

Abstract
General anesthesia (GA) can produce analgesia (loss of pain) independent of inducing loss of consciousness, but the underlying mechanisms remain unclear. We hypothesized that GA suppresses pain in part by activating supraspinal analgesic circuits. We discovered a distinct population of GABAergic neurons activated by GA in the mouse central amygdala (CeAGA neurons). In vivo calcium imaging revealed that different GA drugs activate a shared ensemble of CeAGA neurons. CeAGA neurons also possess basal activity that mostly reflect animals’ internal state rather than external stimuli. Optogenetic activation of CeAGA potently suppressed both pain-elicited reflexive and self-recuperating behaviors across sensory modalities, and abolished neuropathic pain-induced mechanical (hyper-)sensitivity. Conversely, inhibition of CeAGA activity exacerbated pain, produced strong aversion, and cancelled the analgesic effect of low-dose ketamine. CeAGA neurons have widespread inhibitory projections to numerous affective pain-processing centers. Our study points to CeAGA as a potential powerful therapeutic target for alleviating chronic pain.
INTRODUCTION
One of the main effects of general anesthesia (GA) is analgesia, or loss of pain perception, which is critical for making surgery and other invasive medical procedures more humane. While GA is well known to induce the loss of consciousness, it is often assumed that analgesia is a consequence of the unconscious brain that cannot perceive pain. However, numerous cases of intraoperative awareness in which patients were aware of surgeons’ conversations and other events in the operating room, but nevertheless did not feel pain suggest that there could be a specific analgesic pathway that is independent of the loss of consciousness effects of GA drugs1,2. Furthermore, ketamine, a commonly used GA drugs, at low sub-anesthetic dose, is used by anesthesiologists as an analgesic without inducing unconsciousness, and with minimal physiological impact 3–5. Previously, it was thought that low-dose GA drugs induce analgesia through blocking peripheral nociceptive transmission in the dorsal horn of the spinal cord; however, patients treated in this manner can still perceive stimuli in the absence of pain, which is not consistent with this idea3,6. These observations suggest that low-dose GA analgesia may act at levels above the spinal cord (supraspinal centers) that dissociate pain perception from the detection of noxious sensory stimuli. The locations of such centers and identities of such neurons remain unknown. We hypothesized that GA-induced analgesia involves activation of a specific central pain-suppression circuit. Here we tested our hypothesis and discovered that GA indeed activates a distinct population of GABAergic neurons in the central amygdala. We further discovered that these cells have profound analgesic effects that suppress both the sensory and affective aspects of pain processing in both naïve animals and in models of acute and chronic neuropathic pain and are required for the pain-relieving effect of low-dose ketamine.
RESULTS
Identification of CeAGA neurons that are activated by isoflurane and ketamine anesthetics
To begin to test our hypothesis, we used the immediate early gene Fos (encoded by the c-fos gene) as a marker for recently activated neurons and searched for neurons that remained strongly Fos+ after exposure to 2 hours of isoflurane/oxygen GA (exposure to oxygen alone was used as controls). This experiment revealed three clusters of Fos+ neurons under isoflurane but not oxygen: one in the central amygdala (CeA), one in the oval division of the bed nucleus of the stria terminalis (ovBNST), and one in the supraoptic nucleus (SON) (Fig. 1a and Extended Data 1a,b,c). We recently showed that the GA-activated SON neurons promote slow-wave sleep and extend GA duration, i.e this cluster of cells are related to the sedative aspects of GA7. The functions of GA-activated ovBNST neurons are not yet known. Here we focus on GA-activated Fos+ cells in the CeA (Fig. 1a).
Figure 1 |. Ensembles of neurons in the central amygdala (CeA) are activated by general anesthesia (GA).
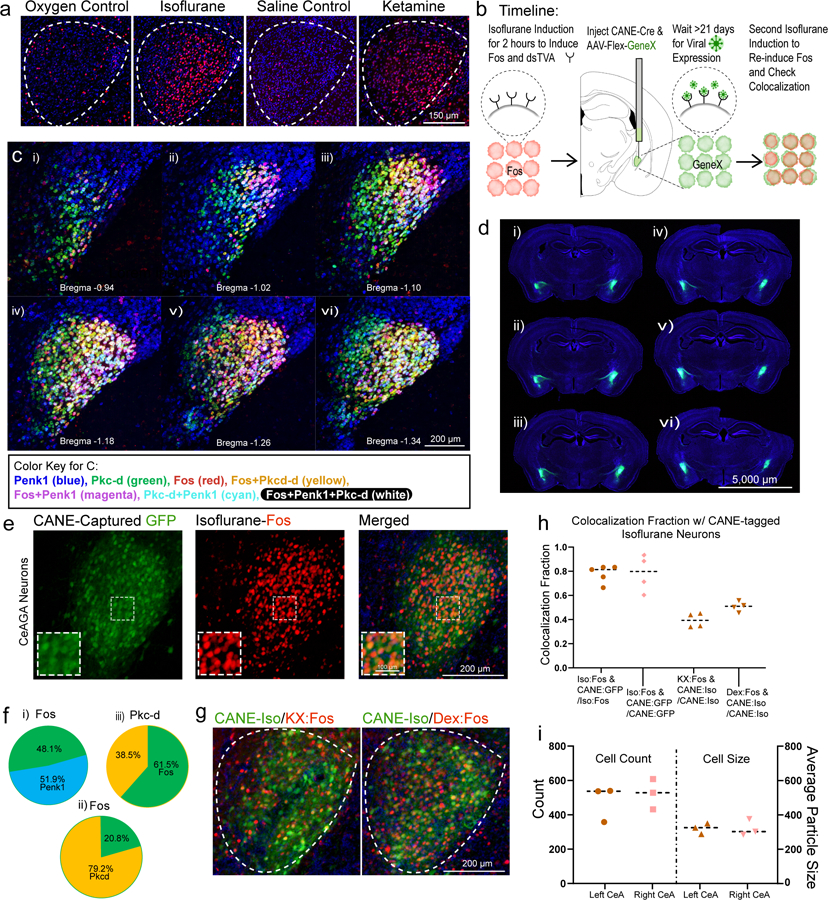
a, Representative images of Fos+ neurons in the CeA from exposure to oxygen control, isoflurane, saline control, and ketamine/xylazine injection. Repeated experiments for n=3 biologically independent samples.
b, Schematic of CANE capturing of Fos+ CeAGA neurons followed by a second exposure to isoflurane GA to re-induce Fos.
c, Representative images of three-color hybridization chain reaction experiments examining the expression of enkephalin (Penk1) and protein kinase C-delta (Pkc-d) with Fos+ CeAGA neurons (induced by isoflurane). Bregma, −0.94 mm to −1.34 mm. Repeated experiments for n=3 biologically independent samples.
d, Six sequential coronal brain sections from one representative mouse containing bilateral CeAGA captured neurons (CANEISO-GFP). Repeated experiments for n=5 biologically independent samples.
e, Representative image of captured CeAGA neurons (green – GFP) from first isoflurane exposure, Fos+ activation from second isoflurane exposure (red) and their merged image showing colocalization (orange cells). Repeated experiments for n=5 biologically independent samples.
f, (i-ii), Percentage of Penk1 and Pkc-d overlap over Fos+ CeAGA neurons, and (iii) Fos+ CeAGA neurons over total Pkc-d neurons (n=3 biologically independent samples).
g, Representative images of CANEGFP captured CeAGA neurons versus Fos+ neurons in the CeA induced by ketamine/xylazine and dexmedetomidine. Repeated experiments for n=4 biologically independent samples.
h, Quantification of the fraction of Fos+ neurons induced by isoflurane (0.776 ± 0.065), ketamine/xylazine (0.390 ± 0.051), and dexmedetomidine (0.505 ± 0.036) over the total Fos+ CeAGA neurons (induced by isoflurane) and CANEISO-GFP over isoflurane-induced Fos+ neurons (0.780 ± 0.132) (n=4 biologically independent samples).
i, Quantification of averaged Fos+ CeAGA neurons in the left (486.33 ± 77.31) and right CeA (527 ± 65.82) and their respective average particle size (321.62 ± 23.26 left, 321.86 ± 35.75 right) (n=3 biologically independent samples). Numbers represent the sum of cell counts from six serial 80µm sections containing CeA (but only a single focal plane (6µm) per section was counted).
Notably, similar to isoflurane, intra-peritoneal administration of ketamine/xylazine (but not saline control) also induced Fos+ expression in a subset of CeA cells (Fig. 1a), hereafter referred to as CeAGA neurons. This observation initially came as a surprise, because CeA is well known to be activated by painful stimuli and plays important functions in processing fear and pain 8–12. Furthermore, CeA is known to contain molecularly and functionally heterogeneous populations of neurons8,9,13. We therefore first characterized CeAGA cells using several molecular markers. Exposing vGat-Cre::Rosa-stop-GFP mice, in which all GABAergic cells are labeled with GFP, to isoflurane GA, revealed that CeAGA neurons are all GABAergic cells (Extended Data 1d). Using two-color fluorescent in situ hybridization or two-color immunofluorescence, we found that CeAGA neurons largely do not overlap with neurons expressing somatostatin (Sst), prodynorphin (Pdyn), Neurotensin (NTS), or CGRP-receptor (Calcrl) (Extended Data 1e–i). While we did not observe any candidate genes that are specifically expressed by all CeAGA neurons, we discovered that some of these cells co-express pre-enkephalin (Penk, here we use the commonly adopted name Penk1) or protein kinase c delta (Prkcd, here we use the commonly adopted name Pkc-d); but only a subset of the total Penk1+ or Pkc-d+ neurons are Fos+ CeAGA. Specifically, using a three-color HCR in situ hybridization method10, we found that Penk1 is widely expressed by cells both in CeA and in nearby striatum and extended amygdala. Pkc-d+ neurons located in the anterior and medial part of the CeA do not overlap with CeAGA neurons, while in the posterior part of the CeA, they partially overlap with CeAGA neurons (Fig. 1c). Collectively, of the total CeAGA neurons, 51.9±7.8% express Penk1, while 79.2±12.6% express Pkc-d (Fig. 1f (i–ii)). Of the total Pkc-d+ cells, 61.5±14.5% are isoflurane-induced Fos+ cells (Fig. 1f (iii)). The partial overlap of Penk1+ or Pkc-d+ cells with CeAGA indicated that GA activated a heterogeneous population of CeA cells.
Selective capturing of isoflurane-activated CeAGA neurons using CANE revealed a shared ensemble of GA-activated neurons in CeA
Since we did not identify any molecular marker genes that can be used to specifically label all CeAGA cells, we sought to label and manipulate them using a Fos based viral-genetic method (Capturing Activated Neuronal Ensembles, or CANE)11,12. CANE uses the FosTVA knock-in mice and engineered viruses (CANE-lentivirus, or CANE-LV) to express desired transgenes in Fos+ cells. The selectivity and efficiency of CANE capturing CeAGA was determined through a two-bout anesthesia paradigm (Fig. 1b). First, we co-injected CANE-LV-Cre and AAV-Flex-GFP into the CeA at 2 hours after isoflurane GA induction to express GFP in CeAGA neurons (Fig. 1b,d,e). Second, 3 weeks later, the same animals were re-exposed to isoflurane GA and sacrificed for Fos immunostaining (Fig. 1e). Note that previous studies revealed certain lateralization of CeA14–16. However, we found that isoflurane GA activated similar number of neurons with similar cell body sizes in both left and right CeA (Fig. 1i), and we could use CANE to label CeAGA neurons equally well on both sides (one representative example mouse is shown in Fig. 1d). Furthermore, serial sections through the forebrain revealed selective capturing CeAGA cells without ectopic labeling in the nearby basolateral amygdala or along the injection path (Fig. 1d). On average, 71.0±18.3% of the total Fos+ cells were CANEISO-GFP+, and 77.60±6.5% CANEISO-GFP+ cells re-expressed Fos from the second exposure to isoflurane (n=5 mice) (Fig. 1h). This result indicated that CANE is sufficiently specific and efficient at capturing CeAGA neurons.
Next, we asked whether CANE-labeled isoflurane-activated neurons could induce Fos by other anesthetics. We exposed mice labeled with CANEISO-GFP to either ketamine/xylazine (K/X) (n=4) or dexmedetomidine (Dex) (n=4) anesthesia. About 39.0±7.1% and 51.0±8.1% of CANEISO-GFP+ cells re-expressed Fos in response to K/X or Dex, respectively (Fig. 1g,h), suggesting there exist a shared ensemble of CeA neurons activated by different GA drugs. Since isoflurane reliably induces Fos+ in the largest population of CeA neurons, we therefore in all subsequent experiments used CANE in conjunction with isoflurane anesthesia to capture CeAGA neurons.
In vivo imaging of CeAGA neurons in responses to isoflurane or ketamine anesthesia
To characterize the in vivo activity of CeAGA neurons during GA induction and maintenance in freely moving mice, we performed in vivo calcium imaging experiments. We used CANE to express the calcium indicator GCaMP6m in CeAGA neurons by co-injecting CANE-LV-Cre and AAV2/1-CAG-Flex-GCaMP6m into the CeA of FosTVA mice after 2 hours of isoflurane GA (Fig. 2a, Extended Data 2a). Subsequently, we recorded the calcium dynamics of these cells when we re-exposed mice to isoflurane using a gradient refractive index (GRIN) lens coupled to a miniaturized integrated fluorescence microscope and processed using the MIN1PIPE method17 (Fig. 2a, left panel). Importantly, re-exposure of isoflurane activated many CANE-GCaMP6m captured CeAGA neurons (Fig. 2b and Supplementary Video 1). To characterize the activity patterns, we sorted neurons based on the ratio of an individual neuron’s mean fluorescence during isoflurane exposure (0 – 20 min) to its baseline (awake state, −4 – 0 min) (583 neurons, Fig. 2c). This analysis revealed that during isoflurane GA, 89.7% (523/583) of imaged neurons exhibited increased fluorescence (iso.-active neurons, cyan rectangle in Fig. 2c), while a much smaller minority (10.3%, 60/583), showed decreased fluorescence (iso.-suppressed neurons, yellow rectangle in Fig. 2c). The small number of isoflurane-suppressed cells might be the result of non-specific labeling due to background Fos expression at the time of CANE capturing.
Figure 2 |. Activity patterns of CANEISO-GCaMP6m captured CeAGA neurons during isoflurane and ketamine GA.
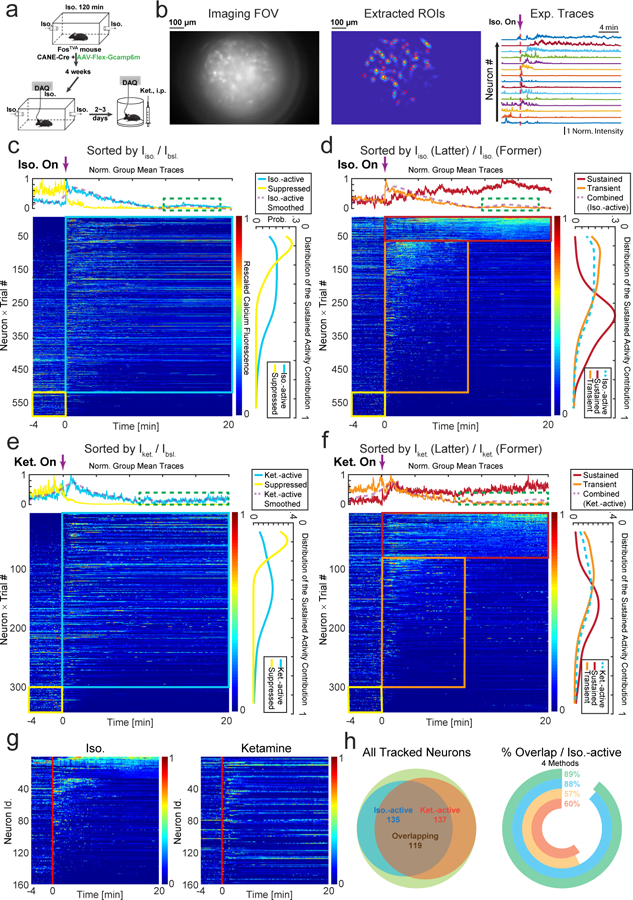
a, Calcium imaging recording of CANEISO-GCaMP6m captured CeAGA neurons during isoflurane or ketamine induced general anesthesia (GA).
b, Left, an example frame from the raw calcium imaging video. Middle, extracted regions of interest (ROIs) footprints superimposed on the max intensity projection. Right, normalized calcium fluorescence traces of the neurons during isoflurane induced GA. Norm. Intensity, normalized calcium signals were rescaled to 0–1.
c, Activity patterns of CANEISO-GCaMP6m captured CeAGA neurons sorted by the ratio of individual neuron’s mean activity during isoflurane exposure (0 – 20 min) to its baseline activity (awake state, −4 – 0 min). Cyan and yellow rectangle indicates iso.- active, and iso.-suppressed neurons separately. Top panel, the average fluorescence traces of each group of neurons. Right panel, distribution of contributions to the sustained activity from each group of neurons. Green dash rectangle indicates a subgroup of iso.-active neurons remaining activated under anesthesia.
d, Two subpopulations of iso.-active neurons sorted by the ratio of individual neuron’s mean activity during the last 10 minutes (10 – 20 min) of isoflurane exposure to its first 10 minutes (0 – 10 min). Red and orange rectangle indicates isoflurane-sustained and isoflurane-transient neurons separately.
e, Activity patterns of CANEISO-GCaMP6m captured CeAGA neurons in responses to ketamine sorted by the ratio of individual neuron’s mean activity after ketamine injection to its baseline activity. Cyan and yellow rectangle indicates ketamine-active, and ketamine-suppressed neurons, respectively.
f, Two subpopulations of ketamine-active neurons sorted by the ratio of individual neuron’s mean activity during the last 10 minutes of ketamine to its first 10 minutes. Red and orange rectangle indicates ketamine-sustained and ketamine-transient neurons separately. Arrows, time when isoflurane or ketamine was administered.
g, Calcium activity of CANEISO-GCaMP6m captured CeAGA neurons tracked across isoflurane and ketamine sessions (9 mice × 1trial). Neurons are aligned by the sustained activity from isoflurane GA.
h, Left, summary of overlap using effectiveness corrected measurement based on d and f. Green, total number of same-cell tracked neurons across sessions (N=160). Yellow, Iso.–active neurons. Orange, Ket.–active neurons. Right, the percentage of neurons activated by both ketamine and isoflurane among all Iso.–active neurons, calculated using four methods based on the ratios between the post- and pre-stimulus activity. Green, effective time; Cyan, effective mean versus mean activity; Yellow, mean activity; Orange, effective mean activity.
