

Abstract
Asbestos is still a social burden worldwide as a carcinogen causing malignant mesothelioma. Whereas recent studies suggest that local iron reduction is a preventive strategy against carcinogenesis, little is known regarding the cellular and molecular mechanisms surrounding excess iron. Here by differentially using high-risk and low-risk asbestos fibers (crocidolite and anthophyllite, respectively), we identified asbestos-induced mutagenic milieu for mesothelial cells. Rat and cell experiments revealed that phagocytosis of asbestos by macrophages results in their distinctive necrotic death; initially lysosome-depenent cell death and later ferroptosis, which increase intra- and extra-cellular catalytic Fe(II). DNA damage in mesothelial cells, as assessed by 8-hydroxy-2′-deoxyguanosine and γ-H2AX, increased after crocidolite exposure during regeneration accompanied by β-catenin activation. Conversely, β-catenin overexpression in mesothelial cells induced higher intracellular catalytic Fe(II) with increased G2/M cell-cycle fraction, when p16INK4A genomic loci localized more peripherally in the nucleus. Mesothelial cells after challenge of H2O2 under β-catenin overexpression presented low p16INK4A expression with a high incidence of deletion in p16INK4A locus. Thus, crocidolite generated catalytic Fe(II)-rich mutagenic environment for mesothelial cells by necrotizing macrophages with lysosomal cell death and ferroptosis. These results suggest novel molecular strategies to prevent mesothelial carcinogenesis after asbestos exposure.
Keywords: Asbestos, Iron, Ferroptosis, Lysosomal cell death, p16CDKN2A
Graphical abstract
Highlights
-
•
We identified asbestos-induced mutagenic milieu for mesothelial cells, using high- and low-risk asbestos fibers.
-
•
Crocidolite causes necrotic death in macrophages, initially lysosome-depenent cell death and later ferroptosis.
-
•
-Catenin overexpression in regenerative mesothelial cells in mutagenic milieu provides multiple risks for carcinogenesis.
Abbreviations
- ANOVA
analysis of variance
- CAS
CRISPR-associated proteins
- CTSB
cathepsin B
- DAPI
4′,6-diamidino-2-phenylindole
- DFO
deferoxamine
- DIM
differential interference contrast microscope
- DMT1
divalent metal transporter 1
- DSB
double-strand break
- FAC
ferric ammonium citrate
- FACS
fluorescent activated cell sorter
- FBS
fetal bovine serum
- FISH
fluorescent in situ hybridization
- FITC
fluorescein isothiocyanate
- FL
full length
- FSC
forward scatter
- GFP
green fluorescent protein
- GGT
γ-glutamyltransferase
- GPX
glutathine peroxidase
- GSEA
Gene Set Enrichment Analysis
- HNE
4-hydroxy-2-nonenal
- LDCD
lysosome-dependent cell death
- LDH
lactate dehydrogenase
- LMP
lysosomal membrane permealization
- MHC
major histocompatibility complex
- MM
malignant mesothelioma
- NOX
NADPH oxidase
- 8-OHdG
8-hydroxy-2′-deoxyguanosine
- PEI
polyethyleneimine
- PL
peritoneal lavage
- PRE
post-transcriptional regulatory element
- ROS
reactive oxygen species
- SSC
saline sodium citrate
- SSC
side scatter
- Tf
transferrin
- TfR1
transferrin receptor 1
1. Introduction
Asbestos, a natural fibrous nanomaterial [1], has been abundantly used worldwide for various industrial purposes, but induces distinct respiratory pathologies, including pulmonary fibrosis, lung cancer and malignant mesothelioma (MM) decades after exposure. Thus, asbestos is still a huge social burden worldwide [[2], [3], [4]]. Asbestos-associated carcinogenesis has been studied from two distinct standpoints, direct and/or indirect effects [5]. Direct effects suggest that asbestos fibers are phagocytosed by mesothelial cells, reach inside the nucleus and generate mutations almost physically [[6], [7], [8]]. On the other hand, indirect effects hypothesize that asbestos fibers localize inside macrophages and that oxidative stress from frustrated phagocytosis induces mesothelial genetic alterations [9] through activation of Nalp3 inflammasome [10] and tissue remodeling processes [11].
Pathogenicity of asbestos fibers has been associated with iron [4,[12], [13], [14]], as exemplified by asbestos body in histology [15]. Intraperitoneal injection of ferric saccharate induces MM in wild-type rats [16,17], and more recently continued oxidative stress via Fenton reaction [18] in the kidney of wild-type rats leads to early p16INK4A deletion [19], resulting in renal cell carcinoma [20,21]. These reports suggest that excess iron plays a role in carcinogenesis [22,23]. Macrophages play a crucial role in iron regulation, including iron recovery in spleen from senescent red blood cells. Thus, macrophage dysfunction may contribute to carcinogenesis through iron-dependent oxidative stress. However, little is known concerning the molecular mechanisms of asbestos-induced mesothelial carcinogenesis surrounding excess iron.
Regarding the mutation spectrum collected using human samples, ~70% of MM exhibits p16INK4A homozygous deletion [4,[24], [25], [26], [27], [28]], which is nowadays used for the pathological diagnosis of MM [29,30]. The rat model also provides the same deletion [31]. Here we for the first time demonstrate that ceaseless macrophage necrotic death, through lysosomal cell death and ferroptosis [32,33], generates catalytic Fe(II)-rich stromal mutagenic microenvironment for mesothelial cells and further that the associated activation of β-catenin is advantageous in mesothelial cells to obtain deletion of p16INK4A via increased fraction of G2/M phase, increased intracellular catalytic Fe(II) and juxta-nuclear membrane position of p16INK4A genomic loci.
2. Materials and methods
2.1. Animal experiments
Asbestos-induced peritonitis model was produced as previously described [31]. Briefly, male Fischer-344 rats (SLC Japan, Shizuoka, Japan; 6 wks old; 140–150 g body weight) were injected intraperitoneally (ip) with a suspension of 10 mg asbestos (crocidolite [UICC], anthophyllite [34]). We collected peritoneal lavage (PL; 30 ml physiological saline, containing 200 μM Tris amine 2-pyridyl methyl [Sigma-Aldrich] for stabilization of Fe[II]). Mesothelial cells were collected from rat mesentery as previously described [35]. We dissected organs for histological analysis at autopsy after euthanasia. The animal experiment committee of Nagoya University Graduate School of Medicine approved the experiments.
2.2. Cell culture
Met5A human mesothelial cell line (ATCC), LP9 human mesothelial cell line (Coriell Institute), Met5A-derived stable cell lines expressing Fucci [36], mouse RAW264 and human THP1 macrophage cell lines and HEK293T cell line (RIKEN Cell Bank, Tsukuba, Japan) were grown under standard sterile cell culture conditions (37 °C, humidified atmosphere, 5% CO2) in RPMI1640 (Wako, Osaka, Japan), containing 10% fetal bovine serum (FBS; Biowest, #S1780; Nuaillé, France) and 1% antibiotic-antimycotic (Gibco). Cells were used within two months after thawing. In the co-culture experiments, THP-1 or RAW264 cells were differentiated in 6 transwell inserts (membrane pore size of 0.4 μm, Corning, #3450). Macrophages and Met5A cells were co-cultured in CO2 incubator with RPMI1640 medium with 1% FBS for 48 h prior to incubation with or without asbestos.
2.3. Microarray-based gene expression analysis
Microarray analysis was perfomed, using Agilent SuperPrint G3 Rat GE Ver 2.0 8x60 K Microarray slides (G4858A#74036, Agilent Technologies, Santa Clara, CA) and Low Input Quick Amp Labeling kit, using Agilent's one-color Microarray-based Gene Expression Analysis protocol (5190-2305, Agilent Technologies). Total RNA was extracted from rat mesothelium 1 wk after crocidolite injection. Amplified cRNA was labeled with Cy3-CTP. Hybridization was done in a hybridization oven at 65 °C for 17 h with rotation. Microarray slides were then washed with GE wash buffer and scanned. Data were analyzed with GeneSpring FX 13.1 software (Agilent Technologies).
2.4. GSEA (gene set enrichment analysis)
Based on the entire microarray profiles using predefined gene sets, GSEA exploits novel computational methods [37,38]. Pre-defined gene set in the Molecular Signatures Database (MSigDB) was selected for analyses; the C5 set, a Gene Ontology (GO) molecular function gene set derived from the Molecular Function Ontology database in MSigDB was used. The gene sets included in the analysis were limited to those that contained between 10 and 500 genes. Permutation was conducted 1000 times according to default-weighted enrichment statistics and by using a signal-to-noise metric to rank genes according to their differential expression levels across the no-treatment and crocidolite (1 wk) groups. Significant gene sets were defined as those with a nominal P-value <0.05.
2.5. Cloning
Cloning was performed using chemically competent E. coli, derived from Stbl3 Competent Cells (C737303, Thermo Fischer). Correct cloning and integration into target vectors were confirmed with sequencing.
2.6. Cloning of components for endogenous β-catenin
CMV promoter of pcDNA3-EGFP (Addgene) was replaced by Cag promoter (PX461, #48140, addgene) to enhance promoter activity in mesothelial cells. Cag-pcDNA3-EGFP construct was generated, using a general protocol with restriction enzymes. Insert products were obtained by PCR amplification. After ligation of these components, the product was used for transformation, from which plasmids were recovered and verified by sequencing.
