

Abstract
Objective
Juvenile dermatomyositis (DM) is a heterogeneous systemic immune‐mediated vasculopathy. This study was undertaken to 1) identify inflammation/endothelial dysfunction–related biomarker profiles reflecting disease severity at diagnosis, and 2) establish whether such biomarker profiles could be used for predicting the response to treatment in patients with juvenile DM.
Methods
In total, 39 biomarkers related to activation of endothelial cells, endothelial dysfunction, and inflammation were measured using multiplex technology in serum samples from treatment‐naive patients with juvenile DM from 2 independent cohorts (n = 30 and n = 29). Data were analyzed by unsupervised hierarchical clustering, nonparametric tests with correction for multiple comparisons, and Kaplan‐Meier tests with Cox proportional hazards models for analysis of treatment duration. Myositis‐specific antibodies (MSAs) were measured in the patients’ serum using line blot assays.
Results
Severe vasculopathy in patients with juvenile DM was associated with low serum levels of intercellular adhesion molecule 1 (Spearman's rho [rs] = 0.465, P = 0.0111) and high serum levels of endoglin (rs = −0.67, P < 0.0001). In the discovery cohort, unsupervised hierarchical clustering analysis of the biomarker profiles yielded 2 distinct patient clusters, of which the smaller cluster (cluster 1; n = 8) exhibited high serum levels of CXCL13, CCL19, galectin‐9, CXCL10, tumor necrosis factor receptor type II (TNFRII), and galectin‐1 (false discovery rate <0.0001), and this cluster had greater severity of muscle disease and global disease activity (each P < 0.05 versus cluster 2). In the validation cohort, correlations between the serum levels of galectin‐9, CXCL10, TNFRII, and galectin‐1 and the severity of global disease activity were confirmed (rs = 0.40–0.52, P < 0.05). Stratification of patients according to the 4 confirmed biomarkers identified a cluster of patients with severe symptoms (comprising 64.7% of patients) who were considered at high risk of requiring more intensive treatment in the first 3 months after diagnosis (P = 0.0437 versus other cluster). Moreover, high serum levels of galectin‐9, CXCL10, and TNFRII were predictive of a longer total treatment duration (P < 0.05). The biomarker‐based clusters were not evidently correlated with patients’ MSA serotypes.
Conclusion
Results of this study confirm the heterogeneity of new‐onset juvenile DM based on serum biomarker profiles. Patients with high serum levels of galectin‐9, CXCL10, TNFRII, and galectin‐1 may respond suboptimally to conventional treatment, and may therefore benefit from more intensive monitoring and/or treatment.
Introduction
Juvenile dermatomyositis (DM) is a rare systemic immune‐mediated disease with an incidence of 2–4 cases per million per year 1. The clinical presentation is heterogeneous, as patients can develop a spectrum of symptoms, ranging from the typical symptoms of proximal skeletal muscle weakness and pathognomonic skin rash, to involvement of vital organs such as the lungs, heart, brain, and intestines 2. The clinical heterogeneity of juvenile DM has been linked to myositis‐specific autoantibodies (MSAs), the levels of which may distinguish distinct clinical phenotypes and could be prognostic for the disease course and the need for second‐line therapy 3. Despite this disease heterogeneity, current treatment guidelines are not yet adapted to subgroup‐specific needs 4. Stratification of patients, e.g., into high‐risk or low‐risk groups, may facilitate the development of personalized monitoring and treatment strategies.
In addition to inflammation of the muscles and skin, vasculopathy is an important hallmark of juvenile DM 5, 6. The disease is characterized by a loss of capillaries, morphologic changes to the endothelium, endothelial cell activation, and small vessel angiopathy 5, 6, 7, 8. Complement and immune complexes are involved in its pathogenesis, but a disturbed balance between angiostatic and angiogenic factors also plays a role 9, 10, 11. The degree of vasculopathy is correlated with the expression of interferon (IFN)–inducible angiostatic chemokines 11, indicating that vascular injury may be related to the IFN signature 5. This IFN signature has been identified in the serum and many cell types, including endothelial cells, from patients with juvenile DM 11, 12, 13, 14, 15, 16, 17.
The degree of vasculopathy is clinically relevant: pathologic changes in nailfold capillaries are associated with clinical disease activity 18, and prominent vascular injury evident in muscle biopsy tissue was found to be associated with severe clinical presentation and outcomes 19, 20. Recently, Gitiaux et al identified a subgroup of juvenile DM patients with severe disease, based on trajectories of clinical parameters during follow‐up 19. Juvenile DM in these patients was characterized by severe muscle weakness, frequent limb edema, gastrointestinal involvement, higher myopathologic scores (e.g., capillary dropout), and low remission rates 19. Most of these manifestations could be related to vasculopathy 19.
Herein we used a minimally invasive biomarker‐based approach to identify juvenile DM patients, both from a discovery cohort and from an independent validation cohort, who had a severe disease course already present at diagnosis. The biomarker panel analyzed included previously described and novel markers related to endothelial cell activation, endothelial dysfunction, and IFN‐driven inflammation 5, 17. Specifically, we investigated whether biomarker profiles were linked to vasculopathy, disease severity, and frequency of MSA subtypes, and whether the levels of these biomarkers might be predictive of the response to induction therapy and the time required to attain drug‐free remission (DFR).
Patients and Methods
Participants
For this study, 59 patients meeting the Bohan and Peter criteria for definite or probable juvenile DM 21, 22, from Chicago, Illinois for the discovery cohort (n = 30) and from Utrecht, The Netherlands and Singapore for the validation cohort (n = 25 and n = 4), were included before the start of treatment, between March 2004 and June 2018. Scores on the Childhood Myositis Assessment Scale (CMAS) (scale 0–52, or scale 0–49 for ages 4–5 years) 23 were recorded in both cohorts as a measure of muscle disease activity. Disease Activity Scores (DAS) for the muscle (DAS‐M) (scale 0–11) and skin (DAS‐S) (scale 0–9), and global DAS for total disease activity (DAS‐T) (scale 0–20) were recorded in the discovery cohort 24, while in the validation cohort, the physician's global assessment of disease activity (PhGA) (scale 0–10) 25 and the cutaneous assessment tool (CAT) for juvenile DM (scale 0–116) 26 were used.
Intensification of treatment was defined as the addition of new immunosuppressive medication or an increase in the dose of a previous medication. Time to attainment of DFR was defined as the time (in months) between the date at the start of immunosuppressive treatment and the date at the time of withdrawal of all immunosuppressive drugs. In the discovery cohort, treatment regimens were individualized, while in the validation cohort, treatment regimens were based on the SHARE (Single Hub and Access point for pediatric Rheumatology in Europe) recommendations 4.
End row loops (ERLs), as a proxy for the severity of vasculopathy 27, were assessed by nailfold capillaroscopy. Standardized digital nailfold images from 8 fingers, excluding the thumbs, were obtained, and findings were analyzed by counting the ERLs in a 3‐mm segment, yielding the average count of ERLs/mm. The lower limit of normal for this value is 7 ERLs/mm (interquartile range 7.61–8.94), as previously reported 27. To define severe vasculopathy, a cutoff ERL score of <4 was used, based on the median value for all abnormal ERL scores in the cohort (abnormal ERL defined as a score of <7; n = 27). ERLs were only assessed in the discovery cohort.
In addition, serum samples from all patients were assessed for the frequency of MSAs. Serum MSAs were measured by line blot assay (Euroimmun DL 1530‐1601‐4 G/5001‐4 G/6401‐4 G).
This study was approved by the institutional ethics committees of the involved centers: the UMC Utrecht (METC approval no. 15‐191), the SingHealth centralized Institutional Review Board (IRB) (CIRB approval no. 2014/083/E), and the Ann & Robert H. Lurie Children's Hospital of Chicago (IRB approval nos. 2001‐11715 and 2010‐14117). The study was conducted in patients in accordance with the Declaration of Helsinki. Age‐appropriate written informed consent was obtained prior to inclusion of each patient in the study.
Analysis of a biomarker panel in patients’ serum
Serum was spun down and stored at −80°C within 4 hours after collection. In total, 39 biomarkers related to endothelial cell activation, endothelial dysfunction, and inflammation were simultaneously measured using multiplex technology (xMAP; Luminex) in 50 μl of serum, as described previously 28. The markers were selected based on previously noted associations with endothelial cell activation, endothelial dysfunction, or inflammation and/or juvenile DM pathogenesis.