Among the isoflurane-active neurons, we found subsets of neurons that had very low baseline activity but were significantly activated by isoflurane (neurons located at the top of the sorted heat map in Fig. 2c), while other subsets had spontaneous baseline activity and were further activated following isoflurane exposure. We re-sorted the neurons by comparing their mean activity during the last 10 minutes (10 – 20 min) of isoflurane exposure to those during the first 10 minutes (0 – 10 min) (Fig. 2d). This comparison revealed two subpopulations: 11.8% (69/583) neurons showed persistent-firing during the whole GA process (iso.-sustained neurons, red rectangle in Fig. 2d), and 77.9% (454/583) neurons showed transient-firing (with increased activity only at 0–10 min of isoflurane exposure, iso.-transient neurons, orange rectangle in Fig. 2d). This result suggested that transient activation by isoflurane was sufficient to induce and/or maintain Fos expression in CeAGA neurons that enabled CANE-based capturing of these cells. We later tested whether low-concentration isoflurane (0.5%) would result in more persistently activated neurons (see later in the paper). All 12 imaged mice contained both sustained and transient isoflurane-active neurons across different imaging sessions (Extended data 2d). In addition, we also applied several different quantitative analysis methods (see Methods for details) to further characterize CeAGA responses across individuals and across anesthesia sessions. All analyses revealed that the majority of CANEISO-GCaMP6m captured CeAGA neurons were reactivated by isoflurane (Extended data 2f). Note that the heterogeneous activity pattern of CeAGA neurons is consistent with the heterogeneity revealed by our molecular characterizations (i.e. partial overlap with Penk1 and Pkc-d, Fig. 1c, f).
We next asked whether the CANE-GCaMP6m captured isoflurane-active neurons could also respond to K/X induced GA (ketamine, 100mg/kg, xylazine, 10mg/kg) in vivo. 89.4% of CANEISO-GCaMP6m neurons had increased calcium signals after K/X injection (304/340 were ketamine-active neurons and 36/340 were ketamine-suppressed neurons, Fig. 2e). Ketamine-active neurons also show either persistent (23.2%, 79/340 neurons) or transient (66.2%, 225/340 neurons) activation after ketamine administration (Fig. 2f). The percentages of ketamine-transiently versus sustainably activated neurons from individual mice are shown (in Extended data 2e). Additional characterizations using other quantitative methods showed the distributions of ketamine-responsive CeAGA neurons based on these different measurements (Extended data 2g).
Furthermore, using a previously described method for registering images and tracking cells across days18 with some modifications (see Methods), we managed to track 160 neurons (captured using CANEISO-GCaMP6m) between isoflurane-GA and ketamine-GA imaging sessions (same cells across days from 9 mice, Fig. 2g). 88.15% (119/135) of tracked isoflurane-activated neurons were also activated by ketamine (Fig. 2h) using the criterion applied in Fig. 2c and e. When analyzing the activity profiles of these 160 neurons using other methods and criteria (see Methods), even with the most stringent criteria, we still found that 57% of isoflurane-activated neurons were also activated by ketamine (Fig. 2h, right panel). Note that the percentage dual-activated neurons obtained with calcium imaging is higher than that obtained using Fos staining (Fig. 1h, g), perhaps due to the fact that neuronal firing does not always lead to Fos expression. Taken together, these in vivo imaging results strongly supported the existence of a shared ensemble of CeAGA neurons that can be activated by both isoflurane and ketamine GA.
Activity of the majority of CeAGA neurons is suppressed by stress
The transient activation of subsets of CeAGA neurons upon isoflurane infusion or ketamine administration raised the question whether such activities are stress responses. To test this, we subjected mice for 90min of restraint stress. Stress strongly induced Fos expression in the basolateral amygdala (BLA) but only moderately activated Fos in the CeA (Extended data 3b). We next captured CeAGA neurons with CANEISO-tdTomato, and subsequently subjected the same mice to 90min restraint stress (Extended data 3a). We found that only 12±2% CeAGA-tdTomato neurons co-localized with Fos+ stress-activated neurons, and 27±2% stress-activated neurons co-localized with CeAGA-tdTomato neurons (N=3, Extended data 3b). We further imaged the in vivo calcium activity of CANEISO-GCaMP6m captured CeAGA neurons in response to restraint stress (282 neurons, Extended data 3a). Activity of most of CANEISO-GCaMP6m neurons were suppressed during restraint with only a small number of cells exhibited stress-induced activation (Extended data 3c, d). Furthermore, since CANE captured cells containing a small proportion of isoflurane-suppressed neurons, to further characterize the stress responses, we also applied the same-cell tracking18 and identified neurons that were imaged during both isoflurane-GA and restraint stress sessions (172 tracked same neurons). Using two different measures to categorize their responses to isoflurane (see Methods), we confirmed that the majority of isoflurane-activated CeAGA neurons were inhibited by stress (Extended data 3e–3h).
Optogenetic manipulation of CeAGA neurons did not induce fear-like behaviors or change the gross brain state
Since CeA is generally known as a key center in the fear circuit, we therefore examined whether activation of CeAGA neurons induced anxiety- and fear-like behavior. To do this, we expressed either the optogenetic neuronal activator channelrhodopsin19 (CeAGA-ChR2) or the optogenetic silencer enhanced archaerhodopsin 3.020 (CeAGA-eArch) in CeAGA cells using CANE (after 2 hours of isoflurane, examples of post hoc fiber tracts are shown in Extended data 2c). Mice with CANE-mediated GFP expression in CeAGA neurons (CeAGA-GFP) were used as controls. We subjected CeAGA-GFP, CeAGA-ChR2 and CeAGA-eArch mice to the open field (OF) and the elevated plus maze (EPM) tests with or without photo-illumination. Neither optogenetic activation nor inhibition of CeAGA neurons induced any fear-like freezing or fleeing or cornering behaviors in the OF (Supplementary Video 2, Extended data 4a, b), or changed the mouse’s behavior in the EPM (Extended data 4a, c, d). Specifically, optogenetic manipulation of CeAGA neurons did not increase the time that animals spent in the outer perimeter of the OF or in the closed arm of the EPM (Extended data 4b, d). We further examined whether activation of CeAGA neurons altered the gross brain state by comparing electroencephalogram (EEG) recorded in the frontal and parietal cortex in the absence or presence of optogenetic activation of CeAGA neurons in CeAGA-ChR2 mice (n=3). The results showed that activating CeAGA neurons have no observable effects on the EEG power spectrums (Extended Data 4e, f).
Optogenetic activation or silencing of CeAGA neurons bidirectionally and potently altered nocifensive reflexes across sensory modalities
Next, we asked whether these CeAGA neurons play a role in regulating nociception sensitivity in naïve mice. We subjected the three groups of mice (CeAGA-GFP control, CeAGA-ChR2 and CeAGA-eArch) to a battery of commonly used mechanical, thermal, or cold sensory test. For mechanical sensitivity, a range of von Frey filaments (0.008–1.0 g) or an electronic von Frey device (50 g/5 sec) was used to stimulate either the face (with all whiskers intact) (Fig. 3a) or the hind paw, respectively. We assessed the reflexive withdrawal responses to stimuli applied to left or right side (in randomized order for different animals) with or without light-illumination in all three groups of animals (CeAGA-GFP, n=7; CeAGA-ChR2, n=8; or CeAGA-eArch, n=7). For face von Frey tests, we recorded the average number of withdrawals out of 10 trials using von Frey filaments (0.008 to 1.0 gram). The differences between conditions with and without light-illumination were calculated (Fig. 3b, all exact P-values for all figures are provided in Supplementary Table 1). Without light, all three groups of mice (CeAGA-GFP, CeAGA-ChR2, CeAGA-eArch) had no withdrawal response to filaments from 0.008 to 0.04 grams applied to the face on either side; mice began to show various numbers of head withdrawal at 0.16g, and by 1.0g, all animals responded reliably in all trials. Light illumination of the right CeA in control CeAGA-GFP mice (N=7) did not induce any statistically meaningful changes in withdrawal frequencies across the entire force range (Fig. 3b, green line, Extended data 5a). By contrast, in CeAGA-ChR2 mice (N=8), ChR2-activation of right CeAGA cells significantly reduced the amount of head withdrawals for both the ipsilateral and contralateral mechanical stimulations in the 0.16–0.60g range (Fig. 3b, blue line, Extended data 5a). The opposite effects were observed in CeAGA-eArch group (N=7), photo-silencing of right CeAGA rendered mice hypersensitive and responded to innocuous filaments applied to the face especially on the ipsilateral side (Fig. 3b, purple line, Extended data 5a). Similar bidirectional modulation of paw responses to electronic von Frey tests were observed: ChR2-activating CeAGA markedly reduced hind paw mechanical sensitivity (on both ipsilateral and contralateral side), whereas eArch-silencing of CeAGA significantly reduced the force needed to elicit the paw withdrawal reflex for the ipsilateral hind paw (Fig. 3c). Since the effects of eArch-mediated silencing CeAGA were much stronger on the ipsilateral side, we expressed eArch bilaterally in CeAGA using CANE in a few mice (n=4). Stimulating the face of these mice with the 0.04g filament normally failed to elicit any responses without photo-silencing; however, bilateral photo-silencing of CeAGA elicited withdrawal and even defensive responses to this normally below-detection threshold filament in all 4 mice (Supplementary Video 3).
Figure 3 |. Activation or inhibition of CeAGA neurons bi-directionally modulated pain-related behaviors in naïve mice and acute pain models.
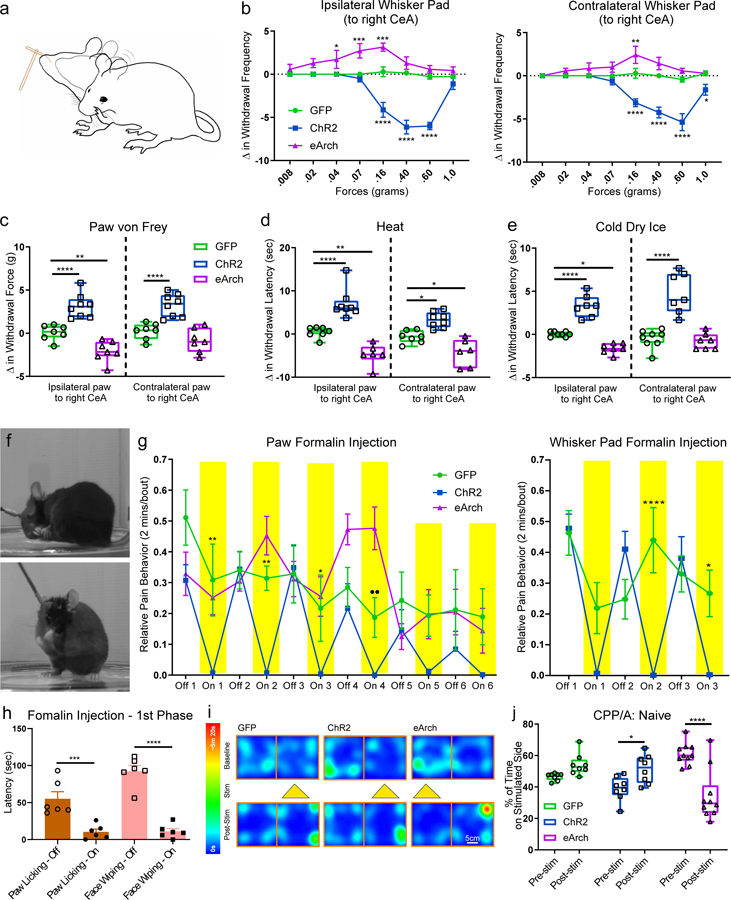
a, Schematic of the responses to von Frey filaments applied to the whisker pad, including head withdraw and face wiping.
b, Quantification of optogenetic manipulation of CeAGA neurons induced changes in the withdrawal frequency to 8 different von Frey filaments in the contralateral and ipsilateral whisker pad (to the right CeA). (Ipsilateral, control, n=7 animals (0 ± 0 (.008g), 0 ±0 (.02g), 0 ± 0 (.04g), 0 ± 0 (.07g), 0.29 ± 0.57 (.16g), 0.14 ± 0.26 (.40g), −0.29 ± 0.29 (.60g), −0.29 ± 0.18 (1.0g)), ChR2, n=8 animals (0 ± 0 (.008g), 0 ± 0 (.02g),0 ± 0 (.04g), −0.5 ± 0.38 (.07g), −4.13 ± 0.85 (.16g), −6.13 ± 0.79 (.40g), −6.0 ± 0.42 (.60g), −1.13 ± 0.64 (1.0g)), eArch, n=7 animals 0.57 ± 0.57 (.008g), 1.29 ± 0.52 (.02g), 1.71 ± 1.04 (.04g), 2.71 ± 0.84 (.07g), 3.14 ± 0.46 (.16g), 1.29 ± 0.7 (.40g), 0.57 ± 0.43 (.60g), 0.43 ± 0.43 (1.0g)); two-way ANOVA; ****P<0.0001, ***P<0.001, and *P<0.05; Contralateral, control, n=7 animals (0 ± 0 (.008g), 0 ± 0 (.02g), 0 ± 0 (.04g), 0 ± 0 (.07g), 0.29 ± 0.57 (.16g), 0 ± 0.22 (.40g), −0.43 ± 0.30 (.60g), 0.29 ± 0.29 (1.0g)), ChR2, n=8 animals (0 ± 0 (.008g), 0 ± 0 (.02g), 0 ± 0 (.04g), −0.63 ± 042 (.07g), −3.13 ± 0.44 (.16g), −4.25 ± 0.62 (.40g), −5.38 ± 0.98 (.60g), −1.63 ± 0.63 (1.0g)), eArch, n=7 animals (0 ± 0 (.008g), 0.57 ± 0.30 (.02g), 0.86 ± 0.63 (.04g), 1.0 ± 0.58 (.07g), 2.43 ± 1.0 (.16g), 1.43 ± 0.72 (.40g), 0.57 ± 0.30 (.60g), 0.29 ± 0.29 (1.0g)); two-way ANOVA; ****P<0.0001, **P<0.01, and *P<0.05; F14,152=11.40 (ipsi) F14,152=8.680 (contra).
c, Optogenetic manipulation of CeAGA induced changes in the withdrawal threshold in response to electronic von Frey applied to the paw (control, n=7 animals (0.07 ± 0.33g (ipsi), 0.21 ± 0.35g (contra)), ChR2, n=8 animals (3.12 ± 0.49g (ipsi), 3.23 ± 0.47g (contra)), eArch, n=7 animals (−2.09 ± 0.46g (ipsi), −0.86 ± 0.53g (contra)); two-way ANOVA; ****P<0.0001, **P<0.01; F2,38=49.51). Data are mean ± s.e.m..
d and e, Quantification of the optogenetics induced change in withdrawal latency (t2-t1) for Hargreaves heat (control, n=7 animals (0.42 ± 0.47s (ipsi), −0.67 ± 0.54s (contra)), ChR2, n=8 animals (7.0 ± 1.19s (ipsi), 3.28 ± 0.64s (contra)), eArch, n=6 animals (−4.76 ± 1.02s (ipsi), −4.33 ± 1.29s (contra)); two-way ANOVA; ****P<0.0001, **P<0.01, and *P<0.05; F2,36=57.56) and Cold dry ice test (control, n=8 animals (0.04 ± 0.10s (ipsi), −0.51 ± 0.38s (contra)), ChR2, n=7 animals (3.29 ± 0.48s (ipsi), 4.76 ± 0.88 (contra)), eArch, n=7 animals (−1.62 ± 0.22s (ipsi), −0.81 ± 0.35s (contra)); two-way ANOVA; ****P<0.0001, *P<0.05; F2,38=71.37). Data are mean ± s.e.m..
f, Example images of coping behaviors such as licking hind paw (top) or wiping whisker pad (bottom) to separate injections of formalin on different days.
g, Quantification of self-caring behaviors (total licking duration (sec)) per 2-minute bins with off and on light stimulation. (Hind paw formalin injection, control, n=8 animals (0.51 ± 0.09 (off1), 0.31 ± 0.12 (on1), 0.34 ± 0.06 (off2), 0.31 ± 0.04 (on2), 0.33 ± 0.09 (off3), 0.22 ± 0.11 (on3), 0.28 ± 0.06 (off4), 0.19 ± 0.06 (on4), 0.24 ± 0.09 (off5), 0.19 ± 0.07 (on5), 0.21 ± 0.13 (off6), 0.19 ± 0.09 (on6)); ChR2, n=9 animals (0.31 ± 0.05 (off1), 0.01 ± 0.01 (on1), 0.34 ± 0.06 (off2), 0.01 ± 0 (on2), 0.35 ± 0.07 (off3), 0 ± 0 (on3), 0.22 ± 0.08 (off4), 0 ± 0 (on4), 0.15 ± 0.07 (off5), 0.01 ± 0.01 (on5), 0.08 ± 0.06 (off6), 0 ± 0 (on6)); eArch, n=7 animals (0.33 ± 0.07 (off1), 0.25 ± 0.05 (on1), 0.30 ± 0.03 (off2), 0.45 ± 0.06 (on2), 0.31 ± 0.06 (off3), 0.25 ± 0.06 (on3), 0.47 ± 0.05 (off4), 0.48 ± 0.07 (on4), 0.13 ± 0.04 (off5), 0.20 ± 0.08 (on5), 0.20 ± 0.07 (off6), 0.14 ± 0.07 (on6)); two-way repeated measure ANOVA; **P<0.01, ••P<0.01 and *P<0.05; F5,75=4.81; Whisker pad formalin injection, control, n=7 animals (0.46 ± 0.07 (off1), 0.22 ± 0.08 (on1), 0.25 ± 0.06 (off2), 0.44 ± 0.11 (on2), 0.33 ± 0.06 (off3), 0.27 ± 0.08 (on3)), ChR2, n=9 animals 7animals (0.48 ± 0.05 (off1), 0.01 ± 0.01 (on1), 0.41 ± 0.06 (off2), 0 ± 0 (on2), 0.38 ± 0.07 (off3), 0 ± 0 (on3)); two-way repeated measure ANOVA; ****P<0.0001, *P<0.05; F5,70=8.825). Data are mean ± s.e.m..
h, Quantification of total wiping and licking behaviors during the first phase after formalin injection comparing off and on stimulation (2 minute bins with off and on light stimulation, total 6 minutes of stimulation) (ChR2, n=6 animals (55.00 ± 21.56s (licking-off), 10.33 ± 8.6s5 (licking-on), 92.33 ± 17.84s (wiping-off), 11.00 ± 8.33s (wiping-on)); one-way ANOVA; ***P<0.001 and ****P<0.0001; F3,20=33.55).
i, Example heat map of CPP/CPA experiment.
j, Quantification of the percent of time (%) naïve mice spent on the stimulated side (control, n=8 animals (46.52 ± 0.80 (pre), 54.16 ± 2.45 (post), ChR2, n=8 animals (39.05 ± 2.70 (pre), 51.54 ± 3.20 (post), eArch, n=10 animals (61.16 ± 2.17 (pre), 34.39 ± 5.11 (post),; two-way repeated measure ANOVA; ****P<0.0001 and *P<0.05; F2,23=29.66). Data are mean ± s.e.m..