2.7. shRNA knockdown of β-catenin
shRNA was used to knock down β-catenin expression in Met5A cells. β-catenin shRNA vector (pLKO.1 shβ-catenin) was constructed according to the protocol of pLKO.1 puro vector (#8453, Addgene). pLKO.1 shβ-catenin or scramble (Supplementary Table 3), packaging plasmid pCMV-VSV-G-RSV-Rev (RDB04393, RIKEN BRC) and pCAG-HIV-gp (RDB04394, RIKEN BRC) were co-transfected into the 293T cells, by using the polyethyleneimine (PEI; 23966-1, Polyscience, Inc.) according to the manufacturer's protocol and culture medium was replaced 3 h after transfection. Met5A cells were seeded in 12-well plates and transduced in RPMI1640, containing 10% FBS. The cells were reseeded in 6-well plates 12 h after incubation and selected using 250 ng/ml puromycin for 7 days. Knockdown efficiency was confirmed with western blotting.
2.8. Transient overexpression of β-catenin
1 × 106 Met5A cells were seeded in a 6-well plate 1 day before transfection. 1 μg of the plasmid construct of full length-β-catenin and Δ89β-catenin were diluted in 300 μl OptiMEM (Thermo Fischer) and prepared for transfection with 3 μg of PEI with 10 min incubation. The same transfection mix was prepared with an empty vector as a control (mock, pcDNA3-EGFP; #13031, Addgene). The transfection mixtures were added to the cells at a confluence of 80–90%. On the next day, the medium was exchanged to fresh medium. After 48 h incubation, these cells were used for imaging, FACS, and western blotting analyses.
2.9. LDH cytotoxicity assays
Cytotoxicity was evaluated with a LDH-cytotoxicity detection kit (Roch Diagnostics). Met5A and THP1 cells were seeded in a 96-well tissue culture plate at a density of 1.5 × 104 cells per well and incubated with medium containing 10% FBS. Prior to crocidolite exposure, we exchanged the medium to 1% FBS with or without iron (ferric ammonium citrate, FAC; Sigma) at different concentrations per well. After 48–72 h, the supernatant was used for LDH assay (cytotoxicity), and cells were used for WST assay (cell viability). The absorbance of the samples was calculated at each wavelength with a plate reader (PowerScan4 or Multiscan FC). Cytotoxicity was calculated using the formula in the kit protocol.
2.10. Cell proliferation assay
A proliferation kit (WST; Cell Count Reagent SF, Nacalai Tesque, Kyoto, Japan) was used to check cell viability. The assay used highly water-soluble tetrazolium salt. Met5A cells were seeded in a 96-well plate at a density at 1.0 × 104 cells per well and incubated with RPMI1640 medium with 1% FBS. After 48 h, 10 μl of crocidolite suspension at different concentration per well were added. After 24, 48 or 72 h, 10 μl of Cell Count Reagent SF was added to medium and incubated for 2.5 h at 37 °C under protection from light. Absorbance at 450 nm was measured with a microplate reader.
2.11. In vitro stimulation of macrophage
For stimulation of macrophage in vitro, RAW 264 or THP1 cells were plated at 1 × 106 cells/dishes in 6-well plate in the presence of phorbol 12-myristate 13-acetate (PMA, 10 μM). After 12 h, cells were stimulated either with IFN-γ (100 ng/ml, PeproTech), IL-4 (20 ng/ml, PeproTech), LPS (10 ng/ml, Sigma), IgG (Jackson ImmunoResearch) coated wells + LPS (100 ng/ml) or TGF-β (0.5 ng/ml, R&D Systems) as previously described [39,40]. After stimulation, these cells were used for q-PCR, western blotting, cell death assay and transmission electron microscopy.
2.12. Flow cytometry (FACS)
Inflammatory cells were collected from rat peritoneal lavage (PL) as described [41]. The harvested cells were rinsed with centrifugation (200 × g, 3 min); the supernatant was used for detection of extracellular catalytic Fe(II) with plate reader whereas the pellet was used for FACS analysis. SiRhoNox-1 solution (final concentration 2 μM) [[42], [43], [44]] and Hoechst 33342 (Thermo Fischer) were added to the tubes containing cells, followed by incubation in the dark at 37 °C. The cell analysis and sorting were carried out, using FACS Aria II (BD Biosciences). Hoechst 33342-negative cells were removed as dead cells. As reported previously [41], we identified macrophage-subset with forward and side scatters, followed by an analysis on catalytic Fe(II). Fluorescent signals were measured at 370 nm (450/50) for Hoechst33342 and at 633 nm (660/40) for SiRhoNox-1. All flow cytometric data were analyzed with FACS Diva version 8.0.1.
2.13. Transmission electron microscopy
Stimulated THP1 cells as described were plated at 1 × 106 cell/dish in 35-mm tissue culture dishes. These cells were exposed to asbestos (15 μg/cm2) by incubating for 24–48 h. Cells were fixed with 2.5% glutaraldehyde (TAAB) in 0.1 M Sorenson's buffer (0.1 M H2PO4, 0.1 M HPO4 [pH 7.2]) for at least 1.5 h, and then treated with 1% OsO4 in 0.1 M Sorenson's buffer. After dehydration through an ethanol series, cells were embedded in Epon-812 (TAAB). A thin section was cut on an MT-7000 ultra-microtome, stained with 1% uranyl acetate and 0.4% lead citrate and then examined under a JEOL JEM-1400 EXII electron microscope (JEOL, Tokyo, Japan). Pictures were taken on an ORCA-HR digital camera (Hamamatsu Photonics, Hamamatsu, Japan) at 5000–50,000-fold magnification and measurements were performed, using the AMT Image Capture Engine.
2.14. Western blot analysis
Cells were lysed with a lysis buffer (1 M NaCl, 50 mM Tris-HCl, 0.1% SDS, pH 8.0, 0.5% sodium deoxycholate and 1% NP-40) supplemented with a protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Roche). Protein extraction, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or Tricine SDS-PAGE were performed as previously described [45]. The antibodies used are described in the Supplementary Table 1.
2.15. Quantitative real-time PCR
Total RNA was isolated from cultured cells using RNeasy plus kit (Qiagen). cDNA was synthesized from 4 μg of RNA using SuperScript III cDNA synthesis kit (Thermo Scientific). qPCR was performed in triplicates with 1 μl diluted cDNA (1:5) using SYBR green Fas mix (Thermo Scientific) on an iCycler RT-PCR detection system (BioRad). Expression was normalized to Actb or Gapdh. Expression levels were measured, using primers purchased from Thermo Fischer Scientific, and all the primer sequences are described in the Supplementary Table 2.
2.16. FISH analysis
For detecting p16INK4adeletion in the rat mesothelial cells, bacterial artificial chromosome clone RNB1-039C01 (p16INK4a as a test probe) and RNB1-266I03 (rat chromosome 5 centromeric region as a reference probe) were extracted and used for FISH analysis by labeling with biotin-16dUTP (TAKARA) or digoxigenin-11-dUTP (Enzo life Science) in general nick translation methods. The size of the labeled probe was confirmed by agarose gel electrophoresis to be 200 to 400 bp. After ethanol precipitation (20 μl of products, 50 μl of 4 M ammonium acetate, 2.0 μl of 25 μg/μl salmon sperm DNA [Wako], 10 μl of 4 μg/μl of E.coli tRNA and 500 μl of ethanol) at −80 °C for 30 min, the labeled probe was dissolved in formamide. Rat Cot-1 DNA (Chromosome Science Lab) was added (20 μg/μl) to the hybridization mixture to minimize background signal. Stock slides were completely air-dried, followed by pepsin treatment (10 μg/ml in 10 mM HCl, pH 2.0) at 37 °C. Hybridization was performed at 37 °C overnight, followed by washing with 2 × SSC at 42 °C at room temperature, sequentially. Finally, avidin-FITC (Vector; 1:100 dilution) or anti-digoxigenin antibody (Vector; Dylight594-labeled anti-Digoxigenin/Digoxin, 1:200 dilution) and DAPI was applied for nuclear counterstaining.
2.17. Fucci stably expressing mesothelial cells (fMet5A)
The genes encoding orange or green Fucci indicators were amplified with PCR from the pFucci-G1 orange vector or pFucci-S/G2/M green vector (MBL Life Sciences) and cloned into FUGW modified lentiviral vector (#14883, Addgene) with EcoRI and XbaI. Met5A cells were transduced by lentivirus encoding orange Fucci (G1-phase) and green Fucci (S/G2/M-phase) and stable cell lines were cloned by limiting dilution method.
2.18. Cas FISH
Cas9 plasmid construct (pHAGE-TO-nls-st1dCas9-3nls-3XTagBFP2, #64512, Addgene) and sgRNA Expression vector (pLH-stsgRNA3.1; #64118, Addgene) were subcloned as previously described [46]. For imaging, cells were grown on 35 mm glass-bottom dishes (3911-035; Iwaki, Shizuoka, Japan). Then, 150 ng of dCas9 plasmid and 750 ng of sgRNA plasmid were cotransfected using PEI, after which the cells were incubated for another 48 h. Fluorescent signals were measured at 488 nm (510/20) for Fucci-S/G2/M and 540 nm (570/20) and 370 nm (450/50) for BFPCas9 with a confocal microscope (LSM880, Carl Zeiss; Oberkochen, Germany). sgRNA sequences are described in Supplementary Table 4.