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 7.0, SPSS version 21 (IBM), or R version 3.5.1 (CRAN). Values below the detection limit were imputed as being 0.5 times the lowest measured value. For Spearman's rank correlation (rs) analysis, imputed values were excluded. Cutoff values for receiver operating characteristic (ROC) curves were based on the Youden's index. For comparisons between 2 groups, the Mann‐Whitney U test was used for continuous variables and the Fisher's exact test was used for categorical variables, with correction for multiple comparisons based on the false discovery rate (FDR), where applicable. P values less than 0.05 or an FDR less than 0.05 were considered statistically significant.
For unsupervised clustering by principal component analysis (PCA) and heatmap analysis with hierarchical clustering (Euclidian/Ward), data were mean‐centered and scaled per marker. Five markers (plasminogen activator inhibitor 1, fibronectin, oncostatin M, E‐selectin, and thrombomodulin) had >30% of values below the limit of detection and were therefore excluded from the clustering analyses. For time until reaching DFR, a Kaplan‐Meier estimator (dichotomized at the median value) and Cox proportional hazards model with log‐transformed data were used, along with the log‐rank test.
Results
Patient characteristics
The median age of the patients in the discovery and validation cohorts was 5.1 years and 7.0 years, respectively (Table 1). The majority of patients in both cohorts were female, with a significantly higher frequency of female patients in the discovery cohort compared to the validation cohort (86.7% versus 62.1%; P = 0.039) and slightly higher frequency of white patients (77% versus 76%). Moreover, the majority of patients in each cohort were white (77% in the discovery cohort versus 76% in the validation cohort). The duration of untreated disease was higher in the discovery cohort (median 5.9 months versus 3.2 months in the validation cohort; P = 0.02), and treatment intensification was needed in 57% of the discovery cohort, compared to 29% of the validation cohort (P = 0.038). Muscle disease activity was similar in both cohorts (median CMAS 33 in each). Muscle enzyme levels were higher in the validation cohort.
Table 1.
Baseline characteristics of the juvenile dermatomyositis patients from the discovery and validation cohorts*
| Discovery cohort (Chicago, IL) (n = 30) | Validation cohort (Netherlands and Singapore) (n = 25 and n = 4) | P | |
|---|---|---|---|
| Age at sampling, median (IQR) years | 5.1 (3.7–8.6) | 7 (3.9–12.1) | 0.192 |
| Sex, no. (%) female | 26 (86.7) | 18 (62.1) | 0.039 |
| Ethnicity, % white/Hispanic/African American/Asian | 77/20/3/0 | 76/0/10/14 | 0.012 |
| Duration of untreated disease, median (IQR) months | 5.9 (3.6–14.8) | 3.2 (1.4–9.1) | 0.020 |
| Intensification of treatment in first 3 months, no. (%) | 17 (56.7) | 8 (28.6) | 0.038 |
| Disease activity at diagnosis | |||
| CMAS (scale 0–52 or 0–49 for ages 4–5 years), median (IQR) | 33 (23.5–44)† | 33 (15–40.5)‡ | 0.334 |
| PhGA, median (IQR) (scale 0–10) | – | 5.8 (4.0–7.7)‡ | NA |
| DAS total, median (IQR) (scale 0–20) | 12 (9.4–13.3) | – | NA |
| DAS muscle, median (IQR) (scale 0–11) | 6 (3.8–8) | – | NA |
| DAS skin, median (IQR) (scale 0–9) | 5 (5–6.3) | – | NA |
| CAT score, median (IQR) (scale 0–116) | – | 5 (3.5–11) | NA |
| Muscle enzyme levels at diagnosis, median (IQR) IU/liter | |||
| CK | 131 (88–680) | 510 (138–4,357) | 0.053 |
| LDH | 364 (270–460)† | 497 (358–837)‡ | 0.010 |
| AST | 46 (35–80) | 65 (41–307)§ | 0.118 |
| ALT | 26 (17–43)† | 45 (23–112) | 0.158 |
| Myositis‐specific antibodies, no. (%) | |||
| Negative | 14 (46.7) | 16 (55.2) | 0.800 |
| MDA‐5 | 2 (6.7) | 2 (6.9) | 1.000 |
| Mi‐2 | 3 (10) | 0 (0) | 0.237 |
| NXP‐2 | |||
| Total | 2 (6.7) | 5 (17.2) | 0.254 |
| Strongly positive | 1 (3.3) | 3 (10.3) | |
| Weakly positive | 1 (3.3) | 2 (6.9) | |
| SAE‐1 | 0 (0) | 2 (6.9) | 0.237 |
| TIF‐1γ | |||
| Total | 9 (30.0) | 4 (13.8) | 0.209 |
| TIF‐1γ only | 8 (26.7) | 3 (10.3) | |
| TIF‐1γ + PL‐7 | 0 (0) | 1 (3.4) | |
| TIF‐1γ + PL‐12 | 1 (3.3) | 0 (0) | |
| Initial therapy after diagnosis, no. (%) | |||
| Oral Pred. | 1 (3.3) | 0 (0.0) | 1.000 |
| MTX + oral Pred. | 2 (6.7) | 0 (0.0) | 0.492 |
| IVMP + oral Pred. | 1 (3.3) | 1 (3.4) | 1.000 |
| IVMP + oral Pred. + MTX | 19 (63.3) | 20 (69.0) | 0.7847 |
| IVMP + oral Pred. + IVIG | 0 (0.0) | 1 (3.4) | 0.492 |
| IVMP + oral Pred. + MTX + IVIG | 1 (3.3) | 0 (0.0) | 1.000 |
| IVMP + oral Pred. + MTX + HCQ | 6 (20.0) | 7 (24.1) | 0.761 |
P values were calculated by Mann‐Whitney U test for continuous variables, and by Fisher's exact test for categorical variables (comparisons of 2 categories) or chi‐square test (comparisons of >2 categories). Myositis‐specific antibodies were measured by line blot assay. Normalization of muscle enzyme levels to the age‐ and center‐specific upper limits of normal did not change the results. IQR = interquartile range; CMAS = Childhood Myositis Assessment Scale; PhGA = physician's global assessment of disease activity; NA = not applicable; DAS = Disease Activity Score; CAT = cutaneous assessment tool (for juvenile dermatomyositis); CK = creatine kinase; LDH = lactate dehydrogenase; AST = aspartate aminotransferase; ALT = alanine aminotransferase; MDA‐5 = melanoma differentiation–associated protein; NXP‐2 = nuclear matrix protein 2; SAE‐1 = small ubiquitin‐like modifier‐1 activating enzyme; TIF‐1γ = transcription intermediary factor 1γ; Pred. = prednisone; MTX = methotrexate; IVMP = intravenous methylprednisolone; IVIG = intravenous immunoglobulin; HCQ = hydroxychloroquine.
Data not reported for 1 patient.
Data not reported for 4 patients.
Data not reported for 2 patients.
In both cohorts, MSAs were detected in ~50% of patients. Although none of the MSAs differed significantly in their frequency, anti–transcription intermediary factor 1γ (anti–TIF‐1γ) and anti–Mi‐2 antibodies were more frequent in the discovery cohort compared to the validation cohort (for anti–TIF‐1γ, 30% versus 14%; for anti–Mi‐2, 10% versus 0%), whereas anti–nuclear matrix protein 2 (anti–NXP‐2) and anti–small ubiquitin‐like modifier‐1 activating enzyme (anti–SAE‐1) antibodies were more frequent in the validation cohort compared to the discovery cohort (for anti–NXP‐2, 17% versus 7%; for anti–SAE‐1, 7% versus 0%). The frequency of anti–melanoma differentiation–associated protein 5 (anti–MDA‐5) antibodies was equal in both cohorts (each 7%), despite a higher frequency of children with an Asian background in the validation cohort. The initial treatment that was started after diagnosis was similar between the cohorts (Table 1).
Association between biomarker profiles and muscle disease activity
To investigate the heterogeneity of biomarker profiles in treatment‐naive juvenile DM patients, we performed unsupervised hierarchical clustering in the discovery cohort, which led to splitting the patients into 2 distinct clusters, comprising 8 patients (cluster 1) and 22 patients (cluster 2). Compared to the larger cluster (cluster 2), the smaller cluster (cluster 1) stood out because of the patients’ significantly higher serum levels of CXCL13, CCL19, galectin‐9 (Gal‐9), tumor necrosis factor receptor type II (TNFRII), Gal‐1, CXCL10, CXCL9, interleukin‐18 (IL‐18), chitinase‐3–like protein 1 (YKL‐40), CCL2, CCL4, vascular endothelial growth factor (VEGF), E‐selectin, intercellular adhesion molecule 1 (ICAM‐1), and CCL18 and the patients’ significantly lower serum levels of fetuin (FDR <0.05) (Figures 1A and B).
