

Abstract
Recent years have seen an intense period of research on the functions of miRNAs, recently discovered key regulators of gene expression that act through suppression of translation of target mRNAs. Several hundred miRNAs have been identified in humans, and these show characteristic expression patterns, depending on tissue type, cell type or environmental stimuli. Like other types of cancer, the brain tumor glioblastoma shows a distinct miRNA expression signature, and a number of recent studies have linked these miRNA alterations to key hallmarks of glioblastoma including proliferation, survival, invasion, angiogenesis and stem cell-like behavior. These studies have opened the door to the possibility of utilizing miRNAs or miRNA antagonists as therapeutic agents for the treatment of brain tumors.
Keywords: glioblastoma, miRNA, therapeutics
Glioblastoma multiforme (GBM) represents the most common and aggressive intrinsic primary brain tumor in adults. GBM presents unique challenges to therapy due to its location, aggressive biological behavior and diffuse infiltrative growth. Despite the development of new surgical and radiation techniques, and the use of multiple antineoplastic drugs, a cure for malignant gliomas remains elusive [1]. The lack of efficacy of current treatments reflects at least in part the resistance of GBM cells to cytotoxic agents in vivo [2]. Moreover, the short interval for tumor recurrence in GBM patients suggests that tumor cells are able to overcome treatment without major damage. There is a recognized need for new approaches based on increased understanding of the biological and molecular nature of these tumors. Numerous molecular events have been identified in high-grade gliomas, including amplification of the EGF receptor (EGFR), deletion of PTEN, deletion of p16INK4A/p14ARF and TP53 mutation [3]. Recent important developments include the discovery of miRNAs, which are being increasingly linked with cancer [4].
miRNAs
miRNAs belong to a family of small noncoding ssRNA molecules (~21 nucleotides) that are a crucial component of post-transcriptional gene regulation [5,6]. It is estimated that 1000 or more miRNAs may exist in the human genome, with the majority of protein-coding genes regulated by them [7]. It is now thought that the number of genes under regulation by miRNAs