2.19. Statistical analysis
Statistical analyses were performed using one-way analysis of variance (ANOVA) and an unpaired t-test with GraphPad Prism 5 software. Differences were considered significant when p < 0.05. The data are expressed as the means ± SEM (N = 3–10) unless otherwise specified. All the experiments were repeated at least three times.
3. Results
3.1. Only high-risk asbestos (crocidolite) induces iron-rich stromal milieu
Previously we showed in rats that crocidolite (blue asbestos) is highly carcinogenic causing MM [31] whereas a minor asbestos, anthophyllite, did not induce MM in our experiments (low-risk) [34]. To elucidate the cellular mechanism toward iron-rich stromal microenvironment, we first performed histological analysis in rats injected intraperitoneally either with high- or low-risk asbestos fibers. Four weeks after the asbestos injection, chronic granulomatous peritonitis as a reaction to foreign material was present in both the cases with mesothelial cells on granuloma and fibrosis, instead of attaching on the basement membrane. Polarizer analysis revealed that asbestos fibers were inside CD68-positive macrophages and not in mesothelial cells (Fig. 1a; Supplementary Fig. 1a and b). Of note, we observed necrotic death of macrophages only in the crocidolite group (Fig. 1b and c). 4-Hydroxy-2-nonenal (HNE) is a major product of lipid peroxidation [47,48] and is one of the most sensitive markers for Fenton reaction [47,49]. We detected HNE-modified proteins in both the granuloma, and quantified immunopositivity was significantly higher in the crocidolite granuloma than that of anthophyllite (Fig. 1d and e). Thus, we speculated that macrophages engulfing asbestos are responsible for chronic inflammation with generation of reactive oxygen species (ROS). In comparison to macrophages in peritoneal fluid (Supplementary Fig. 1a and b), macrophages in granuloma highly expressed M2-specific markers, such as CD163 and MHC II [50]. Both of them, whether crocidolite or anthophyllite, expressed NOX2, a major ROS producer in macrophages [51] (Supplementary Fig. 1a–c). However, intra-granuloma macrophages significantly accumulated iron only after crocidolite exposure (Fig. 1f and g). Peritoneal macrophages, iNOS-positive and M1 subtype (Supplementary Fig. 1a), collected iron most among all the inflammatory cells, and catalytic Fe(II) [44] was significantly increased intracellularly and extracellularly only after crocidolite exposure (Fig. 1h, i, j; Supplementary Fig. 2). These data suggest that stromal iron overload is a feature of high-risk asbestos (crocidolite), resulting from macrophage necrosis (Supplementary Fig. 1c).
Fig. 1.

Only high-risk asbestos generates iron-rich stromal milieu via macrophage necrosis.
(a) Peritoneal histology of rat model 4 wks after intraperitoneal injection of crocidolite, a high-risk asbestos; HE, hematoxylin & eosin staining; Masson trichrome staining; polarizer detects asbestos fibers (bar = 50 μm). (b, c) Macropahge necrosis is observed only in granuloma by crocidolite but not by anthophyllite, a low-risk asbestos (bar = 20 μm). (d, e) Deposition of 4-hydroxy-2-nonenal (HNE)-modified proteins is significantly higher in the peritoneal stroma after crocidolite exposure than anthophyllite exposure. (f, g) Iron deposition by Berlin blue staining is significantly higher after crocidolite exposure than anthophyllite exposure. (h, i, j) Macrophage fraction in peritoneal lavage as well as lavage fluid shows significantly higher catalytic Fe(II) 4 wks after crocidolite exposure than anthophyllite exposure. SSC, side scatter; FSC, forward scatter; area by black line, macrophage fraction; PL, peritoneal lavage. Catalytic Fe(II) was detected by SiRhoNox-1 (means ± SEM; N ≧ 4). Refer to text for details. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.2. Crocidolite induces lysosome-dependent cell death in the initial phase and ferroptosis in the end phase in macrophages
To classify crocidolite-induced necrosis, we selected a macrophage cell line (THP-1 stimulated by PMA) for further analysis. We found that additional iron treatment decreased the cell viability of macrophages after asbestos exposure and enhanced necrotic death (Fig. 2a and b). We then performed a transmission electron microscopy to investigate which type of necrosis is induced by crocidolite. Crocidolite reached phagolysosome 24 h after exposure and permeabilized the phagolysosomal membrane whereas anthophyllite did not permeabilize it (Fig. 2c). Furthermore, we observed mitochondrial shrinkage after crocidolite exposure (Fig. 2d and e). Cathepsin B, a lysosomal enzyme, if present in nucleus or cytosol, causes lysosome-dependent cell death (LDCD) [33,52]. We found cathepsin B localized to nucleus and cytosol of the macrophages engulfing crocidolite, which was enhanced with additional iron (Fig. 2f and g). Here, time-lapse analysis revealed that ROS surge in phagolysosome was followed by cytosolic increase in ROS and lysosomal increase in catalytic Fe(II) within a time-course of 120 min (Supplementary Fig. 4a–c). These results suggest that ROS originates not only from frustrated phagocytosis [11] but from LDCD of macrophages.
Fig. 2.

Crocidolite induces macrophage necrosis via lysosome-dependent cell death at an early phase and later via ferroptosis.
(a, b) Cell viability (WST) and necrosis ratio (LDH) assays reveal higher toxicity of crocidolite than anthophyllite for THP1 macropahge in the presence of excess iron. Crocidolite or anthophyllite asbestos was exposed to THP1 macrophage cells in the presence of 10 μg/ml ferric ammonium citrate (FAC) for 48 h. (c) Transmission electron microscopy of M0-state THP1 macrophage exposed to asbestos for 24 h. Crocidolite causes lysosomal rupture whereas anthopyllite fibers are confined inside lysosome (bar = 5 μm). (d, e) Transmission electron microscopy of mitochondrial damage in THP1 after crocidolite exposure. Significantly shorter mitochondria are observed (bar = 200 nm). NT, no-treatment. (f, g) Nuclear localization of lysosomal cathepsin B (CTSB) in IL-4 stimulated THP1 cells after exposure to crocidolite (25 μg/cm2) and 100 μg/ml FAC. White arrowhead, nucleus; cathepsin B, red; nucleus, cyan (bar = 50 μm). (h) Time-course immunoblot analysis reveals the involvement of ferroptosis. IL-4 stimulated THP1 was exposed to crocidolite (25 μg/cm2). (i) Immunoblot analysis of IL-4 stimulated THP1 72 h after crocidolite exposure (25 μg/cm2) in the presence of various inhibitors. Necrostatin-1; (necroptosis inhibitor, 40 μM), Ferrostain-1; DFO, deferoxamine (ferroptosis inhibitors; 500 nM and 400 μM, respectively), zVAD-FMK (apoptosis inhibitor, 184 μM), z-FA-FMK (lysosome-dependent cell death inhibitor, 20 μM), Pepstatin-A (autophagic cell death inhibitor, 20 μg/ml). (J) LDH assay in IL-4 stimulated THP1 exposed to crocidolite in the same condition as in (I) (means ± SEM; N ≧ 6). Refer to text for details. . (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
From these observations, we speculated the involvement of ferroptosis [32] in crocidolite-induced macropahge cell death. Recent studies imply that cell death regulated by certain signal pathways in the initial phase may result in multiple forms of necrosis, ending in the rupture of plasma membrane [53]. To further elucidate the type of regulated necrosis, we performed protein analyses. Whereas ferroptosis- and necroptosis-associated proteins presented no significant changes in the initial phase of macrophage death by crocidolite, protein levels of Lamp2 and CathepsinB increased time-dependently (Fig. 2h). Thus, crocidolite induced LCDC in macrophages at the initial-phase. At the end-phase 96 h after crocidolite exposure, Lamp2 and Cathepsin B increased (LDCD); GPX4 and TfR1 decreased and ferritin increased (ferroptosis); RIP1 and MLKL increased (necroptosis). Whereas necroptosis inhibitor (necrostatin-1) and ferroptosis inhibitor (ferrostatin-1 and desferal [DFO]) partially suppressed the macrophage detachment and death, caspase inhibitor (z-VAD-FMK), cathepsin B inhibitor (z-FA-FMK) and protease inhibitor (pepstatin A) were not effective (Fig. 2i and j; Supplementary Fig. 3a). These data suggest that macrophage necrosis ended in ferroptosis at the end phase.
3.3. Wnt/β-catenin pathway is activated during mesothelial regeneration
We next examined the biological responses of mesothelial cells after crocidolite exposure. In a rat model of intraperitoneal injection, we found mesothelial cells continuously synthesizing DNA (c-Myc-, Ki-67-, BrdU-positive cells) on the peritoneal surface (Fig. 3a and b). This regenerative activity of mesothelial cells continued at least from 24 h to 2 weeks after crocidolite exposure, when regenerative mesothelial cells were on the granuloma capturing crocidolite (Fig. 3a and b). What kinds of signaling pathways are activated during this mesothelial regeneration is still poorly understood [54,55]. To clarify this, we performed gene set enrichment analysis (GEO Accession No. GSE149285), using rat mesothelial cells collected from mesentery after exposure to crocidolite (Supplementary Fig. 5a–c). We discovered the association of Wnt/β-catenin pathway with mesothelial regeneration (Fig. 3c and d). Expression of CTNNB1 (catenin β1) was significantly increased in mesothelial cells 1 week after crocidolite exposure (Fig. 3e), which was confirmed with immunohistochemistry and mRNA analyses (Fig. 3b, e). Other overexpressed genes included c-Met and Tgfbr1.