Figure 1.
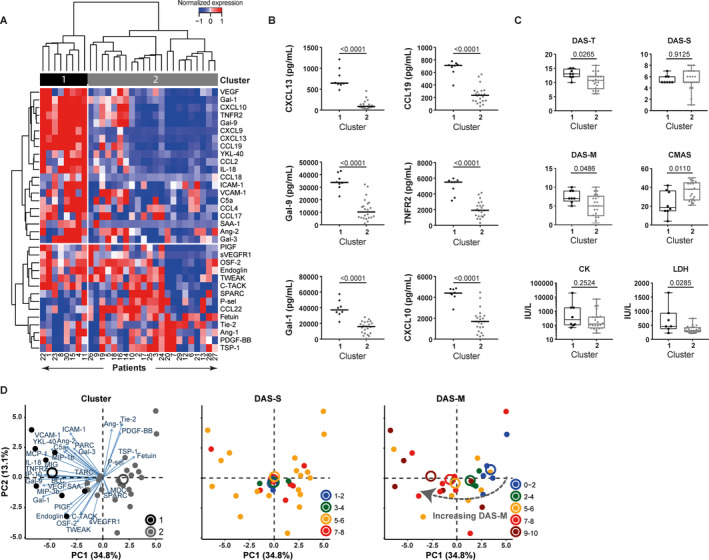
Association of heterogeneous biomarker profiles with differences in clinical disease activity in patients with juvenile dermatomyositis (DM). A panel of biomarkers for endothelial dysfunction and inflammation was measured by multiplex assay in the serum of 30 treatment‐naive juvenile DM patients (discovery cohort). A, Unsupervised hierarchical clustering (by Euclidian distance and Ward's method) of 30 patients in the discovery cohort based on serum levels of 34 biomarkers (mean‐centered and scaled values) yielded 2 distinct patient clusters (clusters 1 and 2). Values at the bottom represent unique patient identifiers (not ranked). B, Serum levels of the 6 markers most significantly different between cluster 1 (n = 8) and cluster 2 (n = 22) were compared by Mann‐Whitney U test, with correction for multiple comparisons based on the false discovery rate. Symbols represent individual patients; horizontal lines show the median. C, Clinical scores for global Disease Activity Score (DAS‐T), skin Disease Activity Score (DAS‐S), muscle Disease Activity Score (DAS‐M), Childhood Myositis Assessment Scale (CMAS) score, and muscle enzyme levels (creatine kinase [CK] and lactate dehydrogenase [LDH]) were compared between the 2 clusters. P values were determined by Mann‐Whitney U test. Results are shown as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the 10th and 90th percentiles. D, Principal components (PC1 and PC2) analysis based on the 34 mean‐centered markers shows patients stratified by cluster, DAS‐S scores, and DAS‐M scores. Circles with different colors represent clusters 1 and 2 or ranges of DAS‐S and DAS‐M scores. Closed circles represent individual patients, while open circles represent cluster centers. VEGF = vascular endothelial growth factor; Gal‐1 = galectin‐1; TNFRII = tumor necrosis factor receptor type II; YKL‐40 = chitinase‐3–like protein 1; IL‐18 = interleukin‐18; ICAM‐1 = intercellular adhesion molecule 1; VCAM‐1 = vascular cell adhesion molecule 1; SAA1 = serum amyloid A 1; Ang‐2 = angiopoietin‐2; PIGF = placental growth factor; sVEGFR1 = soluble VEGF receptor 1; OSF‐2 = periostin; TWEAK = TNF‐related weak inducer of apoptosis; C‐TACK = cutaneous T cell–attracting chemokine; SPARC = secreted protein acidic and rich in cysteine; P‐sel = P‐selectin; Tie‐2 = angiopoeitin‐1 receptor; PDGF‐BB = platelet‐derived growth factor BB; TSP‐1 = thrombospondin‐1.
To assess whether distinct biomarker profiles corresponded to specific clinical profiles, we compared disease characteristics between the clusters (Figure 1C). Total disease activity (median DAS‐T score) was significantly higher in cluster 1 than in cluster 2 (P = 0.0265), which was attributable to significantly higher muscle disease activity in cluster 1 (median DAS‐M score, P = 0.0486; median CMAS score, P = 0.011) (Figure 1C). Skin disease activity scores and creatine kinase levels in the muscle were comparable, but patients in cluster 1 had higher lactate dehydrogenase (LDH) levels in the muscle (P = 0.0285).
Multidimensional PCA identified muscle disease activity as an important factor explaining the variance in biomarker profiles, and confirmed that patients with the highest DAS‐M score spatially overlapped with cluster 1 (Figure 1D). ERL scores were similar between the clusters. Levels of 12 of the 16 biomarkers that were differentially expressed between the clusters correlated with the DAS‐T, DAS‐M, or CMAS scores. These included CXCL13, CCL19, Gal‐9, TNFRII, Gal‐1, CXCL10, CXCL9, IL‐18, YKL‐40, CCL2, CCL4, and ICAM‐1 (|rs| = 0.35–0.67, P < 0.05) (Table 2). High serum levels of these markers may therefore identify a subgroup of patients with more severe disease.
Table 2.
Spearman's rank correlations of serum biomarker levels with clinical disease activity scores in juvenile dermatomyositis patients in the discovery and validation cohorts*
| Discovery cohort | Validation cohort | ||||
|---|---|---|---|---|---|
| DAS‐T (n = 30) | DAS‐M (n = 30) | CMAS (n = 29) | PhGA (n = 25) | CMAS (n = 25) | |
| CXCL13 | 0.538† | 0.539† | −0.476† | NS | NS |
| CCL19 | 0.541† | 0.553† | −0.497† | NS | NS |
| Gal‐9 | 0.519† | 0.496† | −0.428‡ | 0.403‡ | NS |
| TNFRII | 0.518† | 0.489† | −0.377‡ | 0.439‡ | NS |
| Gal‐1 | 0.471† | 0.495† | NS | 0.486‡ | NS |
| CXCL10 | 0.505† | 0.458‡ | −0.397‡ | 0.445‡ | NS |
| CXCL9 | 0.432‡ | NS | NS | NS | NS |
| IL‐18 | 0.503† | 0.415‡ | −0.490† | NS | NS |
| YKL‐40 | 0.465† | 0.524† | −0.667§ | NS | NS |
| CCL2 | 0.557† | 0.566† | −0.532† | NS | NS |
| CCL4 | 0.388‡ | NS | −0.396‡ | NS | NS |
| VEGF | NS | NS | NS | NS | NS |
| E‐selectin | NS | NS | NS | NS | NS |
| ICAM‐1 | NS | NS | −0.515† | NS | NS |
| Fetuin | NS | NS | NS | NS | NS |
| CCL18 | NS | NS | NS | NS | NS |
| sVEGFR‐1¶ | 0.422‡ | 0.427‡ | NS | NS | −0.454‡ |
Spearman's rank correlation coefficients are shown for those biomarkers whose serum levels were significantly different between cluster 1 and cluster 2. For biomarkers with out‐of‐range values (i.e., below the detection limit), the values were imputed (for CXCL9 and E‐selectin), and imputed values were excluded from the analysis to prevent skewing of the data. DAS‐T = Disease Activity Score for total disease activity; DAS‐M = Disease Activity Score for the muscle; CMAS = Childhood Myositis Assessment Scale; PhGA = physician's global assessment of disease activity; NS = not significant; Gal‐9 = galectin‐9; TNFRII = tumor necrosis factor receptor type II; IL‐18 = interleukin‐18; YKL‐40 = chitinase 3–like protein 1; VEGF = vascular endothelial growth factor; ICAM‐1 = intercellular adhesion molecule 1.
P < 0.01.
P < 0.05.
P < 0.0001.
This marker, soluble VEGF receptor 1 (sVEGFR‐1), only identifies patients in cluster 1 in the validation cohort, but not in the discovery cohort.
Association of biomarker profiles with vasculopathy
We next assessed which of the 39 markers showed a direct correlation with ERL scores (discovery cohort, n = 29). ERL scores correlated negatively with the serum levels of endoglin (rs = −0.67, P < 0.0001) as well as thrombospondin‐1 (TSP‐1) and VEGF (each rs = −0.415, P = 0.0252), and correlated positively with the serum levels of ICAM‐1 (rs = 0.465, P = 0.0111) (Figure 2A). Patients with an ERL score of <4, indicating severe vasculopathy, had significantly higher serum endoglin levels and lower serum ICAM‐1 levels compared to patients with an ERL score of ≥4 (P = 0.012 and P = 0.0079, respectively) (Figure 2B).
