiratory supercomplexes [97,98], mammalian Complex I [99,100], and ATP synthase [101]. As such, there are not to my knowledge any structures of mitochondrial proteins resolved in the presence of MLCL. Such structures would give invaluable insights as to the molecular detail of how MLCL interacts with mitochondrial proteins, and how that may differ from interaction with CL, although the obvious problem might be that with less tight association, MLCL is less likely to be resolved in any structures. Similar advances in cryo-electron tomography have led to visualisation of membrane proteins in intact mitochondria with unprecedented resolution [102]. However, again, to my knowledge, this technique has not been applied to mitochondria of people with BTHS, or more generally, cells with a high MLCL:CL ratio. Cryo-electron tomography would allow us to understand further how certain distinctive mitochondrial proteins (such as Complex I, ATP synthase, and cytochrome bc1) are organised in the disrupted cristae of BTHS mitochondria, and in particular, how the arrangement of respiratory supercomplexes is altered.
Mass spectroscopy has, as discussed above [37,51], provided invaluable insight into MLCL–protein interactions. New techniques that allow for mass spectroscopy of proteins ejected directly from native mitochondrial membranes have also recently been published [103]. Although such an approach may require some fine tuning [104,105], this methodology could potentially be used to look at the mitochondria of people with BTHS and would allow further understanding of the extent of MLCL–protein interaction and effects of MLCL accumulation [106].
Computational techniques, in particular molecular dynamics simulations, have also seen significant development [107]. Molecular dynamics simulations allow multiple lipid interaction sites to be identified and characterised [108,109], and new methodologies mean that relative free energies of lipid binding can be assessed [110]. Molecular dynamics simulations have been used successfully to investigate CL interactions with mitochondrial [111–115] and bacterial [116–118] proteins, although, to my knowledge, have yet to be extended to investigate MLCL. Simulations of MLCL embedded in mitochondrial membranes containing mitochondrial proteins would demonstrate differences between interaction sites of MLCL and CL and enable the calculation of MLCL vs. CL binding free energies. Coarse-grained molecular dynamics simulations [119] of crowded mitochondrial membranes (e.g. [120,121]) will further help to show how protein crowding impacts on MLCL–protein interactions. We now have a good indication of where CL would bind in yeast supercomplexes [97,98] (Figure 3). Given the increasing sophistication of molecular dynamics simulations and their success in reproducing the effect of membrane curvature on MLCL and CL localisation [19], it appears that the use of simulations could give an exciting insight into the hypothesis that MLCL is less favourably sequestered than CL in respiratory supercomplexes, and the nature of MLCL–protein interactions more broadly.
Figure 3. Cryo-electron microscopy structure of a yeast supercomplex.

A yeast supercomplex (PDB ID: 6HUY [98]) containing a dimer of cytochrome bc1 (or Complex III; in white), two monomers of cytochrome c oxidase (Complex IV; grey), with resolved cardiolipin lying in the protein–protein interfaces (cardiolipin is shown as spheres: carbon atoms in yellow; oxygen, red; phosphorous, brown).
Such advances will help to shed light on possible therapies both for BTHS, a disease specifically caused defective CL remodelling leading to defective CL content and accumulation of MLCL as well as the much broader category of pathologies where CL content is altered.
Competing Interests
The author declares that there are no competing interests associated with this manuscript.
Perspectives
A marked increase in the MLCL:CL ratio is the hallmark of people with BTHS, a genetic disease caused solely by defective CL remodelling. People with BTHS have mitochondria displaying defective oxidative phosphorylation and abnormal cristae structure, causing multi-system disorder with potentially life-limiting effects.
It has been shown that MLCL associates less tightly with many inner membrane proteins, and this finding underlies an explanation for how an increase in the MLCL:CL ratio affects people with BTHS. However, the nature of MLCL interaction with mitochondrial proteins is unclear.
Structural, analytical, and computational techniques, used in combination, are poised to provide answers.
Abbreviations
- BTHS
Barth syndrome
- CL
cardiolipin
- MLCL
monolysocardiolipin
Funding
A.L.D is supported by the Biotechnology and Biological Sciences Research Council, [grant number BB/R00126X/1], and Pembroke College, Oxford (BTP Fellowship).
Open Access
Open access for this article was enabled by the participation of University of Oxford in an all-inclusive Read & Publish pilot with Portland Press and the Biochemical Society under a transformative agreement with JISC.
Author Contributions
A.L.D. wrote and edited the manuscript.