We next performed the Hargreaves heat and cold dry ice tests (Fig. 3d,e). Light illumination in control CeAGA-GFP group did not alter animals’ reflexive withdrawal behaviors in responses to heat or dry ice. Notably, photo-activation or photo-silencing of CeAGA neurons during exposure to heat and dry ice significantly increased or decreased, respectively, the withdrawal latency compared to no-light conditions or to the control groups (Fig. 3d,e with ipsilateral side having stronger effects). Taken together, these experiments showed that activation and silencing of CeAGA neurons can bi-directionally and potently modulate nocifensive reflex responses in naïve mice across multiple somatosensory modalities.
Optogenetic activation or silencing of CeAGA neurons bidirectionally and potently regulated pain-elicited self-caring behaviors
Besides reflexive defensive behaviors, more sustained pain also invoke self-caring responses such as licking and wiping, and these behaviors are thought to be indicative of pain perception. A recent study showed that there exist separate neural circuits for mediating reflexive versus self-caring/recuperative behaviors21. We injected formalin into either the hind paw or the whisker pad in CeAGA-GFP (control), CeAGA-ChR2, and CeAGA-eArch mice and tested mice with or without light illumination of the CeA. Formalin causes a biphasic response: the first phase of the acute pain that usually lasts a few minutes, and the second phase inflammatory pain that starts around ~20 minutes after formalin injection and lasts 10–15 minutes22,23. Mice exhibit intense licking of the hind paw or wiping of the whisker pad during the second phase (Fig. 3f,g)22,23. At the start of the second phase, we first video recorded animals’ behavior for two minutes and then illuminated the CeAGA neurons every two minutes for three to six times. Remarkably, ChR2-activation of CeAGA neurons (unilaterally) completely abolished paw-licking or face-wiping behavior regardless of whether the right or left paw or whisker pad was injected with formalin, as compared with the persistent self-recuperating behaviors of CeAGA-GFP mice that were unaffected by light (Fig. 3g). As soon as CeAGA was activated, the mice stopped licking (Supplementary Video 4, Fig. 3g, left) or wiping (Supplementary Video 5, Fig. 3g, right) and simply walked around the cage. In the initial 2 minutes following light stimulation, control CeAGA-GFP mice also decreased face-wiping time (Fig. 3g, right). This may be due to either the initial distraction of the light or other unknown effects of light on CeA neurons. ChR2-activation of CeAGA neurons also drastically reduced licking and wiping behavior in the first phase of acute pain induced by formalin injection (N=6, Fig. 3h). Note that activating CeAGA neurons did not have any effect on mating behaviors as measured by the number of ultrasonic vocalization syllables and the duration of anogenital sniffing/mounting (Extended data 5c–d). Thus, the ceasing of the self-caring behaviors upon ChR2-activation of CeAGA neurons is unlikely due to the suppression of general motivation.
Formalin-induced self-caring behaviors gradually subside after ~10 minutes in the second phase, because inflammation responses decrease over time, but the affected area is likely still sensitive. We asked what happened if we optogenetically silenced CeAGA neurons during this period in the hind paw formalin test. Initially in the beginning of the second phase, animals with eArch-silencing of CeAGA neurons behaved similarly to control CeAGA-GFP mice, perhaps because mice already licked constantly at early stages (Fig. 3g, On 1 and 2); and after several bouts of inhibiting CeAGA at early stages, mice started licking paw even when light was off (Fig. 3g, Off 4). Interestingly, at later stages (>8 minutes into the second phase) when mice exhibited low levels of paw-licking, photo-silencing of CeAGA-eArch mice immediately re-elicited robust licking toward the inflamed paw (Supplementary Video 6, Fig. 3g). Hence, when CeAGA neurons are silenced, animals appeared to perceive the subsided injury as becoming intense or painful again. Taken together, these experiments showed that activation and silencing of CeAGA neurons can bi-directionally and potently regulate pain-elicited intentional self-caring behaviors.
Partial lateralization of CeAGA functions
Most of the experiments described above were performed by manipulating either the right or both the right and left CeAGA neurons. We next tested whether there could be a lateralization of CeAGA functions by activating only the left CeAGA cells. In CeAGA-ChR2LEFT mice (N=6), ChR2-activation of left CeAGA reduced the amount of paw withdrawals for both the ipsilateral and contralateral paw in the von Frey test (in the 1.0–2.0g force range, Extended data 6a). Surprisingly, in the face von Frey test, activation of left CeAGA neurons did not produce any observable changes in head withdrawal responses (Extended data 6b). On the other hand, for cold and heat, as well as paw-formalin and face-formalin tests (n=7 CeAGA-ChR2LEFT mice), ChR2-activation of left CeAGA cells increased the withdrawal latencies (Extended data 6c), and drastically reduced licking and wiping behaviors in in all tests (Extended data 6d). Thus, except for the partial lateralization of the role for suppressing facial mechanical sensitivity to the right side, activation of left CeAGA neurons produced similar results as those of the right CeAGA.
In vivo imaging of CeAGA neuron activity during sensory tests
The findings that optogenetic silencing of CeAGA neurons rendered CeAGA-eArch mice hypersensitive to mechanical, heat, and cold stimuli and exacerbated pain-elicited caring behaviors raised the possibility that the basal level (on-going) activity of CeAGA neurons function to prevent behavioral hypersensitivity. We next asked whether the spontaneous activity of CeAGA neuron would change in response to noxious stimuli in awake behaving mice. We performed in vivo calcium imaging of CANEISO-GCaMP6m captured CeAGA neurons while performing heat, cold, or von Frey stimuli in awake behaving mice (Fig. 4b). To avoid any potential photo-damage caused by prolonged imaging, different sensory tests were performed on different days with at least 1-day interval between the tests. The animals’ behaviors were tracked using video recordings (Fig. 4a,c, see Methods).
Figure 4 |. In vivo calcium imaging of CeAGA activities in sensory tests.
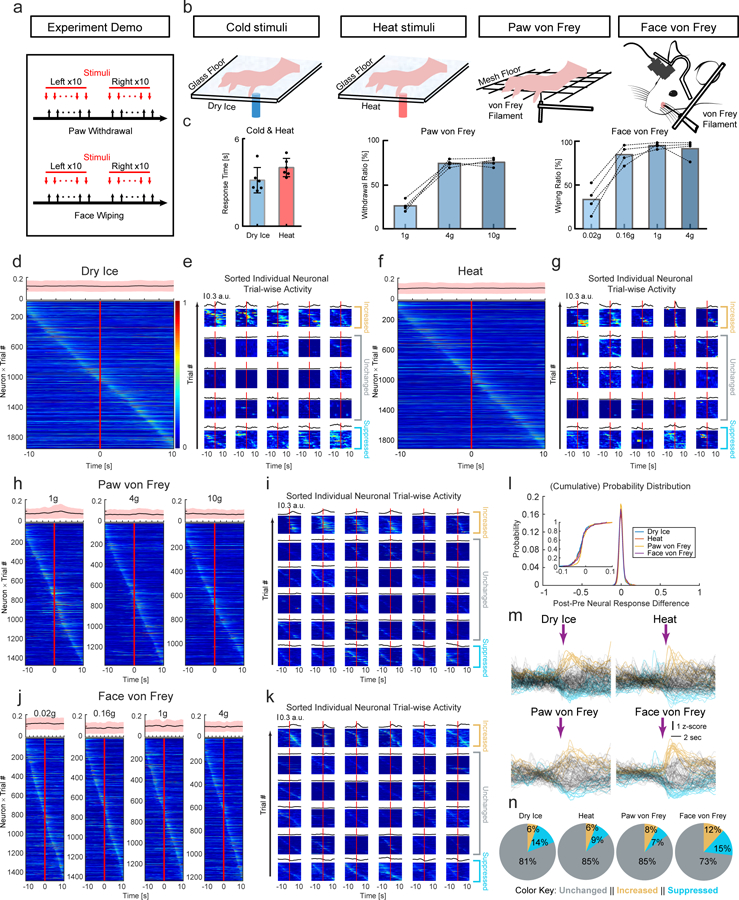
a, Scheme of nociceptive stimuli on the mice hind paws and facial pads. Red arrows, the onset of stimuli. Black arrows, the onset of withdrawal reflex responses.
b, Diagram showing calcium imaging of CeAGA neurons during cold, heat, graded von Frey stimuli applied to the hind paws in freely moving mice, or von Frey stimuli applied to the whisker pads in head-fixed mice.
c, Left, paw withdrawal latencies in response to dry ice (3.16 ± 0.36s) and heat stimuli (4.02 ± 0.26s) (n=6 animals). Middle, the percentage (%) of paw withdrawal in response to each filament of von Frey stimuli (n=4 animals (26.25 ± 3.17% (1g), 75.00 ± 2.08% (4g), 76.25 ± 2.43% (10g)). Right, the percentage of face wiping in response to each filament of von Frey stimuli (n=4 animals, 35.00 ± 8.42% (0.02g), 88.75 ± 5.54% (0.16g), 99.50 ± 1.71% (1.0g), 95.75 ± 5.31% (4.0g)). Data are mean ± s.e.m..
d, f, h and j, Neuronal activity patterns during d, cold stimuli, f, heat stimuli, h, von Frey stimuli to paws, and j, von Frey stimuli to facial pads sorted by neurons peak responses timing (from −10 to +10 sec, 0 is the onset of response, for those trials without response in the von Frey stimuli, 0 is the withdraw of von Frey filament). Top of each heatmap, averaged population activity. Thick lines indicated mean and shaded areas indicated s.e.m..
e, g, i and k, Three patterns of representative individual neuron responses plotted for all the trials to e, g, i and k sorted by neurons peak responses timing (from −10 to +10 sec). Mean response of each neuron was plotted on the top of each corresponding heatmap. l, Probability distribution (inset, cumulative probability distribution) of neural response differences between post- and pre-response to calcium signal intensity during the 4 sensory tests.
m, Averaged calcium traces of individual neuron’s responses to noxious stimuli. Individual trial response was plotted in either black (unchanged neurons), blue (suppressed neurons) and yellow (activated neurons).
n, Percentage of 3 response types of CeAGA neurons during 4 sensory tests.
In vivo calcium imaging revealed that as a population, there were no apparent time-locked changes in CeAGA neurons activity with regard to either the onset of the heat, cold, or von Frey stimuli (Extended data 7), or to the onset of withdrawal reflex responses or the withdrawal of the stimuli by experimenter (Fig. 4d,f,h,j). The on-going activity of individual CeAGA neurons was distributed across the trial period, and the averaged population activity was flat (average is shown on top of each heat map), suggesting CeAGA neuron activity was uncorrelated and unresponsive to acutely applied sensory stimuli.
We also analyzed activity of individual CeAGA neurons. In all tests, neurons could be classified into three groups 1) activity-increased, 2) activity-unchanged (non-responsive to stimuli), and 3) activity-suppressed (see heat maps of many example cells in Fig. 4e,g,i,k, averaged traces shown in Fig. 4m, and percentages of the three types of neurons in each test shown in Fig. 4n). For each neuron, we also computed the difference between post- and pre-stimulus calcium signal intensity and plotted the distributions across 4 sensory tests (Fig. 4l). In all cases, the majority of imaged neurons did not alter their activity upon noxious stimuli applications in agreement with the averaged population activity data. These results suggest that the on-going activity of CeAGA neurons may largely reflect the animal’s internal state, rather than the immediate experience of noxious stimuli. Consistent with the imaging result, we found that CANE-GFP captured CeAGA neurons had minimal overlap with formalin-pain activated Fos+ CeA-nociceptive neurons (Extended data 8a).
Silencing CeAGA neurons is aversive in the absence of noxious stimuli
An essence of the pain experience is aversion, or the perceived negative affect. If the observed on-going CeAGA activity indeed functions to prevent abnormal hypersensitivity to non-noxious stimuli, silencing these neurons would lead to allodynia in an otherwise innocuous environment and therefore could be aversive for mice. To test this, we subjected CeAGA-GFP, CeAGA-ChR2, and CeAGA-eArch mice to Conditioned Place Preference/Aversion (CPP/CPA) tests over a period of six days. On day 1, animals were left to freely explore a 2-chamber box. On day 2 and 4, CeAGA-GFP and CeAGA-ChR2 animals received photo-illumination of the CeAGA when they were in the less preferred chamber, and received no stimulation in the preferred chamber on day 3 and 5. CeAGA-eArch animals received photo-illumination in the preferred chamber. On day 6, animals were re-tested for their preference. Both CeAGA-GFP and CeAGA-ChR2 mice moderately preferred the photo-illuminated side, however, the preference was only statistically significant for CeAGA-ChR2 mice (Fig. 3i,j). Notably, in CeAGA-eArch animals, silencing of CeAGA dramatically changed their preference leading to significant avoidance of the chamber where they had experienced photo-silencing (Fig. 3i,j). Thus, silencing CeAGA neurons induced a strong place aversion in mice in the absence of noxious stimuli.
Activation of CeAGA neurons strongly reduced nociception-related behaviors in a chronic neuropathic pain model
The next key question is whether activating CeAGA neurons can suppress mechanical hypersensitivity in chronic neuropathic pain conditions. We subjected CeAGA-GFP and CeAGA-ChR2 mice to a chronic orofacial neuropathic pain model in which the infraorbital nerve (IoN) was ligated on the right side of the face, referred to as chronic constriction injury (CCI-IoN, Fig. 5a). This injury caused persistent sensitization of the whisker pad on the IoN-ligated side to non-noxious tactile stimuli 24. In face von Frey test, without photo-stimulation, CCI-IoN mice started to show withdrawal responses to the normally innocuous 0.02 g force on both sides (Fig. 5b, blue curve deviates from control green curve at 0.02g in both sides indicating that activating CeAGA reduced the hypersensitive withdrawal responses to the innocuous filaments, also see Extended data 5b). This is consistent with the known mechanical hypersensitivity, or allodynia, induced by neuropathic pain, including the so-called mirror pain on the uninjured side (Fig. 5b, right)25,26. Remarkably, unilateral ChR2-activation of CeAGA neurons (right CeAGA) dramatically reduced the withdrawal responses on the injured side (right side), as many animals simply showed no responses at all, even to 1.0g stimulation (Fig. 5b, left blue line and Supplementary Video 7), whereas illuminating control CeAGA-GFP animals did not alter their sensitivity to the entire range of filaments (Fig. 5b, green line and Supplementary Video 8). Note that in the naïve condition and for the un-injured side, a 1.0g von Frey stimulus applied to the face elicited consistent withdrawal reflexes even with ChR2-activation of CeAGA (Fig. 3b). By contrast, under CCI-IoN, activation of CeAGA revealed that the injured side was impaired at responding to 1.0g (i.e. hypo-sensitive at periphery), thereby suggesting that mechanical hypersensitivity in neuropathic pain conditions mostly result from abnormal central processing.
Figure 5 |. Activation of CeAGA neurons strongly reduced nociception-related behaviors in chronic constriction injury model and drove CPP.