Figure 2.
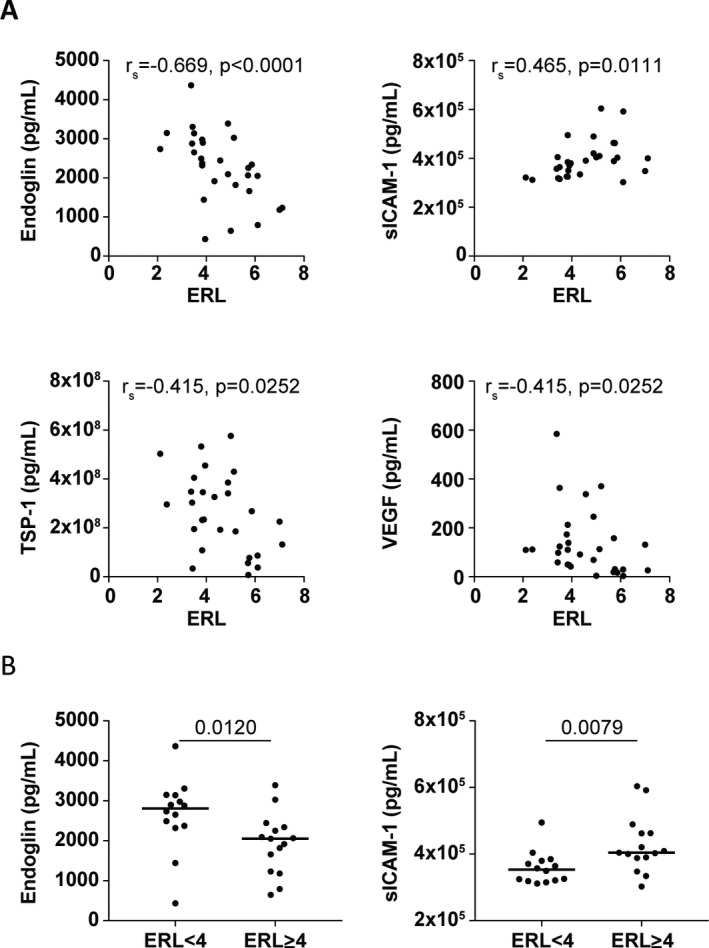
Correlation of biomarker levels with end row loop (ERL) scores. A panel of biomarkers for endothelial dysfunction and inflammation was measured by multiplex assay in the serum of 30 treatment‐naive juvenile dermatomyositis (DM) patients (discovery cohort). ERL scores (a count of ERLs/mm in the fingers, as a measure of the severity of vasculopathy) were assessed by nailfold capillaroscopy. A, Spearman's rank correlations (rs) were determined to assess the correlation between serum biomarker levels and ERL scores in 29 juvenile DM patients. B, Serum levels of endoglin and soluble intercellular adhesion molecule 1 (sICAM‐1) were assessed in juvenile DM patients stratified according to low ERL scores (defined as <4) and high ERL scores (defined as ≥4). Symbols represent individual patients; horizontal lines show the median. Values above the graphs are P values, determined by Mann‐Whitney U test. TSP‐1 = thrombospondin‐1; VEGF = vascular endothelial growth factor.
With regard to serum levels of endoglin, the area under the ROC curve (AUC) for identifying patients with low ERL scores (indicative of severe vasculopathy) relative to high ERL scores was 0.771 (P = 0.0129). An optimal cutoff value of 2,286 pg/ml for the endoglin level yielded a sensitivity of 85.7% and specificity of 73.3% for identifying patients with a low ERL score. With regard to serum levels of ICAM‐1, the AUC for identifying patients with a low ERL score was 0.786 (P = 0.0088), and a cutoff value of 386,425 pg/ml yielded a sensitivity of 85.7% and specificity of 80%. The combination of the 2 markers in a prediction model improved the AUC to 0.833 (P = 0.0023), but did not yield a higher sensitivity (79%) or specificity (80%) than that with ICAM‐1 alone. The presence of low serum levels of ICAM‐1 (and/or high serum levels of endoglin) may thus be suitable to identify patients with severe vasculopathy.
Validation of biomarker and clinical profiles in an independent cohort
To validate the association between biomarker profiles and clinical disease, the same biomarker panel was measured in an independent validation cohort of patients with juvenile DM (n = 29). The 16 markers that identified patients with severe (muscle) disease activity in the discovery cohort were again assessed for correlations with clinical disease activity in the validation cohort. Four markers correlated significantly with clinical disease activity in both the discovery and validation cohorts: Gal‐9, TNFRII, Gal‐1, and CXCL10 (rs = 0.40–0.52 for correlations with the DAS‐T and PhGA scores) (Table 2 and Figures 3A–D). Reciprocal analysis of the 2 cohorts using the same criteria, i.e., 1) clustering of the validation cohort by all markers, 2) selection of markers identifying the more severely affected cluster, and 3) subsequent assessment of correlations with clinical disease parameters, yielded the same 4 markers (Gal‐9, TNFRII, Gal‐1, and CXCL10), as well as soluble VEGF receptor 1 (sVEGFR‐1) (Table 2). Since sVEGFR‐1 was not one of the markers identifying cluster 1 in the discovery cohort, it was not included as an additional marker in subsequent analyses.
Figure 3.
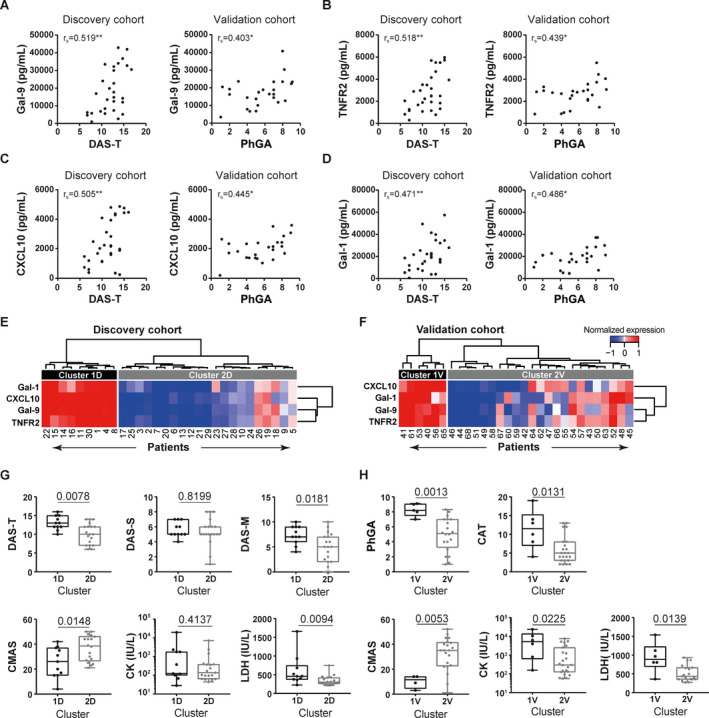
Identification of galectin‐9 (Gal‐9), tumor necrosis factor receptor type II (TNFRII), CXCL10, and Gal‐1 as biomarkers for stratification of patients with severe juvenile dermatomyositis (DM). A panel of biomarkers for endothelial dysfunction and inflammation was measured by multiplex assay in the serum of 59 treatment‐naive juvenile DM patients. A–D, Spearman's rank correlations (rs) were determined to assess the correlations of serum levels of Gal‐9 (A), TNFRII (B), CXCL10 (C), and Gal‐1 (D) with global disease activity as measured by the global Disease Activity Score (DAS‐T) in the discovery (D) cohort and the physician's global assessment of disease activity (PhGA) in the validation (V) cohort. * = P < 0.05; ** = P < 0.01. E and F, Unsupervised hierarchical clustering of juvenile DM patients in the discovery cohort (E) and validation cohort (F) was performed according to the normalized serum expression values for Gal‐9, TNFRII, CXCL10, and Gal‐1. Values at the bottom represent unique patient identifiers (not ranked). G and H, Clinical measures of disease activity were compared between cluster 1 and cluster 2 (the same clusters as identified in E and F) in the discovery cohort (G) and validation cohort (H). Results are shown as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the 10th and 90th percentiles. Values above the graphs are P values, determined by Mann‐Whitney U test. DAS‐S = Disease Activity Score for the skin; DAS‐M = Disease Activity Score for the muscle; CMAS = Childhood Myositis Assessment Scale (score); CK = creatine kinase; LDH = lactate dehydrogenase; CAT = cutaneous assessment tool for juvenile DM. Color figure can be viewed in the online issue, which is available at: http://onlinelibrary.wiley.com/doi/10.1002/art.41236/abstract.