References
- 1.Schlame M. and Xu Y. (2020) The function of tafazzin, a mitochondrial phospholipid-lysophospholipid acyltransferase. J. Mol. Biol. 712, 136486 10.1016/j.jmb.2020.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horvath S.E. and Daum G. (2013) Lipids of mitochondria. Prog. Lipid Res. 52, 590–614 10.1016/j.plipres.2013.07.002 [DOI] [PubMed] [Google Scholar]
- 3.Luévano-Martínez L.A. (2015) The chimeric origin of the cardiolipin biosynthetic pathway in the Eukarya domain. Biochim. Biophys. Acta 1847, 599–606 10.1016/j.bbabio.2015.03.005 [DOI] [PubMed] [Google Scholar]
- 4.Valianpour F., Mitsakos V., Schlemmer D., Towbin J.A., Taylor J.M., Ekert P.G. et al. (2005) Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J. Lipid Res. 46, 1182–1195 10.1194/jlr.M500056-JLR200 [DOI] [PubMed] [Google Scholar]
- 5.Paradies G., Paradies V., De Benedictis V., Ruggiero F.M. and Petrosillo G. (2014) Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 1837, 408–417 10.1016/j.bbabio.2013.10.006 [DOI] [PubMed] [Google Scholar]
- 6.Ikon N. and Ryan R.O. (2017) Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta 1859, 1156–1163 10.1016/j.bbamem.2017.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrosillo G., Ruggiero F.M. and Paradies G. (2003) Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J. 17, 2202–2208 10.1096/fj.03-0012com [DOI] [PubMed] [Google Scholar]
- 8.Chu C.T., Ji J., Dagda R.K., Jiang J.F., Tyurina Y.Y., Kapralov A.A. et al. (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205 10.1038/ncb2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramachandran R. (2018) Mitochondrial dynamics: the dynamin superfamily and execution by collusion. Semin. Cell Dev. Biol. 76, 201–212 10.1016/j.semcdb.2017.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ban T., Ishihara T., Kohno H., Saita S., Ichimura A., Maenaka K. et al. (2017) Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 19, 856–863 10.1038/ncb3560 [DOI] [PubMed] [Google Scholar]
- 11.Pennington E.R., Funai K., Brown D.A. and Shaikh S.R. (2019) The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim. Biophys. Acta 1864, 1039–1052 10.1016/j.bbalip.2019.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye C., Shen Z. and Greenberg M.L. (2016) Cardiolipin remodeling: a regulatory hub for modulating cardiolipin metabolism and function. J. Bioenerg. Biomembr. 48, 113–123 10.1007/s10863-014-9591-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Hafidi M., Correa F. and Zazueta C. (2020) Mitochondrial dysfunction in metabolic and cardiovascular diseases associated with cardiolipin remodeling. Biochim. Biophys. Acta 1866, 165744 10.1016/j.bbadis.2020.165744 [DOI] [PubMed] [Google Scholar]
- 14.Beltrán-Heredia E., Tsai F.-C., Salinas-Almaguer S., Cao F.J., Bassereau P. and Monroy F. (2019) Membrane curvature induces cardiolipin sorting. Commun. Biol. 2, 225 10.1038/s42003-019-0471-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyd K.J., Alder N.N. and May E.R. (2017) Buckling under pressure: curvature-based lipid segregation and stability modulation in cardiolipin-containing bilayers. Langmuir 33, 6937–6946 10.1021/acs.langmuir.7b01185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson B.A., Ramanathan A. and Lopez C.F. (2019) Cardiolipin-dependent properties of model mitochondrial membranes from molecular simulations. Biophys. J. 117, 429–444 10.1016/j.bpj.2019.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elías-Wolff F., Lindén M., Lyubartsev A.P. and Brandt E.G. (2019) Curvature sensing by cardiolipin in simulated buckled membranes. Soft Matter 15, 792–802 10.1039/C8SM02133C [DOI] [PubMed] [Google Scholar]
- 18.Powell G.L. and Marsh D. (1985) Polymorphic phase behavior of cardiolipin derivatives studied by 31P NMR and X-ray diffraction. Biochemistry 24, 2902–2908 10.1021/bi00333a013 [DOI] [PubMed] [Google Scholar]
- 19.Boyd K.J., Alder N.N. and May E.R. (2018) Molecular dynamics analysis of cardiolipin and monolysocardiolipin on bilayer properties. Biophys. J. 114, 2116–2127 10.1016/j.bpj.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahlberg M. (2007) Polymorphic phase behavior of cardiolipin derivatives studied by coarse-grained molecular dynamics. J. Phys. Chem B 111, 7194–7200 10.1021/jp071954f [DOI] [PubMed] [Google Scholar]
