

Abstract
Systemic sclerosis (SSc) is an autoimmune rheumatic disease with heterogeneous clinical manifestations and variable disease course in which the severity of pathology dictates the disease prognosis and course. Among autoimmune rheumatic diseases, SSc carries the highest mortality, but there are exciting new therapeutic targets that appear to halt the progression of SSc manifestations such as skin or lung fibrosis. In selected patients, high-intensity regimens with autologous stem cell transplantation can favorably modify the disease course. From what was once thought to be an untreatable disease, targeted therapies have now changed the outlook of SSc to a treatable disease. Our review discusses the targeted therapies that are modifying the outlook of selected organ involvement and creating opportunities for the future. We also define a framework for low disease activity in SSc.
Introduction
Systemic sclerosis (SSc) is a rare disease characterized by vasculopathy and fibrosis in the skin and internal organs (1). The proposed pathophysiology is a triad of vascular damage with endothelial dysfunction, dysregulation of innate and adaptive immunity, and widespread fibrosis in multiple organs (2, 3). The mortality in SSc is higher than any other rheumatic disease (4, 5).
Contrary to rheumatoid arthritis (RA), the concept and use of disease-modifying therapies (DMT) that attenuate or reverse pathology and clinical impact are not currently applied to SSc. The notion of disease modification in SSc has now advanced to reality based on the data from recent clinical trials in SSc. Autologous hematopoietic stem cell transplantation (HSCT) trials in dcSSc have demonstrated survival benefit including meaningful improvement in skin, lung fibrosis, and health-related quality of life (HRQoL) (6–9). In this review, we discuss specific treatments that have modified the course of organ-specific manifestations and start the conversation on defining low disease activity in SSc.
What is disease-modifying therapy?
We borrow the concept of DMT from the use of disease modifying anti-rheumatic drugs (DMARDs) and biological response modifiers in RA. In the past three decades, the management of RA has evolved from symptom management to the implementation of DMARDs and/or biological response modifiers. The early institution of DMARDs or biological response modifiers induces clinical remission, reduces the frequency of relapse, abrogates joint damage, preserves physical function, improves HRQoL, and prevents long-term disability (10). Similarly, we can conceptualize DMT in SSc as therapies or medication regimens, that positively impact the disease course by stabilizing and potentially improving in organ function. This, in turn, improves HRQoL and reduces morbidity and mortality (11).
Natural history of the disease
The understanding of the natural history of SSc disease process is vital to the concept of DMT in the context of timing and patient selection. The early clinical features include Raynaud’s phenomenon (RP) and gastro-esophageal reflux disease (12). Skin fibrosis is a pathological hallmark of the disease and is frequently preceded by puffy and swollen fingers. Patients with puffy fingers, definite RP, typical nailfold capillary changes, and the presence of an SSc specific antibodies, can be diagnosed with Very Early Diagnosis of Systemic Sclerosis (VEDOSS) (13, 14). Thereafter, patients may progress to one of three clinical disease subsets based on the extent of skin involvement.
Those patients with skin involvement restricted to the limbs distal to the elbows or knees, with or without face involvement, are classified as limited cutaneous SSc (lcSSc). Those patients with distal as well as proximal involvement (including torse), who are classified as diffuse cutaneous SSc (dcSSc). A small subset of patients without skin involvement but who have scleroderma-specific antibodies and internal organ involvement, termed systemic sclerosis sine scleroderma (15–17). This differentiation is important as dcSSc is associated with higher morbidity and mortality, mainly due to more severeand/or progressive internal organ involvement (18). However, this differentiation of the clinical phenotypes is an over-simplification of the disease process.
The biology of SSc is complex, heterogeneous, and dynamic, with sequentially overlapping features of inflammation, autoimmunity, tissue injury, and fibrosis. Skin thickness is generally progressive within the first 3-years after the start of RP in dcSSc, but there is individual variability (15, 16). The extent and severity of skin involvement in dcSSc generally level off by years 4–5 and then clinically appears to improve both via deremodeling and atrophy(19). Only a minority of patients have a new emergence of progressive cutaneous involvement after five years of disease onset. There is an increased risk of the onset of internal organ involvement during the progressive skin phase. For example, in dcSSc, most of the internal organ involvement (lung, renal, cardiac, and gastrointestinal) occurs in the first 3–5 years of the disease onset (Figure 1)(16). In the early phase, internal organ involvement – although clinically silent – may evolve at the same time as progressive skin disease in the early phase. There are exceptions– for example, pulmonary arterial hypertension (PAH) in generally a later complication and is more common in lcSSc (20). Separately or overlapping with pulmonary hypertension, lung fibrosis can set in. Fibrosis can advance in a self-perpetuating manner and may not be driven solely by an immune-mediated process (21).
Figure 1: The usual timing of organ-specific manifestations in systemic sclerosis.
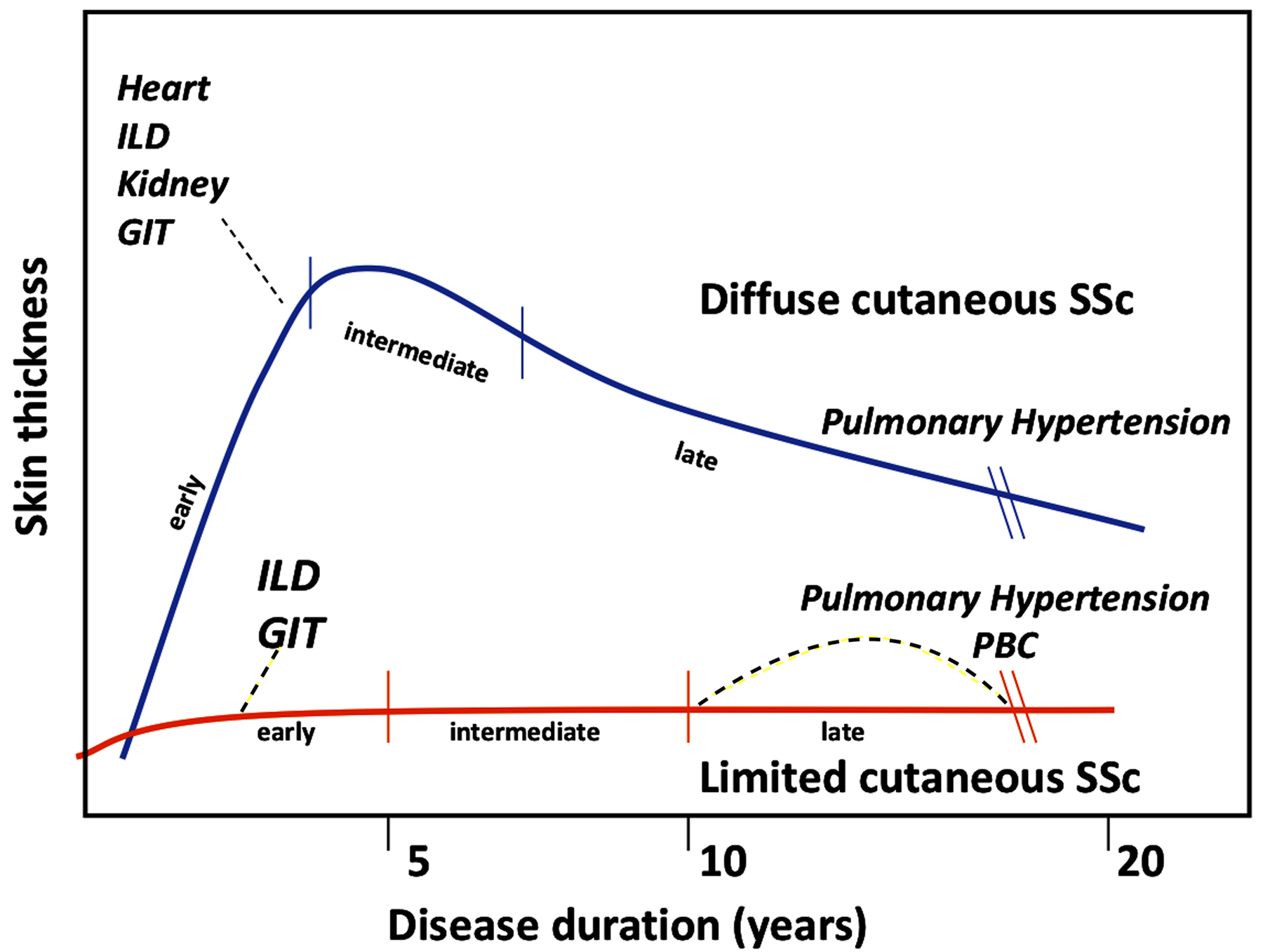
ILD Interstitial Lung Disease, GIT gastrointestinal tract, PBC Primary Biliary Cirrhosis
(Adapted from Steen V, Medsger TA. Systemic Sclerosis: Lippincott Williams & Wilkins; 1996)
In our opinion, SSc can be conceptualized as a family of similar diseases, an idea that is supported by molecular subsets identified by whole genome gene expression profiling, it has distinct clinical and serological features and recognized phases within some of the sub-types (22). The delayed emergence of new organ involvement and gradual progression of the disease provides clinicians with a realistic opportunity to impede disease progression and change disease course.