We next examined whether these 4 markers would be able and sufficient to identify severely affected patients. Hierarchical clustering of the 2 cohorts based on these 4 markers yielded 2 distinct patient clusters in each cohort (clusters of 9 patients and 21 patients in the discovery cohort, and clusters of 6 patients and 23 patients in the validation cohort) (Figures 3E and F). Indeed, in both cohorts, the smaller subgroup of patients with high levels of Gal‐9, TNFRII, Gal‐1, and CXCL10 (cluster 1D and cluster 1V from the discovery and validation cohorts, respectively) had significantly higher total disease activity and muscle disease activity scores compared to patients in cluster 2. Patients in cluster 1D had higher DAS‐T scores (P = 0.0078), higher DAS‐M scores (P = 0.0181), and lower CMAS scores (P = 0.0148) compared to patients in cluster 2D, and patients in cluster 1V had higher PhGA scores (P = 0.0013) and lower CMAS scores (P = 0.0053) compared to patients in cluster 2V (Figures 3G and H). Skin disease activity did not differ between the clusters in the discovery cohort, but in the validation cohort, skin disease activity (CAT score) was significantly higher in cluster 1V than in cluster 2V (P = 0.0131). In both cohorts, LDH levels were higher in cluster 1 than in cluster 2 (P < 0.05 in each cohort). Thus, the combination of high serum levels of Gal‐9, TNFRII, Gal‐1, and CXCL10 may be sufficient to identify a subgroup of patients with severe global and muscle disease.
To assess the potency of each individual marker for identifying severely affected patients, patients were stratified into severe disease (those in the >75th percentile of PhGA, DAS‐T, and DAS‐M scores or those in the <25th percentile of CMAS scores) and nonsevere (muscle or global) disease, and the AUCs of the ROC curves were determined for each marker. TNFRII had the highest AUC for identifying patients with severe muscle disease (AUC 0.80) and severe global disease (AUC 0.73), with a sensitivity of 80% and 69% and specificity of 82% and 76%, respectively, at a cutoff level of 3,010 pg/ml for levels of TNFRII. Due to the high correlations between the markers (rs = 0.76–0.95), a combined model with these markers was not constructed. Therefore, it can be concluded that high serum levels of Gal‐9, TNFRII, Gal‐1, and CXCL10 can identify severely affected patients at the time of diagnosis, with TNFRII being the best indicator of severe disease.
Association of biomarker profiles with MSAs
Since MSA serotypes were previously linked to disease phenotypes, including (muscle) disease severity 7, 8, we compared the frequencies of MSAs (according to MSA category) between the biomarker‐based clusters. The cohorts were combined into a single cohort to yield a sufficient number of patients per MSA category. Clustering by Gal‐9, CXCL10, TNFRII, and Gal‐1 expression produced 2 clusters of patients (combined cohort clusters 1C and 2C) (Figure 4A).
Figure 4.
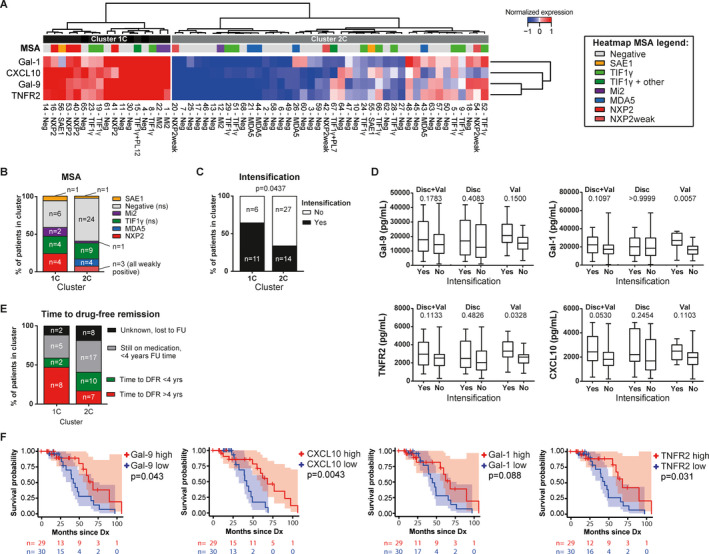
Identification of galectin‐1 (Gal‐1), CXCL10, Gal‐9, and tumor necrosis factor receptor type II (TNFRII) as biomarkers for patient stratification and for prognosis of response to therapy. A panel of biomarkers for endothelial dysfunction and inflammation was measured by multiplex assay in the serum of 59 treatment‐naive juvenile dermatomyositis (DM) patients. A, Unsupervised hierarchical clustering of 59 juvenile DM patients in the discovery and validation cohorts combined (C) was performed according to the normalized serum expression values for Gal‐9, CXCL10, TNFRII, and Gal‐1 and status of each myositis‐specific antibody (MSA) serotype. The heatmap legend indicates the different MSA serotypes according to different colors. Values at the bottom represent unique patient identifiers (not ranked). B, Cumulative frequencies of MSA serotypes were compared between clusters 1C and 2C. P values for cluster 1C versus cluster 2C, as determined by chi‐square test, were not significant (NS) for the MSA‐negative and anti–transcription intermediary factor 1γ (anti–TIF1γ)–positive patients. C, Cumulative frequencies of patients needing intensification of treatment within the first 3 months after diagnosis were compared between cluster 1C and cluster 2C. Due to missing data on intensification of treatment for 1 patient in cluster 2C, the analysis was performed after exclusion of this patient. P values were determined by Fisher's exact test. D, The capacity of serum levels of Gal‐9, CXCL10, TNFRII, and Gal‐1 to differentiate patients requiring intensification of treatment was assessed in the combined cohort and in the discovery (Disc) and validation (Val) cohorts separately. Results are shown as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the 10th and 90th percentiles. Values above the graphs are P values, determined by Mann‐Whitney U test. E, Cumulative frequencies of patients 1) with time to attainment of drug‐free remission (DFR) of >4 years, 2) with time to attainment of DFR of <4 years, 3) who were still receiving medication at <4 years of follow‐up (FU), or 4) whose medication use was unknown and who were lost to follow‐up were compared between cluster 1C and cluster 2C. F, Kaplan‐Meier curves show the months from diagnosis (Dx) until achievement of remission while not receiving immunosuppressive treatment among patients stratified according to the median serum levels of Gal‐9, CXCL10, Gal‐1, and TNFRII (high levels [n = 29] versus low levels [n = 30]). Shaded areas show the confidence intervals. Values below the plots are the number of patients at risk in the high‐level (red) and low‐level (blue) groups at each time point. P values were determined by log‐rank test. Neg = negative; NXP‐2 = nuclear matrix protein 2; SAE‐1 = small ubiquitin‐like modifier‐1 activating enzyme; MDA‐5 = melanoma differentiation–associated protein 5. Color figure can be viewed in the online issue, which is available athttp://onlinelibrary.wiley.com/doi/10.1002/art.41236/abstract.
Although none of the MSAs differed significantly in frequency between these 2 clusters, MSA‐negative patients were relatively more frequent in cluster 2C than in cluster 1C (57% versus 35%) (Figures 4A and B). Frequencies of patients with anti–TIF‐1γ antibodies were similar between the clusters (21% in cluster 1C and 23% in cluster 2C). The numbers of patients with anti–SAE‐1, anti–Mi‐2, anti–NXP‐2, and anti–MDA‐5 antibodies were too small to statistically compare their frequencies between clusters, but it was notable that all 4 patients with anti–MDA‐5 autoantibodies were in cluster 2C. Moreover, all patients with strong expression of anti–NXP‐2 antibodies were in cluster 1C, whereas patients with weak anti–NXP‐2 expression were present only in cluster 2C. Since weak antibody positivity may sometimes indicate a false‐positive measurement, this might imply a difference in anti–NXP‐2 frequencies between cluster 1C and cluster 2C. The findings thus suggest that MSA serotypes were not directly linked to the biomarker‐based patient clusters, but that patients with strong anti–NXP‐2 positivity may be more likely to be in the severe disease cluster, characterized by high biomarker levels, whereas the opposite may be true for patients with anti–MDA‐5 antibodies.
Prognostic value of biomarkers for suboptimal response to initial treatment
We next assessed whether high serum levels of Gal‐9, CXCL10, TNFRII, and/or Gal‐1 could be prognostic for a suboptimal response to initial treatment. The frequency at which intensification of treatment was required within the first 3 months after diagnosis was significantly higher in cluster 1C than in cluster 2C (64.7% versus 34.1%; P = 0.0437) (Figure 4C), suggesting that, indeed, high biomarker levels may identify an “at risk” group of patients who might require intensification of their initial treatment.