- 21.Kates M., Syz J.Y., Gosser D. and Haines T.H. (1993) pH-dissociation characteristics of cardiolipin and its 2′-deoxy analogue. Lipids 28, 877–882 10.1007/BF02537494 [DOI] [PubMed] [Google Scholar]
- 22.Haines T.H. and Dencher N.A. (2002) Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett. 528, 35–39 10.1016/S0014-5793(02)03292-1 [DOI] [PubMed] [Google Scholar]
- 23.Olofsson G. and Sparr E. (2013) Ionization constants pKa of cardiolipin. PLoS One 8, e73040 10.1371/journal.pone.0073040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sathappa M. and Alder N.N. (2016) The ionization properties of cardiolipin and its variants in model bilayers. Biochim. Biophys. Acta 1858, 1362–1372 10.1016/j.bbamem.2016.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kooijman E.E., Swim L.A., Graber Z.T., Tyurina Y.Y., Bayır H., Kagan V.E. et al. (2017) Magic angle spinning 31P NMR spectroscopy reveals two essentially identical ionization states for the cardiolipin phosphates in phospholipid liposomes. Biochim. Biophys. Acta 1859, 61–68 10.1016/j.bbamem.2016.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dahlberg M., Marini A., Mennucci B. and Maliniak A. (2010) Quantum chemical modeling of the cardiolipin headgroup. J. Phys. Chem. A 114, 4375–4387 10.1021/jp9110019 [DOI] [PubMed] [Google Scholar]
- 27.Planas-Iglesias J., Dwarakanath H., Mohammadyani D., Yanamala N., Kagan V.E.E.E. and Klein-Seetharaman J. (2015) Cardiolipin interactions with proteins. Biophys. J. 109, 1282–1294 10.1016/j.bpj.2015.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Houtkooper R.H., Rodenburg R.J., Thiels C., van Lenthe H., Stet F., Poll-The B.T. et al. (2009) Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal. Biochem. 387, 230–237 10.1016/j.ab.2009.01.032 [DOI] [PubMed] [Google Scholar]
- 29.Gonzalvez F., D'Aurelio M., Boutant M., Moustapha A., Puech J.-P., Landes T. et al. (2013) Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta 1832, 1194–1206 10.1016/j.bbadis.2013.03.005 [DOI] [PubMed] [Google Scholar]
- 30.Oemer G., Koch J., Wohlfarter Y., Alam M.T., Lackner K., Sailer S. et al. (2020) Phospholipid acyl chain diversity controls the tissue-specific assembly of mitochondrial cardiolipins. Cell Rep. 30, 4281–4291.e4 10.1016/j.celrep.2020.02.115 [DOI] [PubMed] [Google Scholar]
- 31.Schlame M., Ren M., Xu Y., Greenberg M.L. and Haller I. (2005) Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 138, 38–49 10.1016/j.chemphyslip.2005.08.002 [DOI] [PubMed] [Google Scholar]
- 32.Kiebish M.A., Han X., Cheng H., Chuang J.H. and Seyfried T.N. (2008) Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: lipidomic evidence supporting the Warburg theory of cancer. J. Lipid Res. 49, 2545–2556 10.1194/jlr.M800319-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tyurina Y.Y., Poloyac S.M., Tyurin V.A., Kapralov A.A., Jiang J., Anthonymuthu T.S. et al. (2014) A mitochondrial pathway for biosynthesis of lipid mediators. Nat. Chem. 6, 542–552 10.1038/nchem.1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sparvero L.J., Amoscato A.A., Fink A.B., Anthonymuthu T., New L.A., Kochanek P.M. et al. (2016) Imaging mass spectrometry reveals loss of polyunsaturated cardiolipins in the cortical contusion, hippocampus, and thalamus after traumatic brain injury. J. Neurochem. 139, 659–675 10.1111/jnc.13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlame M. and Greenberg M.L. (2017) Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta 1862, 3–7 10.1016/j.bbalip.2016.