Why is disease-modifying therapy a challenge in SSc?
Many challenges exist in demonstrating disease-modifying effects in SSc patients. First, the disease is heterogeneous with different patterns of evolution amongst the clinical subsets, as already outlined (5, 23–25). Patients usually present with predominately vasculopathic complications [such as RP, digital ulcers (DU), PAH, scleroderma renal crisis (SRC), and gastrointestinal involvement], predominately fibrotic complications (such as skin fibrosis, joint involvement, lung fibrosis, and cardiac fibrosis), or a combination of these. Within each cutaneous subgroup, there is heterogeneity in the internal organ involvement(18). Second, there are molecular differences in the skin gene expression data in patients with a similar phenotype. One such formulation identified four subsets based on skin gene expression data: normal-like, inflammatory, fibroproliferative, and limited (22, 26). These subsets identify patients at risk for internal organ involvement, such as ILD, and response to current therapies (26, 27). Measuring gene expression subsets in clinical trials, and possibly even in routine clinical care, may in the near future, breakdown and clarify patient heterogeneity, and provide a window through which to understand and predict patient response to therapy. Third, the predictors of disease status at a specific time point (incidence or severity of organ-based complications; largely influenced by autoantibodies) may differ from predictors of disease progression (28, 29).
Unlike the disease activity score (DAS-28), clinical disease activity index (CDAI), or others in RA, we lack reliable tools with which we can define the achievement of remission in SSc. In dcSSc, modified Rodnan skin score, and recently, a combined responder index in dcSSc (ACR CRISS - a composite end point that captures cardio-pulmonary-renal involvement and change in mRSS, health assessment questionnaire-disability index [HAQ-DI], patient global assessment, physician global assessment, and forced vital capacity [FVC] % predicted) are used as outcomes to measure the efficacy of drugs (30). These measures have not been validated in lcSSc and may not perform well (31). Further, clinical heterogeneity of the disease does not allow precise definition of global disease activity. Composite scores like the revised European Scleroderma Research Group Activity Index have been proposed, but have not been widely accepted in the evaluation of disease activity(28). Novel approaches for assessing disease activity in SSc are currently under development(32).
Are there currently DMTs for SSc?
Despite the limitations in disease activity measurement in SSc, treatment approaches directed toward specific biologic targets appear to be positively influencing outcomes in SSc (Supplementary material, Table 1). This concept can be approached by categorizing SSc manifestations into vasculopathy, immunological or inflammatory involvement, and tissue fibrosis.
Vasculopathy
The predominant vascular complications in SSc are Raynaud’s phenomenon (RP), PAH, SRC, and DU. The morbidity and mortality are high in patients with PAH and SRC. RP and DU are chronic complications that can limit hand function, increase morbidity and disability (33) and impact HRQoL. The pathophysiologic mechanisms in SSc vasculopathy are characterized by initial vascular endothelial injury and dysfunction followed by vessel wall remodeling with intimal and medial thickening, leading to luminal narrowing, vascular stiffness, and tissue hypoxia (34).
One of the relevant vasculopathic manifestation, which is associated with significant mortality and morbidity in SSc patients, is PAH. The prevalence of PAH measured by right heart catheterization in large cohorts of SSc patients ranges from 5 to 12% (35, 36). SSc-PAH is associated with a worse outcome compared to idiopathic PAH because there are non-PAH related factors in SSc like coexistent ILD associated PH, pulmonary venoocclusive disease, SSc myocardial disease, and later age of onset (37, 38). Greater emphasis has been put on early screening and detection of SSc-PAH with composite algorithms, allowing for the earlier institution of PAH-specific therapy(39–41). There is a growing body of evidence that this approach may improve the morbidity outcomes although the effect on long term mortality is unclear(42). The lower incidence of SSc-PAH in patients on dihydropyridine calcium antagonists offers a tantalizing glimpse into the potential disease-modifying actions of fairly modest vasodilator therapy on long-term outcomes in SSc (43). There are multiple approved therapies for the management of PAH that target one of the three pathogenic pathways: (1) endothelin antagonists; (2) nitric oxide (NO) /soluble guanylate cyclase (GC) agonists/ stimulators; and (3) prostacyclin analogs (44). High quality randomized controlled trials have shown that upfront or sequential combination therapies delay time to clinical worsening in PAH patients. Similar approaches with combination therapies have suggested efficacy in SSc-PAH. In a recent meta-analysis, combination therapy targeting PAH conferred therapeutic efficacy compared with monotherapy in patients with SSc-PAH. There is a 27% reduction in clinical worsening (pooled relative risk of 0.73, 95% confidence interval (CI) [0.60–0.89], p=0.002) and a probable improvement of exercise capacity in these patients (45). Recent trial of rituximab in SSc-PAH showed trends of benefit on functional status (six munute walk test) and pulmonary vascular resistence vs. placebo (NCT01086540) and there is an ongoing trial of tocilizumab (NCT02676947)] in the background of currently approved therapies (46).
In SSc, common and burdensome vascular manifestations include RP and DU. RP can be the harbinger of SSc and usually precedes tissue fibrosis(47). RP is a manifestation of abnormal cutaneous vessel function involved in thermal regulation of blood flow (48). The presence of RP and the loss of normal regulation of cutaneous vascular tone is often a predictor of developing SSc, although it is not specific, cannot be used alone as a predictor, and may be long delayed (47, 49). DU are a significant cause of morbidity with around 50% of SSc patients developing DUs during their disease history (18). DU can be a sporadic phenomenon; but, for some patients, they are recurrent, continuous, and/or refractory (50). DUs can lead to significant disability in the form of impaired hand function and increased pain, loss of employment, and medical complications like gangrene, cellulitis, osteomyelitis, and digital amputation. Progress has been made in secondary prevention, although with mixed results. PDE-5 inhibitors, especially sildenafil, can reduce the frequency of RP attacks in SSc(51). A recent RCT comparing the use of oral sildenafil (20 mg three times daily) to placebo favored sildenafil in significantly decreasing the number of DUs at week-12, but did not meet the primary end-point of time to healing(52). In SSc patients with refractory and recurrent DUs, bosentan (an endothelin-1 receptor antagonist; 62.5 mg twice a day for four weeks, then 125 mg twice a day) can reduce the number of new DUs in those with >4 previous DUs, without any effect on healing existing DUs (53, 54). Intravenous prostanoid therapy improves DU healing and reduces the number of new DUs. In two multicenter, double-blind, randomized trials, i.v. prostanoid therapy (iloprost 0.5–2.0 ng/kg/min over six hours, on consecutive 5 days) was associated with significant improvement in the frequency of RP attacks and greater DU healing (55, 56).