Although this combination of biomarkers could identify “at risk” patients, the individual biomarkers showed only a trend toward being higher in patients needing intensification of treatment in the combined cohort (for CXCL10, P = 0.053; for Gal‐1, P = 0.1097; for Gal‐9, P = 0.1783; for TNFRII, P = 0.1133) (Figure 4D). In the validation cohort, higher serum levels of Gal‐1 and TNFRII potently identified patients needing intensification of treatment (for Gal‐1, P = 0.0057; for TNFRII, P = 0.0328) (Figure 4D). The results of the corresponding ROC curves are available from the corresponding author upon request. Again, due to the high correlation between the biomarkers, a combined model of these markers was not constructed. Thus, the combination of the 4 markers—Gal‐9, CXCL10, TNFRII, and Gal‐1—may identify patients who would be considered at risk for a suboptimal response to induction therapy.
Prognostic value of biomarkers for predicting time to attainment of DFR
Finally, we examined whether patients with high serum levels of Gal‐9, CXCL10, TNFRII, and/or Gal‐1 at diagnosis would need a longer time to attain DFR. At the time of analysis, 24 (41%) of 59 patients had attained DFR (median follow‐up time 4.0 years). Of the 10 patients in cluster 1C with a known time until DFR or with more than 4 years of follow‐up, 8 (80%) were still receiving medication after 4 years, whereas this was the case for only 7 (41%) of 17 patients in cluster 2C (P < 0.05) (Figure 4E).
Kaplan‐Meier survival analysis with dichotomization of the single biomarkers into “high” and “low” serum levels according to the median values showed that patients with high serum levels of CXCL10, Gal‐9, or TNFRII at diagnosis needed a significantly longer time to attain DFR than patients with low biomarker levels (P < 0.05) (Figure 4F), although some cross‐over of the Kaplan‐Meier curves was evident, indicating that confounding factors may play a role, and the confidence intervals overlapped.
In Cox proportional hazards models, high biomarker levels showed a trend toward conferring a higher risk of “not getting off treatment” as compared to low biomarker levels (for Gal‐9, hazard ratio [HR] 0.32, P = 0.06; for TNFRII, HR 0.24, P = 0.08; for CXCL10, HR 0.39, P = 0.1; for Gal‐1, HR 0.41, P = 0.1). Patients with an ERL score of <4, indicating severe vasculopathy, also showed a trend toward needing longer time to attain DFR (Kaplan‐Meier log‐rank P = 0.069). Taken together, these results indicate that patients with high serum levels of Gal‐9, CXCL10, or TNFRII at diagnosis may be at risk for a longer disease course.
Discussion
Herein we have shown that juvenile DM is a heterogeneous disease, not only in the clinical presentation, but also in the biomarker profiles at diagnosis. In 2 independent cohorts of patients with juvenile DM, we were able to identify a subgroup of patients, constituting approximately one‐third of the study population, who exhibited high serum levels of Gal‐9, CXCL10, TNFRII, and Gal‐1 and higher muscle and global disease activity, who were considered at risk of requiring intensification of initial treatment, and who experienced a longer time to reach DFR. These biomarkers could thus be used to identify a severely affected subgroup at diagnosis, and may be prognostic for the disease course. The biomarker‐based clusters were not evidently correlated with MSA serotypes, but patients with strong anti–NXP‐2 positivity were more likely to be in the subgroup with high serum levels of Gal‐9, CXCL10, TNFRII, and Gal‐1 at diagnosis.
Our results are consistent with the findings from a study by Gitiaux et al, in which approximately one‐third of patients were identified as being severely affected 19. In our study, the severely affected subgroup of patients were not identified as having more severe vasculopathy based on their ERL scores. Moreover, the vasculopathy‐associated biomarkers endoglin, ICAM‐1, TSP‐1, and VEGF did not aid in the identification of this subgroup. However, the observed correlations of VEGF and endoglin serum levels with severe vasculopathy are consistent with the findings of a recent study in which both proteins were shown to be up‐regulated in lesions with active capillary injury in the muscle of patients with juvenile DM 7. Endoglin and VEGF were also found to be expressed in the muscle of patients with DM in other studies 29, 30. Soluble endoglin has more widely been described as an antiangiogenic molecule and marker for vasculopathy 31. TSP‐1 was previously suggested to be an antiangiogenic regulator 32 with a vasculopathic role in patients with juvenile DM (in particular, patients with the TNF‐308A allele) 33. We found a positive association between the serum ICAM‐1 levels and ERL scores in the present study. Since endothelial activation is one of the hallmarks of juvenile DM–associated vasculopathy 34, and soluble ICAM‐1 levels are correlated with endothelial surface ICAM‐1 expression 35, this finding is difficult to explain in the context of the disease. Future studies may provide insights into the underlying biologic processes.
Two of the 4 identified severity markers, Gal‐9 and CXCL10, are known IFN‐related proteins 36, 37. The correlation between severe muscle disease activity and high levels of IFN‐related markers or the IFN signature (in blood and biopsy samples) has been previously demonstrated in juvenile DM patients at the time of disease onset 13, 38, 39 and during follow‐up 16. A higher IFN signature was also associated with a longer time to reach clinically inactive disease 39. The serum levels of Gal‐9, CXCL10, and TNFRII were shown to specifically correlate with muscle disease activity during follow‐up in our cohorts of patients with juvenile DM, and Gal‐9 and CXCL10 serum levels were recently validated as biomarkers for disease activity in patients with juvenile DM 17, 40. Moreover, patients experiencing a disease flare within the first year after the start of treatment had higher levels of Gal‐9 and CXCL10 at diagnosis 40. TNFRII levels were found to be high in adult DM patients with a high type I IFN score, and neutralization of the IFN signature by an anti‐IFNα monoclonal antibody resulted in decreased levels of TNFRII, suggesting that TNFRII may also be related to IFN‐driven inflammation 41.
The increased expression of circulating IFN‐inducible proteins in severely affected patients further supports the pathogenic role of IFNs in juvenile DM immunopathology. Gal‐1 has not been previously linked to DM, but degeneration of injured muscles induces high expression of Gal‐1, and its expression may increase muscle regeneration in experimental models 42, 43. In addition, Gal‐1 is an antiviral effector molecule expressed by endothelial cells and a negative regulator of both T cell recruitment to the endothelium and transendothelial migration 44, 45, 46.
Consistent with our results indicating that muscle disease activity was more pronounced in the severely affected patient cluster, the anti–NXP‐2 antibody serotype was previously associated with more severe muscle disease, whereas anti–MDA‐5–positive patients were less likely to have muscle weakness 3. In contrast with the findings from previous studies, we did not observe high levels of IFN‐related markers in patients with anti–MDA‐5 antibodies 47.
As opposed to muscle disease activity, skin disease activity was not related to any of the markers or marker profiles in the 2 cohorts. Possible explanations for this finding could be that skin disease activity showed relatively low variance across patients (mainly in the discovery cohort), or that muscle disease had a dominant effect on the biomarker profiles (as also suggested by the PCA plots in Figure 1D), which could overrule any moderate correlations with skin disease.
Whereas in the validation cohort, Gal‐1 and TNFRII serum levels were higher in patients requiring intensification of treatment, this was not the case in the discovery cohort. This difference could not be explained by differences in the initial treatment approach, as the approaches were similar between the cohorts. We could speculate that the standardized treatment regimen in the validation cohort (as opposed to the more individualized strategy employed in the discovery cohort) led to a homogeneous/well‐defined group of patients receiving intensification of treatment—with certain specific characteristics conforming to the applied protocol. This homogeneity could be reflected in their biomarker profiles. In addition, the difference may be related to the longer duration of untreated disease in the discovery cohort compared to the validation cohort.
Juvenile DM is a very rare disease, which hampers the collection of large sample numbers for study purposes. A unique strength of our study is that we were able to perform biomarker profiling in 2 large (given the rarity of the disease), independent cohorts of treatment‐naive patients. We were thereby able to analyze the unmodified disease signatures in different patients without possible treatment effects on biomarker profiles. Although the cohorts showed some differences in baseline characteristics (e.g., duration of untreated disease, MSA frequencies, requirement of treatment intensification, ethnicity), which could have conferred bias in their biomarker profiles, we were able to validate our findings, showing that Gal‐9, TNFRII, Gal‐1, and CXCL10 are robust markers for the identification of a subgroup of patients with severe disease, even in cohorts with different patient characteristics. A limitation of this study is the difference in the methods used to record disease activity between the 2 cohorts, which was attributable to historical and regional differences in the data collection methods. However, the PhGA and DAS‐T scores, as well as the CAT and DAS‐S scores, have been shown to correlate strongly 48.