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beranek A., Rechberger G., Knauer H., Wolinski H., Kohlwein S.D. and Leber R. (2009) Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J. Biol. Chem. 284, 11572–11578 10.1074/jbc.M805511200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Y., Anjaneyulu M., Donelian A., Yu W., Greenberg M.L., Ren M. et al. (2019) Assembly of the complexes of oxidative phosphorylation triggers the remodeling of cardiolipin. Proc. Natl Acad. Sci. U.S.A. 116, 11235–11240 10.1073/pnas.1900890116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zachman D.K., Chicco A.J., McCune S.A., Murphy R.C., Moore R.L. and Sparagna G.C. (2010) The role of calcium-independent phospholipase A2 in cardiolipin remodeling in the spontaneously hypertensive heart failure rat heart. J. Lipid Res. 51, 525–534 10.1194/jlr.M000646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buckland A.G., Kinkaid A.R. and Wilton D.C. (1998) Cardiolipin hydrolysis by human phospholipases A2 the multiple enzymatic activities of human cytosolic phospholipase A2. Biochim. Biophys. Acta 1390, 65–72 10.1016/S0005-2760(97)00170-7 [DOI] [PubMed] [Google Scholar]
- 40.Hsu Y.H., Dumlao D.S., Cao J. and Dennis E.A. (2013) Assessing phospholipase A2 activity toward cardiolipin by mass spectrometry. PLoS One 8, e59267 10.1371/journal.pone.0059267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu G.Y., Ho Moon S., Jenkins C.M., Li M., Sims H.F., Guan S. et al. (2017) The phospholipase iPLA2 is a major mediator releasing oxidized aliphatic chains from cardiolipin, integrating mitochondrial bioenergetics and signaling. J. Biol. Chem. 292, 10672–10684 10.1074/jbc.M117.783068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Y., Malhotra A., Ren M., Schlame M. and Procedures E. (2006) The enzymatic function of tafazzin. J. Biol. Chem. 281, 39217–39224 10.1074/jbc.M606100200 [DOI] [PubMed] [Google Scholar]
- 43.Abe M., Hasegawa Y., Oku M., Sawada Y., Tanaka E., Sakai Y. et al. (2016) Mechanism for remodeling of the acyl chain composition of cardiolipin catalyzed by Saccharomyces cerevisiae tafazzin. J. Biol. Chem. 291, 15491–15502 10.1074/jbc.M116.718510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlame M., Xu Y. and Ren M. (2017) The basis for acyl specificity in the tafazzin reaction. J. Biol. Chem. 292, 5499–5506 10.1074/jbc.M116.769182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao J., Liu Y., Lockwood J., Burn P. and Shi Y. (2004) A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 279, 31727–31734 10.1074/jbc.M402930200 [DOI] [PubMed] [Google Scholar]
- 46.Taylor W.A. and Hatch G.M. (2003) Purification and characterization of monolysocardiolipin acyltransferase from pig liver mitochondria. J. Biol. Chem. 278, 12716–12721 10.1074/jbc.M210329200 [DOI] [PubMed] [Google Scholar]
- 47.Miklas J.W., Clark E., Levy S., Detraux D., Leonard A., Beussman K. et al. (2019) TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat. Commun. 10, 4671 10.1038/s41467-019-12482-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia C., Fu Z., Battaile K.P. and Kim J.J.P. (2019) Crystal structure of human mitochondrial trifunctional protein, a fatty acid β-oxidation metabolon. Proc. Natl Acad. Sci. U.S.A. 116, 6069–6074 10.1073/pnas.1816317116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor W.A., Mejia E.M., Mitchell R.W., Choy P.C., Sparagna G.C. and Hatch G.M. (2012) Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS One 7, e48628 10.1371/journal.pone.0048628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren M., Xu Y., Erdjument-Bromage H., Donelian A., Phoon C.K.L., Terada N. et al. (2019) Extramitochondrial cardiolipin suggests a novel function of mitochondria in spermatogenesis. J. Cell Biol. 218, 1491–1502 10.1083/jcb.201808131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Y., Phoon C.K.L., Berno B., D'Souza K., Hoedt E., Zhang G. et al. (2016) Loss of protein association causes cardiolipin degradation in Barth syndrome. Nat. Chem. Biol. 12, 641–647 10.1038/nchembio.2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vreken P., Valianpour F., Nijtmans L.G., Grivell L.A., Plecko B., Wanders R.J. et al. (2000) Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 279, 378–382 10.1006/bbrc.2000.3952 [DOI] [PubMed] [Google Scholar]
- 53.Schlame M., Towbin J.A., Heerdt P.M., Jehle R., DiMauro S. and Blanck T.J.J. (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 51, 634–637 10.1002/ana.10176 [DOI] [PubMed] [Google Scholar]