Fig. 3.

Mesothelial regeneration after crocidolite-induced damage accompanies activation of Wnt/β-catanin signaling pathway.
(a) Intraperitoneal administration of crocidolite induces progressive peritonitis in rats. Arrowhead, crocidolite fibers. (b) Corresponding histology of peritoneum to (a) with immunohistochemistry (β-catenin, c-Myc, Ki-67 and BrdU). (c, d) Collected rat mesothelial cells were used for microarray analysis. Gene-set enrichment analysis (GSEA) revealed significant upregulation of β-catenin signaling pathway. (e) qRT-PCR analysis of selected regeneration-associated genes (β-catenin, MET, EGFR and TGFβR1) in rat mesothelial cells collected from mesentery (means ± SEM; N ≧ 4). Refer to text for details. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.4. Hydrogen peroxide source from macrophages is not limited to NOX2
To evaluate the ROS source in macrophages, we used THP-1 cells after PMA stimulation and analyzed NOX2 expression, a major source of H2O2 in macrophages [56,57]. Both the peritoneal and intra-granuloma macrophages expressed NOX2, which was not significantly different after exposure either to crocidolite or anthophyllite (Supplementary Fig. 1d–f). However, the amounts of H2O2 in the extracellular fluid were significantly increased only after crocidolite exposure when the macrophages were stimulated either with IFN-γ or IL-4 (Supplementary Fig. 1f). Macrophages in granuloma expressed CD163 and MHC II, categorized as IL-4-stimulated macrophages (Supplementary Fig. 1c). Production of H2O2 increased not only in IFN-γ group (M1 subtype) but also in IL-4 group (M2a subtype), whereas Nox2 expression was low (Supplementary Fig. 1d–f). These results suggest that the source of extracellular H2O2 is not limited to NOX2.
3.5. Necrotic macrophages induce iron-dependent oxidative damage to distant mesothelial cells
Necrotizing macrophages produced extracellular H2O2 (Supplementary Fig. 1f). To determine whether crocidolite indirectly induces mesothelial cytotoxicity via injured macrophages, we performed in vitro experiments, using double chamber co-culture system between mesothelial cells and macrophages (Fig. 4a). H2O2 from macrophage necrosis indirectly caused lipid peroxidation in mesothelial cells (Fig. 4b–d). 8-Hydroxy-2′-deoxyguanosine (8-OHdG) is a marker of oxidative DNA damage [[58], [59], [60]]. We observed that nuclear 8-OHdG in mesothelial cells increased in the same system in an iron concentration-dependent manner (Fig. 4e and f). Furthermore, in rat experiments, nuclear 8-OHdG was observed in mesothelial cells sitting on granuloma and fibrosis only after crocidolite exposure (Fig. 4g and h).
Fig. 4.

Macrophage ferroptosis via crocidolite causes oxidative damage in distant mesothelial cells.
(a) Scheme of coculture system for Met5A mesothelial (bottom) and RAW264 macropahge (top) cells. (b–d) Macrophage ferroptosis via crocidolite in the presence of iron (FAC) induces lipid peroxidation (LPO; green color by Click-iT Lipid peroxidation kit) iron dose-dependently, which further causes lipid peroxidation in distant mesothelial cells. (e, f) In the same co-culture system, nuclear 8-hydroxy-2′-deoxyguanosine (8-OHdG) increased in an iron dose-dependent manner (8-OHdG, green; Hoechst33342, cyan; DIM, differential interference contrast microscope). (g, h) Significantly increased nuclear 8-OHdG signals in mesothelial cells in rats 4 wks after crocidolite i.p. injection in comparison to anthophyllite (means ± SEM; N ≧ 4). Refer to text for details. . (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.6. Stabilization of β-catenin orchestrates iron metabolism through the cell cycle
To elucidate the association between β-catenin and iron metabolism in mesothelial cells especially from the viewpoint of the cell cycle, we used β-catenin overexpression system (Fig. 5a). β-Catenin overexpression in mesothelial cells significantly increased intracellular catalytic Fe(II) in a dose-dependent manner in comparison to the mock transfection group (Fig. 5b and c). Lithium Chloride (LiCl) inhibits GSK-3β which works for degradation of β-catenin through phosphorylation to Ser33, 37 and Thr41 [61]. We confirmed an increase in β-catenin expression by LiCl or hWnt with time-dependence. Simultaneously, we observed a gradual increase in expression of transferrin receptor 1 (TfR1) (Fig. 5d and e). Phosphorylation of β-catenin at Ser675 (pβ-catenin-S675) induces β-catenin stabilization and nuclear translocation, starting transcriptional activity for proliferation. The cells with nuclear β-catenin accumulation revealed significantly increased catalytic Fe(II). We categorized the immunostaining pattern of β-catenin into 4 different types (low expression, [plasma] membrane, nuclear and high expression) in increasing orders (Fig. 5f and g). These results suggest the association of TfR1 expression with β-catenin-dependent Fe(II) uptake. Furthermore, we found that β-catenin expression and catalytic Fe(II) were linear in correlation with DNA amount per cell (Fig. 5h and i). We also found that TfR1 expression and pβ-catenin-S675 are in linear correlation. Thus, overexpressed β-catenin (Δ89 as degradation recognition site-deficient β-catenin) contributes to and accommodate an iron-rich status in mesothelial cells with increased ferritin, an iron storage protein (Fig. 5j) whereas β-catenin knockdown decreased iron import and storage (Fig. 5j). In summary, β-catenin overexpression led to increased intracellular catalytic Fe(II), which was at the highest at G2/M phase.
Fig. 5.

β-Catenin overexpression increases catalytic Fe(II) in mesothelial cell by modifying iron metabolism and cell cycle.
(a) Scheme of β-catenin overexpression system as a fusion protein with GFP. FL, full-length; Δ89-β-catenin, N-terminal 89-amino acids were deleted for stabilization of β-catenin. PRE, post-transcriptional regulatory element. (b, c) Catalytic Fe(II) (SiRhoNox-1 fluorescent probe, magenta) was significantly increased in β-catenin overexpressed Met5A mesothelial cell. (d, e) Immunoblot analysis of iron metabolism and β-catenin; LiCl (20 mM) and hWnt1 (20 ng/ml) for β-catenin activation. (f, g) Imaging for catalytic Fe(II) and β-catenin in FL-β-catenin (GFP) overexpressed Met5A cells. Expression pattern of β-catenin was classified into 4 types as follows from low to high; low expression, (plasma) membrane localization, nuclear localization and high expression. Catalytic Fe(II) levels were β-catenin dose-dependent in (g). (h, i) FACS analysis of β-catenin (GFP; green) and catalytic Fe(II) (SiRhoNox-1, magenta). Cell cycle was separated by DNA amount (Hoechst33342, blue) into G1, S and G2/M phases in FACS analysis. G2/M phase reveals high β-catenin expression and high catalytic Fe(II). (j) Immunoblotting analysis for the association of β-catenin levels with iron metabolism in Met5A cells. β-Catenin overexpression on the left increases intracellular iron and its knockdown on the right decreases it (NT, no treatment; Scr, scrambled; sh β-cat: β-catenin knock down; means ± SEM; N ≧ 4). Refer to text for details. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.7. β-Catenin-overexpressing mesothelial cells suffer iron-dependent DNA double-strand breaks
DNA double-strand breaks (DSBs) is a major event during the production process of carcinogenic mutations and deletions. DSBs recruit γH2AX to the DSB foci [60]. In in vitro analysis using Met5A mesothelial cells, γH2AX expression was induced in an iron-dependent manner in the presence of H2O2 (Fig. 6a). Immunohistochemical analysis revealed that γ-H2AX immunopositivity is significantly higher in cells with high β-catenin levels, which were often associated with mitosis (Fig. 6b and c). Upon β-catenin stabilization with LiCl, γH2AX levels further increased in β-catenin-overexpressed mesothelial cells in the presence of iron and H2O2 (Fig. 6d). On the peritoneal surface of granuloma in rats, regenerative mesothelial cells strongly showed DSBs after crocidolite exposure whereas those after anthophyllite exposure did not (Fig. 6e).
Fig. 6.

β-Catenin overexpression promotes induction of DNA double-strand breaks in oxidatively stressed mesothelial cells.