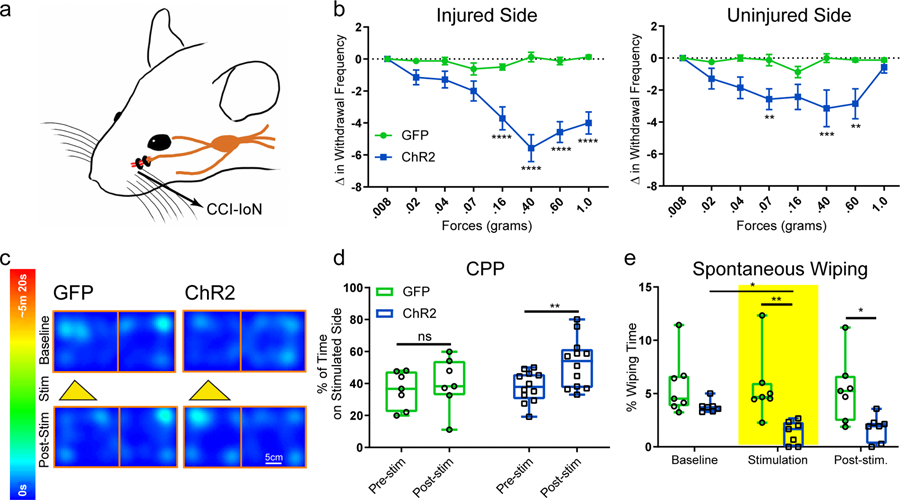
a, Schematic of the site of chronic constriction injury of the Infraorbital Nerve.
b, Quantification of activating CeAGA induced changes in withdrawal frequency to 8 different von Frey filaments in the injured and non-injured side of the whisker pad after IoN-CCI. (Injured side, control, n=8 animals (0 ± 0 (.008g), −0.13 ± 0.13 (.02g), −0.13 ± 0.23 (.04g), −0.63 ± 0.38 (.07g), −0.50 ± 0.19 (.16g), 0.13 ± 0.30 (.40g), −0.13 ± 0.23 (.60g), 0.13 ± 0.13 (1.0g)); ChR2, n=7 animals (0 ± 0 (.008g), −1.14 ± 0.46 (.02g), −1.29 ± 0.52 (.04g), −2.00 ± 0.62 (.07g), −3.71 ± 0.71 (.16g), −5.57 ± 0.84 (.40g), −4.57 ± 0.65 (.60g), −4.00 ± 0.69 (1.0g)); two-way ANOVA; ****P<0.0001; F7,104=10.74; Un-Injured side, control, n=8 animals (0 ± 0 (.008g), −0.25 ± 0.16 (.02g), 0 ± 0.19 (.04g), −0.13 ± 0.35 (.07g), −0.88 ± 0.35 (.16g), 0 ± 0.27 (.40g), −0.13 ± 0.13 (.60g), −0.13 ± 0.13 (1.0g)); ChR2, n=7 animals (0 ± 0 (.008g), −1.29 ± 0.64 (.02g), −1.86 ± 0.67 (.04g), −2.57 ± 0.65 (.07g), −2.43 ± 0.78 (.16g), −3.14 ± 1.14 (.40g), −2.86 ± 0.94 (.60g), −0.57 ± 0.37 (1.0g)); two-way ANOVA; **P<0.01 (0.07g), ***P<0.001 (0.40g), **P<0.01 (0.60g) ; F7,104=2.393). Data are mean ± s.e.m..
c, Example heat map of CPP/CPA experiment. Control and ChR2 mice received stimulation on non-preferred side.
d, Quantification of the percent (%) of time spent on the stimulated side after chronic constriction injury (control, n=7 animals (35.92 ± 4.38 (pre), 40.17 ± 6.09 (post), ChR2, n=12 animals (37.87 ± 2.76 (pre), 52.74 ± 4.48 (post); two-way ANOVA; **P<0.01, ns, P=0.80; F1,17=7.185). Data are mean ± s.e.m..
e, Quantification of percent (%) of time eliciting spontaneous wiping after chronic constriction injury before, during and after light stimulation (control, n=7, ChR2, n=7 animals; 7 minutes baseline no light (5.75 ± 1.07 (control), 3.79 ± 0.22 (ChR2), followed by 5 minutes of light-stimulation (5.61 ± 1.20 (control), 1.33 ± 0.42 (ChR2),, and another 7 minutes post-stimulation (5.37 ± 1.16 (control), 1.73 ± 0.46 (ChR2),; two-way repeated measures ANOVA; **P<0.01, *P<0.05; F1,12=11.98). Data are mean ± s.e.m..
We also performed conditioned place preference test in CCI-IoN animals. Photo-activation of CeAGA neurons in CeAGA-ChR2, but not in control CeAGA-GFP mice, produced a place preference memory for the light-activated chamber presumably due to pain-relief received in that chamber (Fig. 5c,d). Furthermore, CCI-IoN animals exhibited spontaneous asymmetric face-wiping of the injured side, an indicator of their perceiving spontaneous pain. ChR2-activation of CeAGA neurons in CeAGA-ChR2 mice (but not photo-illumination of the CeA in CeAGA-GFP mice) significantly reduced the total wiping duration in CCI-IoN mice (Fig. 5e), and there was also a lasting effect in the post-stimulation period (Fig. 5e). Taken together, activation of the CeAGA can potently suppress pain-related behaviors in the chronic neuropathic pain model.
CeAGA neurons project broadly to many pain-processing centers in the brain to suppress their activity
To further understand how CeAGA neurons exert their analgesic effects, we traced the axonal projections of CeAGA neurons in CeAGA-GFP mice. The potential downstream targets of CeAGA neurons included: the prefrontal cortex (pre-limbic and cingulate), the nucleus accumbens (NAc), the dorsal medial striatum, the insular cortex (Ins), the bed nucleus of stria terminalis (BNST), the basolateral amygdala (BLA), the cortical amygdaloid nucleus (PMCo, PLCo), the temporal association cortex (TeA), the ectorhinal cortex (Ect), entorhinal cortex (Ent), the subthalamic and peri-subthalamic region (SubTh), the posterior intralaminar nucleus of the thalamus, the ventrolateral periaquaductal grey (PAG), the parabrachial nucleus (PBN), the reticular nucleus (RT, mostly the intermediate RT), and the nucleus of the solitary tract (SolT) (Fig. 6). Many of these regions had previously been indicated in processing sensory or emotional aspects of pain27–30. Notably, most previous studies did not find a direct projection from CeA to prefrontal cortex. Future studies with more precise molecular-marker based CeAGA labeling method will be needed to validate this connection. The contralateral side of the NAc, CPu, insular, BLA, and TeA/Ect were also innervated, albeit more sparsely compared to the ipsilateral equivalent (Fig. 6). Such bilateral projections could explain the bilateral effects with ipsilateral bias of activating CeAGA unilaterally (Fig. 3). Since BLA is immediately adjacent to CeA where the CeAGA cells are labeled, the long exposure time resulted in saturated fluorescent image of CeA and BLA region (Fig. 6, panel 5). A lower exposure at higher magnification showed CeAGA-axons but not BLA cell bodies were labeled (Extended data 8b). Representative images of CeAGA axon projections in each of the target areas from multiple different mice and the quantifications of the averaged projection densities are shown (Extended data 9).
Figure 6 |. Whole brain mapping of axonal projections from CeAGA neurons.
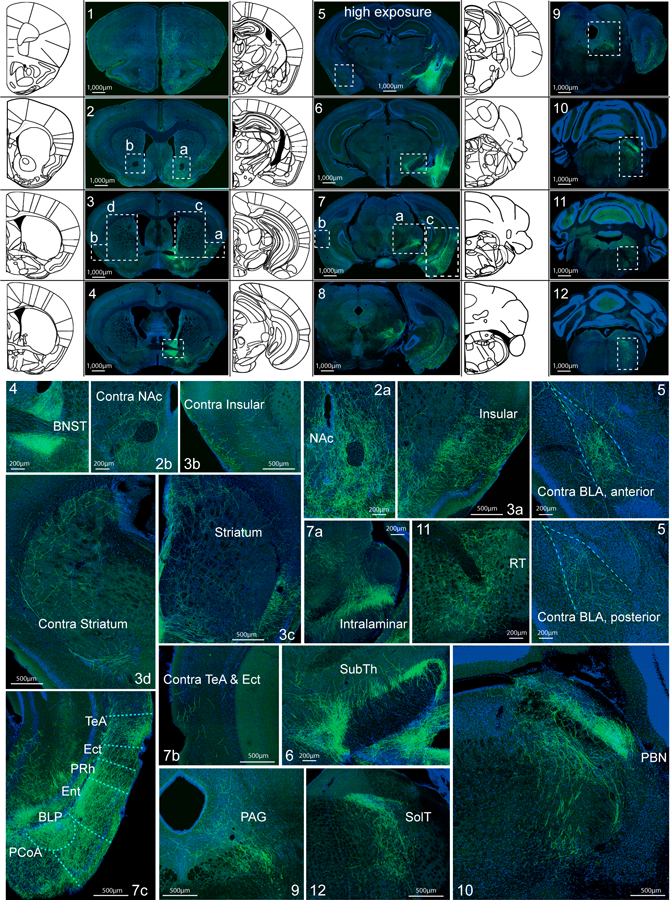
Top half: Coronal schematic next to example coronal slices. Boxes indicate the location of high-magnification zoomed in view of axonal projections. Bottom half: In sequential order: 1, cortex; 2a, nucleus accumbens (NAc) 2b, contralateral NAc; 3a, Insular, 3b, contralateral insular; 3c, striatum, 3d, contralateral striatum; 4, bed nucleus stria terminalis (BNST); 5, contralateral basal amygdala (BLA, top, anterior; bottom, posterior); 6, subthamalic nucleus (SubTh), 7a, posterior intralaminar nucleus of thalamus; 7b, contralateral temporal association cortex (TeA); 7c, TeA, ectorhinal cortex (Ect), perirhinal cortex (PRh), entorhinal cortex (Ent), posterior basal lateral amygdala (BLP), posterior cortical amygdala nucleus (PCoA); 8, posterior TeA, Ect, Ent, and midbrain reticular nucleus (RR); 9, periaqueductal grey (PAG); 10, parabrachial nucleus (PBN); 11, rostral reticular formation (RT); and 12, nucleus of solitary tract (SolT) and caudal intermediate reticular formation. Repeated experiments for n=5 biologically independent samples.
Furthermore, we noted that many of CeAGA neurons’ axonal targets contained formalin-pain induced Fos+ neurons (white-colored nuclei in Fig. 7a,c, these areas are the same brain regions shown in Fig. 6). Considering that CeAGA neurons are GABAergic, in principle, they are in ideal position to potently suppress the activities of these pain-activated neurons through their projections. Indeed, CeAGA-ChR2 mice that received bilateral optogenetic activation of CeAGA (N=3) showed significantly reduced number of Fos+ neurons in all CeAGA target regions (Compare Fig. 7b,d, to Fig. 7a,c, quantification in 7e). Thus, CeAGA neurons can potently inhibit neural activation across numerous pain-processing centers in the brain.
Figure 7 |. Activation of CeAGA neurons reduced formalin-induced activity to all CeAGA target regions and silencing CeAGA blocked the analgesic effect of low- dose ketamine.
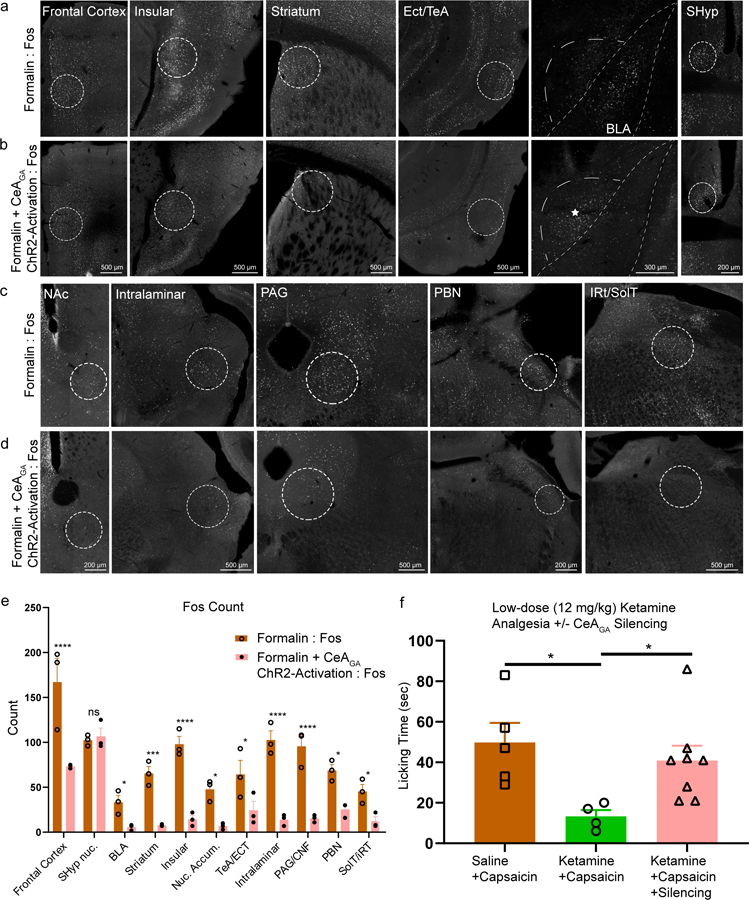
a, c, Representative images of strong Fos+ expression induced by formalin. The regions shown here are all CeAGA targets revealed in Figure 6 including: frontal cortex, insular, striatum, ectorhinal (Ect) and temporal association cortex (TeA), basalateral amygdala (BLA), nucleus accumbens (NAc), intralaminar, periaqueductal grey (PAG), parabrachial nucleus (PBN), and intermediate reticular nucleus (iRT) and solitary tract (SolT). Note that the septohypothalamic nucleus (SHyp) do not receive projections from CeAGA neurons and serve as a negative control.
b, d, Representative images of reduced Fos+ expression with ChR2 activation of CeAGA neurons (captured with CANE under isoflurane) after formalin injection in those same brain regions as a, c. Note that * signifies the CeAGA cells activated under ChR2. Fig. 7a-d were repeated for n=3 biologically independent samples.
e, Quantification of averaged Fos+ cell count in areas that receive CeAGA neuron projections induced by formalin (a, c) or formalin plus ChR2 activation of CeAGA (b, d). Cell count represents the total number of cells from sections containing each of the regions but using only a single focal plane from each section for counting. (Formalin Fos, n=3 biologically independent samples; Formalin + CeAGA-ChR2 Fos, n=3 biologically independent samples; two-way ANOVA; ****P<0.0001 (FC, Ins, Intralaminar, PAG), ***P<0.001 (striatum), *P<0.05* (BLA, NAc, TeA/ECT, PBN, SolT, ns, P>0.999); F1,44=186.8).
f, Quantification of total licking time (sec) in response to capsaicin injection into the paw with co-administration (i.p.) of saline or low-dose (12 mg/kg) ketamine with or without optogenetic silencing of CeAGA neurons that were captured with CANE under isoflurane (Saline +Capsaicin, n=5 animals (40.80 ± 24.1s); Ketamine +Capsaicin, n=4 animals (12.25 ± 6.94s); Ketamine +Capsaicin +Silencing, n=8 animals (34.75 ± 12.93s); one-way ANOVA with unpaired t-test, two-tailed; *P<0.05; F2,14=4.447).
Activity of CeAGA neurons is required for the analgesic effect of low-dose ketamine
We started this study by hypothesizing that the analgesic effect of general anesthetics is separable from the GA-induced loss-of-consciousness, especially at low GA drug concentrations. Indeed, when we imaged the in vivo activity of CANEISO-GCaMP6m neurons, captured with 1.5% isoflurane, we found that more neurons became sustainably activated in response to the 0.5% isoflurane (total of 106 same neurons tracked between 1.5% and 0.5% isoflurane sessions, Extended data 10 a,b). Since it is difficult to perform behavioral tests under low dose isoflurane in the gas/induction chamber, we turned to low-dose ketamine, which is clinically known to have an analgesic effect. Previous studies showed that at 12mg/kg, ketamine can reduce pain behavioral responses in mice31. We tracked the activity of 69 same neurons across three imaging sessions: 1.5% isoflurane, regular-dose ketamine (100mg/kg), and low-dose ketamine (12mg/kg), and found that most of tracked CANEISO-captured CeAGA neurons can be activated in all three sessions including low-dose ketamine (Extended data 10c,d).
To test whether the activity of CeAGA neurons are required for the analgesic effect of low-dose ketamine, we subjected CeAGA-eArch mice (n=8) to the capsaicin test. After injection of capsaicin (2 μg/10 μL) into the paw, the saline (i.p.) group (n=5) elicited robust paw licking. Low-dose ketamine significantly reduced the duration of licking (n=4) (Fig. 7f). Importantly, when CeAGA neurons were optogenetically silenced (silenced for first and last 5min out of the total 15min) in the capsaicin+ketamine conditions, mice licked the paw for similar amount of time as that in capsaicin+saline conditions, hence the analgesic effect of ketamine was abolished (Fig. 7f). The result revealed that the activity of CeAGA neurons is required for the pain-relieving effect of low-dose ketamine.
DISCUSSION
The existence of a central analgesic system was first postulated based on a seminal study published by H.K. Beecher in 1946, a physician served in US army during World War II, who showed that badly wounded soldiers, although alert and not in shock, did not report pain or in need for medication32. This finding triggered the subsequent search for the brain’s internal analgesic system. Furthermore, the clinically important phenomenon called placebo analgesia also suggests the existence of a central pain suppression system33–35, but the exact circuits carrying out the placebo effect are unknown. Over the past decades, important progresses were made that delineated a midbrain periaqueductal grey-brainstem-spinal descending pain modulation pathway36–38, however whether forebrain contains a central analgesic center and how might it work remain unknown. Here, starting with the hypothesis that GA induced analgesia is an active process, we discovered and delineated CeAGA neurons as a key node in the central nervous system that are activated by GA, function to potently suppress pain responses in both acute and chronic conditions, project to numerous pain-processing centers, and mediate the analgesic effects of general anesthetics (that is independent of their sedative effect).
Interestingly, CeA has previously been implicated as a critical structure for both stress- and placebo-induced analgesia34,39,40, and lesions of CeA abolishes such analgesia in rats41. However, due to the multi-functional nature of CeA which includes regulating innate and learned fear, pain (both pro-pain and anti-pain), and appetitive/feeding related behaviors9,14,42–44 as well as its molecular heterogeneity13, it was unknown what subsets of CeA neurons are involved in analgesia. It is also unknown whether stress and placebo employ different or same CeA neurons for analgesia. Note that stress is a double-edged sword, i.e. stress can either enhance or reduce pain depending on different contexts39,45. In our study, most of CeAGA neurons are suppressed by restraint stress (Extended data 3), suggesting that CeAGA may not be involved in stress-induced analgesia (SIA). Furthermore, activating CeAGA neurons had no effect on fear- or anxiety-like behaviors (Extended Data 4), whereas SIA is often induced by fear and is considered as part of the fight or flight responses39,45. Hence, it is possible that CeA neurons involved in fear responses are more likely to be involved in SIA. Along this line, Sst-expressing CeA neurons, which are non-overlapping with CeAGA neurons revealed here and are known to mediate the fear-induced freezing responses, were shown to attenuate pain-responses in a recent study46. However, there was a small number of CeAGA neurons that were activated by stress (Extended data 3). Thus, we cannot rule out the possibility that strong stresses, such as acute life-threatening conditions, may activate more CeAGA neurons to produce analgesia. It will also be interesting to test in the future whether CeAGA is involved in placebo analgesia.