- 54.Ji J., Baart S., Vikulina A.S., Clark R.S.B., Anthonymuthu T.S., Tyurin V.A. et al. (2015) Deciphering of mitochondrial cardiolipin oxidative signaling in cerebral ischemia-reperfusion. J. Cereb. Blood Flow Metab. 35, 319–328 10.1038/jcbfm.2014.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bione S., D'Adamo P., Maestrini E., Gedeon A.K., Bolhuis P.A. and Toniolo D. (1996) A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12, 385–389 10.1038/ng0496-385 [DOI] [PubMed] [Google Scholar]
- 56.Schlame M. and Ren M. (2006) Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 580, 5450–5455 10.1016/j.febslet.2006.07.022 [DOI] [PubMed] [Google Scholar]
- 57.Xu Y., Sutachan J.J., Plesken H., Kelley R.I. and Schlame M. (2005) Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab. Invest. 85, 823–830 10.1038/labinvest.3700274 [DOI] [PubMed] [Google Scholar]
- 58.Christodoulou J., McInnes R.R., Jay V., Wilson G., Becker L.E., Lehotay D.C. et al. (1994) Barth syndrome: clinical observations and genetic linkage studies. Am. J. Med. Genet. 50, 255–264 10.1002/ajmg.1320500309 [DOI] [PubMed] [Google Scholar]
- 59.Barth P.G., Van den Bogert C., Bolhuis P.A., Scholte H.R., van Gennip A.H., Schutgens R.B.H. et al. (1996) X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): Respiratory-chain abnormalities in cultured fibroblasts. J. Inherit. Metab. Dis. 19, 157–160 10.1007/BF01799418 [DOI] [PubMed] [Google Scholar]
- 60.Wang G., McCain M.L., Yang L., He A., Pasqualini F.S., Agarwal A. et al. (2014) Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 20, 616–623 10.1038/nm.3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McKenzie M., Lazarou M., Thorburn D.R. and Ryan M.T. (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469 10.1016/j.jmb.2006.06.057 [DOI] [PubMed] [Google Scholar]
- 62.Acehan D., Xu Y., Stokes D.L. and Schlame M. (2007) Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Invest. 87, 40–48 10.1038/labinvest.3700480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Acehan D., Malhotra A., Xu Y., Ren M., Stokes D.L. and Schlame M. (2011) Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys. J. 100, 2184–2192 10.1016/j.bpj.2011.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van Werkhoven M.A., Thorburn D.R., Gedeon A.K. and Pitt J.J. (2006) Monolysocardiolipin in cultured fibroblasts is a sensitive and specific marker for Barth syndrome. J. Lipid Res. 47, 2346–2351 10.1194/jlr.D600024-JLR200 [DOI] [PubMed] [Google Scholar]
- 65.Bowron A., Honeychurch J., Williams M., Tsai-Goodman B., Clayton N., Jones L. et al. (2015) Barth syndrome without tetralinoleoyl cardiolipin deficiency: a possible ameliorated phenotype. J. Inherit. Metab. Dis. 38, 279–286 10.1007/s10545-014-9747-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kulik W., Van Lenthe H., Stet F.S., Houtkooper R.H., Kemp H., Stone J.E. et al. (2008) Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome. Clin. Chem. 54, 371–378 10.1373/clinchem.2007.095711 [DOI] [PubMed] [Google Scholar]
- 67.Fry M. and Green D.E. (1981) Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J. Biol. Chem. 256, 1874–1880 PMID: [PubMed] [Google Scholar]
- 68.Sharpley M.S., Shannon R.J., Draghi F. and Hirst J. (2006) Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry 45, 241–248 10.1021/bi051809x [DOI] [PubMed] [Google Scholar]
- 69.Pfeiffer K., Gohil V., Stuart R.A., Hunte C., Brandt U., Greenberg M.L. et al. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 10.1074/jbc.M308366200 [DOI] [PubMed] [Google Scholar]
- 70.Dale M.P. and Robinson N.C. (1988) Synthesis of cardiolipin derivatives with protection of the free hydroxyl: its application to the study of cardiolipin stimulation of cytochrome c oxidase. Biochemistry 27, 8270–8275 10.1021/bi00421a042 [DOI] [PubMed] [Google Scholar]