(a) DNA double-strand breaks evaluated by γ-H2AX in Met5A and 293T cells exposed to oxidative stress for 2 h by Fenton reaction (0–100 μg/ml FAC and 200 μM H2O2). (b, c) Association of FL-β-catenin expression with γ-H2AX in Met5A cells under exposure to H2O2 (200 μM, 2 h; green, β-catenin-GFP; magenta, γH2AX; bar = 5 μm). (d) Immunoblot analysis of γH2AX in various conditions to modify β-catenin expression and oxidative stress. Transfected Met5A cells are exposed to iron (FAC, 100 μg/ml, 2 h), LiCl (20 mM, 6 h) and/or H2O2 (200 μM, 2 h). (e) Immunohistochemistry of γH2AX in rat mesothelium on the granuloma after exposure either to anthophyllite or crocidolite (4 wks; red arrow heads, γH2AX-positive; black arrow head: γH2AX-negative; bar = 50 μm). Quantification of γH2AX positive cells on the right side (means ± SEM; N ≧ 4). . (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.8. Asbestos microenvironment induces p16INK4A deletion in repairing mesothelial cells
Nuclear localization of the genomic loci is critical for the deletion process because catalytic Fe(II)-dependent reactive species attacks DNA more from the nuclear periphery and less to the center [62]. We hypothesized that the p16INK4A deletion events occur in the early stage of mesothelial carcinogenesis. To detect nuclear localization of p16INK4A in each cell cycle, we used cell cycle indicator (Fucci) [36] and CasFISH system which can visualize a target gene using labeled-dCas9 (deficient in DNA cutting effect) [46] (Fig. 7a). We found that p16INK4A loci were localized to central nuclei at G1 phase whereas the localization was altered to the near nuclear membrane at G2/M phase (Fig. 7a and b). By the FISH (fluorescent in situ hybridization) analysis, we observed mainly two types of nuclear signals, namely, no deletion and hemizygous deletion (Fig. 7c). Iron- and H2O2-rich environment with β-catenin stabilization by LiCl was necessary to observe significant hemizygous deletion in the p16INK4A loci in Met5A cells (Fig. 7d). We then studied the expression of p15INK4B and p16INK4A in H2O2 or asbestos survivor cells to confirm the inactivation of p16INK4A gene locus (Fig. 7e). H2O2 challenge decreased both P15 and P16 expression in mesothelial cells (Fig. 7f). p15INK4B and p16INK4A gene loci are close each other and p15INK4B fulfills backup of p16INK4A function [63]. Thus, we speculate that oxidative stress induced deficiency of both p15INK4B and p16INK4A.
Fig. 7.

p16INK4A loci are more nuclear peripheral at G2/M phase and its allele is lost accompanied by low p16INK4A expression after oxidative stress under high β-catenin expression.
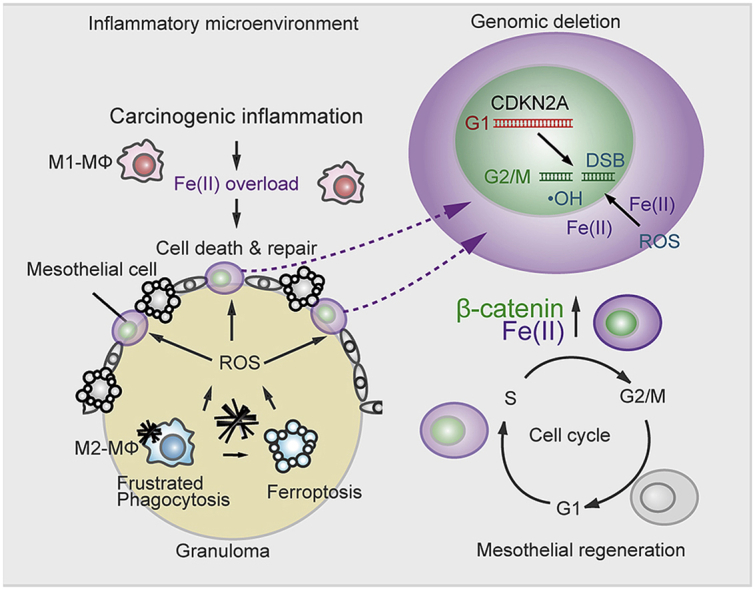
(a, b) dCas FISH analysis for p16INK4A loci in Met5A cells shows their more peripheral presence at G2/M phase in comparison to G1 phase; red, G1 phase; green, G2/M phase; white, p16INK4A locus (bar = 5 μm). (c, d) FISH analysis in rat peritoneal mesothelial cell reveals hemizygous deletion 24 h after incubation with FAC (100 μg/ml, 2 h), LiCl (20 mM, 6 h) and H2O2 (200 μM, 2 h). p16INK4A, green (FITC); chromosome No. 5 centromere, red (Dyelight 594) are detected in the nuclei (blue, DAPI). Each FISH signals were counted for deletion score; no deletion, p16INK4A/centromere ratio = ~1.0, heterozygous deletion, signals of p16INK4A/centromere ratio = ~0.5. (bar = 5 μm) (e, f) Scheme of culture system for mesothelial survivor analysis using Met5A or LP-9 human mesothelial cells. Immunoblot analysis in Met5A and LP9 cells for p15INK4B and p16INK4A after exposure of H2O2 (25 μM), asbestos (25 μg/cm2) for 3 days. After 2 wks of recovery, survived cells were used for protein detection analysis. (g) Graphical abstract illustrating the key concepts on the iron-rich stromal mutagenic milieu after exposure to crocidolite (left). Mesothelial regeneration after exposure to crocidolite accompanies β-catenin overexpression, which increases the risk of p16INK4A deletion via multiple mechanisms, such as increased G2/M phase fraction, higher intracellular catalytic Fe(II) and peripheral transfer of p16INK4A genomic loci in the nucleus (means ± SEM; N ≧ 4). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
We previously showed that anthophyllite, a minor iron-containing asbestos, presents a low risk for mesothelial carcinogenesis [34]. Based on the results, here we revealed for the first time, with differential animal experiments between high-risk and low-risk asbestos exposure (crocidolite and anthophyllite, respectively), that iron excess-induced mutagenic milieu is established for mesothelial carcinogenesis. Mesothelial cells cover somatic cavities as a layer of flat cells [15]. We discovered that the only mesothelial cells exposed to high-risk asbestos are sandwiched by this iron-rich milieu not only from the stromal side but also from the cavity side. The cavity side was full of catalytic Fe(II) and the stromal side showed massive macrophage necrosis as granuloma with lipid peroxidation. We further found that the H2O2 source from macrophages was not limited to NOX2. It is of note that an additional source of ROS from macrophages exposed to crocidolite was recently identified as γ-glutamyltransferase (GGT), whose expression is indeed induced by crocidolite [64]. Interestingly, macrophagic GGT-dependent ROS production has been recently recognized as the main source of oxidative stress during neuroinflammation, in which oxidative injury and clinical signs could be largely suppressed by inhibiting GGT activity [65]. Then, we studied the precise mechanisms for regulated macrophage necrosis by crocidolite.
Lysosomal damage by asbestos was critical for the massive killing of macrophages. Nuclear transfer of cathepsin B was evident in the case of crocidolite in the presence of excess iron in THP1 cells. Mitochondria were also significantly injured, which requires further investigation. At a later phase, decreased GPX4 level presumably via degradation, increased ferritin level and partial recovery with ferrostain-1 and deferoxamine (DFO) were observed, indicating the involvement of ferroptosis [32] in crocidolite-induced macrophage necrosis. TfR1 showed a controversial results with the inhibitors used, but we think that decrease in TfR1 after ferrostatin-1 means the efforts to normalize high iron as a non-tumours cell and that the increase in TfR1 after DFO suggest a response after iron depletion by DFO. In summary, stromal iron excess was derived from necrotized macrophages, at an early phase by lysosome-dependent cell death and later by ferroptosis. We cannot completely rule out the possibility of minor overlap of other regulated cell death, such as apoptosis, pyroptosis or necroptosis [66,67].
Macrophage plays a central role in iron metabolism, including heme-iron turnover in senescent red blood cells, hepcidin regulation and lipocalin system [68]. Additionally, lysosome is one of the most Fe(II)-rich organella [41], and iron should go through lysosomal/endosomal membrane protein (divalent metal transporter 1: DMT1) for intracellular use [69]. Thus, the lysosomal membrane permeabilization (LMP) by crocidolite disturbs cellular iron metabolism, and macrophage cell death disseminates accumulated iron, eventually leading to iron excess in granuloma. It is critical that the macrophages cannot eliminate not only asbestos but also iron. Furthermore, the cells which collect necrotic debris are also macrophages. These vicious cycles induce continuous necrotic death and establishes catalytic Fe(II)-dependent stromal mutagenic environment.
Then, we focused on the mesothelial cells and screened with expressional microarray for characteristic signalling during regeneration of crocidolite-induced mesothelial injury. β-Catenin was the most significant among the positive results, including c-Met and Tgfbr1. Double chamber culture system demonstrated a distant injurious effect of crocidolite-exposed macrophages to mesothelial cells, which revealed oxidative damage with lipid peroxidation and an increase in 8-OHdG. A rat model also confirmed this.
Finally, we sought for possible links between β-catenin signal activation and p16INK4A deletion, a largest target gene of MM carcinogenesis [4,[24], [25], [26], [27], [28]]. We previously reported a positive autocrine loop between β-catenin and connective tissue growth factor in asbestos-induced rat MM [70, 71]. There are three major merits in the activation of β-catenin signaling for mesothelial carcinogenesis. First, it is associated with regeneration. Thus, continued cell cycles with a higher fraction of G2/M phase increase mutation rate. Second, β-catenin modifies iron metabolism, increasing intracellular catalytic Fe(II) dose-dependently with β-catenin expression and especially at G2/M phase. This is consistent with a previous report, dictating the association between iron metabolism and Wnt/β-catenin pathway [72]. However, we for the first time analyzed the catalytic Fe(II) in association with cell cycle. Third, unexpectedly, genomic loci for p16INK4A are directed toward nuclear periphery at G2/M phase. Indeed, deletion of p16INK4A was demonstrated in mesothelial cells in the presence of high iron and H2O2 under high β-catenin expression. These results suggest that the inflammatory and mutagenic microenvironment by crocidolite selectively induced DNA DSBs in mesothelial progenitor or repairing cells, leading eventually to selective p16INK4A deletion.