Both our molecular marker analyses and our in vivo imaging studies revealed that CeAGA neurons contain a heterogeneous population of neurons. We found that CeAGA neurons do not express Sst, Pdyn, and Nts, however, a subset of CeAGA neurons expresses Penk1 or Pkc-d (but Penk1+ and Pkc-d+ neurons represent a larger population of CeA neurons). We should note that Pkc-d+ CeA neurons located in the capsular part of CeA (CeC) are involved in processing noxious and other aversive signals and can elicit defensive behaviors9,46–48; whereas Pkc-d+ CeA neurons in the later division (CeL) inhibit defensive behaviors8,49 and inhibit feeding in response to positive state such as satiety9,49. We found that many CeAGA neurons expressing Pkc-d and that activating CeAGA neurons inhibited pain-responses, exactly opposite to the effect of activating Pkc-d+ capsular-CeA neurons46. Future work is needed to find additional marker to separate anti-versus pro-nociception Pkc-d cells. Neither Pkc-d nor Penk1 nor both markers could define CeAGA. While it is possible that a specific subset of CeAGA neurons exert most of the analgesic function, it is also possible that the entire heterogenous ensemble of CeAGA neurons as delineated by Fos+ is required to work together to suppress pain. We observed wide-spread projections of CeAGA neurons to many regions activated by painful stimuli (Fig. 6). It is likely that different subpopulations of CeAGA innervate only a subset of these pain-processing centers, and the whole populations of CeAGA are needed to carry out the full analgesic functions.
We also want to highlight the finding in the neuropathic orofacial pain model, in which nerve injury is well known to cause mechanical hypersensitivity. Under the conditions of CeAGA neurons activation, we found that the injured side was significantly less-responsive, i.e hypo-sensitive, to the normally painful 1.0g von Frey filament (Fig. 5). This result is consistent with the fact that ligation injury damaged the nerve and thereby impair the peripheral endings’ ability to detect mechanical stimuli. The finding strongly suggested that the commonly observed tactile allodynia in neuropathic pain is likely due to altered central processing of signals transduced by spared uninjured sensory fibers. Activation of CeAGA neurons can suppresses such abnormal central processing. We further observed that all of the CeAGA target regions contain neurons strongly activated by painful stimuli, and remarkably all such pain-elicited activity can be suppressed by concurrent optogenetic stimulation of CeAGA (Fig. 7). Interestingly, CeAGA neurons do not project to sensory thalamus, or to primary or secondary somatosensory cortex (data not shown), suggesting these neurons likely function to dissociate pain perception from sensation, which was exactly the observed effect of low-dose ketamine on human patients3–5.
At present, we do not yet know how general anesthetics induce transient or sustained activation of CeAGA neurons. We also do not know whether persistently activating CeAGA neurons will be non-addictive. Nevertheless, our work raises the exciting possibility of harnessing the power of this endogenous analgesic system to relieve chronic pain. Future work aimed at identifying small molecular compounds that can specifically activate these powerful analgesic neurons without the sedative effects of GA drugs could be developed into the next generation pain killers.
METHODS
Animal statement
All experiments were conducted according to protocols approved by The Duke University Institutional Animal Care and Use Committee.
Animals
Adult male and female (more than 8 weeks old) FosTVA mice (Jackson Laboratory, stock 027831) were used for all experiments. Mice were housed in the vivarium with 12-hour light and dark cycle and were given food and water ad libitum. vGAT-IRES-cre mice (Jackson Laboratory, stock 016962) were used for some immunohistochemistry experiments.
Viruses
CANE-LV-Cre (titer, 5×108 ifu/mL; CANE-LV envelope [Addgene Plasmid #86666]) were produced as previously described. Various AAVs were co-injected with CANE-LV-Cre: AAV2/1-CAG-Flex-GFP (UNC Vector Core), AAV2/1-CBA-Flex-ChR2-mCherry (UPenn Vector Core), AAV2/1-hSyn-ChR2(H134R)-eYFP (UPenn Vector Core and Addgene), AAV2/1-Ef1a-DIO-eArch-eYFP (Addgene), AAV2/1-CAG-Flex-GCaMP6m (Addgene).
Surgical procedures
Viral delivery.
To capture and express desired transgenes in CeAGA neurons, FosTVA mice were anesthetized with isoflurane (1.5% isoflurane, 0.75% oxygen) for two hours (to induce Fos expression in CeAGA) in a chamber before mice were transported to a stereotaxic frame (David Kopf Instruments) and small craniotomies were created over the target region. The coordinates of CeA used relative to bregma were: AP = 1.15 or 1.20 +/− 0.05 mm, ML = 2.83 or 2.86 +/− 0.02 mm, DV = −4.17 or −4.22 +/− 0.03 mm. The CANE-LV-Cre and Cre-dependent AAV were mixed (1:1) prior to injection. 1 µl total was delivered at a rate of 60 nl/min per injection and left for 10 minutes post injection for efficient diffusion of the virus.
Optic fiber implantation.
After viral injection, an optical fiber (200 µm core diameter, ThorLabs) was inserted 300 µm above the injection site and secured using Metabond (Parkell) and dental cement. During post hoc immunohistochemistry, viral expression, site of injection and insertion of optical fiber was confirmed; animals with failed expression or off target optical fiber placement were excluded from all analysis.
EEG/EMG and CeAGA optogenetic experimental procedure.
Three stainless steel screws were placed on the left frontal, left parietal, and right cerebellar cortex as EEG electrodes and two headmount-coupled stainless steel leads (#8201-SS, Pinnacle technology Inc) were inserted into bilateral neck muscles as EMG electrodes. For optogenetic activation, one optical fiber was inserted on top of right CeA. All three EEG electrodes were further connected onto the headmount (#8201-SS, Pinnacle technology Inc). EEG and EMG were recorded with Sirenia Acquisition (Pinnacle technology Inc) at 1000 Hz. For optogenetic experiments, after 5 min of recorded EEG baseline in the recording chamber, three laser trains (20 Hz, 20 ms, 2 min on followed by 2 min off, ~4 mW from the fiber tip, 473-nm Blue Laser) were given per experimental animal.
GRIN lens implantation and baseplate attachment.
GRIN lens (7.3 × 0.6 mm, Inscopix) was implanted according to Inscopix instructions. A holder (Inscopix, Gripper Part ID: 1050–002199) was used to lower the miniature microscope with baseplate onto the top of the GRIN lens until the GCaMP6m fluorescence was visible under the illumination from the miniscope’s LED. Subsequently, the baseplate was fixed to the skull with dental cement darkened with carbon powder to prevent external light from contaminating the imaging field-of-view. A cover (Inscopix, Part ID: 1050–002193) was attached to the baseplate to protect the microendoscope.
Chronic Constriction Injury of the Infraorbital Nerve (CCI-IoN).
Animals were anesthetized with ketamine/xylazine and a small incision (~ 0.35 cm) parallel to the midline was made starting at the caudal end of the third row of whiskers toward the ipsilateral orbit. The superficial fascia was gently separated to expose the infraorbital nerve (IoN) trunk at its distal segment outside the orbital cavity. Two chromic ligatures (6–0, Angiotech) were loosely tied around the distal part of the IoN (1 mm apart). The wound was checked for hemostasis and the incision was closed with three 5–0 silk sutures (Angiotech).
In vivo optogenetic activation or silencing
Animals with optical fiber implants were connected to an optical patch cable (ThorLabs) coupled to either a 473 nm or 561 nm laser (Opto Engine LLC). Light pulse was controlled by a pulse generator (Master 8 or AIM-2 Optogenetic Interface). 473 nm laser was applied in pulsed mode (~3.5 mW/mm2, 20 Hz, 20 ms pulse width) to animals that expressed ChR2 in CeAGA and their respective GFP controls, while 561 nm laser was applied in continuous mode (~15 mW/mm2) to animals that expressed eArch in CeAGA and their respective GFP controls.
Immunohistochemistry
To detect general anesthesia-activated neurons, animals were anesthetized with isoflurane, or ketamine/xylazine, or dexmedetomidine, for two hours then transcardially perfused with 10% sucrose in cold phosphate buffer saline (PBS, pH 7.4) followed by 4% cold paraformaldehyde (PFA) fixation solution. To detect restraint stress-activated neurons, mice were placed in the restrainer for 90 min (TV-150, Braintree scientific Inc). All brains were post-fixed in PFA overnight at 4°C, cryoprotected in 30% sucrose PBS solution for 2–3 days at 4°C, frozen in O.C.T compound (Tissue-Tek, Sakura), and then stored at −80°C until sectioning. Floating brain sections (80 µm) were stained using standard immunofluorescent protocol. The primary antibody used were: goat anti-Fos (Santa Cruz Biotechnology, sc520g, 1:300)50, rabbit anti-Fos (Cell Signaling, #2250, 1:1500)51, anti-Neurotensin (ImmunoStar, 20072)52. The secondary antibody used are: Alexa Fluor 488 donkey anti-goat (Jackson ImmunoResearch, 705-545-147, 1:500)53, Alexa Fluor 647 anti-rabbit (Jackson ImmunoResearch, 711-605-152, 1:500)54, and Cy3 donkey anti-goat (Jackson ImmunoResearch, 705-165-147, 1:500)55 (see Life Sciences Reporting Summary).
Fluorescent in situ hybridization
For each brain collected after one hour of isoflurane anesthesia, 8–10 slices (60 µm thick) containing the central amygdala (CeA) were collected and in situ was performed as described previously11. Penk1, Pkc-d, Pdyn, Sst, and Fos probes were the same as the ones used by the Allen Brain Atlas. FITC-labeled Fos probe was paired with Dig-labeled Penk1, Pkc-d, Pdyn, or Sst probes to analyze the co-localization of Fos to these markers.
For 3-color HCR in situ hybridization to examine Fos, Penk1, and Pkc-d expression:
HCR in situ were performed as described10. Probes were ordered from Molecular Instruments. For each brain collected after one hour of isoflurane anesthesia, 8–10 ((60 µm thick) containing the central amygdala (CeA) were collected and hybridization chain reaction in situ was performed. On day 1, collected brain slices were exposed to probe hybridization buffer with HCR Probe Set. On day 2, brain slices were washed with probe wash buffer, received amplification buffer and amplifier. On day 3, brain slices were counter stained with DAPI and mounted. Penk1 (488 nm), Pkc-d (647 nm), and Fos (546 nm), probes were used to examine any overlaps between these markers.
Histological image acquisition and quantification
Image Acquisition.
Brain slices were visualized with a laser scanning confocal microscope (Zeiss 700). Entire brain slices were imaged at 10x resolution, while the entire CeA was imaged at 20x resolution using z-stack (~30 µm). Cells were manually quantified for co-localization and total Fos count between Fos expressing neurons and respective markers by a researcher blinded to the samples. For each animal, six slices were averaged for the entire CeA region before averaging percentages across all animals.
Projection Average Intensity Values.
The regions of interest/borders for each region were drawn according to the Allen Brain Atlas. The mean, or average intensity value was calculated for each region using the histogram function in Photoshop. The mean value was recorded across all samples and an average was computed across three samples.
Behavioral Tests
Formalin injection.
Formalin (Sigma, 37%) was diluted to 4% with PBS and 10 µl was unilaterally injected into the top of the hind paw or the whisker pad to induce inflammatory pain. The animals displayed self-recuperating behavior dependent on injection site such as licking of the hind paw or wiping of the whisker pad. Formalin injection induced 2 distinct phases of acute pain. Self-recuperating behavior was video recorded immediately after injection. During the first and second phase, optogenetic stimulation was initiated for 2 minutes-on and -off periods for 3 (first phase, 6 minutes stimulation) and 6 times (second phase, 12 minutes stimulation). Self-recuperating behavior were recorded and analyzed.
Spontaneous Wiping behavior.
After CCI-IoN, animals exhibited spontaneous wiping behavior as a result of the injury. Animals were placed into a clear cylindrical chamber and attached to the patch cable. Each animal was placed in the chamber and video recorded for 19 minutes (7-minute baseline, 5-minute stimulation, and 7-minute post-stimulation). Optogenetic illumination was turned on during the middle 5-min period and total time exhibiting spontaneous wiping behavior was measured.
Low-dose Ketamine Analgesia experiments.
Mice were injected with 12 mg/kg of ketamine and placed back into their home cage for six minutes. Then mice were given an injection of 10 ul of 0.2 μg/μl capsaicin (capsaicin was diluted with from a 10 µg/µl stock to 0.2 µg/µl in saline with 4% ethanol and 4% Tween-80). Mice were immediately video recorded for 15 minutes in a cylinder plexiglass container. Mice with optogenetic silencing received silencing during the first (0–5 min) and last (10–15 min) 5-minute bins. The total licking time was computed from the entire 15 minutes of recording as the animals exhibited self-recuperating behavior by licking the hind paw.
Hargreaves heat test.
To examine thermal sensitivity, animals were placed in the Hargreaves test enclosure for 5–10 minutes to acclimate. The guiding lines were used to position the infrared emitter/detector directly underneath the plantar region of the hind paws. The I.R. intensity was set to 40. The reaction time was recorded. Each animal received 3 trials per hind paw, each applied at least 30 seconds apart.
Cold dry ice test.
To examine cold sensitivity, animals were placed in a 13 × 13 × 20 cm chamber that was raised 30.5 cm above the floor and left to acclimate for 5–10 minutes. The floor of the chamber was a 0.80 mm thick polycarbonate sheet. Dry ice were pounded into a fine powder and packed tightly into a ½ inch diameter syringe in order to form a cylindrical shape. Dry ice was pushed out of the syringe and centered on the plantar region of the hind paws. The syringe was promptly removed after the animal displayed a withdrawal reflex. Withdrawal behavior was video recorded. Each animal received 3 trials per hind paw, each applied at least 10 seconds apart.
von Frey test on the face.
To assess mechanical sensitivity, animals received 10 repeated application (10–20 seconds apart) of various von Frey filaments with increasing forces (.008 to 1 gram) to the whisker pads (with all whiskers kept intact). In the CCI-IoN model, filaments were applied near the ligation injury. Withdrawal Threshold was defined as the first filament that induced withdrawal latency over 50% of the 10 repeated applications.
von Frey test on the paw.
To assess mechanical sensitivity, animals received 10 repeated application (10–20 seconds apart) of various von Frey filaments with increasing forces (0.40g to 4.0g) to the hind paws.
Electronic von Frey test on the hind paws.
The electronic von Frey (eVF) test was used exclusively for the hind paws. The eVF rigid tip was applied to the plantar region of the hind paws and the withdrawal threshold was recorded. The calibration was set at reaching 50 grams in 5 seconds. Each animal received 3 trials per hind paw, each applied at least 10–20 seconds apart.
Open Field test.
To assess locomotion and anxiety-like behavior, animals were placed in a 30 × 30 cm box. Each animal was placed in the center of the box and locomotion activity and freezing behavior was recorded for a total of 15 minutes. Animals were attached to the patch cable and optogenetic illumination was turned on during the middle 5-min period. ANY-Maze software (Stoelting Co.) was used to create a 15 × 15 cm center zone within the box and also used to analyze the video recording.
Elevated Plus Maze test.
To assess anxiety-like behavior to optogenetic illumination of CeA, mice were placed in an elevated plus maze for 15 minutes. Animals were attached to the patch cable and received optogenetic illumination during the middle 5-minute period. Locomotion and time spent in the open and closed arms were analyzed and recorded using ANY-Maze software.
Conditioned Place Preference/Aversion test.
To determine if optogenetic activation or silencing of CeAGA induced a place preference or aversion, animals were placed in a 2-chamber 20 × 40 cm box and attached to a patch cable. Each chamber contained two distinct visual patterns of horizontal and vertical stripes. On day 1, animals were placed in the center of the box and left to explore the box without illumination for 10 minutes. The preference of the animal was determined by the chamber the animal spent more time in. For the next 4 days (day 2–5), animals received illumination on day 3 and 5, and no illumination on day 2 and 4. On the last day (day 6), animals could explore freely the 2 chambers to determine the new place preference without illumination. ChR2-expressing animals received illumination on the side it did not prefer for 22 minutes with 2 min on and off periods, while eArch-expressing animals received illumination on the side it did prefer for 10 minutes continuously. This test was designed to bias against the animal’s natural preference. ANY-Maze software was used to analyze the time spent in each chamber.
In a separate experiment, CCI-IoN animals expressing ChR2 or GFP controls were also placed in the same 2-chamber box to determine if optogenetic activation of CeAGA altered the natural place preference in a state of chronic pain.
Ultrasonic Vocalization and Courtship Behavior.
Male animals were placed in a clear cylindrical chamber with estrus female BL6 animals for 90 sec (baseline), then male animals received optogenetic stimulation for 90 sec. Male ultrasonic vocalizations were recorded with an ultrasonic microphone (CM16/CMPA-P48; Avisoft-Bioacoustics) and analyzed with MUPET56. Courtship behavior was defined as anogenital sniffing and mounting. Behavior was video recorded, and the differences of total syllables elicited during ultrasonic vocalizations and the duration of courtship behavior with and without light stimulation were quantified.
In Vivo Calcium Imaging and Image Data Analyses
Order of calcium imaging recording in behavioral experiments.
After the baseplate attachment, each of the CANEISO-GCaMP6m CeAGA mouse was acclimated to handling and attachment of the Inscopix miniature microscope for 10–15 min per day for 3–5 days. Doric TTL Plus Generators (Doric, OTPG_4) was used to trigger and synchronize behavioral video recordings with calcium recordings.
Part1, anesthesia and noxious stimuli test.
We first imaged neural activities during re-exposure to isoflurane induced anesthesia (Day 1, 5min baseline followed by 20 min of 1.5% isoflurane mixed with oxygen). On day 3, we imaged the neurons during ketamine/xylazine induced anesthesia (5min baseline followed by i.p. injection of ketamine (100mg/kg) /xylazine (10mg/kg) and imaged for 20 min continuously). Day 5 to day 8, we imaged the neurons before, during, and after applications of noxious stimuli (dry ice, laser heat, von Frey on hind paw and von Frey on facial pad), with 1 day interval between each sensory test experiment. 2 days after, we repeated imaging of neural activities in some mice during 1.5% isoflurane (day 10) and ketamine /xylazine (day 12) induced anesthesia. 7 mice underwent imaging under anesthesia, 6 of them underwent cold and heat stimuli, 4 of them underwent von Frey stimuli on the hind paws and facial pads.
Part2, anesthesia and stress.