A major life-threatening vasculopathic manifestations of SSc is SRC(57). SRC is a rare complication that affects 2–15% of patients with SSc (11% of dcSSc and 4% of lcSSc patients) (36). SRC typically presents in patients with early, rapidly progressive, dcSSc often with anti-RNA polymerase III antibodies(58). The prognosis of SRC substantially improved in the 1980s with the introduction of angiotensin-converting-enzyme inhibitors (ACE-i) for rapid blood pressure control, with additional antihypertensive agents as required(57). In a prospective analysis of 108 patients with SRC in a single center, patients on ACE-i [captopril (n=47) and enalapril (n = 8)] had significantly better survival rate at 1 year (76%) and 5 years (66%) compared to patients, not on ACE-i (1 year 15% and 5 years is 10%)(57). In another prospective trial, 145 patients with SRC treated with ACE-i demonstrated survival rates of 90% and 85% at five and eight years, respectively, after the onset of SRC(59). Furthermore, treatment with ACE-i decreased the need for permanent dialysis(16). Overall, current patient survival is 70–82% at one year but decreases to 50–60% at five years despite dialysis support.
In summary, there are therapies available for vasculopathy that have disease-modifying effects, including improved HRoL, morbidity, and survival. These effects are well demonstrated for SRC and PAH with unequivocal benefits in clinical trials.
Immuno-inflammatory involvement
The concept of ablating an autoreactive immune system followed by its replacement with a self-tolerant one, also called HSCT, has been successfully explored in SSc (7, 8). Oral or pulse i.v. cyclophosphamide (CYC) in symptomatic established SSc-ILD has a significant, although modest beneficial effect on lung function, thickening of the skin, dyspnea, and HRQoL (60, 61); it has no impact on long term survival (62, 63). Three major prospective trials were initiated to examine the role of HSCT in SSc treatment - ASTIS (7), ASSIST (8) and SCOT(6). These studies compared autologous HSCT (± radiation) to various i.v. CYC regimens. All studies included early dcSSc patients with moderate-to-severe skin thickness and internal organ involvement (lung involvement largely accounted for the vast majority of the cases). The patients were those predicted to rapidly progress. Although there were substantial differences in the study design among these trials, the results of the three studies allow one to draw valid conclusions regarding the effect of HSCT in early SSc patients with progressive skin and/or lung involvement. The following are the notable observations in the transplant arms: 1) clinically meaningful improvement in the skin thickness, 2) overall stabilization of lung function, 3) clinically meaningful improvement in the HRQoL, 4) overall survival benefit although higher short term serious adverse events in the ASTIS and SCOT Trials and higher mortality in the transplant arm during the first year post-transplant in the ASTIS trial, and 5) SSc heart disease (myocardial involvement and PAH) seems to be the main driver of transplantation-related mortality (6–8, 64).
In summary, HSCT trials provide clear evidence of immune-mediated pathogenesis in SSc and documents long-term, clinically important, disease modification in early aggressive disease.
Tissue fibrosis
Three important manifestations of tissue fibrosis include skin fibrosis, interstitial lung disease, and myocardial fibrosis.
Skin fibrosis is a cardinal manifestation and is seen in most SSc patients although a small minority have no skin involvement (systemic sclerosis sine scleroderma)(17, 65). Skin fibrosis is associated with significant morbidity due to pruritis, DU, skin tightness, and skin ulcers at other sites and markedly decreased function. A rapidly progressive phenotype of skin fibrosis is associated with higher mortality due to progressive internal organ involvement (66). Recently, immunosuppressive therapies such as CYC, mycophenolate mofetil (MMF), and biological response modifiers such as abatacept and tocilizumab have been evaluated for their effects on skin thickening in dcSSc. Based on the data from Scleroderma Lung Studies I and II (SLS I and II), CYC and MMF clinically and meaningfully improved mRSS in dcSSc compared to placebo (67). In a recent RCT, abatacept (vs placebo) had a clinical meaningful change in the ACR-CRISS despite non-significant change in mRSS. Decline in mRSS over 12 months was clinically and significantly higher in abatacept vs. placebo for the Inflammatory and normal-like skin gene expression subsets(68). In another RCT, subcutaneous tocilizumab trended to improve mRSS but also highlighted a marked heterogeneity in individual response (69).
SSc-ILD is present in 70–80% of patients with SSc and approximately 20–25% develop symptomatic ILD (70, 71). ILD is the leading cause of death in SSc and accounts for over one-third of SSc-related deaths(25). Immunosuppressive therapies have been consistently explored for the treatment of SSc-ILD, with different results. In the SLS I, patients with SSc-ILD received oral CYC or matching placebo for 12-months and were followed, double-blind, for an additional 12-months(60). At the end of 12-months, significant, albeit modest, treatment effects of CYC vs. placebo were observed on FVC and total lung capacity (TLC), but not on DLCO. The effect on FVC persisted at 18-months in the CYC group (although CYC was no longer being given) but was no longer present at 24-months. Additionally, CYC improved dyspnea, HRQoL, and functional ability. CYC treatment did not change long-term survival, not surprising as it was only given for 1 year (63). In SLS II, patients with SSc-ILD were randomized to receive either daily oral MMF at 3 grams/day for 24-months or daily oral CYC for 12-months (followed by placebo for 12-months)(72). No significant differences were observed in the long-term survival or organ failure for patients randomized by CYC versus MMF. In a recently conducted long-term follow-up of patients in SLS I and II, the majority of patients died of complications related to SSc; respiratory failure from end-stage lung disease was one of the leading causes of death(63). In phase III clinical data, IL-6 inhibition in early SSc with elevated CRP led to stabilization of FVC% in the tocilizumab group vs. a clinically meaningful decline in the placebo group over 48 weeks [treatment difference of 4.2%; p = 0.0002] (69). The mean [SD] FVC% was 82.1% [14.8%] at baseline which highlights the benefit of treating patients with subclinical ILD with high-risk features (early dcSSc, and elevated CRP). Rituximab (RTX) therapy in SSc has shown promising effects on both ILD and skin thickening. A recent open-label, randomized, controlled trial of RTX (1000 mg × 2 doses) vs. monthly pulse CYC analyzed a population of 60 early, treatment naïve, anti-SCL-70+, dcSSc patients with ILD (73). The RTX group improved their FVC% at the end of 6 months [RTX group +5.8% vs. CYC group −1.2%]. The data, overall, suggest that targeted biological therapies may have disease-modifying effect in ILD with preservation of lung function(69, 74).
Recently, in a 52-week placebo-controlled RCT, treatment with nintedanib - a tyrosine kinase inhibitor - slowed the progression of the FVC decline in SSc-ILD and led to approval by the Food and Drug Administration (75). The adjusted annual rate of decline in FVC was lower in the nintedanib-treated group than in the placebo-treated group (difference, 41.0 ml per year, P = 0.04) although no clinical benefits for other manifestations of SSc, dyspnea, or function were observed. Overall, about half of the patients were on baseline MMF; these patients had lower FVC decline if in the placebo group and lower magnitude of the nintedanib treatment effect on FVC. The rate of gastrointestinal adverse events was higher in the nintedanib than in the placebo group. Currently, there is an ongoing double-blind RCT (SLS III) comparing the combination of MMF with pirfenidone (an anti-fibrotic agent approved for idiopathic pulmonary fibrosis) vs. MMF alone in the treatment of SSc-ILD (NCT03221257).
Cardiac involvement is marked by myocardial fibrosis and has been reported in >50% of autopsies (76). It is frequently encountered in SSc patients, is often asymptomatic, and is associated with higher mortality (23, 36, 58). Alteration in heart rhythm with hemodynamically significant arrhythmias, including ventricular tachycardia, is associated with high mortality. Apart from medical therapy for systolic heart failure, other supportive measures like implantable cardioverter defibrillator, dual chamber pacing, or cardiac transplantation may be necessary.