Further validation of these data in a large prospective cohort will allow for the construction of prediction models that could be used to predict those patients who will require intensification of treatment or to predict the length of time to attainment of DFR, possibly combining one or more of the biomarkers with clinical parameters (such as age at onset and clinical disease activity). We speculate that patients in the “at risk” group could benefit from more intensive monitoring during induction treatment in order to detect suboptimal response to therapy in an early phase. This early detection would promote a swift intensification of treatment. Future studies will have to point out whether more aggressive or targeted initial treatment (e.g., with JAK inhibition or anti‐IFN antibodies) could also be a treatment option in these patients 49, 50. Considering the longer time to remission, it may be useful to discuss with patients and parents the expectations of treatment length and the possible effect on cumulative medication dose, as these may have implications for the long‐term outcomes.
In conclusion, the results of this study underline the clinical and serologic heterogeneity of juvenile DM. Moreover, the identification of high serum levels of the biomarkers Gal‐9, TNFRII, CXCL10, and Gal‐1 in patients with juvenile DM would suggest that these markers may be useful tools for identifying severely affected patients who could be at risk of a suboptimal response to standard immunosuppressive treatment.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. van Wijk and van Royen‐Kerkhof had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Wienke, Pachman, van Wijk, van Royen‐Kerkhof.
Acquisition of data
Wienke, Pachman, Morgan, Yeo, Amoruso, Hans, Kamphuis, Hoppenreijs, Armbrust, van den Berg, Hissink Muller, Gelderman, Arkachaisri, van Wijk, van Royen‐Kerkhof.
Analysis and interpretation of data
Wienke, Pachman, van Wijk, van Royen‐Kerkhof.
Additional Disclosures
Author Gelderman is an employee of Sanquin Diagnostic Services.
Acknowledgments
We thank the pediatric rheumatology research group at Lurie Children's Hospital, Chicago, which is supported by the CureJM Foundation, and KK Women's and Children's Hospital, Singapore, for kindly providing the samples and the extensive clinical expertise with which to perform this study. We thank the Dutch juvenile DM network, and especially Annette van Dijk‐Hummelman and Ellen Schatorjé, for their help and support in the collection and inclusion of patients and samples. We also thank the Luminex core facility for performing all biomarker measurements. A special thanks goes to the board members of the patient group Myositis of the VSN (Vereniging Spierziekten Nederland), the Stichting KAISZ (Kinderen met Auto‐ImmuunSysteemZiekten), and the Bas Stichting (Dutch juvenile DM patient organization) for their explicit and continuous support for our biomarker research in juvenile DM, as well as the CureJM Foundation, which has enabled the Chicago Center to amass the biobank data for this study.
The Chicago Juvenile Dermatomyositis (DM) Registry/Biorepository was supported by the Cure JM Foundation and the National Institute of Nursing Research, NIH (grant NR‐012692). The Dutch juvenile DM cohort was supported by the Princess Beatrix Fund, the BAS Foundation, Innovatiefonds Zorgverzekeraars, and the Cure JM Foundation. The Singaporean juvenile DM cohort was supported by the Singapore Ministry of Health's National Medical Research Council (grants NMRC‐CG‐M003‐2017, NMRC‐STaR‐020‐2013, MOHIAFCAT2‐2‐08, and NMRC‐TA‐0059‐2017). Dr. Pachman's work was supported by the NIH (grant 1R43‐AR‐073547‐01).
Drs. van Wijk and van Royen‐Kerkhof contributed equally to this work.
Dr. Pachman has received research support from ReveraGen Biopharma. No other disclosures relevant to this article were reported.
All data and protocols from the biomarker cohort study are available to the scientific community upon reasonable request.
References
- 1. Meyer A, Meyer N, Schaeffer M, Gottenberg J‐E, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford) 2015;54:50–63. [DOI] [PubMed] [Google Scholar]
- 2. Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore) 2013;92:25–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tansley SL, Simou S, Shaddick G, Betteridge ZE, Almeida B, Gunawardena H, et al. Autoantibodies in juvenile‐onset myositis: their diagnostic value and associated clinical phenotype in a large UK cohort. J Autoimmun 2017;84:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Enders FB, Bader‐Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM, et al. Consensus‐based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis 2017;76:329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wienke J, Deakin CT, Wedderburn LR, van Wijk F, van Royen‐Kerkhof A. Systemic and tissue inflammation in juvenile dermatomyositis: from pathogenesis to the quest for monitoring tools [review]. Front Immunol 2018;9:2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Papadopoulou C, McCann LJ. The vasculopathy of juvenile dermatomyositis. Front Pediatr 2018;6:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baumann M, Gumpold C, Mueller‐Felber W, Schoser B, Haberler C, Loescher WN, et al. Pattern of myogenesis and vascular repair in early and advanced lesions of juvenile dermatomyositis. Neuromuscul Disord 2018;28:973–85. [DOI] [PubMed] [Google Scholar]
- 8. Kim E, Cook‐Mills J, Morgan G, Sredni ST, Pachman LM. Increased expression of vascular cell adhesion molecule 1 in muscle biopsy samples from juvenile dermatomyositis patients with short duration of untreated disease is regulated by miR‐126. Arthritis Rheum 2012;64:3809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goncalves FG, Chimelli L, Sallum AM, Marie SK, Kiss MH, Ferriani VP. Immunohistological analysis of CD59 and membrane attack complex of complement in muscle in juvenile dermatomyositis. J Rheumatol 2002;29:1301–7. [PubMed] [Google Scholar]
- 10. Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med 1986;314:329–34. [DOI] [PubMed] [Google Scholar]
- 11. Fall N, Bove KE, Stringer K, Lovell DJ, Brunner HI, Weiss J, et al. Association between lack of angiogenic response in muscle tissue and high expression of angiostatic ELR‐negative CXC chemokines in patients with juvenile dermatomyositis: possible link to vasculopathy. Arthritis Rheum 2005;52:3175–80. [DOI] [PubMed] [Google Scholar]
- 12. Gitiaux C, Latroche C, Weiss‐Gayet M, Rodero MP, Duffy D, Bader‐Meunier B, et al. Myogenic progenitor cells exhibit type I interferon–driven proangiogenic properties and molecular signature during juvenile dermatomyositis. Arthritis Rheumatol 2018;70:134–45. [DOI] [PubMed] [Google Scholar]
- 13. O'Connor KA, Abbott KA, Sabin B, Kuroda M, Pachman LM. MxA gene expression in juvenile dermatomyositis peripheral blood mononuclear cells: association with muscle involvement. Clin Immunol 2006;120:319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rice GI, Melki I, Fremond M‐L, Briggs TA, Rodero MP, Kitabayashi N, et al. Assessment of type I interferon signaling in pediatric inflammatory disease. J Clin Immunol 2017;37:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med 2017;214:1547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reed AM, Peterson E, Bilgic H, Ytterberg SR, Amin S, Hein MS, et al. Changes in novel biomarkers of disease activity in juvenile and adult dermatomyositis are sensitive biomarkers of disease course. Arthritis Rheum 2012;64:4078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bellutti Enders F, van Wijk F, Scholman R, Hofer M, Prakken BJ, van Royen‐Kerkhof A, et al. Correlation of CXCL10, tumor necrosis factor receptor type II, and galectin 9 with disease activity in juvenile dermatomyositis. Arthritis Rheumatol 2014;66:2281–9. [DOI] [PubMed] [Google Scholar]
- 18. Schmeling H, Stephens S, Goia C, Manlhiot C, Schneider R, Luthra S, et al. Nailfold capillary density is importantly associated over time with muscle and skin disease activity in juvenile dermatomyositis. Rheumatology (Oxford) 2011;50:885–93. [DOI] [PubMed] [Google Scholar]