Calcium imaging recordings of restraint stress (8 min baseline followed by 8 min restriction of AIMSTM Rodent restraint bags, and later 8 min release from the restriction), and isoflurane anesthesia (1.5%, 5min baseline followed by 20 min of 1.5% isoflurane mixed with oxygen) were done separately within one day (Day 1). Then, day 3, we imaged neurons during low dose ketamine (5min baseline followed by i.p. injection of ketamine (12 mg/kg) and imaged for 20 min continuously), and regular dose ketamine on day 4 (5min baseline followed by i.p. injection of ketamine (100 mg/kg) and imaged for 20 min continuously). On day 6, we imaged neurons during low dose isoflurane (0.5%, 5min baseline followed by 20 min of 0.5% isoflurane mixed with oxygen). Mice moved around under low dose ketamine and low dose isoflurane administration. On day 8, we repeated imaging recordings of restraint stress and isoflurane (1.5%) induced anesthesia separately. 5 mice went through this series of imaging.
Imaging data analysis:
All the calcium imaging data were processed from the raw video by MIN1PIPE17, where videos underwent background subtraction, movement correction, automatic seeds selection and ROI separation. The extracted ROIs were then manually inspected again by experienced researchers to ensure only the most reliable units were included. The traces of the refined set of ROIs were then rescaled to the same range (between 0 and 1 with arbitrary unit).
For the isoflurane and ketamine experiments, the refined traces were aligned according to the delivery moment of the anesthetics, and the window from 4 min prior to (baseline, −4 – 0 min) to 20 min post (isoflurane or ketamine, 0–20 min) the delivery moment was selected to perform data analysis. The aligned traces were sorted by the average calcium fluorescent intensity ratio of all the units, first sorted by (isoflurane or ketamine period) / (baseline) (Fig. 2 c and e), and then sorted by (last 10 min of isoflurane or ketamine) / (first 10 min of isoflurane/ketamine) (Fig. 2 d and f). The sorted traces were roughly classified into isoflurane- or ketamine- active and suppressed group (Fig. 2 c and e), or sustained, transient and suppressed groups (Fig. 2 d and f, Extended Data 3 a and b), respectively, where the average trace of each group was calculated. To further examine the finer dynamic property of different groups, the percentage contribution of the last 10 min to the overall 24 min duration of each trace was calculated. The distribution of the sustained activity and transient activity in percentage contribution was then calculated from each group or each mouse
To evaluate the isoflurane and ketamine neural response, we in general applied 4 approaches including direct calculation and corrected calculation (Fig. 2h, right panel, Extended Data 3 c and d). Direct calculation method is computing the ratio of the mean activity for stimulus ON (isoflurane or ketamine delivery) and OFF (baseline) periods, indicated by Mean/Mean, where activity in the stimulus ON period might be underestimated due to the changes of brain dynamics during the long imaging time window over anesthesia (20 min). Therefore, we also calculated the corrected (effective) mean intensity (Eff.) for the ON and/or OFF periods (Eff./ Eff. or Eff. / Mean) before computing the ON/OFF activity ratio, by computing only the average activity during the time when the calcium intensity is at least two median absolute deviation above the overall mean. In addition, we calculated the effective active duration of individual neurons. Neurons with effective active time over certain amount of time (1 min) after stimulus ON (anesthesia onset) could be considered as activated by the stimulus (Eff. time).
In the sensory stimuli application experiments (Fig. 4), for every stimulus applied in these experiments, a trial was defined with a window of 10 s before and 10 s after the stimulus. All the traces from the same experimental condition were epoched based on the trials, and were then pooled together and sorted by the peak activity latency within the trial. The mean activity trace was calculated by averaging across the epoched traces of all neurons and all trials. For each individual neuron, the sorted traces from all trials of the same condition were further pooled together to show the neuronal response to the stimuli. To investigate the neuronal response change, the pooled traces were averaged and the overall activity difference between the 5 s post-response average and the 5 s pre-response average was calculated for all individual neurons, based upon which the empirical probability distribution was computed across neurons. To group the neurons based on the response types, the activity difference of individual neurons was compared with a threshold of two times of the standard deviation of the averaged neuronal activity in that trial. If the activity difference was higher than the threshold, the neuron was considered to be in the activity increased group; if the activity difference was lower than the negative of the threshold, the neuron was considered to be in the activity suppressed group; otherwise the neuron was classified as being in the unchanged group. The proportion of each group was then calculated.
In the restraint stress experiment (Extended data 3), 1st 8 min was recorded for baseline spontaneous activity (−8 – 0 min), 2nd 8 min (0 – 8 min) was recorded when mice were under restraint, and 3rd 8 min (8 – 16 min) was recorded when mice released from the restraint. To further quantify the stress neural responses and compared with their responses to isoflurane (as shown in Extended data 3e–h), we computed the corrected/effective mean calcium intensity for the stress period and two baseline periods. Only the time points when signal intensity was over two median absolute deviation above the mean were considered as effective time points. Then we calculated the ratio between the corrected mean intensities for each neuron, and label the neuron to be stress activated if the ratio is above 1 while stress suppressed if the ratio is below 1. To further have a more conservative calculation, we excluded the neurons with maximum intensity below 0.1 (normalized intensity) over the whole duration. The remaining neurons were labeled as the robust group, and used for later cross-analysis with isoflurane-responses.
Same cell tracking (cross day analysis) of CeAGA calcium activity:
To register neurons across days from different calcium imaging recordings sessions of the same mouse, we adapted CellReg method with Log Demons registration method used in MIN1PIPE17,18. In brief, all the extracted ROIs from each session to be tracked were collected, and then the modified CellReg was applied. Only the ROIs that were reliably tracked were used for subsequent analyses.
EEG analysis.
The EEG signal was first epoched to laser on (2 min) and off (2 min) periods, and the power spectrum was calculated using FFT. The power spectrum was then normalized to the full frequency band on which each epoch is valid, and only the band [0, 20Hz] was extracted to further compute the average trace across the 9 epochs (N= 3 mice, 3 repetitions in each mouse).
Statistics
All statistical analyses were performed in GraphPad Prism 8. Behavior data was analyzed with 2-way ANOVA followed by Tukey, Sidak, and Dunnett’s post hoc multiple comparison test when appropriate. All t-tests were performed as two-tailed. Significance levels are indicated as follows: *: P<0.05, **:P<0.01, ***: P<0.001, and ****: P<0.0001, and precise P-values are given when appropriate in Supplementary Table 1. Sample sizes were determined based on previous publications in the lab and common practice in animal behavior experiments11,12,57,58 (see Life Sciences Reporting Summary). We did not observe any sex-dependent differences in all our experiments, therefore results from males and females were grouped together and analyzed together. Data distribution was assumed to be normal but this was not formally tested, instead all graphs contain individual data points, and mean±s.e.m..
Randomization and Data Collection
Animals were randomly assigned to various treatment groups to receive either GFP control, channelrhodopsin, or enhanced archaerhodopsin. Data analysis were performed blind to the conditions of the experiment by a different experimenter. All animals were used as data points, and animals were only excluded from analysis if they were incorrectly targeted virally.
Data and Code Availability Statement
All raw data described in this study are available from the corresponding authors upon request. All codes described in this study are available from the corresponding authors upon request. The MIN1PIPE code for processing miniscope imaging (correspondence should be addressed to Jinghao Lu, jinghao.lu@duke.edu) is available at: https://github.com/JinghaoLu/MIN1PIPE
Extended Data
Extended Data Fig. 1. Isoflurane general anesthesia activates different neuronal ensembles and molecular marker analysis of CeAGA neurons.
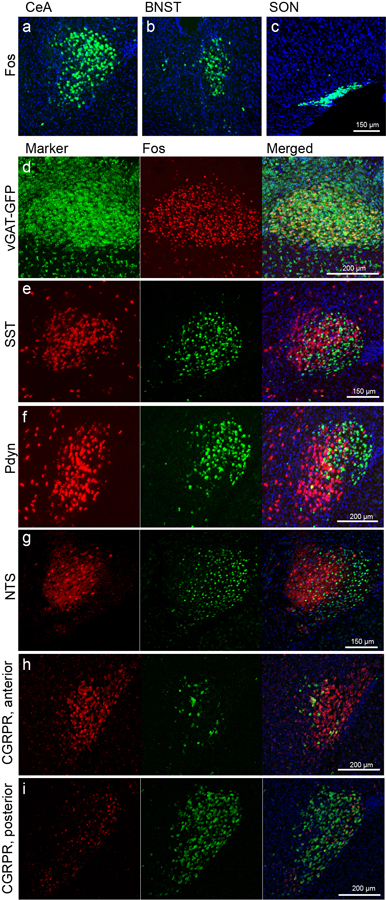
General anesthesia activated the a, central amygdala (CeA), b, bed nucleus of stria terminalis (BNST), and c, super optic nucleus (SON). Representative images two-color experiments examining the expression of various markers in Fos+ CeAGA neurons (induced by isoflurane). d, CeAGA neurons are a subset of GABAergic (vGAT) neurons in the central amygdala and all CeAGA express vGAT-GFP. e-i, CeAGA neurons have minimal to no overlap with e, somatostatin (SST), f, prodynorphin (Pdyn), g, neurotensin (NTS), and h-i, calcitonin gene-related peptide receptor (CGRPR) expressing neurons in CeA.
Extended Data Fig. 2. Representative post hoc histology and session distributions of activity of CANE-captured isoflurane-activated CeAGA neurons in response to isoflurane or ketamine.
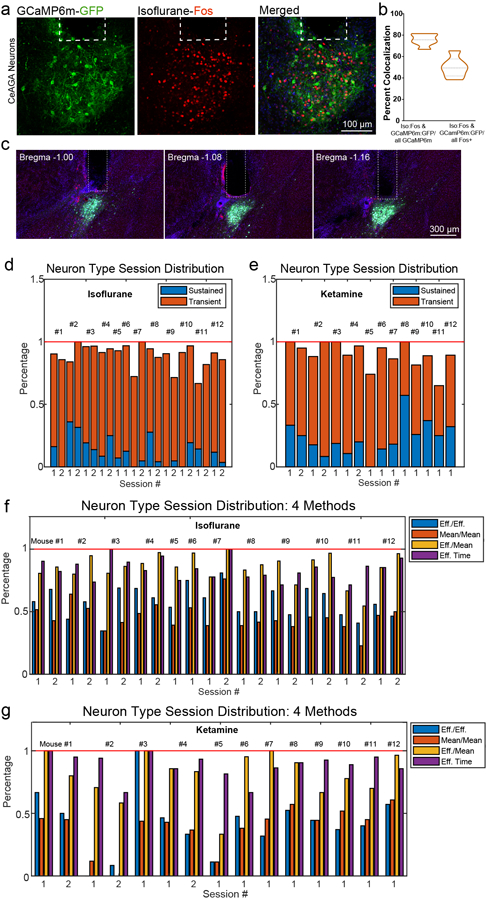
a, Representative images of CANE-GCaMP6m+ neurons (green) and isoflurane-activated Fos+ neurons (red) and their overlap. Dotted box showing the placement of the GRIN lens in CeA. b, Quantification of the percent colocalization of Iso:Fos/all GCaMP6m (75.45±5.04%) and GCaMP6m:GFP/all Fos (50.1±8.70%) (n=6 animals). Data are mean ± s.e.m. c, Three consecutive representative images of post hoc histology showing optic fiber tract into the CeA from bregma −1.00 mm to −1.16 mm. Optic fiber diameter is 200 µm. d, Session-wise percentage distribution of iso.-sustained and iso.-transient neurons from Figure 2d. e, Session-wise percentage distribution of ketamine-sustained and ketamine-transient neurons from Figure 2f. f and g, Session-wise percentage distribution of the isoflurane-activated neurons (f), or the ketamine-activated neurons (g), calculated using four methods calculated based on the ratio between the post- and pre-stimulation (i.e. anesthetic administration) activity. Cyan, Eff./Eff., mean activity of (post-stim) effective time / mean activity of (pre-stim) effective time. Orange, Mean/Mean, mean activity of all the (post-stim) time /mean activity of all the (pre-stim) time. Yellow, Eff./Mean, mean activity of (post-stim) effective time / mean activity of all the (pre-stim) time. Purple, Eff. time, total (post-stim) effective time. “Effective time” refers to the time points of a neural trace whose intensity is two median absolute deviation above its mean. Only these time points were considered to compute the effective mean. See Methods for details of the 4 methods. Pre-, pre-stimuli, awake state (−4 – 0 min). Post-, post-stimuli, isoflurane or ketamine (0–20 min).
Extended Data Fig. 3. CANE-captured CeAGA neurons are mostly inhibited by stress.
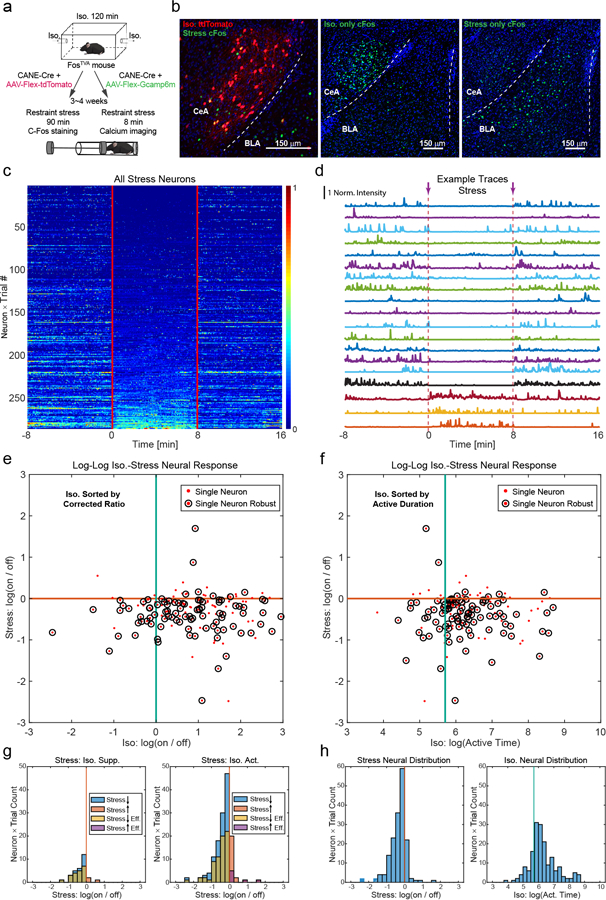
a, Schematic of CANE captured isoflurane-activated CeAGA neurons followed by a second exposure to restraint stress (left, for Fos expression, right, for calcium imaging). b, Left, representative images of CANEISO-tdTomato neurons (red) and stress-activated Fos+ neurons (green) and their overlap. (n=3 animals). Right, representative images of isoflurane-only activated Fos+ neurons (green, Repeated experiments n=5 biologically independent samples.) and compared to stress-only activated Fos+ neurons (green, Repeated experiments n=3 biologically independent samples.) in the CeA. c, Heatmap, activity patterns of CANEISO-GCaMP6m captured CeAGA neurons in stress experiment sorted by the average activity during stress period (−8 – 0 min, pre-stress; 0 – 8 min, restraint stress; 8 −16 min, post-stress. 282 neurons from 5 mice × 2 trials). A small number of neurons (in the bottom of the heatmap) were activated by stress. d, Example traces of CANEISO-GCaMP6m captured CeAGA neurons in stress experiment showing both stress-inhibited and stress-activated neurons. Norm. Intensity: normalized calcium signals rescaled to 0–1. e, Scatter plots of the tracked same neurons based on isoflurane and stress related activity patterns. Each dot is calculated based on effectiveness corrected activity ratio between the post- and pre-stimulus periods for isoflurane-stimulus and stress-stimulus, separately. Corrected ratio, mean activity of (post-) effective time / mean activity of (pre-) effective time. Single dots represent individual neurons in the logarithmic scale coordinates, and the circled dots represent robustly firing neurons, with the maximum intensity of each neuron in the whole duration exceeding a threshold. f, Same plots with isoflurane responses calculated by actively firing time exceeding a threshold (1 min). Active duration, total (post-) effective time of effective moment>1 min. “Effective time” refers to the time points of a neural trace whose intensity is two median absolute deviation above its mean. Only these time points were considered to compute the mean. g, Neuron count summary of e. Left, Neuron count distribution of activity of isoflurane-suppressed neurons during stress. Right, Neural count distribution of activity of isoflurane-activated neurons during stress. h, Neural count distribution of f. Left, marginal count distribution of activity of CANEISO-GCaMP6m captured CeAGA neurons during stress. Right, marginal count distribution of activity of CANEISO-GCaMP6m captured CeAGA neurons during isoflurane GA. (282 total neurons from 5 mice × 2 trials.)
Extended Data Fig. 4. Manipulation of CeAGA neurons did not induce anxiety-like or fear-like behavior or change the gross brain state.