- 71.Laage S., Tao Y. and McDermott A.E. (2015) Cardiolipin interaction with subunit c of ATP synthase: solid-state NMR characterization. Biochim. Biophys. Acta 1848, 260–265 10.1016/j.bbamem.2014.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou M., Morgner N., Barrera N.P., Politis A., Isaacson S.C., Matak-Vinkovic D. et al. (2011) Mass spectrometry of intact V-Type ATPases reveals bound lipids and the effects of nucleotide binding. Science 334, 380–385 10.1126/science.1210148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eble K.S., Coleman W.B., Hantgan R.R. and Cunningham C.C. (1990) Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J. Biol. Chem. 265, 19434–19440 PMID: 2147180 [PubMed] [Google Scholar]
- 74.Pebay-Peyroula E., Dahout-Gonzalez C., Kahn R., Trézéguet V., Lauquin G.J.-M. and Brandolin G. (2003) Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426, 39–44 10.1038/nature02056 [DOI] [PubMed] [Google Scholar]
- 75.Robinson N.C., Zborowski J. and Talbert L.H. (1990) Cardiolipin-depleted bovine heart cytochrome c oxidase: binding stoichiometry and affinity for cardiolipin derivatives. Biochemistry 29, 8962–8969 10.1021/bi00490a012 [DOI] [PubMed] [Google Scholar]
- 76.Hoffmann B., Stöckl A., Schlame M., Beyer K. and Klingenberg M. (1994) The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. J. Biol. Chem. 269, 1940–1944 PMID: [PubMed] [Google Scholar]
- 77.Liu J., Durrant D., Yang H.S., He Y., Whitby F.G., Myszka D.G. et al. (2005) The interaction between tBid and cardiolipin or monolysocardiolipin. Biochem. Biophys. Res. Commun. 330, 865–870 10.1016/j.bbrc.2005.03.048 [DOI] [PubMed] [Google Scholar]
- 78.Esposti M D., Cristea I.M., Gaskell S.J., Nakao Y. and Dive C. (2003) Proapoptotic Bid binds to monolysocardiolipin, a new molecular connection between mitochondrial membranes and cell death. Cell Death Differ. 10, 1300–1309 10.1038/sj.cdd.4401306 [DOI] [PubMed] [Google Scholar]
- 79.Pennington E.R., Sullivan E.M., Fix A., Dadoo S., Zeczycki T.N., DeSantis A. et al. (2018) Proteolipid domains form in biomimetic and cardiac mitochondrial vesicles and are regulated by cardiolipin concentration but not monolyso-cardiolipin. J. Biol. Chem. 293, 15933–15946 10.1074/jbc.RA118.004948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kantari C. and Walczak H. (2011) Caspase-8 and Bid: caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta 1813, 558–563 10.1016/j.bbamcr.2011.01.026 [DOI] [PubMed] [Google Scholar]
- 81.Lutter M., Fang M., Luo X., Nishijima M., Xie X. and Wang X. (2000) Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat. Cell Biol. 2, 754–756 10.1038/35036395 [DOI] [PubMed] [Google Scholar]
- 82.Gong X.-M., Choi J., Franzin C.M., Zhai D., Reed J.C. and Marassi F.M. (2004) Conformation of membrane-associated proapoptotic tBid. J. Biol. Chem. 279, 28954–28960 10.1074/jbc.M403490200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oh K.J., Barbuto S., Meyer N., Kim R.-S., Collier R.J. and Korsmeyer S.J. (2005) Conformational changes in BID, a pro-apoptotic BCL-2 family member, upon membrane binding. J. Biol. Chem. 280, 753–767 10.1074/jbc.M405428200 [DOI] [PubMed] [Google Scholar]
- 84.Wang Y. and Tjandra N. (2013) Structural insights of tBid, the caspase-8-activated Bid, and its BH 3 domain. J. Biol. Chem. 288, 35840–35851 10.1074/jbc.M113.503680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Esmann M., Powell G.L. and Marsh D. (1988) Spin label studies on the selectivity of lipid-protein interaction of cardiolipin analogues with the Na+/K+-ATPase. Biochim. Biophys. Acta 941, 287–292 10.1016/0005-2736(88)90190-3 [DOI] [PubMed] [Google Scholar]
- 86.Laskowski M., Augustynek B., Kulawiak B., Koprowski P., Bednarczyk P., Jarmuszkiewicz W. et al. (2016) What do we not know about mitochondrial potassium channels? Biochim. Biophys. Acta 1857, 1247–1257 10.1016/j.bbabio.2016.03.007 [DOI] [PubMed] [Google Scholar]