In summary, for fibrosis, data suggests that the improvement in skin involvement may not be an achievable end point in trials at present due to an insensitive measurement tool, difficulty in defining sufficiently uniform entry criteria into trials, and due to individual heterogeneity. However, fibrosis in other organs, particularly the lungs may be amenable to treatment with biologics and recently – a tyrosine kinase inhibitor.
Other unmet needs
There are other disabling manifestations in SSc where the pathogenesis is poorly understood and/ or don’t have validated outcome measures. The gastrointestinal tract is involved in up to 95% of patients with SSc and is a presenting feature in about 10% of patients(77). It causes substantial morbidity and is responsible for 6% to 12% of mortality in SSc patients. Calcinosis, characterized by the deposition of insoluble calcium salts in the skin and subcutaneous tissue, is seen in about 25% of patients with SSc (78). In SSc, arthritis and joint contractures (small and large joints) are commonly seen in about one-third of patients, with large joint contractures predictive of mortality (79, 80). Telangiectasias, while themselves harmless in the skin, can be a major source of body image dissatisfaction in addition to being harbingers of pulmonary vascular disease that could make them valuable markers of disease progression. They may also be a source of GI bleeding leading to potential for increased morbidity (81). These manifestations are often unaccounted as a disease outcome in pharmacological trials and need to be included in future trials with consistent ways to measure the treatment outcome.
What should modification of disease course look like today and how to measure it?
Ideal DMT should halt the progression of the disease and hopefully induce remission, and preferably also reverse some of the major organ complications, as seen in the recent trials with HSCT on fibrotic complications (Figure 2). It is reasonable to expect DMT to stabilize organ function without any further worsening of other domains.
Figure 2: The long-term impact of ideal disease-modifying therapy in predominately fibrotic phenotype on SSc outcomes (in comparison to HSCT).
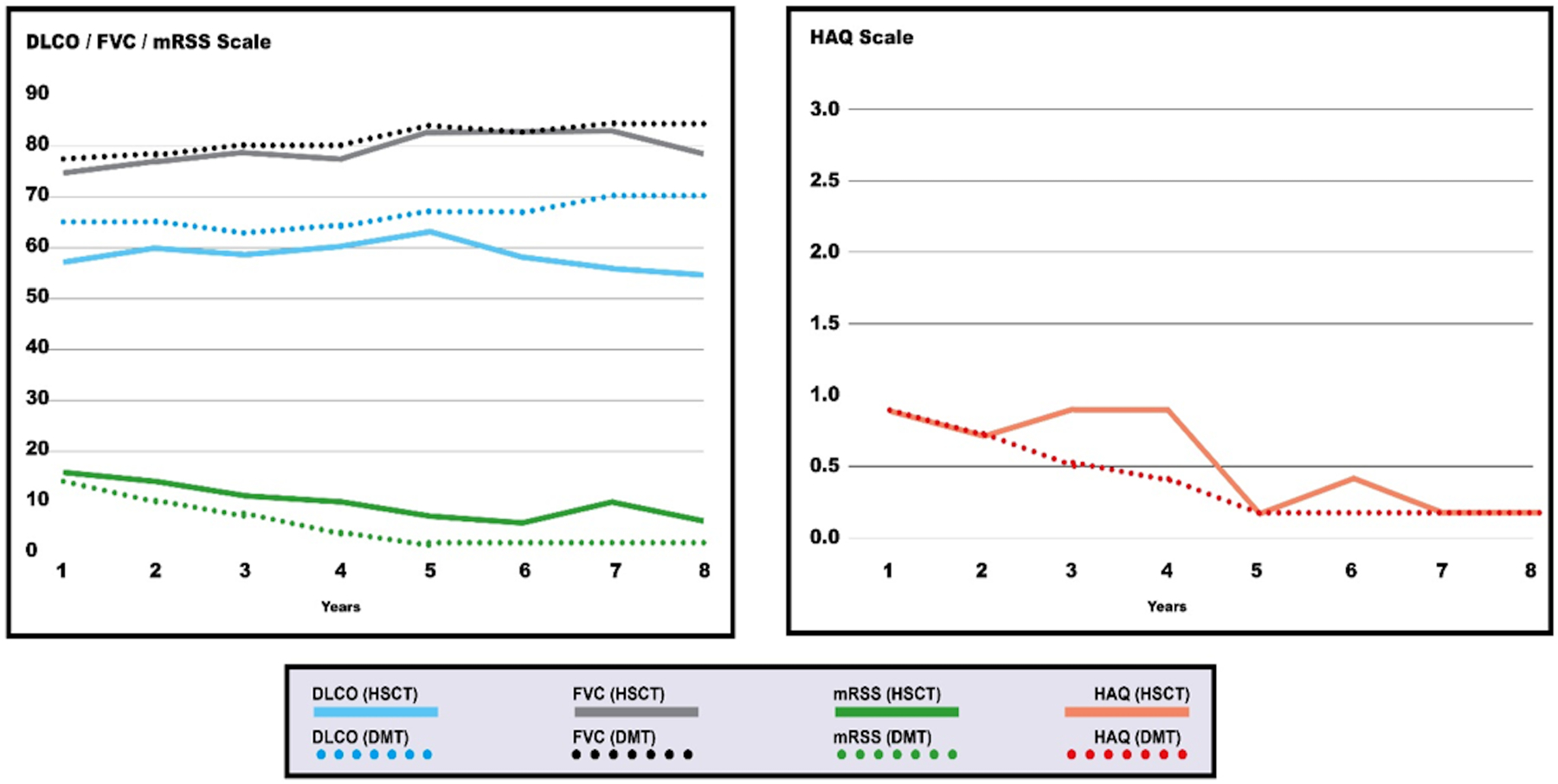
DMT Disease Modifying Therapy, HSCT Hematopetic Stem Cell Therapy, DLCO diffusion capacity for carbon monoxide, FVC forced vital capacity, MRSS Modified Rodnan Skin Score, HAQ Health Assessment Questionnaire
Reliable, valid, and responsive outcome measures are needed to assess the effect of DMT. Based on the RCTs conducted for key clinical manifestations in SSc (shown in Supplementary material Table 1) lessons have been learned about outcome measures. mRSS – a measure of skin thickness – has shown natural regression, despite enrichment for early disease and/or elevated acute phase reactants at baseline (68, 69, 82). Combined measures of response, analogous to such measures in RA, may be a way forward. In the RCTs of abatacept and tocilizumab in dcSSc, mRSS was not able to separate active therapies from placebo but there were statistically significant and clinically meaningful improvements in the ACR-CRISS, a combined measure designed to capture the global or holistic evaluation in early SSc. In tocilizumab, ACR CRISS was driven by improvement in FVC% whereas the HAQ-DI and physician global assessments were statistically significant in the abatacept trial. ACR-CRISS core set measures should be included in forthcoming clinical trials. Another example is the global rank composite score used in the SCOT trial, which utilized a hierarchical combined measure of response. In SSc-ILD, a combination of objective measures (FVC, DLCO, and lung imaging scores of fibrosis) and patient reported measure of dyspnea showed responses and, in combination, could be utilized to increase sensitivity and discrimination. At this point FVC is an approvable regulatory endpoint (60, 61). In PAH, recent successes have been achieved with clinically meaningful endpoints such as time to clinical worsening, which is a combined endpoint influenced by morbidity (such as worsening of 6 minute walk distance (6MWD), worsening of New York Heart Association (NYHA) Class, requirement of additional PAH therapy, and hospitalizations due to PAH) or all-cause mortality is an approvable endpoint in PAH(83).
How should we define remission and low disease activity (LDA) in SSc?