In conclusion, our findings highlight the importance of catalytic Fe(II)-dependent mutagenic stromal milieu in mesothelial genomic alterations in that asbestos-induced indirect damage affects regenerative mesothelia with β-catenin-dependence. We discovered that mesothelial activation of β-catenin pathway during regeneration brings multiple advantages for their p16INK4A deletion. Our results would be helpful for establishing strategies to prevent MM in people already exposed to asbestos. In addition to iron modulation [13,14] and anti-inflammation [73] as already suggested, β-catenin pathway would be another molecular target to prevent mesothelial carcinogenesis.
Author contributions
F.I. and S.T. contributed to conception and design of the work and drafted the work; F.I., I.Y., Y.M., K.N., S.I., T.H., H.N., N.,K., Y.O, and S.A. contributed to acquisition and analysis of the data; S.T. substantially revised the manuscript.
Declaration of competing interest
The authors declare no conflict of interest to present.
Acknowledgments
This work was supported, in part, by JST CREST (Grant Number JPMJCR19H4) and JSPS Kakenhi (Grant Number JP17H04064, JP18J13646, JP19H05462 and JP20H05502). Electron microscopic analyses were supported by Koji Itakura, Division of Medical Research Engineering, Nagoya University Graduate School of Medicine. We thank Nobuaki Misawa (Department Pathology and Biological Responses, Nagoya University Graduate School of Medicine) for excellent technical assistance.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101616.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Gualtieri A.F., Andreozzi G.B., Tomatis M., Turci F. Iron from a geochemical viewpoint. Understanding toxicity/pathogenicity mechanisms in iron-bearing minerals with a special attention to mineral fibers. Free Radic. Biol. Med. 2019;133:21–37. doi: 10.1016/j.freeradbiomed.2018.07.023. [DOI] [PubMed] [Google Scholar]
- 2.WHO, IARC . IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. A Review of Human Carcinogens; Part C: Arsenic, Metals, Fibres, and Dusts, Lyon, France. 2012. Asbestos (chrysotile, amosite, crocidolite, tremolite, actinolite, and anthophyllite) pp. 219–309. [PMC free article] [PubMed] [Google Scholar]
- 3.Allen L.P., Baez J., Stern M.E.C., Takahashi K., George F. Trends and the economic effect of asbestos bans and decline in asbestos consumption and production worldwide. Int. J. Environ. Res. Publ. Health. 2018;15(3):531. doi: 10.3390/ijerph15030531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toyokuni S. Iron addiction with ferroptosis-resistance in asbestos-induced mesothelial carcinogenesis: toward the era of mesothelioma prevention. Free Radic. Biol. Med. 2019;133:206–215. doi: 10.1016/j.freeradbiomed.2018.10.401. [DOI] [PubMed] [Google Scholar]
- 5.Toyokuni S. Mechanisms of asbestos-induced carcinogenesis. Nagoya J. Med. Sci. 2009;71(1-2):1–10. [PMC free article] [PubMed] [Google Scholar]
- 6.Sincock A., Seabright M. Induction of chromosome changes in Chinese hamster cells by exposure to asbestos fibres. Nature. 1975;257(5521):56–58. doi: 10.1038/257056a0. [DOI] [PubMed] [Google Scholar]
- 7.Pricejones M.J., Gubbings G., Chamberlain M. The genetic-effects of crocidolite asbestos - comparison of chromosome-abnormalities and sister-chromatid exchanges. Mutat. Res. 1980;79(4):331–336. [Google Scholar]
- 8.Jiang L., Nagai H., Ohara H., Hara S., Tachibana M., Hirano S., Shinohara Y., Kohyama N., Akatsuka S., Toyokuni S. Characteristics and modifying factors of asbestos-induced oxidative DNA damage. Cancer. Sci. 2008;99:2142–2151. doi: 10.1111/j.1349-7006.2008.00934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donaldson K., Slight J., Hannant D., Bolton R.E. Increased release of hydrogen peroxide and superoxide anion from asbestos-primed macrophages. Effect of hydrogen peroxide on the functional activity of alpha 1-protease inhibitor. Inflammation. 1985;9(2):139–147. doi: 10.1007/BF00917586. [DOI] [PubMed] [Google Scholar]
- 10.Dostert C., Petrilli V., Van Bruggen R., Steele C., Mossman B.T., Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donaldson K., Murphy F.A., Duffin R., Poland C.A. Asbestos, carbon nanotubes and the pleural mesothelium: a review of the hypothesis regarding the role of long fibre retention in the parietal pleura, inflammation and mesothelioma. Part. Fibre Toxicol. 2010;7:5. doi: 10.1186/1743-8977-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toyokuni S. Role of iron in carcinogenesis: cancer as a ferrotoxic disease. Cancer. Sci. 2009;100(1):9–16. doi: 10.1111/j.1349-7006.2008.01001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagai H., Okazaki Y., Chew S.H., Misawa N., Yasui H., Toyokuni S. Deferasirox induces mesenchymal-epithelial transition in crocidolite-induced mesothelial carcinogenesis in rats. Cancer. Prev. Res. 2013;6:1222–1230. doi: 10.1158/1940-6207.CAPR-13-0244. [DOI] [PubMed] [Google Scholar]
- 14.Ohara Y., Chew S.H., Shibata T., Okazaki Y., Yamashita K., Toyokuni S. Phlebotomy as a preventive measure for crocidolite-induced mesothelioma in male rats. Cancer. Sci. 2018;109(2):330–339. doi: 10.1111/cas.13460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oury T.D., Sporn T.A., Roggli V.L., editors. Pathology of Asbestos-Associated Diseases. third ed. Springer; Berlin/Heidelberg: 2014. [Google Scholar]
- 16.Okada S., Hamazaki S., Toyokuni S., Midorikawa O. Induction of mesothelioma by intraperitoneal injections of ferric saccharate in male Wistar rats. Br. J. Cancer. 1989;60:708–711. doi: 10.1038/bjc.1989.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Q., Akatsuka S., Yamashita Y., Ohara H., Nagai H., Okazaki Y., Takahashi T., Toyokuni S. Homozygous deletion of CDKN2A/2B is a hallmark of iron-induced high-grade rat mesothelioma. Lab. Invest. 2010;90:360–373. doi: 10.1038/labinvest.2009.140. [DOI] [PubMed] [Google Scholar]
- 18.Toyokuni S. The origin and future of oxidative stress pathology: from the recognition of carcinogenesis as an iron addiction with ferroptosis resistance to non-thermal plasma therapy. Pathol. Int. 2016;66:245–259. doi: 10.1111/pin.12396. [DOI] [PubMed] [Google Scholar]
- 19.Hiroyasu M., Ozeki M., Kohda H., Echizenya M., Tanaka T., Hiai H., Toyokuni S. Specific allelic loss of p16 (INK4A) tumor suppressor gene after weeks of iron-mediated oxidative damage during rat renal carcinogenesis. Am. J. Pathol. 2002;160(2):419–424. doi: 10.1016/S0002-9440(10)64860-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishiyama Y., Suwa H., Okamoto K., Fukumoto M., Hiai H., Toyokuni S. Low incidence of point mutations in H-, K- and N-ras oncogenes and p53 tumor suppressor gene in renal cell carcinoma and peritoneal mesothelioma of Wistar rats induced by ferric nitrilotriacetate. Jpn. J. Cancer. Res. 1995;86:1150–1158. doi: 10.1111/j.1349-7006.1995.tb03308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akatsuka S., Yamashita Y., Ohara H., Liu Y.T., Izumiya M., Abe K., Ochiai M., Jiang L., Nagai H., Okazaki Y., Murakami H., Sekido Y., Arai E., Kanai Y., Hino O., Takahashi T., Nakagama H., Toyokuni S. Fenton reaction induced cancer in wild type rats recapitulates genomic alterations observed in human cancer. PLoS One. 2012;7(8) doi: 10.1371/journal.pone.0043403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toyokuni S. Novel aspects of oxidative stress-associated carcinogenesis. Antioxidants Redox Signal. 2006;8(7-8):1373–1377. doi: 10.1089/ars.2006.8.1373. [DOI] [PubMed] [Google Scholar]
- 23.Toyokuni S., Ito F., Yamashita K., Okazaki Y., Akatsuka S. Iron and thiol redox signaling in cancer: an exquisite balance to escape ferroptosis. Free Radic. Biol. Med. 2017;108:610–626. doi: 10.1016/j.freeradbiomed.2017.04.024. [DOI] [PubMed] [Google Scholar]