- 87.Paumard P., Vaillier J., Coulary B., Schaeffer J., Soubannier V., Mueller D.M. et al. (2002) The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 21, 221–230 10.1093/emboj/21.3.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Strauss M., Hofhaus G., Schröder R.R. and Kühlbrandt W. (2008) Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27, 1154–1160 10.1038/emboj.2008.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Davies K.M., Anselmi C., Wittig I., Faraldo-Gómez J.D. and Kühlbrandt W. (2012) Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl Acad. Sci. U.S.A. 108, 14121–14126 10.1073/pnas.1103621108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Anselmi C., Davies K.M. and Faraldo-Gómez J.D. (2018) Mitochondrial ATP synthase dimers spontaneously associate due to a long-range membrane-induced force. J. Gen. Physiol. 150, 763–770 10.1085/jgp.201812033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Duncan A.L., Robinson A.J. and Walker J.E. (2016) Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Natl Acad. Sci. U.S.A. 113, 8687–8692 10.1073/pnas.1608396113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weber T.A., Koob S., Heide H., Wittig I., Head B., van der Bliek A. et al. (2013) APOOL is a cardiolipin-binding constituent of the Mitofilin/MINOS protein complex determining cristae morphology in mammalian mitochondria. PLoS One 8, e63683 10.1371/journal.pone.0063683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nury H., Dahout-Gonzalez C., Trézéguet V., Lauquin G., Brandolin G. and Pebay-Peyroula E. (2005) Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Lett. 579, 6031–6036 10.1016/j.febslet.2005.09.061 [DOI] [PubMed] [Google Scholar]
- 94.Palsdottir H., Lojero C.G., Trumpower B.L. and Hunte C. (2003) Structure of the yeast cytochrome bc1 complex with a hydroxyquinone anion Qo site inhibitor bound. J. Biol. Chem. 278, 31303–31311 10.1074/jbc.M302195200 [DOI] [PubMed] [Google Scholar]
- 95.Hirata K., Shinzawa-Itoh K., Yano N., Takemura S., Kato K., Hatanaka M. et al. (2014) Determination of damage-free crystal structure of an X-ray-sensitive protein using an XFEL. Nat. Methods 11, 734–736 10.1038/nmeth.2962 [DOI] [PubMed] [Google Scholar]
- 96.Kühlbrandt W. (2014) The resolution revolution. Science 343, 1443–1444 10.1126/science.1251652 [DOI] [PubMed] [Google Scholar]
- 97.Rathore S., Berndtsson J., Marin-Buera L., Conrad J., Carroni M., Brzezinski P. et al. (2019) Cryo-EM structure of the yeast respiratory supercomplex. Nat. Struct. Mol. Biol. 26, 50–57 10.1038/s41594-018-0169-7 [DOI] [PubMed] [Google Scholar]
- 98.Hartley A.M., Lukoyanova N., Zhang Y., Cabrera-Orefice A., Arnold S., Meunier B. et al. (2019) Structure of yeast cytochrome c oxidase in a supercomplex with cytochrome bc1. Nat. Struct. Mol. Biol. 26, 78–83 10.1038/s41594-018-0172-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fiedorczuk K., Letts J.A., Degliesposti G., Kaszuba K., Skehel M. and Sazanov L.A. (2016) Atomic structure of the entire mammalian mitochondrial complex I. Nature 538, 406–410 10.1038/nature19794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Agip A.N.A., Blaza J.N., Bridges H.R., Viscomi C., Rawson S., Muench S.P. et al. (2018) Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 25, 1–9 10.1038/s41594-017-0017-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mühleip A., Mccomas S.E. and Amunts A. (2019) Structure of a mitochondrial ATP synthase with bound native cardiolipin. eLife 8, e51179 10.7554/eLife.51179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Davies K.M., Blum T.B. and Kühlbrandt W. (2018) Conserved in situ arrangement of complex I and III2 in mitochondrial respiratory chain supercomplexes of mammals, yeast, and plants. Proc. Natl Acad. Sci. U.S.A. 115, 3024–3029 10.1073/pnas.1720702115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chorev D.S., Baker L.A., Wu D., Beilsten-Edmands V., Rouse S.L., Zeev-Ben-Mordehai T. et al. (2018) Protein assemblies ejected directly from native membranes yield complexes for mass spectrometry. Science 362, 829–834 10.1126/science.aau0976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hirst J., Kunji E.R.S. and Walker J.E. (2019) Comment on “Protein assemblies ejected directly from native membranes yield complexes for mass spectrometry.”. Science 366, eaaw9830 10.1126/science.aaw9830 [DOI] [PubMed] [Google Scholar]
- 105.Chorev D.S. and Robinson C V. (2019) Response to comment on “Protein assemblies ejected directly from native membranes yield complexes for mass spectrometry.”. Science 366, eaax3102 10.1126/science.aax3102 [DOI] [PubMed] [Google Scholar]