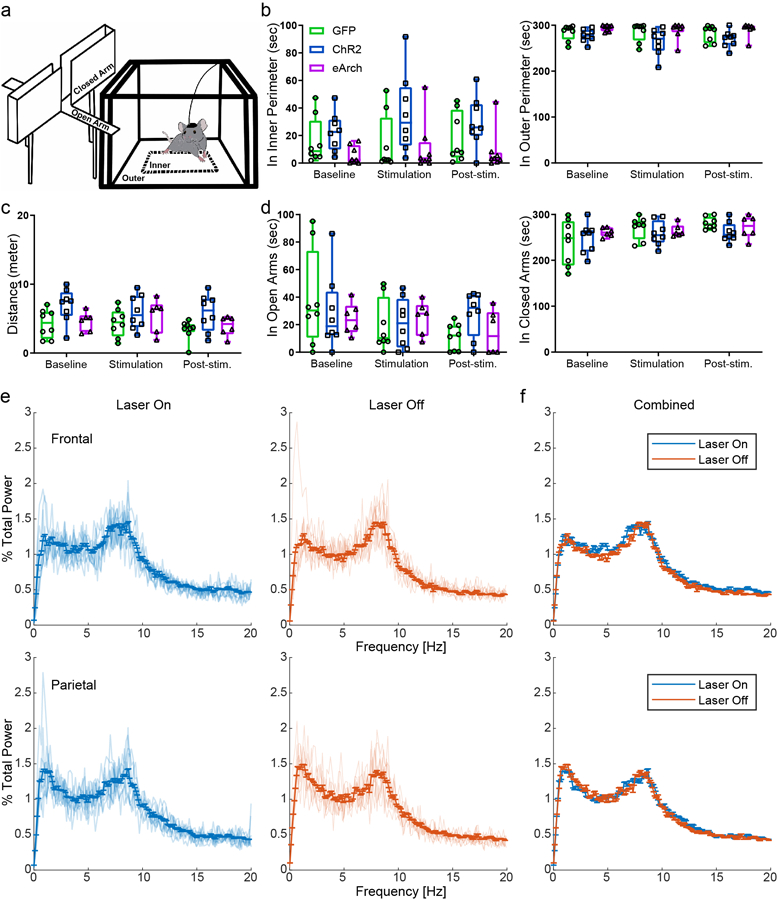
a, Schematics of the Elevated Plus Maze (left) and Open Field (right) apparatus. b, Quantification of total time spent in the inner (GFP: 15.67 ± 5.97s (baseline), 14.18 ± 7.24s (stim), 18.07 ± 6.38s (post); ChR2: 22.59 ± 4.92s (baseline), 35.90 ± 10.22s (stim), 29.73 ± 6.49s (post); eArch: 5.69 ± 2.32s (baseline), 11.01 ± 6.61s (stim), 8.54 ± 5.20s (post)) and outer perimeter (GFP: 284.32 ± 6.01s (baseline), 285.70 ± 7.25s (stim), 281.80 ± 6.39s (post); ChR2: 277.31 ± 4.92s (baseline), 264.00 ± 10.22s (stim), 270.17 ± 6.47s (post); eArch: 294.31 ± 2.32s (baseline), 288.99 ± 6.61s (stim), 291.46 ± 5.20s (post)) of the Open Field Test (control, n=8 animals, ChR2, n=8 animals, eArch, n=6 animals; two-way repeated measures ANOVA; P-value was above 0.05, no significance; F4,42=0.8743 (inner), F4,42=0.8633 (outer)). Data are mean ± s.e.m. c, Quantification of total distance travelled (GFP: 4.27 ± 0.73m (baseline), 4.36 ± 0.72m (stim), 3.36 ± 0.51m (post); ChR2: 6.92 ± 0.87m (baseline), 5.91 ± 0.88m (stim), 5.85 ± 0.95m (post); eArch: 4.59 ± 0.56m (baseline), 5.45 ± 0.99m (stim), 3.91 ± 0.59m (post)), and d, total time spent in the open (GFP: 38.53 ± 12.26s (baseline), 19.19 ± 6.65s (stim), 11.03 ± 3.44s (post); ChR2: 28.61 ± 9.69s (baseline), 21.21 ± 6.31s (stim), 27.91 ± 5.76s (post); eArch (24.33 ± 4.70s (baseline), 24.85 ± 4.98s (stim), 14.35 ± 6.56s (post)) and closed arms (GFP: 239.16 ± 17.28s (baseline), 270.86 ± 8.54s (stim), 280.76 ± 4.61s (post); ChR2: 248.58 ± 11.66s (baseline), 258.80 ± 9.36s (stim), 261.08 ± 7.60s (post); eArch (261.22 ± 4.07s (baseline), 264.45 ± 5.50s (stim), 272.48 ± 10.19s (post)) of the Elevated Plus Maze (control, n=8 animals, ChR2, n=8 animals, eArch, n=8 animals; two-way repeated measures ANOVA; P-value was above 0.05, no significance; F4,38=1.083 (distance), F4,38=1.402 (open arms), F4,38=1.355 (closed arms)). Data are mean ± s.e.m. e, Power spectrum of EEG signals in the frontal and parietal cortex. Left, laser on, right, laser off. f, Overlap of power spectrum of EEG signals in the frontal and parietal cortex from e. The mean spectrum in each condition was calculated from the average across 9 sessions (n= 3 mice, 2 min laser on / 2 min laser off, 3 repetitions in each mouse). The error bar represents the standard error. The power spectrum was normalized.
Extended Data Fig. 5. Manipulations of CeAGA neurons modulated reflexive withdrawal threshold to von Frey filaments and activation of CeAGA neurons did not alter courtship behaviors.
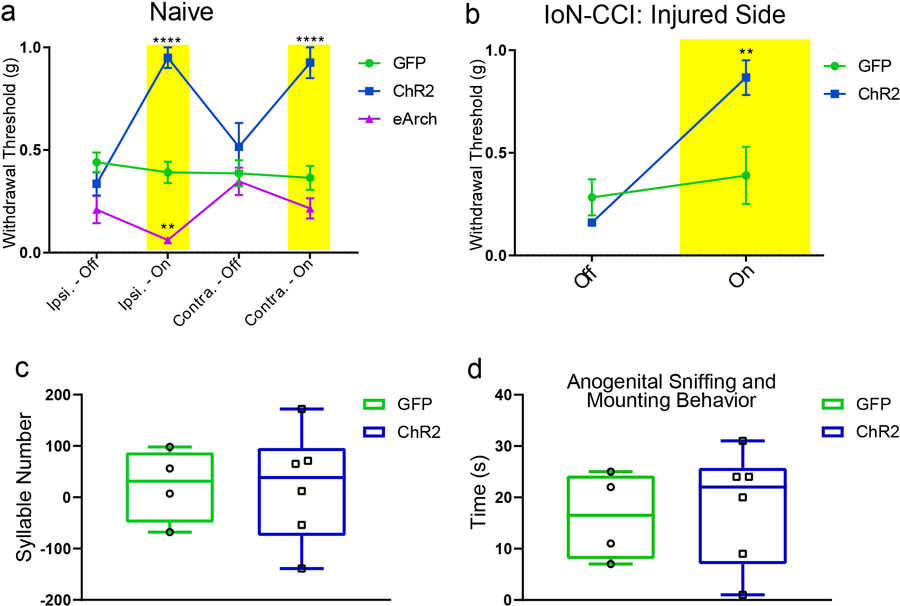
a-b, Quantifications of the withdrawal threshold to von Frey filaments applied to the whisker pad in a, naïve (control, n=9 animals (0.44 ± 0.0g5 (ipsi-off), 0.39 ±0.05g (ipsi-on), 0.39 ±0.06g (contra-off), 0.36 ±0.06g (contra-on)), ChR2, n=8 animals (0.34 ± 0.06g (ipsi-off), 0.95 ±0.05g (ipsi-on), 0.52 ±0.12g (contra-off), 0.93 ±0.08g (contra-on)), eArch, n=7 animals (0.21 ± 0.07g (ipsi-off), 0.06 ± 0.01g (ipsi-on), 0.35 ± 0.07g (contra-off), 0.22 ± 0.05g (contra-on)); two-way ANOVA; ****P<0.0001, **P=0.0042; F6,84=10.80) and b, IoN-CCI mice (control, n=6 animals (0.28 ± 0.09g (off), 0.39 ± 0.14g (on)), ChR2, n=6 animals (0.16 ± 0g (off), 0.87 ± 0.08g (on)); two-way ANOVA; **P=0.0032; F1,20=10.54). c-d, Quantification of light-illumination induced changes in total syllable number of ultrasonic vocalizations (GFP: 23.25 ± 35.65; ChR2: 21.17 ± 44.17), or total duration of anogenital sniffing and mounting behavior (GFP: 16.25 ± 4.31s; ChR2: 18.17 ± 4.53s) in control CeAGA-GFP and CeAGA-ChR2 mice (with light - without light) during 2 min of social interactions (control, n=4 animals, ChR2, n=6 animals; unpaired t-test, two-tailed; P=0.974, F5,3=2.303 (syllable number), P=0.7791, F5,3=1.656 (anogenital sniffing)). Data are mean ± s.e.m.
Extended Data Fig. 6. Activation of the left CeAGA neurons modulated pain-related behaviors in naïve mice and acute pain models.
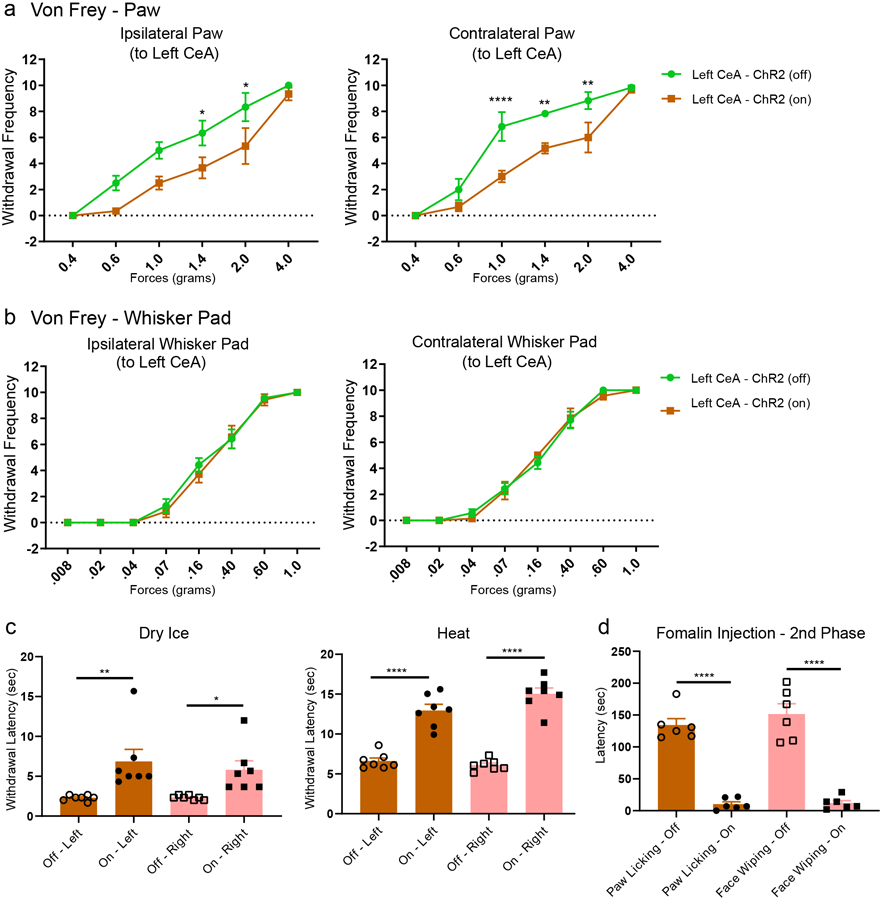
a, Quantification of effects of optogenetic activation of the left CeAGA neurons on the paw withdrawal frequency to six graded von Frey filaments ranging from 0.4 to 4.0 grams applied to the ipsilateral (Off: 0 ± 0 (0.40g), 2.50 ± 0.56 (0.60g), 5.00 ± 0.63 (1.0g), 6.33 ± 0.95 (1.40g), 8.33 ± 1.09 (2.0g), 10 ± 0 (4.0g); On: 0 ± 0 (0.40g), 0.33 ± 0.21 (0.60g), 2.50 ± 0.50 (1.0g), 3.67 ± 0.80 (1.40g), 5.33 ± 1.38 (2.0g), 9.33 ± 0.49 (4.0g)) or contralateral paw (Off: 0 ± 0 (0.40g), 2.00 ± 0.82 (0.60g), 6.83 ± 1.11 (1.0g), 7.83 ± 0.17 (1.40g), 8.83 ± 0.65 (2.0g), 9.83 ± 0.17 (4.0g); On: 0 ± 0 (0.40g), 0.67 ± 0.33 (0.60g), 3.00 ± 0.45 (1.0g),5.0 ± 0.37 (1.40g), 6.0 ± 1.15 (2.0g), 9.67 ± 0.21 (4.0g)) to the left CeA. (Ipsilateral and contralateral, ChR2, n=6 animals; two-way ANOVA; *P=0.0500 (2.0g), *P=0.0217 (1.4g), ****P<0.0001, **P=.0023 (1.4g), **P=.0029 (2.0g); F1,60=20.51 (ipsi), F1,60=28.81 (contra)). b, Quantification of optogenetic activation of the left CeAGA neurons showed that this manipulation did not induce any change in the head withdrawal frequency to eight von Frey filaments ranging from 0.008 to 1.0 gram applied to either the ipsilateral (Off: 0 ± 0 (.008g), 0 ± 0 (0.02g), 0 ± 0 (0.04g), 1.0 ± 0.52 (0.07g), 4.33 ± 0.61 (0.16g), 6.50 ± 0.85 (0.40g), 9.67 ± 0.21 (0.60g), 10.0 ± 0 (1.0g); On: 0 ± 0 (.008g), 0 ± 0 (0.02g), 0 ± 0 (0.04g), 0.67 ± 0.49 (0.07g), 4.17 ± 0.54 (016g), 7.00 ± 0.89 (0.40g), 9.83 ± 0.17 (0.60g), 10.0 ± 0 (1.0g)) or the contralateral whisker pad (Off: 0 ± 0 (.008g), 0 ± 0 (0.02g), 0.50 ± 0.34 (0.04g), 2.50 ± 0.50 (0.07g), 4.67 ± 0.49 (0.16g), 8.0 ± 0.68 (0.40g), 10.0 ± 0 (0.60g), 10.0 ± 0 (1.0g); On: 0 ± 0 (.008g), 0 ± 0 (0.02g), 0.17 ± 0.17 (0.04g), 2.33 ± 0.80 (0.07g), 5.17 ± 0.17 (016g), 8.17 ± 0.79 (0.40g), 9.83 ± 0.17 (0.60g), 10.0 ± 0 (1.0g)) to the left CeA. (Ipsilateral and contralateral, ChR2, n=6 animals; two-way ANOVA; not significant P>0.05; F1,80=0.9205 (ipsi), F1,40=0.000 (contra)). c, Quantification of the optogenetics induced changes in withdrawal latency (sec) to dry ice (2.29 ± 0.33s (off-left), 6.86 ± 3.70s (on-left), 2.33 ± 0.25s (off-right), 5.81 ± 2.75s (on-right)) (ChR2, n=7 animals; one-way ANOVA; **P=0.0045, *P=0.0311; F3,24=6.241) and heat (6.61 ± 0.93s (off-left), 12.94 ± 1.89s (on-left), 6.09 ± 0.65s (off-right), 15.04 ± 1.80s (on-right)) (ChR2, n=7 animals; one-way ANOVA; ****P<0.0001; F3,24=59.93). d, Quantification of total licking and face wiping latency (sec) from left CeAGA neurons optogenetic activation after formalin injection during the second phase of inflammatory pain (134.33 ± 22.88s (paw licking-off), 10.33 ± 8.24s (paw licking-on), 151.67 ± 35.99s (face wiping-off), 12.00 ± 8.74s (face wiping-on)). (ChR2, n=6 animals; one-way ANOVA; ****P<0.0001; F3,20=59.54).
Extended Data Fig. 7. CeAGA activities are not correlated to the onsets of sensory stimuli.
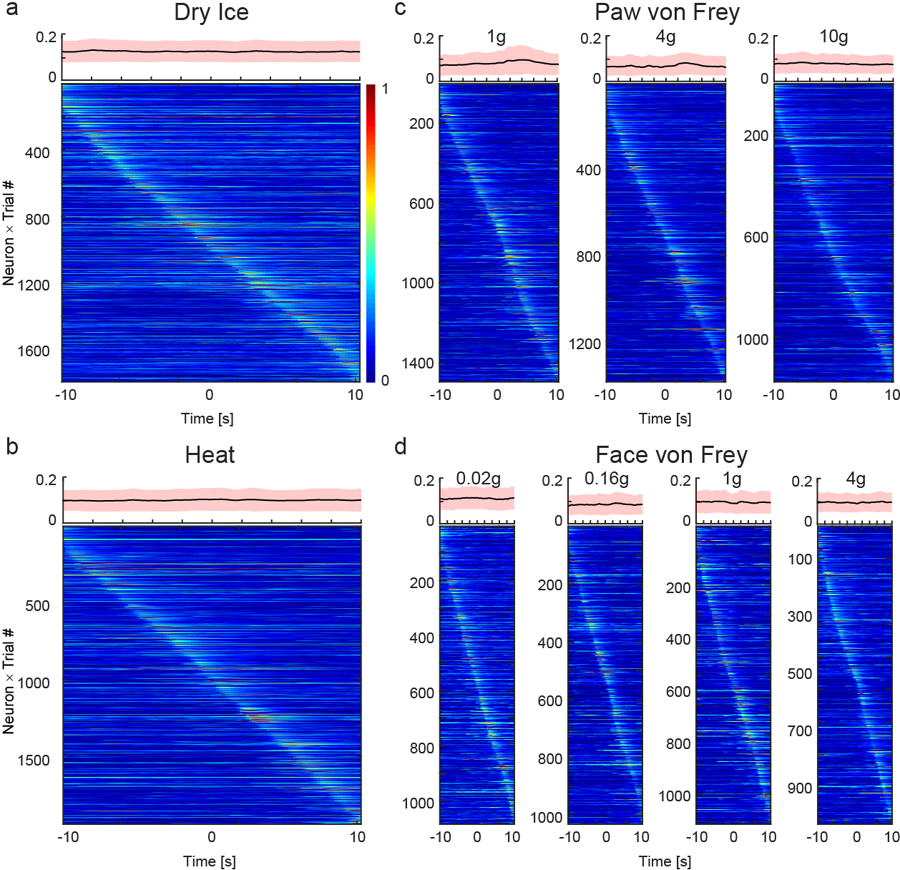
Neuronal activity patterns during a, cold, b, heat, c, paw von Frey, and d, face von Frey tests sorted by neurons peak responses timing: from −10 to +10 sec, with 0 as the onset of stimulus application. Top of each heatmap, averaged population activity from 10 seconds before to 10 seconds after each stimulus onset. Thick lines indicated mean and shaded areas indicated s.e.m.
Extended Data Fig. 8. CeAGA neurons are distinct from pain-activated neurons in CeA and high magnification image of CeAGA neurons projection into the ipsilateral BLA.