- 24.Altomare D.A., Menges C.W., Xu J., Pei J., Zhang L., Tadevosyan A., Neumann-Domer E., Liu Z., Carbone M., Chudoba I., Klein-Szanto A.J., Testa J.R. Losses of both products of the Cdkn2a/Arf locus contribute to asbestos-induced mesothelioma development and cooperate to accelerate tumorigenesis. PLoS One. 2011;6(4) doi: 10.1371/journal.pone.0018828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bott M., Brevet M., Taylor B.S., Shimizu S., Ito T., Wang L., Creaney J., Lake R.A., Zakowski M.F., Reva B., Sander C., Delsite R., Powell S., Zhou Q., Shen R., Olshen A., Rusch V., Ladanyi M. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011;43(7):668–672. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo G., Chmielecki J., Goparaju C., Heguy A., Dolgalev I., Carbone M., Seepo S., Meyerson M., Pass H.I. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer. Res. 2015;75(2):264–269. doi: 10.1158/0008-5472.CAN-14-1008. [DOI] [PubMed] [Google Scholar]
- 27.Bueno R., Stawiski E.W., Goldstein L.D., Durinck S., De Rienzo A., Modrusan Z., Gnad F., Nguyen T.T., Jaiswal B.S., Chirieac L.R., Sciaranghella D., Dao N., Gustafson C.E., Munir K.J., Hackney J.A., Chaudhuri A., Gupta R., Guillory J., Toy K., Ha C., Chen Y.J., Stinson J., Chaudhuri S., Zhang N., Wu T.D., Sugarbaker D.J., de Sauvage F.J., Richards W.G., Seshagiri S. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016;48(4):407–416. doi: 10.1038/ng.3520. [DOI] [PubMed] [Google Scholar]
- 28.Hmeljak J., Sanchez-Vega F., Hoadley K.A., Shih J., Stewart C., Heiman D., Tarpey P., Danilova L., Drill E., Gibb E.A., Bowlby R., Kanchi R., Osmanbeyoglu H.U., Sekido Y., Takeshita J., Newton Y., Graim K., Gupta M., Gay C.M., Diao L., Gibbs D.L., Thorsson V., Iype L., Kantheti H., Severson D.T., Ravegnini G., Desmeules P., Jungbluth A.A., Travis W.D., Dacic S., Chirieac L.R., Galateau-Salle F., Fujimoto J., Husain A.N., Silveira H.C., Rusch V.W., Rintoul R.C., Pass H., Kindler H., Zauderer M.G., Kwiatkowski D.J., Bueno R., Tsao A.S., Creaney J., Lichtenberg T., Leraas K., Bowen J., Network T.R., Felau I., Zenklusen J.C., Akbani R., Cherniack A.D., Byers L.A., Noble M.S., Fletcher J.A., Robertson A.G., Shen R., Aburatani H., Robinson B.W., Campbell P., Ladanyi M. Integrative molecular characterization of malignant pleural mesothelioma. Cancer. Discov. 2018;8(12):1548–1565. doi: 10.1158/2159-8290.CD-18-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hida T., Hamasaki M., Matsumoto S., Sato A., Tsujimura T., Kawahara K., Iwasaki A., Okamoto T., Oda Y., Honda H., Nabeshima K. BAP1 immunohistochemistry and p16 FISH results in combination provide higher confidence in malignant pleural mesothelioma diagnosis: ROC analysis of the two tests. Pathol. Int. 2016;66(10):563–570. doi: 10.1111/pin.12453. [DOI] [PubMed] [Google Scholar]
- 30.Galateau Salle F., Le Stang N., Nicholson A.G., Pissaloux D., Churg A., Klebe S., Roggli V., Tazelaar H., Vignaud J.M., Attanoos R., Beasley M.B., Begueret H., Capron F., Chirieac L., Copin M.C., Dacic S., Danel C., Foulet-Roge A., Gibbs A., Giusiano-Courcambeck S., Hiroshima K., Hofman V., Husain A., Kerr K., Marchevsky A., Nabeshima K., Picquenot J.M., Rouquette I., Sagan C., Sauter J., Thivolet F., Travis W.D., Tsao M.S., Weynand B., Damiola F., Scherpereel A., Pairon J.C., Lantuejoul S., Rusch V., Girard N. New insights on diagnostic reproducibility of biphasic mesotheliomas: a multi-institutional evaluation by the international mesothelioma panel from the MESOPATH reference center. J. Thorac. Oncol. 2018;13:1189–1203. doi: 10.1016/j.jtho.2018.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang L., Akatsuka S., Nagai H., Chew S.H., Ohara H., Okazaki Y., Yamashita Y., Yoshikawa Y., Yasui H., Ikuta K., Sasaki K., Kohgo Y., Hirano S., Shinohara Y., Kohyama N., Takahashi T., Toyokuni S. Iron overload signature in chrysotile-induced malignant mesothelioma. J. Pathol. 2012;228:366–377. doi: 10.1002/path.4075. [DOI] [PubMed] [Google Scholar]
- 32.Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J., Fulda S., Gascon S., Hatzios S.K., Kagan V.E., Noel K., Jiang X., Linkermann A., Murphy M.E., Overholtzer M., Oyagi A., Pagnussat G.C., Park J., Ran Q., Rosenfeld C.S., Salnikow K., Tang D., Torti F.M., Torti S.V., Toyokuni S., Woerpel K.A., Zhang D.D. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., Alnemri E.S., Altucci L., Amelio I., Andrews D.W., Annicchiarico-Petruzzelli M., Antonov A.V., Arama E., Baehrecke E.H., Barlev N.A., Bazan N.G., Bernassola F., Bertrand M.J.M., Bianchi K., Blagosklonny M.V., Blomgren K., Borner C., Boya P., Brenner C., Campanella M., Candi E., Carmona-Gutierrez D., Cecconi F., Chan F.K., Chandel N.S., Cheng E.H., Chipuk J.E., Cidlowski J.A., Ciechanover A., Cohen G.M., Conrad M., Cubillos-Ruiz J.R., Czabotar P.E., D'Angiolella V., Dawson T.M., Dawson V.L., De Laurenzi V., De Maria R., Debatin K.M., DeBerardinis R.J., Deshmukh M., Di Daniele N., Di Virgilio F., Dixit V.M., Dixon S.J., Duckett C.S., Dynlacht B.D., El-Deiry W.S., Elrod J.W., Fimia G.M., Fulda S., Garcia-Saez A.J., Garg A.D., Garrido C., Gavathiotis E., Golstein P., Gottlieb E., Green D.R., Greene L.A., Gronemeyer H., Gross A., Hajnoczky G., Hardwick J.M., Harris I.S., Hengartner M.O., Hetz C., Ichijo H., Jaattela M., Joseph B., Jost P.J., Juin P.P., Kaiser W.J., Karin M., Kaufmann T., Kepp O., Kimchi A., Kitsis R.N., Klionsky D.J., Knight R.A., Kumar S., Lee S.W., Lemasters J.J., Levine B., Linkermann A., Lipton S.A., Lockshin R.A., Lopez-Otin C., Lowe S.W., Luedde T., Lugli E., MacFarlane M., Madeo F., Malewicz M., Malorni W., Manic G., Marine J.C., Martin S.J., Martinou J.C., Medema J.P., Mehlen P., Meier P., Melino S., Miao E.A., Molkentin J.D., Moll U.M., Munoz-Pinedo C., Nagata S., Nunez G., Oberst A., Oren M., Overholtzer M., Pagano M., Panaretakis T., Pasparakis M., Penninger J.M., Pereira D.M., Pervaiz S., Peter M.E., Piacentini M., Pinton P., Prehn J.H.M., Puthalakath H., Rabinovich G.A., Rehm M., Rizzuto R., Rodrigues C.M.P., Rubinsztein D.C., Rudel T., Ryan K.M., Sayan E., Scorrano L., Shao F., Shi Y., Silke J., Simon H.U., Sistigu A., Stockwell B.R., Strasser A., Szabadkai G., Tait S.W.G., Tang D., Tavernarakis N., Thorburn A., Tsujimoto Y., Turk B., Vanden Berghe T., Vandenabeele P., Vander Heiden M.G., Villunger A., Virgin H.W., Vousden K.H., Vucic D., Wagner E.F., Walczak H., Wallach D., Wang Y., Wells J.A., Wood W., Yuan J., Zakeri Z., Zhivotovsky B., Zitvogel L., Melino G., Kroemer G. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aierken D., Okazaki Y., Chew S.H., Sakai A., Wang Y., Nagai H., Misawa N., Kohyama N., Toyokuni S. Rat model demonstrates a high risk of tremolite but a low risk of anthophyllite for mesothelial carcinogenesis. Nagoya J. Med. Sci. 2014;76:149–160. [PMC free article] [PubMed] [Google Scholar]
- 35.Kajiyama H., Kikkawa F., Maeda O., Suzuki T., Ino K., Mizutani S. Increased expression of dipeptidyl peptidase IV in human mesothelial cells by malignant ascites from ovarian carcinoma patients. Oncology-Basel. 2002;63(2):158–165. doi: 10.1159/000063801. [DOI] [PubMed] [Google Scholar]
- 36.Sakaue-Sawano A., Kurokawa H., Morimura T., Hanyu A., Hama H., Osawa H., Kashiwagi S., Fukami K., Miyata T., Miyoshi H., Imamura T., Ogawa M., Masai H., Miyawaki A. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132(3):487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- 37.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Houstis N., Daly M.J., Patterson N., Mesirov J.P., Golub T.R., Tamayo P., Spiegelman B., Lander E.S., Hirschhorn J.N., Altshuler D., Groop L.C. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 38.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 40.Graff J.W., Dickson A.M., Clay G., McCaffrey A.P., Wilson M.E. Identifying functional MicroRNAs in macrophages with polarized phenotypes. J. Biol. Chem. 2012;287(26):21816–21825. doi: 10.1074/jbc.M111.327031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ito F., Nishiyama T., Shi L., Mori M., Hirayama T., Nagasawa H., Yasui H., Toyokuni S. Contrasting intra- and extracellular distribution of catalytic ferrous iron in ovalbumin-induced peritonitis. Biochem. Biophys. Res. Commun. 2016;476(4):600–606. doi: 10.1016/j.bbrc.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 42.Hirayama T., Okuda K., Nagasawa H. A highly selective turn-on fluorescent probe fro iron(II) to visualize labile iron in living cells. Chem. Sci. 2013;4:1250–1256. [Google Scholar]