Based on our current understanding and constraints with testing, disease remission, which we define as the absence of disease activity, may not achievable in the setting of SSc due to the heterogeneity of the disease and few positive trials to this effect. Buoyed by the outcomes in PAH and HSCT trials, it is time to start laying a framework for conceptual definition for LDA in SSc. First, LDA in SSc should be an individual disease state (on or off therapy). Second, LDA when sustained over a period of time should be associated with better outcomes and positive effects on HRQoL (84). The future studies should define the time period that has a favorable impact on outcomes and HRQoL although this will differ based on organ involvement. Third, the distinction between what represents ‘activity’ vs. ‘damage’ is a challenge which currently is an area of investigation(32). Activity is defined as that component of disease severity that is largely reversible and may result in little or no damage in the future. Damage is that component of severity that is largely irreversible. In Figure 3, we lay out preliminary proposal to define low disease activity for the different manifestations in SSc (60, 61, 75, 85–88). These are authors-driven preliminary proposal, influenced by the data from RCTs and observational studies. This proposal will need rigorous testing and validation using a consensus methodology in future studies.
Figure 3: Low disease activity state in systemic sclerosis.
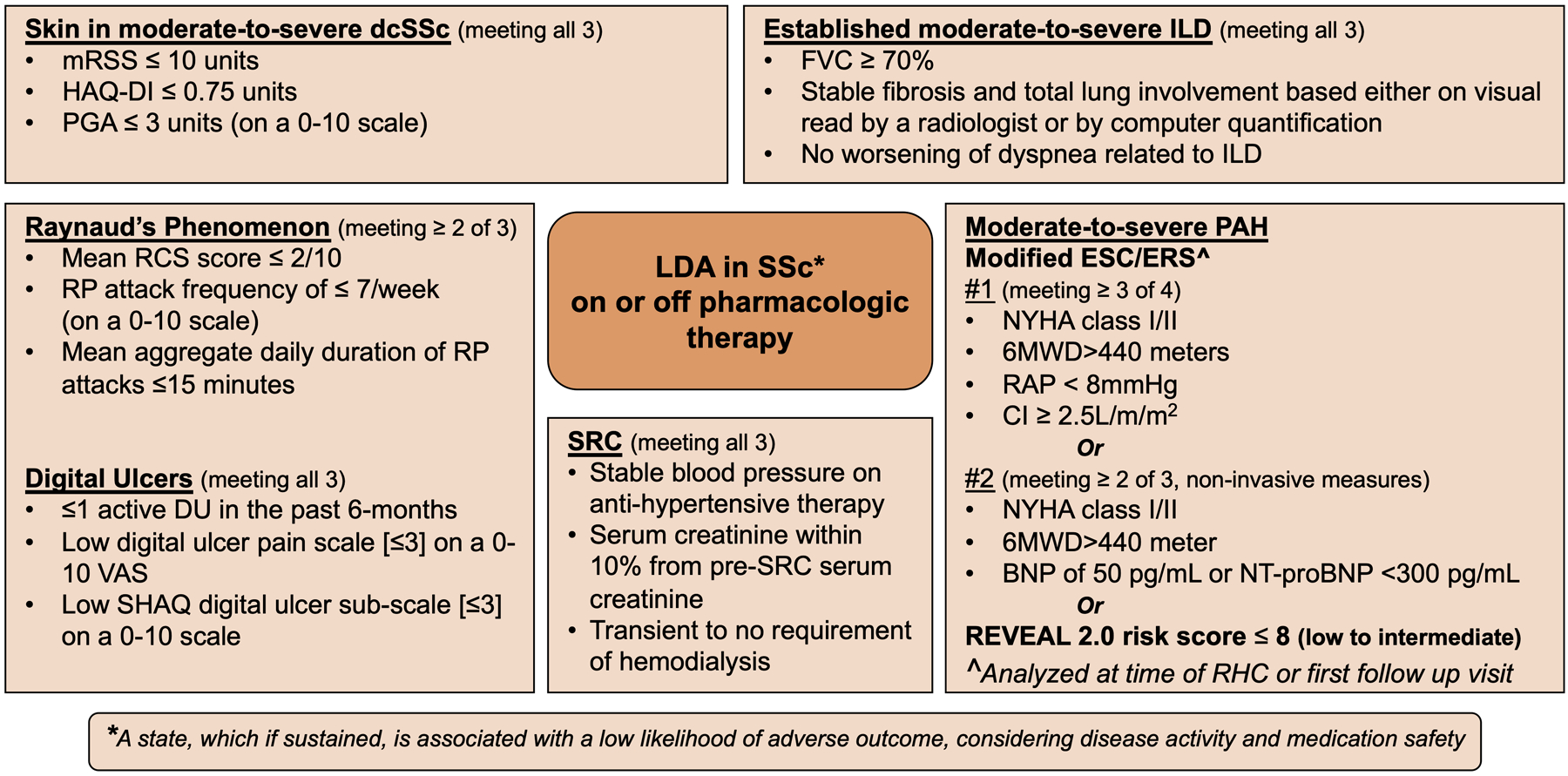
These are author-driven preliminary proposals, influenced by the data from RCTs and observational studies. These proposals will need rigorous testing and validation using a consensus methodology in future studies.
LDA = low disease activity, SSc = systemic sclerosis, mRSS = modified Rodnan skin score, HAQ-DI Health Assessment Questionnaire Disability Index, PGA = patient global assessment, ILD = interstitial lung disease, FVC% pred = forced vital capacity percentage predicted, TLC% pred = total lung capacity percentage predicted, RP = Raynaud’s phenomenon, RCS = Raynaud’s Condition Score, DU = digital ulcers, VAS = visual analog scale, SHAQ = Scleroderma Health Assessment Questionnaire PAH = pulmonary arterial hypertension, PAP = pulmonary arterial pressure, PVR = pulmonary vascular resistance, BNP = brain natriuretic peptide, NT-Pro-BNP = N-terminal pro brain natriuretic peptide, RV = right ventricle, NYHA = New York Heart Association, REVEAL = Registry to Evaluate Early and Long-Term PAH Disease Management, SRC = scleroderma renal crisis
*Chosen arbitrarily as mid-point between P Khanna et al’s estimate of the PASS and Pauling et al’s seasonal data
In conclusion, we summarize the data from recent RCTs, review the outcome measures used in recent RCTs, and propose LDA for different organ involvement in SSc.
Supplementary Material
Acknowledgments
We would like to acknowledge Dr. Thomas A. Medsger Jr, MD (Professor of Medicine, Division of Rheumatology, University of Pittsburgh Medical Center, Pittsburgh, USA) for his thoughtful input on a previous version of the submitted manuscript.
Vivek Nagaraja received honorarium from Eicos Sciences.
Marco-Matucci Cerinic: He has received consulting fees from Bristol-Myers Squibb, Pfizer, Chemomab, Actelion, Janssen, and Lilly.
Daniel Furst has grant/research support from Bristol-Myers Squibb, Corbus, Galapagos GlaxoSmithKline, National Institutes of Health, Pfizer, Sanofi, Talaris, and CSL Behring. He has received consulting fees from Amgen, Bristol-Myers Squibb, Corbus, Galapagos, Novartis, Pfizer, Roche/Genentech, Talaris, CSL Behring, Boehringer Ingelheim. Speakers Bureau: CME only.
Masataka Kuwana has received consulting fees from Actelion, Roche/Chugai, Bayer, Boehringer-Ingelheim, Corbus, and GlaxoSmithKline and serves on the speaker bureaus for Roche/Chugai and Actelion.
Yannick Allanore has received consulting fees from Actelion, Bayer, Bristol-Myers Squibb, Curzion, Boehringer Ingelheim, Inventiva, Roche, and Sanofi.
Christopher Denton has received consulting fees or honoraria from Roche/Genentech, Actelion, GlaxoSmithKline, Sanofi Aventis, Acceleron, Inventiva, CSL Behring, Boehringer Ingelheim, and Bayer.