- 19. Gitiaux C, de Antonio M, Aouizerate J, Gherardi RK, Guilbert T, Barnerias C, et al. Vasculopathy‐related clinical and pathological features are associated with severe juvenile dermatomyositis. Rheumatology (Oxford) 2016;55:470–9. [DOI] [PubMed] [Google Scholar]
- 20. Wargula JC, Lovell DJ, Passo MH, Bove KE, Santangelo JD, Levinson JE. What more can we learn from muscle histopathology in children with dermatomyositis/polymyositis? Clin Exp Rheumatol 2006;24:333–43. [PubMed] [Google Scholar]
- 21. Bohan A, Peter JB. Polymyositis and dermatomyositis: part 1. N Engl J Med 1975;292:344–7. [DOI] [PubMed] [Google Scholar]
- 22. Bohan A, Peter JB. Polymyositis and dermatomyositis: part 2. N Engl J Med 1975;292:403–7. [DOI] [PubMed] [Google Scholar]
- 23. Quiñones R, Morgan GA, Amoruso M, Field R, Huang C‐C, Pachman LM. Lack of achievement of a full score on the childhood myositis assessment scale by healthy four‐year‐olds and those recovering from juvenile dermatomyositis. Arthritis Care Res (Hoboken) 2013;65:1697–701. [DOI] [PubMed] [Google Scholar]
- 24. Bode RK, Klein‐Gitelman MS, Miller ML, Lechman TS, Pachman LM. Disease activity score for children with juvenile dermatomyositis: reliability and validity evidence. Arthritis Rheum 2003;49:7–15. [DOI] [PubMed] [Google Scholar]
- 25. Rider LG, Feldman BM, Perez MD, Rennebohm RM, Lindsley CB, Zemel LS, et al, in coopoeration with the Juvenile Dermatomyositis Disease Activity Collaborative Study Group . Development of validated disease activity and damage indices for the juvenile idiopathic inflammatory myopathies: I. Physician, parent, and patient global assessments. Arthritis Rheum 1997;40:1976–83. [DOI] [PubMed] [Google Scholar]
- 26. Huber AM, Dugan EM, Lachenbruch PA, Feldman BM, Perez MD, Zemel LS, et al. Preliminary validation and clinical meaning of the cutaneous assessment tool in juvenile dermatomyositis. Arthritis Rheum 2008;59:214–21. [DOI] [PubMed] [Google Scholar]
- 27. Christen‐Zaech S, Seshadri R, Sundberg J, Paller AS, Pachman LM. Persistent association of nailfold capillaroscopy changes and skin involvement over thirty‐six months with duration of untreated disease in patients with juvenile dermatomyositis. Arthritis Rheum 2008;58:571–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Jager W, Prakken BJ, Bijlsma JW, Kuis W, Rijkers GT. Improved multiplex immunoassay performance in human plasma and synovial fluid following removal of interfering heterophilic antibodies. J Immunol Methods 2005;300:124–35. [DOI] [PubMed] [Google Scholar]
- 29. Grundtman C, Tham E, Ulfgren A‐K, Lundberg IE. Vascular endothelial growth factor is highly expressed in muscle tissue of patients with polymyositis and patients with dermatomyositis. Arthritis Rheum 2008;58:3224–38. [DOI] [PubMed] [Google Scholar]
- 30. Konttinen YT, Mackiewicz Z, Povilenaite D, Sukura A, Hukkanen M, Virtanen I. Disease‐associated increased HIF‐1, αvβ3 integrin, and Flt‐1 do not suffice to compensate the damage‐inducing loss of blood vessels in inflammatory myopathies. Rheumatol Int 2004;24:333–9. [DOI] [PubMed] [Google Scholar]
- 31. Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 2006;12:642–9. [DOI] [PubMed] [Google Scholar]
- 32. Jiménez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis‐dependent inhibition of neovascularization by thrombospondin‐1. Nat Med 2000;6:41–8. [DOI] [PubMed] [Google Scholar]
- 33. Lutz J, Huwiler KG, Fedczyna T, Lechman TS, Crawford S, Kinsella TR, et al. Increased plasma thrombospondin‐1 (TSP‐1) levels are associated with the TNF α‐308A allele in children with juvenile dermatomyositis. Clin Immunol 2002;103:260–3. [DOI] [PubMed] [Google Scholar]
- 34. Nagaraju K, Rider LG, Fan C, Chen Y‐W, Mitsak M, Rawat R, et al. Endothelial cell activation and neovascularization are prominent in dermatomyositis. J Autoimmune Dis 2006;3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kjærgaard AG, Dige A, Krog J, Tønnesen E, Wogensen L. Soluble adhesion molecules correlate with surface expression in an in vitro model of endothelial activation. Basic Clin Pharmacol Toxicol 2013;113:273–9. [DOI] [PubMed] [Google Scholar]
- 36. Van den Hoogen LL, van Roon JA, Mertens JS, Wienke J, Lopes AP, de Jager W, et al. Galectin‐9 is an easy to measure biomarker for the interferon signature in systemic lupus erythematosus and antiphospholipid syndrome. Ann Rheum Dis 2018;77:1810–4. [DOI] [PubMed] [Google Scholar]
- 37. Antonelli A, Ferrari SM, Giuggioli D, Ferrannini E, Ferri C, Fallahi P. Chemokine (C–X–C motif) ligand (CXCL)10 in autoimmune diseases [review]. Autoimmun Rev 2014;13:272–80. [DOI] [PubMed] [Google Scholar]
- 38. Soponkanaporn S, Deakin CT, Schutz PW, Marshall LR, Yasin SA, Johnson CM, et al. Expression of myxovirus‐resistance protein A: a possible marker of muscle disease activity and autoantibody specificities in juvenile dermatomyositis. Neuropathol Appl Neurobiol 2018;45:410–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moneta GM, Marafon DP, Marasco E, Rosina S, Verardo M, Fiorillo C, et al. Muscle expression of type I and type II interferons is increased in juvenile dermatomyositis and related to clinical and histological features. Arthritis Rheumatol 2019;71:1011–21. [DOI] [PubMed] [Google Scholar]
- 40. Wienke J, Enders FB, Lim J, Mertens JS, van den Hoogen LL, Wijngaarde CA, et al. Galectin‐9 and CXCL10 as biomarkers for disease activity in juvenile dermatomyositis: a longitudinal cohort study and multicohort validation. Arthritis Rheumatol 2019;71:1377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guo X, Higgs BW, Rebelatto M, Zhu W, Greth W, Yao Y, et al. Suppression of soluble T cell‐associated proteins by an anti‐interferon‐alpha monoclonal antibody in adult patients with dermatomyositis or polymyositis. Rheumatology (Oxford) 2014;53:686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cerri DG, Rodrigues LC, Stowell SR, Araujo DD, Coelho MC, Oliveira SR, et al. Degeneration of dystrophic or injured skeletal muscles induces high expression of galectin‐1. Glycobiology 2008;18:842–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chan J, O'Donoghue K, Gavina M, Torrente Y, Kennea N, Mehmet H, et al. Galectin‐1 induces skeletal muscle differentiation in human fetal mesenchymal stem cells and increases muscle regeneration. Stem Cells 2006;24:1879–91. [DOI] [PubMed] [Google Scholar]
- 44. Garner OB, Yun T, Pernet O, Aguilar HC, Park A, Bowden TA, et al. Timing of galectin‐1 exposure differentially modulates Nipah virus entry and syncytium formation in endothelial cells. J Virol 2015;89:2520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Norling LV, Sampaio AL, Cooper D, Perretti M. Inhibitory control of endothelial galectin‐1 on in vitro and in vivo lymphocyte trafficking. FASEB J 2008;22:682–90. [DOI] [PubMed] [Google Scholar]
- 46. He J, Baum LG. Endothelial cell expression of galectin‐1 induced by prostate cancer cells inhibits T‐cell transendothelial migration. Lab Invest 2006;86:578–90. [DOI] [PubMed] [Google Scholar]
- 47. Ono N, Kai K, Maruyama A, Sakai M, Sadanaga Y, Koarada S, et al. The relationship between type 1 IFN and vasculopathy in anti‐MDA5 antibody‐positive dermatomyositis patients. Rheumatology (Oxford) 2019;58:786–91. [DOI] [PubMed] [Google Scholar]
- 48. Campanilho‐Marques R, Almeida B, Deakin C, Arnold K, Gallot N, de Iorio M, et al. Comparison of the utility and validity of three scoring tools to measure skin involvement in patients with juvenile dermatomyositis. Arthritis Care Res (Hoboken) 2016;68:1514–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ladislau L, Suárez‐Calvet X, Toquet S, Landon‐Cardinal O, Amelin D, Depp M, et al. JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis. Brain 2018;141:1609–21. [DOI] [PubMed] [Google Scholar]
- 50. Higgs BW, Zhu W, Morehouse C, White WI, Brohawn P, Guo X, et al. A phase 1b clinical trial evaluating sifalimumab, an anti‐IFN‐α monoclonal antibody, shows target neutralisation of a type I IFN signature in blood of dermatomyositis and polymyositis patients. Ann Rheum Dis 2014;73:256–62. [DOI] [PMC free article] [PubMed] [Google Scholar]