- 106.Robinson C V. (2019) Mass spectrometry: From plasma proteins to mitochondrial membranes. Proc. Natl Acad. Sci. U.S.A. 116, 2814–2820 10.1073/pnas.1820450116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Enkavi G., Javanainen M., Kulig W., Róg T. and Vattulainen I. (2019) Multiscale simulations of biological membranes: the challenge to understand biological phenomena in a living substance. Chem. Rev. 119, 5607–5774 10.1021/acs.chemrev.8b00538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Duncan A.L., Corey R.A. and Sansom M.S.P. (2020) Defining how multiple lipid species interact with inward rectifier potassium (Kir2) channels. Proc. Natl Acad. Sci. U.S.A. 117, 7803–7813 10.1073/pnas.1918387117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hedger G. and Sansom M.S.P. (2016) Lipid interaction sites on channels, transporters and receptors: recent insights from molecular dynamics simulations. Biochim. Biophys. Acta 1858, 2390–2400 10.1016/j.bbamem.2016.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Corey R.A., Vickery O.N., Sansom M.S.P. and Stansfeld P.J. (2019) Insights into membrane protein–lipid interactions from free energy calculations. J. Chem. Theory Comput. 15, 5727–5736 10.1021/acs.jctc.9b00548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jussupow A., Di Luca A. and Kaila V.R.I. (2019) How cardiolipin modulates the dynamics of respiratory complex I. Sci. Adv. 5, eaav1850 10.1126/sciadv.aav1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Arnarez C., Mazat J., Elezgaray J., Marrink S.J. and Periole X. (2013) Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J. Am. Chem. Soc. 135, 3112–3120 10.1021/ja310577u [DOI] [PubMed] [Google Scholar]
- 113.Arnarez C., Marrink S.-J.J. and Periole X. (2013) Identification of cardiolipin binding sites on cytochrome c oxidase at the entrance of proton channels. Sci. Rep. 3, 1263 10.1038/srep01263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hedger G., Rouse S.L., Domański J., Chavent M., Koldsø H. and Sansom M.S.P.P. (2016) Lipid-loving ANTs: molecular simulations of cardiolipin interactions and the organization of the adenine nucleotide translocase in model mitochondrial membranes. Biochemistry 55, 6238–6249 10.1021/acs.biochem.6b00751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duncan A.L., Ruprecht J.J., Kunji E.R.S. and Robinson A.J. (2018) Cardiolipin dynamics and binding to conserved residues in the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta 1860, 1035–1045 10.1016/j.bbamem.2018.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kalli A.C., Sansom M.S.P. and Reithmeier R.A.F. (2015) Molecular dynamics simulations of the bacterial UraA H+-Uracil symporter in lipid bilayers reveal a closed state and a selective interaction with cardiolipin. PLoS Comput. Biol. 11, e1004123 10.1371/journal.pcbi.1004123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gupta K., Donlan J.A.C., Hopper J.T.S., Uzdavinys P., Landreh M., Struwe W.B. et al. (2017) The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 541, 421–424 10.1038/nature20820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Corey R.A., Pyle E., Allen W.J., Watkins D.W., Casiraghi M., Miroux B. et al. (2018) Specific cardiolipin–SecY interactions are required for proton-motive force stimulation of protein secretion. Proc. Natl Acad. Sci. U.S.A. 115, 7967–7972 10.1073/pnas.1721536115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marrink S.J. and Tieleman D.P. (2013) Perspective on the Martini model. Chem. Soc. Rev. 42, 6801–6822 10.1039/c3cs60093a [DOI] [PubMed] [Google Scholar]
- 120.Chavent M., Duncan A.L. and Sansom M.S.P. (2016) Molecular dynamics simulations of membrane proteins and their interactions : from nanoscale to mesoscale. Curr. Opin. Struct. Biol. 40, 8–16 10.1016/j.sbi.2016.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arnarez C., Marrink S.J. and Periole X. (2016) Molecular mechanism of cardiolipin-mediated assembly of respiratory chain supercomplexes. Chem. Sci. 7, 4435–4443 10.1039/C5SC04664E [DOI] [PMC free article] [PubMed] [Google Scholar]