Ganesh Raghu has recived consulting fees from Avalyn, Acceleron Biogen, Blade therapeutics, Bridge Biotherapeutics, BMS, Bellerophan, Fibrogen, Gilead Sciences, IQVIA, Promedior, Nitto, Respivant, Roche-Genentech, Sanofi-Aventis, Veracyte, Zambon, and Boehringer Ingelheim. He has received research grants from the National Institutes of Health.
Vallerie Mclaughlin has received consulting fees from Actelion, Acceleron, Arena, Bayer, Caremark, Gilead, Sonovie, and United Therapeutics.
John Pauling: He has received consulting fees from Actelion pharmaceuticals. Boehringer Ingelheim and Sojournix Pharma. He has received unrestricted research grant from Actelion pharmaceuticals.
Panduranga Rao has no disclosures.
James Seibold has received consulting fees Atlantic, Blade, Eicos, Eiger, Indalo, Mitsubishi, Bayer, Boehringer Ingelheim, Camurus, Corbus, and EMD Serono. He has stock ownership in BriaCell and Pacific Therapeutics. He has paid consultations with DRG and Guidepoint.
Michael Whitfield has received consulting fees from Bristol Myers Squibb and Celdara Medical LLC.
Dinesh Khanna has ownership interest in Eicos Sciences; has received grants from the National Institutes of Health (NIAID and NIAMS); has received consulting fees from Actelion, AstraZeneca, Bayer, Bristol-Myers Squibb, Boehringer Ingelheim, ChemomAb, Corbus, CSL Behring, Cytori, GlaxoSmithKline, Horizon, Pfizer, Regeneron, Roche/Genentech, Sanofi Aventis, and UCB Pharma. Dinesh Khanna is supported by NIH/NIAMS K24 AR063120 and NIH/NIAMS R01 AR-07047.
References:
- 1.Denton CP, Khanna D. Systemic sclerosis. Lancet (London, England). 2017. [DOI] [PubMed] [Google Scholar]
- 2.Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Primers. 2015;1:15002. [DOI] [PubMed] [Google Scholar]
- 3.Varga J, Trojanowska M, Kuwana M. Pathogenesis of systemic sclerosis: recent insights of molecular and cellular mechanisms and therapeutic opportunities. Journal of Scleroderma and Related Disorders. 2017;2(3):137–52. [Google Scholar]
- 4.Nihtyanova SI, Tang EC, Coghlan JG, Wells AU, Black CM, Denton CP. Improved survival in systemic sclerosis is associated with better ascertainment of internal organ disease: a retrospective cohort study. QJM. 2010;103(2):109–15. [DOI] [PubMed] [Google Scholar]
- 5.Elhai M, Meune C, Boubaya M, Avouac J, Hachulla E, Balbir-Gurman A, et al. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis. 2017;76(11):1897–905. [DOI] [PubMed] [Google Scholar]
- 6.Sullivan KM, Goldmuntz EA, Keyes-Elstein L, McSweeney PA, Pinckney A, Welch B, et al. Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma. N Engl J Med. 2018;378(1):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Laar JM, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. 2014;311(24):2490–8. [DOI] [PubMed] [Google Scholar]
- 8.Burt RK, Shah SJ, Dill K, Grant T, Gheorghiade M, Schroeder J, et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet. 2011;378(9790):498–506. [DOI] [PubMed] [Google Scholar]
- 9.Nash RA, McSweeney PA, Crofford LJ, Abidi M, Chen CS, Godwin JD, et al. High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation for severe systemic sclerosis: long-term follow-up of the US multicenter pilot study. Blood. 2007;110(4):1388–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38. [DOI] [PubMed] [Google Scholar]
- 11.St Clair EW, van der Heijde DM, Smolen JS, Maini RN, Bathon JM, Emery P, et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: a randomized, controlled trial. Arthritis Rheum. 2004;50(11):3432–43. [DOI] [PubMed] [Google Scholar]
- 12.van den Hombergh WM, Carreira PE, Knaapen-Hans HK, van den Hoogen FH, Fransen J, Vonk MC. An easy prediction rule for diffuse cutaneous systemic sclerosis using only the timing and type of first symptoms and auto-antibodies: derivation and validation. Rheumatology (Oxford). 2016;55(11):2023–32. [DOI] [PubMed] [Google Scholar]
- 13.van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minier T, Guiducci S, Bellando-Randone S, Bruni C, Lepri G, Czirják L, et al. Preliminary analysis of the very early diagnosis of systemic sclerosis (VEDOSS) EUSTAR multicentre study: evidence for puffy fingers as a pivotal sign for suspicion of systemic sclerosis. Ann Rheum Dis. 2014;73(12):2087–93. [DOI] [PubMed] [Google Scholar]
- 15.Wirz EG, Jaeger VK, Allanore Y, Riemekasten G, Hachulla E, Distler O, et al. Incidence and predictors of cutaneous manifestations during the early course of systemic sclerosis: a 10-year longitudinal study from the EUSTAR database. Ann Rheum Dis. 2016;75(7):1285–92. [DOI] [PubMed] [Google Scholar]
- 16.Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. 2000;43(11):2437–44. [DOI] [PubMed] [Google Scholar]
- 17.LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15(2):202–5. [PubMed] [Google Scholar]
- 18.Walker UA, Tyndall A, Czirjak L, Denton C, Farge-Bancel D, Kowal-Bielecka O, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials And Research group database. Ann Rheum Dis. 2007;66(6):754–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merkel PA, Silliman NP, Clements PJ, Denton CP, Furst DE, Mayes MD, et al. Patterns and predictors of change in outcome measures in clinical trials in scleroderma: an individual patient meta-analysis of 629 subjects with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2012;64(10):3420–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hachulla E, Launay D, Mouthon L, Sitbon O, Berezne A, Guillevin L, et al. Is pulmonary arterial hypertension really a late complication of systemic sclerosis? Chest. 2009;136(5):1211–9. [DOI] [PubMed] [Google Scholar]
- 21.Bhattacharyya S, Midwood KS, Yin H, Varga J. Toll-like receptor-4 signaling drives persistent fibroblast activation and prevents fibrosis resolution in scleroderma. Advances in wound care. 2017;6(10):356–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One. 2008;3(7):e2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tyndall AJ, Bannert B, Vonk M, Airo P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809–15. [DOI] [PubMed] [Google Scholar]
- 24.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66(7):940–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elhai M, Meune C, Avouac J, Kahan A, Allanore Y. Trends in mortality in patients with systemic sclerosis over 40 years: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford). 2012;51(6):1017–26. [DOI] [PubMed] [Google Scholar]
- 26.Hinchcliff M, Huang CC, Wood TA, Matthew Mahoney J, Martyanov V, Bhattacharyya S, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol. 2013;133(8):1979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martyanov V, Whitfield ML. Molecular stratification and precision medicine in systemic sclerosis from genomic and proteomic data. Curr Opin Rheumatol. 2016;28(1):83–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nevskaya T, Baron M, Pope JE, Canadian Scleroderma Research G. Predictive value of European Scleroderma Group Activity Index in an early scleroderma cohort. Rheumatology (Oxford). 2017. [DOI] [PubMed] [Google Scholar]
- 29.Nihtyanova SI, Sari A, Harvey JC, Leslie A, Derrett-Smith EC, Fonseca C, et al. Using autoantibodies and cutaneous subset to develop outcome-based disease classification in systemic sclerosis. Arthritis Rheumatol. 2019. [DOI] [PubMed] [Google Scholar]