- 43.Hirayama T., Tsuboi H., Niwa M., Miki A., Kadota S., Ikeshita Y., Okuda K., Nagasawa H. A universal fluorogenic switch for Fe(ii) ion based on N-oxide chemistry permits the visualization of intracellular redox equilibrium shift towards labile iron in hypoxic tumor cells. Chem. Sci. 2017;8(7):4858–4866. doi: 10.1039/c6sc05457a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirayama T. Fluorescent probes for the detection of catalytic Fe(II) ion. Free Radic. Biol. Med. 2019;133:38–45. doi: 10.1016/j.freeradbiomed.2018.07.004. [DOI] [PubMed] [Google Scholar]
- 45.Schagger H. Tricine-SDS-PAGE. Nat. Protoc. 2006;1(1):16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 46.Ma H.H., Naseri A., Reyes-Gutierrez P., Wolfe S.A., Zhang S.J., Pederson T. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. U.S.A. 2015;112(10):3002–3007. doi: 10.1073/pnas.1420024112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toyokuni S., Uchida K., Okamoto K., Hattori-Nakakuki Y., Hiai H., Stadtman E.R. Formation of 4-hydroxy-2-nonenal-modified proteins in the renal proximal tubules of rats treated with a renal carcinogen, ferric nitrilotriacetate. Proc. Natl. Acad. Sci. U.S.A. 1994;91:2616–2620. doi: 10.1073/pnas.91.7.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog. Lipid Res. 2003;42(4):318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 49.Toyokuni S., Luo X.P., Tanaka T., Uchida K., Hiai H., Lehotay D.C. Induction of a wide range of C2-12 aldehydes and C7-12 acyloins in the kidney of Wistar rats after treatment with a renal carcinogen, ferric nitrilotriacetate. Free Radic. Biol. Med. 1997;22:1019–1027. doi: 10.1016/s0891-5849(96)00489-3. [DOI] [PubMed] [Google Scholar]
- 50.Italiani P., Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. Functional differentiation. Front. Immunol. 2014;5:514. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 52.Aits S., Jaattela M. Lysosomal cell death at a glance. J. Cell Sci. 2013;126(Pt 9):1905–1912. doi: 10.1242/jcs.091181. [DOI] [PubMed] [Google Scholar]
- 53.Vanden Berghe T., Linkermann A., Jouan-Lanhouet S., Walczak H., Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014;15(2):135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 54.Dang C.V. MYC on the path to cancer. Cell. 2012;149(1):22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reinke J.M., Sorg H. Wound repair and regeneration. Eur. Surg. Res. 2012;49(1):35–43. doi: 10.1159/000339613. [DOI] [PubMed] [Google Scholar]
- 56.Cathcart M.K. Regulation of superoxide anion production by NADPH oxidase in monocytes/macrophages: contributions to atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004;24(1):23–28. doi: 10.1161/01.ATV.0000097769.47306.12. [DOI] [PubMed] [Google Scholar]
- 57.Bae Y.S., Lee J.H., Choi S.H., Kim S., Almazan F., Witztum J.L., Miller Y.I. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ. Res. 2009;104(2):210–218. doi: 10.1161/CIRCRESAHA.108.181040. 221p following 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Toyokuni S., Tanaka T., Hattori Y., Nishiyama Y., Ochi H., Hiai H., Uchida K., Osawa T. Quantitative immunohistochemical determination of 8-hydroxy-2'-deoxyguanosine by a monoclonal antibody N45.1: its application to ferric nitrilotriacetate-induced renal carcinogenesis model. Lab. Invest. 1997;76:365–374. [PubMed] [Google Scholar]
- 59.Toyokuni S., Akatsuka S. Pathological investigation of oxidative stress in the post-genomic era. Pathol. Int. 2007;57(8):461–473. doi: 10.1111/j.1440-1827.2007.02127.x. [DOI] [PubMed] [Google Scholar]
- 60.Tubbs A., Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168(4):644–656. doi: 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li V.S.W., Ng S.S., Boersema P.J., Low T.Y., Karthaus W.R., Gerlach J.P., Mohammed S., Heck A.J.R., Maurice M.M., Mahmoudi T., Clevers H. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell. 2012;149(6):1245–1256. doi: 10.1016/j.cell.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 62.Akatsuka S., Aung T.T., Dutta K.K., Jiang L., Lee W.-H., Liu Y.-T., Onuki J., Shirase T., Yamasaki K., Ochi H., Naito Y., Yoshikawa T., Kasai H., Tominaga Y., Sakumi K., Nakabeppu Y., Kawai Y., Uchida K., Yamasaki A., Tsuruyama T., Yamada Y., Toyokuni S. Contrasting genome-wide distribution of 8-hydroxyguanine and acrolein-modified adenine during oxidative stress-induced renal carcinogenesis. Am. J. Pathol. 2006;169:1328–1342. doi: 10.2353/ajpath.2006.051280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krimpenfort P., IJpenberg A., Song J.Y., van der Valk M., Nawijn M., Zevenhoven J., Berns A. p15(Ink4b) is a critical tumour suppressor in the absence of p16(Ink4a) Nature. 2007;448(7156):943–946. doi: 10.1038/nature06084. [DOI] [PubMed] [Google Scholar]
- 64.Corti A., Bonetti J., Dominici S., Piaggi S., Fierabracci V., Foddis R., Pompella A. Induction of gamma-glutamyltransferase activity and consequent prooxidant reactions in human macrophages exposed to crocidolite asbestos. Toxicol. Sci. 2019 doi: 10.1093/toxsci/kfz175. [DOI] [PubMed] [Google Scholar]
- 65.Mendiola A.S., Ryu J.K., Bardehle S., Meyer-Franke A., Ang K.K., Wilson C., Baeten K.M., Hanspers K., Merlini M., Thomas S., Petersen M.A., Williams A., Thomas R., Rafalski V.A., Meza-Acevedo R., Tognatta R., Yan Z., Pfaff S.J., Machado M.R., Bedard C., Rios Coronado P.E., Jiang X., Wang J., Pleiss M.A., Green A.J., Zamvil S.S., Pico A.R., Bruneau B.G., Arkin M.R., Akassoglou K. Transcriptional profiling and therapeutic targeting of oxidative stress in neuroinflammation. Nat. Immunol. 2020;21(5):513–524. doi: 10.1038/s41590-020-0654-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang H.N., Rivera Z., Jube S., Nasu M., Bertino P., Goparaju C., Franzoso G., Lotze M.T., Krausz T., Pass H.I., Bianchi M.E., Carbone M. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc. Natl. Acad. Sci. U.S.A. 2010;107(28):12611–12616. doi: 10.1073/pnas.1006542107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He B., Shi Y.J., Liang Y.Q., Yang A.P., Fan Z.P., Yuan L., Zou X.J., Chang X., Zhang H., Wang X.Q., Dai W.B., Wang Y.G., Zhang Q. Single-walled carbon-nanohorns improve biocompatibility over nanotubes by triggering less protein-initiated pyroptosis and apoptosis in macrophages. Nat. Commun. 2018;9:2393. doi: 10.1038/s41467-018-04700-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Soares M.P., Hamza I. Macrophages and iron metabolism. Immunity. 2016;44(3):492–504. doi: 10.1016/j.immuni.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Torti S.V., Torti F.M. Iron and cancer: more ore to be mined. Nat. Rev. Cancer. 2013;13(5):342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang L., Yamashita Y., Chew S.H., Akatsuka S., Ukai S., Wang S., Nagai H., Okazaki Y., Takahashi T., Toyokuni S. Connective tissue growth factor and β-catenin constitute an autocrine loop for activation in rat sarcomatoid mesothelioma. J. Pathol. 2014;233:402–414. doi: 10.1002/path.4377. [DOI] [PubMed] [Google Scholar]
- 71.Ohara Y., Chew S.H., Misawa N., Wang S., Somiya D., Nakamura K., Kajiyama H., Kikkawa F., Tsuyuki Y., Jiang L., Yamashita K., Sekido Y., Lipson K.E., Toyokuni S. Connective tissue growth factor-specific monoclonal antibody inhibits growth of malignant mesothelioma in an orthotopic mouse model. Oncotarget. 2018;9(26):18494–18509. doi: 10.18632/oncotarget.24892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brookes M.J., Boult J., Roberts K., Cooper B.T., Hotchin N.A., Matthews G., Iqbal T., Tselepis C. A role for iron in Wnt signalling. Oncogene. 2008;27(7):966–975. doi: 10.1038/sj.onc.1210711. [DOI] [PubMed] [Google Scholar]
- 73.Yang H., Pellegrini L., Napolitano A., Giorgi C., Jube S., Preti A., Jennings C.J., De Marchis F., Flores E.G., Larson D., Pagano I., Tanji M., Powers A., Kanodia S., Gaudino G., Pastorino S., Pass H.I., Pinton P., Bianchi M.E., Carbone M. Aspirin delays mesothelioma growth by inhibiting HMGB1-mediated tumor progression. Cell Death Dis. 2015;6 doi: 10.1038/cddis.2015.153. e1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.