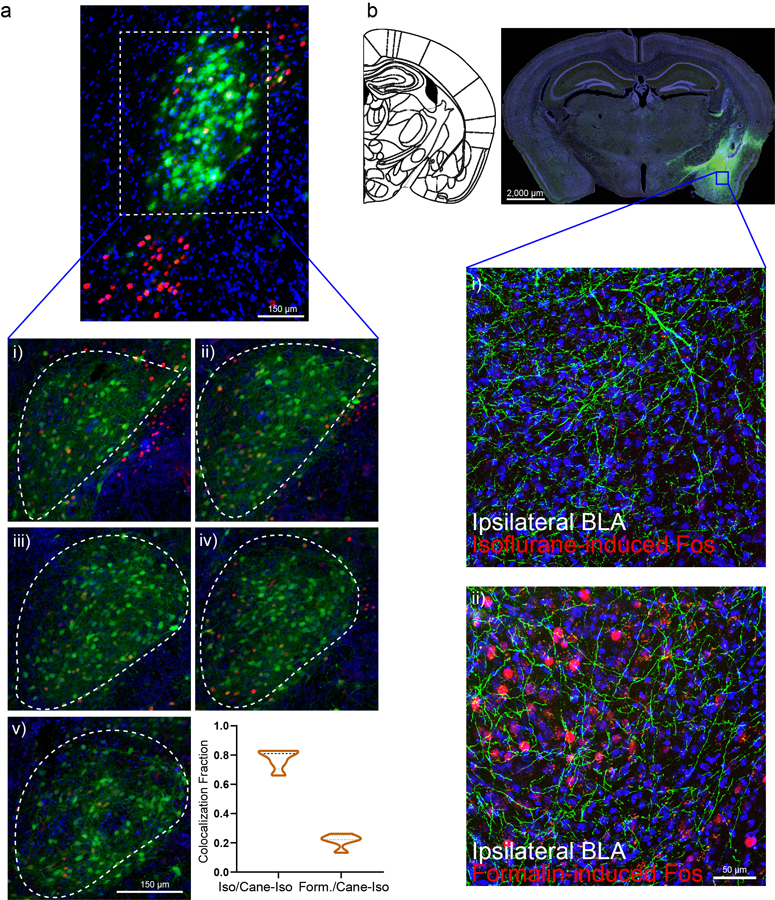
a, CeAGA neurons have minimal co-localization with formalin-induced Fos+ cells. Formalin-activated cells primarily locate in the capsular division of CeA outside the lateral division where CeAGA locate. Insert i)-v), Example of five consecutive slices of the lateral division of CeA showing minimal co-localization with formalin-induced Fos+ cells with CeAGA neurons, and the quantification of fraction of co-colocalization between CANE-captured CeAGA cells and formalin-induced Fos+ cells (n=5 biologically independent samples for each condition) (0.78 ± 0.06 (Iso/Cane-Iso); 0.22 ± 0.04 (Form/Cane-Iso)).b. Coronal schematic next to example coronal slice of low magnification (high exposure) of CANE-GFP labeled CeAGA neurons and their axons with a box around the ipsilateral BLA. Insert i)-ii), High mag images show projections in ipsilateral BLA with top panel showing that isoflurane did not induce Fos+ cells, and bottom panel showing that formalin-pain induced robust Fos+ expression in BLA. (n=3 biologically independent samples).
Extended Data Fig. 9. Consistent axonal projections from CeAGA neurons.
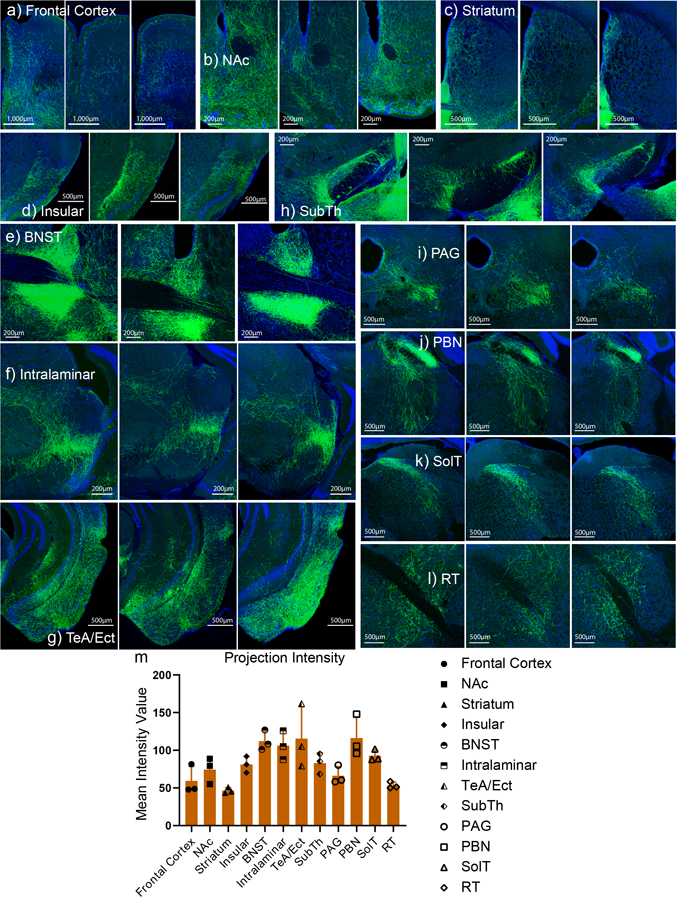
In sequential order: a) frontal cortex, b) nucleus accumbens (NAc), c) striatum, d) insular, e) bed nucleus stria terminalis (BNST), f) intralaminar, g) temporal association cortex (TeA) and ectorhinal cortex (Ect), h) subthalamic nucelus (SubTh), i) periaqueductal grey (PAG), j) parabrachial nucleus (PBN), k) solitary tract (SolT), and l) reticular formation (RT). m) Quantification of the mean intensity value (artificial units) of the axonal projections from each region of interest (ROI) listed above (a-l) (n=3 biologically independent samples) (59.40 ± 15.68 (FC), 74.38 ± 14.18 (NAc), 46.59 ± 3.21 (Striatum), 81.07 ± 8.86 (Ins), 112.09 ± 10.85 (BNST), 106.15 ± 15.69 (Intra), 115.35 ± 34.52 (TeA/Ect), 82.99 ± 11.06 (SubTh), 66.32 ± 9.84 (PAG), 116.29 ± 22.67 (PBN), 92.70 ± 6.27 (SolT), 53.26 ± 3.63 (RT)).
Extended Data Fig. 10. CeAGA neurons are also activated by low dose anesthetics.
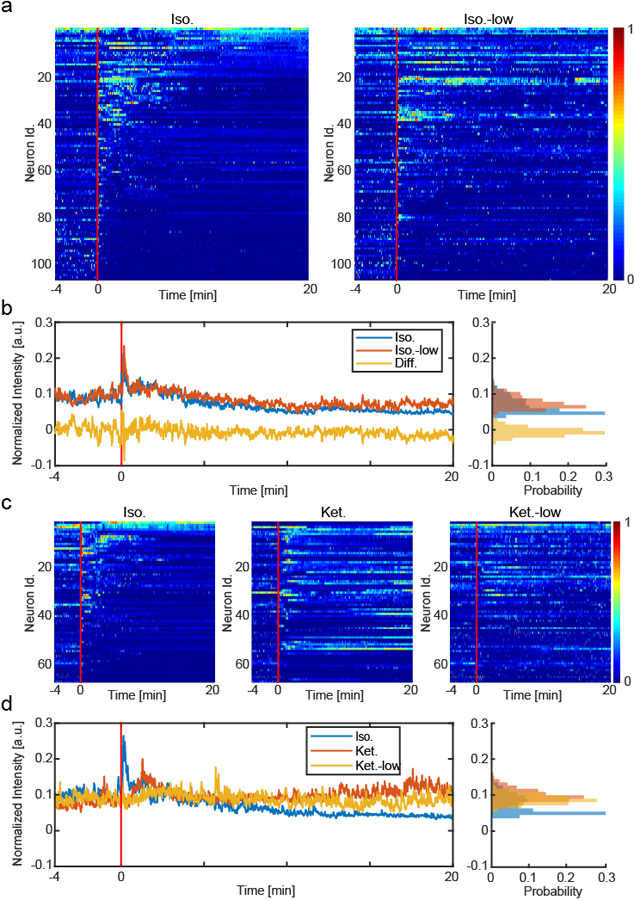
a, Heatmaps, activity patterns of the same neurons tracked in isoflurane (1.5%) and low isoflurane (0.5%) experiments, aligned by isoflurane (1.5%) neural patterns. 106 tracked same neurons from 5 mice × 1 trial. b, Left, mean and difference traces of the population normalized activity in isoflurane and low isoflurane experiments. Right, intensity distribution of the traces. c, Heatmaps, activity patterns of the neurons tracked in isoflurane (1.5%), ketamine (100mg/kg) and low ketamine (12mg/kg) experiments, aligned by isoflurane neural patterns. 69 tracked same neurons from 5 mice × 1 trial. d, Left, mean trace of the population normalized activity in isoflurane, ketamine and low ketamine experiments. Right, intensity distribution of the traces.
Supplementary Material
Acknowledgement
We thank J. Takatoh for helping with optimizing image compression that retains good resolution and for designing color scheme for projection data, and thank V. Prevosto and P. Thompson for helping with statistics. We also thank R. Mooney, Ru-Rong Ji, and members of the Wang lab for critical reading of this manuscript. We thank the Janelia GENIE project for making GCaMP6m sensor available to research community. Correspondence for calcium imaging data-processing codes should be addressed to Jinghao Lu (jinghao.lu@duke.edu). T.H. is supported by the NSF GRFP scholarship. Y.C. is supported by R01 DE027454. This work is supported by an NIH DP1MH103908, the W. M. Keck Foundation grant, an NIH R01 NS109947, and NIH R01 DE029342.
Footnotes
Competing Financial Interests Statement
The authors declare no competing financial interests.
References
- 1.Aranake A et al. Increased risk of intraoperative awareness in patients with a history of awareness. Anesthesiology 119, 1275–1283 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Sebel PS et al. The incidence of awareness during anesthesia: a multicenter United States study. Anesth. Analg 99, 833–9, table of contents (2004). [DOI] [PubMed] [Google Scholar]
- 3.Bergman SA Ketamine: review of its pharmacology and its use in pediatric anesthesia. Anesth Prog 46, 10–20 (1999). [PMC free article] [PubMed] [Google Scholar]
- 4.Sadove MS, Shulman M, Hatano S & Fevold N Analgesic effects of ketamine administered in subdissociative doses. Anesth. Analg 50, 452–457 (1971). [PubMed] [Google Scholar]
- 5.Gorlin AW, Rosenfeld DM & Ramakrishna H Intravenous sub-anesthetic ketamine for perioperative analgesia. J Anaesthesiol Clin Pharmacol 32, 160–167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown EN, Purdon PL & Van Dort CJ General anesthesia and altered states of arousal: a systems neuroscience analysis. Annu. Rev. Neurosci 34, 601–628 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang-Xie L-F et al. A common neuroendocrine substrate for diverse general anesthetics and sleep. Neuron 102, 1053–1065.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haubensak W et al. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature 468, 270–276 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J, Zhang X, Muralidhar S, LeBlanc SA & Tonegawa S Basolateral to central amygdala neural circuits for appetitive behaviors. Neuron 93, 1464–1479.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi HMT et al. Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakurai K et al. Capturing and Manipulating Activated Neuronal Ensembles with CANE Delineates a Hypothalamic Social-Fear Circuit. Neuron 92, 739–753 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez E et al. A craniofacial-specific monosynaptic circuit enables heightened affective pain. Nat. Neurosci 20, 1734–1743 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCullough KM, Morrison FG, Hartmann J, Carlezon WA & Ressler KJ Quantified coexpression analysis of central amygdala subpopulations. Eneuro 5, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sadler KE et al. Divergent functions of the left and right central amygdala in visceral nociception. Pain 158, 747–759 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji G & Neugebauer V Hemispheric lateralization of pain processing by amygdala neurons. J. Neurophysiol 102, 2253–2264 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baas D, Aleman A & Kahn RS Lateralization of amygdala activation: a systematic review of functional neuroimaging studies. Brain Res Brain Res Rev 45, 96–103 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Lu J et al. MIN1PIPE: A Miniscope 1-Photon-Based Calcium Imaging Signal Extraction Pipeline. Cell Rep 23, 3673–3684 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheintuch L et al. Tracking the Same Neurons across Multiple Days in Ca2+ Imaging Data. Cell Rep 21, 1102–1115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyden ES, Zhang F, Bamberg E, Nagel G & Deisseroth K Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci 8, 1263–1268 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Chow BY et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 463, 98–102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang T et al. Identifying the pathways required for coping behaviours associated with sustained pain. Nature 565, 86–90 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunskaar S & Hole K The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain 30, 103–114 (1986). [DOI] [PubMed] [Google Scholar]
- 23.Basbaum AI, Bautista DM, Scherrer G & Julius D Cellular and molecular mechanisms of pain. Cell 139, 267–284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding W et al. An improved rodent model of trigeminal neuropathic pain by unilateral chronic constriction injury of distal infraorbital nerve. J. Pain 18, 899–907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giglio JA & Gregg JM Development of mirror pain following trigeminal nerve injury: a case report and review of neuropathic mechanisms. Gen Dent 66, 27–32 (2018). [PubMed] [Google Scholar]
- 26.Milligan ED et al. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J. Neurosci 23, 1026–1040 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai Y-Q, Wang W, Paulucci-Holthauzen A & Pan ZZ Brain circuits mediating opposing effects on emotion and pain. J. Neurosci 38, 6340–6349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han S, Soleiman MT, Soden ME, Zweifel LS & Palmiter RD Elucidating an Affective Pain Circuit that Creates a Threat Memory. Cell 162, 363–374 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D et al. Functional divergence of delta and mu opioid receptor organization in CNS pain circuits. Neuron 98, 90–108.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corder G et al. An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 363, 276–281 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levin-Arama M et al. Subcutaneous Compared with Intraperitoneal KetamineXylazine for Anesthesia of Mice. J. Am. Assoc. Lab. Anim. Sci 55, 794–800 (2016). [PMC free article] [PubMed] [Google Scholar]
- 32.Beecher HK Pain in men wounded in battle. Bull U S Army Med Dep 5, 445–454 (1946). [PubMed] [Google Scholar]
- 33.Petrovic P, Kalso E, Petersson KM & Ingvar M Placebo and opioid analgesia-- imaging a shared neuronal network. Science 295, 1737–1740 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Benedetti F Placebo and endogenous mechanisms of analgesia. Handb Exp Pharmacol 393–413 (2007). [DOI] [PubMed]
- 35.Eippert F et al. Activation of the opioidergic descending pain control system underlies placebo analgesia. Neuron 63, 533–543 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Basbaum AI & Fields HL Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu. Rev. Neurosci 7, 309–338 (1984). [DOI] [PubMed] [Google Scholar]
- 37.Ossipov MH, Dussor GO & Porreca F Central modulation of pain. J. Clin. Invest 120, 3779–3787 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lau BK & Vaughan CW Descending modulation of pain: the GABA disinhibition hypothesis of analgesia. Curr. Opin. Neurobiol 29, 159–164 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Butler RK & Finn DP Stress-induced analgesia. Prog. Neurobiol 88, 184–202 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Wager TD, Scott DJ & Zubieta J-K Placebo effects on human mu-opioid activity during pain. Proc. Natl. Acad. Sci. USA 104, 11056–11061 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Werka T & Marek P Post-stress analgesia after lesions to the central nucleus of the amygdala in rats. Acta Neurobiol Exp (Wars) 50, 13–22 (1990). [PubMed] [Google Scholar]
- 42.Duvarci S & Pare D Amygdala microcircuits controlling learned fear. Neuron 82, 966–980 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herry C & Johansen JP Encoding of fear learning and memory in distributed neuronal circuits. Nat. Neurosci 17, 1644–1654 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Neugebauer V Amygdala pain mechanisms. Handb Exp Pharmacol 227, 261–284 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parikh D et al. Stress-induced analgesia and endogenous opioid peptides: the importance of stress duration. Eur. J. Pharmacol 650, 563–567 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilson TD et al. Dual and opposing functions of the central amygdala in the modulation of pain. Cell Rep 29, 332–346.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu K et al. The central amygdala controls learning in the lateral amygdala. Nat. Neurosci 20, 1680–1685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cui Y et al. A Central Amygdala-Substantia Innominata Neural Circuitry Encodes Aversive Reinforcement Signals. Cell Rep 21, 1770–1782 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Cai H, Haubensak W, Anthony TE & Anderson DJ Central amygdala PKC-δ(+) neurons mediate the influence of multiple anorexigenic signals. Nat. Neurosci 17, 1240–1248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 50.Venza M et al. The overriding of TRAIL resistance by the histone deacetylase inhibitor MS-275 involves c-myc up-regulation in cutaneous, uveal, and mucosal melanoma. Int. Immunopharmacol 28, 313–321 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Pang D et al. Polyphyllin II inhibits liver cancer cell proliferation, migration and invasion through downregulated cofilin activity and the AKT/NF-κB pathway. Biol. Open 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Faget L et al. Opponent control of behavioral reinforcement by inhibitory and excitatory projections from the ventral pallidum. Nat. Commun 9, 849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zamparo I et al. Axonal odorant receptors mediate axon targeting. Cell Rep 29, 4334–4348.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hara Y et al. Anti-tumor effects of an antagonistic mAb against the ASCT2 amino acid transporter on KRAS-mutated human colorectal cancer cells. Cancer Med 9, 302–312 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simon CM et al. Stasimon contributes to the loss of sensory synapses and motor neuron death in a mouse model of spinal muscular atrophy. Cell Rep 29, 3885–3901.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Segbroeck M, Knoll AT, Levitt P & Narayanan S MUPET-Mouse Ultrasonic Profile ExTraction: A Signal Processing Tool for Rapid and Unsupervised Analysis of Ultrasonic Vocalizations. Neuron 94, 465–485.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y, Chen Y, Liedtke W & Wang F Lack of evidence for ectopic sprouting of genetically labeled Aβ touch afferents in inflammatory and neuropathic trigeminal pain. Mol. Pain 11, 18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y et al. Identifying local and descending inputs for primary sensory neurons. J. Clin. Invest 125, 3782–3794 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data described in this study are available from the corresponding authors upon request. All codes described in this study are available from the corresponding authors upon request. The MIN1PIPE code for processing miniscope imaging (correspondence should be addressed to Jinghao Lu, jinghao.lu@duke.edu) is available at: https://github.com/JinghaoLu/MIN1PIPE