- 30.Khanna D, Berrocal VJ, Giannini EH, Seibold JR, Merkel PA, Mayes MD, et al. The American College of Rheumatology Provisional Composite Response Index for Clinical Trials in Early Diffuse Cutaneous Systemic Sclerosis. Arthritis Rheumatol. 2016;68(2):299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allanore Y Limited cutaneous systemic sclerosis: the unfairly neglected subset. Journal of Scleroderma and Related Disorders. 2016;1(3):241–6. [Google Scholar]
- 32.Baron M, Kahaleh B, Bernstein EJ, Chung L, Clements PJ, Denton C, et al. An interim report of the scleroderma clinical trials consortium working groups. Journal of Scleroderma and Related Disorders. 2018:2397198318783926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mihai C, Landewe R, van der Heijde D, Walker UA, Constantin PI, Gherghe AM, et al. Digital ulcers predict a worse disease course in patients with systemic sclerosis. Ann Rheum Dis. 2016;75(4):681–6. [DOI] [PubMed] [Google Scholar]
- 34.Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. 2013;65(8):1953–62. [DOI] [PubMed] [Google Scholar]
- 35.Avouac J, Airo P, Meune C, Beretta L, Dieude P, Caramaschi P, et al. Prevalence of pulmonary hypertension in systemic sclerosis in European Caucasians and metaanalysis of 5 studies. J Rheumatol. 2010;37(11):2290–8. [DOI] [PubMed] [Google Scholar]
- 36.Nihtyanova SI, Schreiber BE, Ong VH, Rosenberg D, Moinzadeh P, Coghlan JG, et al. Prediction of pulmonary complications and long-term survival in systemic sclerosis. Arthritis Rheumatol. 2014;66(6):1625–35. [DOI] [PubMed] [Google Scholar]
- 37.Lefevre G, Dauchet L, Hachulla E, Montani D, Sobanski V, Lambert M, et al. Survival and prognostic factors in systemic sclerosis-associated pulmonary hypertension: a systematic review and meta-analysis. Arthritis Rheum. 2013;65(9):2412–23. [DOI] [PubMed] [Google Scholar]
- 38.Sobanski V, Giovannelli J, Lynch BM, Schreiber BE, Nihtyanova SI, Harvey J, et al. Characteristics and Survival of Anti-U1 RNP Antibody-Positive Patients With Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Arthritis Rheumatol. 2016;68(2):484–93. [DOI] [PubMed] [Google Scholar]
- 39.Coghlan JG, Denton CP, Grunig E, Bonderman D, Distler O, Khanna D, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis. 2014;73(7):1340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khanna D, McLaughlin V. Screening and Early Detection of Pulmonary Arterial Hypertension in Connective Tissue Diseases. It Is Time to Institute It! Am J Respir Crit Care Med. 2015;192(9):1032–3. [DOI] [PubMed] [Google Scholar]
- 41.Young A, Nagaraja V, Basilious M, Habib M, Townsend W, Gladue H, et al. Update of screening and diagnostic modalities for connective tissue disease-associated pulmonary arterial hypertension. Semin Arthritis Rheum. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mihai C, Antic M, Dobrota R, Bonderman D, Chadha-Boreham H, Coghlan JG, et al. Factors associated with disease progression in early-diagnosed pulmonary arterial hypertension associated with systemic sclerosis: longitudinal data from the DETECT cohort. Ann Rheum Dis. 2018;77(1):128–32. [DOI] [PubMed] [Google Scholar]
- 43.Steen V, Medsger TA Jr. Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum. 2003;48(2):516–22. [DOI] [PubMed] [Google Scholar]
- 44.Klinger JR, Elliott G, Levine DJ, Bossone E, Duvall L, Fagan K, et al. Therapy for Pulmonary Arterial Hypertension in Adults 2018: Update of the CHEST Guideline and Expert Panel Report. Chest. 2019. [DOI] [PubMed] [Google Scholar]
- 45.Pan J, Lei L, Zhao C. Comparison between the efficacy of combination therapy and monotherapy in connective tissue disease associated pulmonary arterial hypertension: a systematic review and meta-analysis. Clinical and experimental rheumatology. 2018. [PubMed] [Google Scholar]
- 46.Nicolls M, Badesch D, Chung L, Domsic R, Medsger T, Pinckney A, et al. Safety and Efficacy of B-cell Depletion with Rituximab for the Treatment of Systemic Sclerosis-associated Pulmonary Arterial Hypertension in a Multi-center NIH Clinical Trial ARTHRITIS & RHEUMATOLOGY; 2019: WILEY 111 RIVER ST, HOBOKEN 07030–5774, NJ USA; 2019. [Google Scholar]
- 47.Avouac J, Fransen J, Walker UA, Riccieri V, Smith V, Muller C, et al. Preliminary criteria for the very early diagnosis of systemic sclerosis: results of a Delphi Consensus Study from EULAR Scleroderma Trials and Research Group. Ann Rheum Dis. 2011;70(3):476–81. [DOI] [PubMed] [Google Scholar]
- 48.Flavahan NA. A vascular mechanistic approach to understanding Raynaud phenomenon. Nat Rev Rheumatol. 2015;11(3):146–58. [DOI] [PubMed] [Google Scholar]
- 49.Koenig M, Joyal F, Fritzler MJ, Roussin A, Abrahamowicz M, Boire G, et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: a twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum. 2008;58(12):3902–12. [DOI] [PubMed] [Google Scholar]
- 50.Matucci-Cerinic M, Krieg T, Guillevin L, Schwierin B, Rosenberg D, Cornelisse P, et al. Elucidating the burden of recurrent and chronic digital ulcers in systemic sclerosis: long-term results from the DUO Registry. Ann Rheum Dis. 2016;75(10):1770–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herrick AL, van den Hoogen F, Gabrielli A, Tamimi N, Reid C, O’Connell D, et al. Modified-release sildenafil reduces Raynaud’s phenomenon attack frequency in limited cutaneous systemic sclerosis. Arthritis Rheum. 2011;63(3):775–82. [DOI] [PubMed] [Google Scholar]
- 52.Hachulla E, Hatron PY, Carpentier P, Agard C, Chatelus E, Jego P, et al. Efficacy of sildenafil on ischaemic digital ulcer healing in systemic sclerosis: the placebo-controlled SEDUCE study. Ann Rheum Dis. 2016;75(6):1009–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Korn JH, Mayes M, Matucci Cerinic M, Rainisio M, Pope J, Hachulla E, et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum. 2004;50(12):3985–93. [DOI] [PubMed] [Google Scholar]
- 54.Matucci-Cerinic M, Denton CP, Furst DE, Mayes MD, Hsu VM, Carpentier P, et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2011;70(1):32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wigley FM, Seibold JR, Wise RA, McCloskey DA, Dole WP. Intravenous iloprost treatment of Raynaud’s phenomenon and ischemic ulcers secondary to systemic sclerosis. J Rheumatol. 1992;19(9):1407–14. [PubMed] [Google Scholar]
- 56.Wigley FM, Wise RA, Seibold JR, McCloskey DA, Kujala G, Medsger TA Jr., et al. Intravenous iloprost infusion in patients with Raynaud phenomenon secondary to systemic sclerosis. A multicenter, placebo-controlled, double-blind study. Ann Intern Med. 1994;120(3):199–206. [DOI] [PubMed] [Google Scholar]
- 57.Steen VD, Costantino JP, Shapiro AP, Medsger TA Jr. Outcome of renal crisis in systemic sclerosis: relation to availability of angiotensin converting enzyme (ACE) inhibitors. Ann Intern Med. 1990;113(5):352–7. [DOI] [PubMed] [Google Scholar]
- 58.Jaeger VK, Wirz EG, Allanore Y, Rossbach P, Riemekasten G, Hachulla E, et al. Incidences and Risk Factors of Organ Manifestations in the Early Course of Systemic Sclerosis: A Longitudinal EUSTAR Study. PLoS One. 2016;11(10):e0163894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steen VD, Medsger TA Jr. Long-term outcomes of scleroderma renal crisis. Ann Intern Med. 2000;133(8):600–3. [DOI] [PubMed] [Google Scholar]
- 60.Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655–66. [DOI] [PubMed] [Google Scholar]
- 61.Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4(9):708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Volkmann ER, Tashkin DP, Li N, Roth MD, Khanna D, Hoffmann-Vold AM, et al. Mycophenolate Mofetil Versus Placebo for Systemic Sclerosis-Related Interstitial Lung Disease: An Analysis of Scleroderma Lung Studies I and II. Arthritis Rheumatol. 2017;69(7):1451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Volkmann ER, Tashkin DP, Sim M, Li N, Goldmuntz E, Keyes-Elstein L, et al. Short-term progression of interstitial lung disease in systemic sclerosis predicts long-term survival in two independent clinical trial cohorts. Ann Rheum Dis. 2019;78(1):122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sullivan K, Pinckney A, Goldmuntz E, Welch B, Khanna D, Simms RW, et al. Myeloablative Autologous Hematopoietic Stem Cell Transplantation for Severe Scleroderma: Long-Term Outcomes 6–11 Years after Entry on a Randomized Study Comparing Transplantation and Cyclophosphamide ARTHRITIS & RHEUMATOLOGY; 2018: WILEY 111 RIVER ST, HOBOKEN 07030–5774, NJ USA; 2018. [Google Scholar]
- 65.Poormoghim H, Lucas M, Fertig N, Medsger TA. Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum. 2000;43(2):444–51. [DOI] [PubMed] [Google Scholar]
- 66.Shand L, Lunt M, Nihtyanova S, Hoseini M, Silman A, Black CM, et al. Relationship between change in skin score and disease outcome in diffuse cutaneous systemic sclerosis: application of a latent linear trajectory model. Arthritis Rheum. 2007;56(7):2422–31. [DOI] [PubMed] [Google Scholar]
- 67.Namas R, Tashkin DP, Furst DE, Wilhalme H, Tseng CH, Roth MD, et al. Efficacy of mycophenolate mofetil and oral cyclophosphamide on skin thickness: Post-hoc analyses from the Scleroderma Lung Study I and II. Arthritis Care Res (Hoboken). 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khanna D, Spino C, Johnson S, Chung L, Whitfield M, Denton CP, et al. Abatacept in Early Diffuse Cutaneous Systemic Sclerosis - Results of a Phase 2 Investigator-Initiated, Multicenter, Double-Blind Randomized Placebo-Controlled Trial. Arthritis Rheumatol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khanna D, Lin CJ, Kuwana M, Allanore Y, Batalov A, Butrimiene I, et al. Efficacy and Safety of Tocilizumab for the Treatment of Systemic Sclerosis: Results from a Phase 3 Randomized Controlled Trial ARTHRITIS & RHEUMATOLOGY; 2018: WILEY 111 RIVER ST, HOBOKEN 07030–5774, NJ USA; 2018. [Google Scholar]
- 70.Launay D, Remy-Jardin M, Michon-Pasturel U, Mastora I, Hachulla E, Lambert M, et al. High resolution computed tomography in fibrosing alveolitis associated with systemic sclerosis. J Rheumatol. 2006;33(9):1789–801. [PubMed] [Google Scholar]
- 71.Hoa S, Bernatsky S, Steele RJ, Baron M, Hudson M. Association between immunosuppressive therapy and course of mild interstitial lung disease in systemic sclerosis. Rheumatology (Oxford). 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4(9):708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sircar G, Goswami RP, Sircar D, Ghosh A, Ghosh P. Intravenous cyclophosphamide vs rituximab for the treatment of early diffuse scleroderma lung disease: open label, randomized, controlled trial. Rheumatology (Oxford). 2018;57(12):2106–13. [DOI] [PubMed] [Google Scholar]
- 74.Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet. 2016;387(10038):2630–40. [DOI] [PubMed] [Google Scholar]
- 75.Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N Engl J Med. 2019;380(26):2518–28. [DOI] [PubMed] [Google Scholar]
- 76.Kahan A, Allanore Y. Primary myocardial involvement in systemic sclerosis. Rheumatology (Oxford). 2006;45 Suppl 4:iv14–7. [DOI] [PubMed] [Google Scholar]
- 77.Forbes A, Marie I. Gastrointestinal complications: the most frequent internal complications of systemic sclerosis. Rheumatology (Oxford). 2009;48 Suppl 3:iii36–9. [DOI] [PubMed] [Google Scholar]
- 78.Herrick AL, Gallas A. Systemic sclerosis-related calcinosis. Journal of Scleroderma and Related Disorders. 2016;1(2):194–203. [Google Scholar]
- 79.Avouac J, Walker U, Tyndall A, Kahan A, Matucci-Cerinic M, Allanore Y, et al. Characteristics of joint involvement and relationships with systemic inflammation in systemic sclerosis: results from the EULAR Scleroderma Trial and Research Group (EUSTAR) database. J Rheumatol. 2010;37(7):1488–501. [DOI] [PubMed] [Google Scholar]
- 80.Clements PJ, Wong WK, Hurwitz EL, Furst DE, Mayes M, White B, et al. The Disability Index of the Health Assessment Questionnaire is a predictor and correlate of outcome in the high-dose versus low-dose penicillamine in systemic sclerosis trial. Arthritis Rheum. 2001;44(3):653–61. [DOI] [PubMed] [Google Scholar]
- 81.Ghrénassia E, Avouac J, Khanna D, Derk CT, Distler O, Suliman YA, et al. Prevalence, correlates and outcomes of gastric antral vascular ectasia in systemic sclerosis: a EUSTAR case-control study. J Rheumatol. 2014;41(1):99–105. [DOI] [PubMed] [Google Scholar]
- 82.Distler O, Allanore Y, Denton C, Kuwana M, Matucci-Cerinic M, Pope J, et al. OP0183 EFFICACY AND SAFETY OF RIOCIGUAT IN PATIENTS WITH EARLY DIFFUSE CUTANEOUS SYSTEMIC SCLEROSIS AND INTERSTITIAL LUNG DISEASE (SSC-ILD): RESULTS FROM THE PHASE IIB RISE-SSC STUDY. Annals of the Rheumatic Diseases. 2019;78(Suppl 2):167-. [Google Scholar]
- 83.Sitbon O, Gomberg-Maitland M, Granton J, Lewis MI, Mathai SC, Rainisio M, et al. Clinical trial design and new therapies for pulmonary arterial hypertension. Eur Respir J. 2019;53(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Vollenhoven R, Voskuyl A, Bertsias G, Aranow C, Aringer M, Arnaud L, et al. A framework for remission in SLE: consensus findings from a large international task force on definitions of remission in SLE (DORIS). Ann Rheum Dis. 2017;76(3):554–61. [DOI] [PubMed] [Google Scholar]
- 85.Khanna PP, Maranian P, Gregory J, Khanna D. The minimally important difference and patient acceptable symptom state for the Raynaud’s condition score in patients with Raynaud’s phenomenon in a large randomised controlled clinical trial. Ann Rheum Dis. 2010;69(3):588–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pauling JD, Reilly E, Smith T, Frech TM. Factors influencing Raynaud’s condition score diary outcomes in systemic sclerosis. The Journal of rheumatology. 2019:jrheum. 180818. [DOI] [PubMed] [Google Scholar]
- 87.Steen VD, Medsger TA. The value of the Health Assessment Questionnaire and special patient-generated scales to demonstrate change in systemic sclerosis patients over time. Arthritis Rheum. 1997;40(11):1984–91. [DOI] [PubMed] [Google Scholar]
- 88.Benza RL, Gomberg-Maitland M, Elliott CG, Farber HW, Foreman AJ, Frost AE, et al. Predicting Survival in Patients With Pulmonary Arterial Hypertension: The REVEAL Risk Score Calculator 2.0 and Comparison With ESC/ERS-Based Risk Assessment Strategies. Chest. 2019;156(2):323–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.