is much higher than the widely quoted estimates of 30%, which may be rather conservative [7]. Following transcription, processing occurs to yield a mature miRNA, which then blocks expression of its target mRNA upon binding the 3′-untranslated region (3′-UTR). An outline of this process is shown in Figure 1 . Complementarity to the ‘seed’ region of the miRNA over 7–8 base pairs at the 5′ end is critical in determining active miRNA target sites. Recent data demonstrate that miRNAs can target regions of the mRNA transcript outside the 3′-UTR just as effectively [8]. Target suppression by miRNAs occurs through both inhibition of translation and cleavage of mRNA targets [9,10]. In this article we will discuss the link between miRNAs and high-grade gliomas, and their emerging therapeutic potential.
Figure 1. miRNA processing.
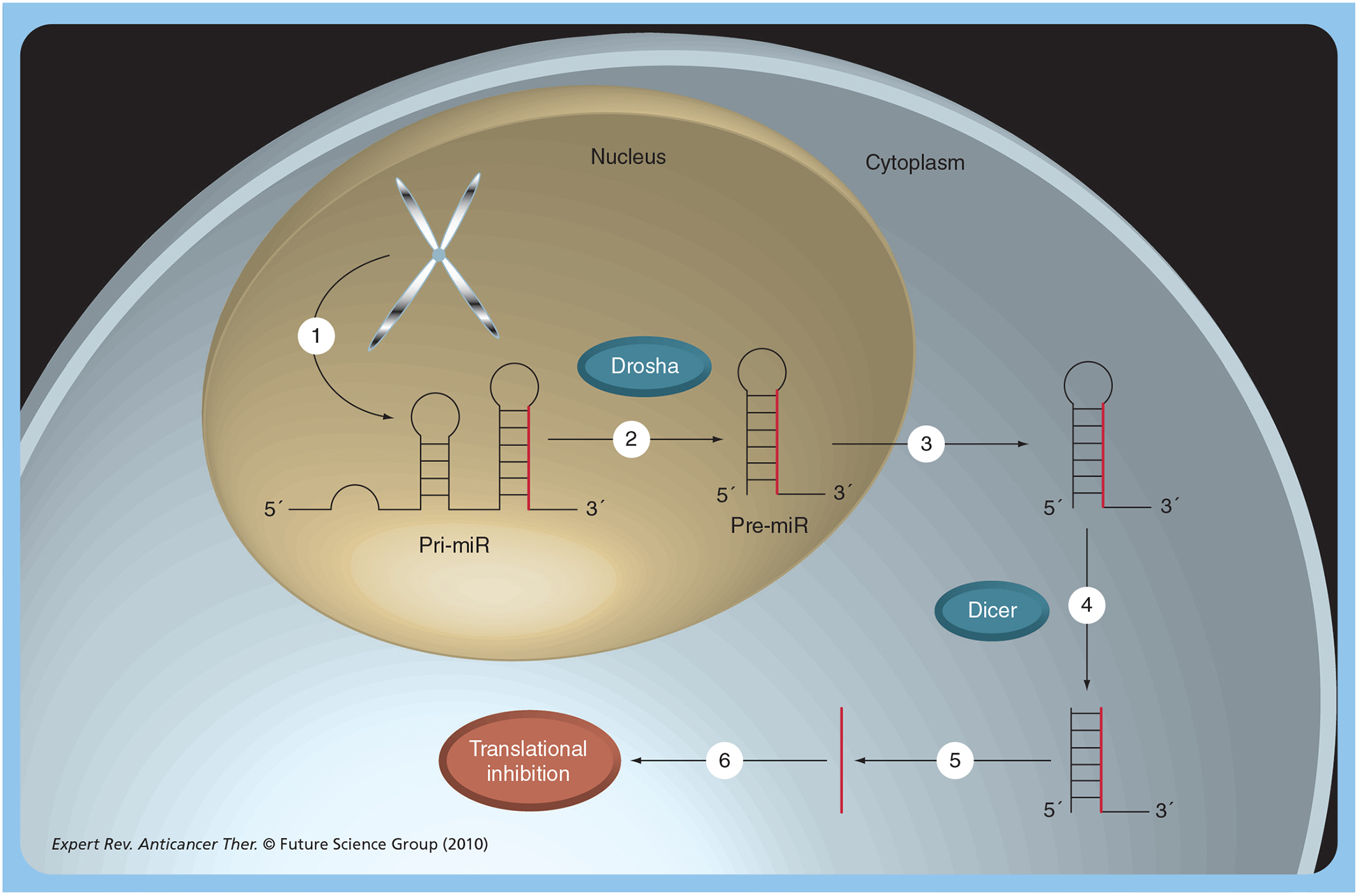
(1) MiRNAs are synthesized in the nucleus by transcription to form the pri-miR transcript. (2) This is processed within the nucleus by a complex containing Drosha to form the hairpin pre-miR. (3) The pre-miR is exported to the cytoplasm by a mechanism involving exportin and the Ran5 GTPase. (4) The pre-miR is further processed, by Dicer, to form the single-stranded pre-miR. This is loaded into an argonaute protein complex known as the RNA-induced silencing complex (RISC). (5) The RISC is guided to target mRNAs due to complementarity with the miRNA. This causes repression of transcription, probably by multiple mechanisms. Pre-miR: miRNA precursor; Pri-miR: Primary miRNA.
miRNAs in GBM
Microarray profiling techniques have identified dozens of miRNAs whose expression is significantly altered in gliomas versus normal brain tissue [11]. Functional data indicate that some of these miRNAs seem to act as oncogenes or ‘oncomirs’ and are expressed at high levels in glioma cells. Other miRNAs are expressed comparatively weakly in glioma cells and may have a tumor suppressor function. It should be noted that the effects of miRNAs are likely the result of modulating effects on multiple different pathways. In fact, miRNA alterations have been shown to have an impact on many key pathways in glioma, as shown in Figure 2. Functional studies of miRNAs in glioblastoma are described later and summarized in Table 1.
Figure 2. miRNA involvement in glioma signaling pathways.
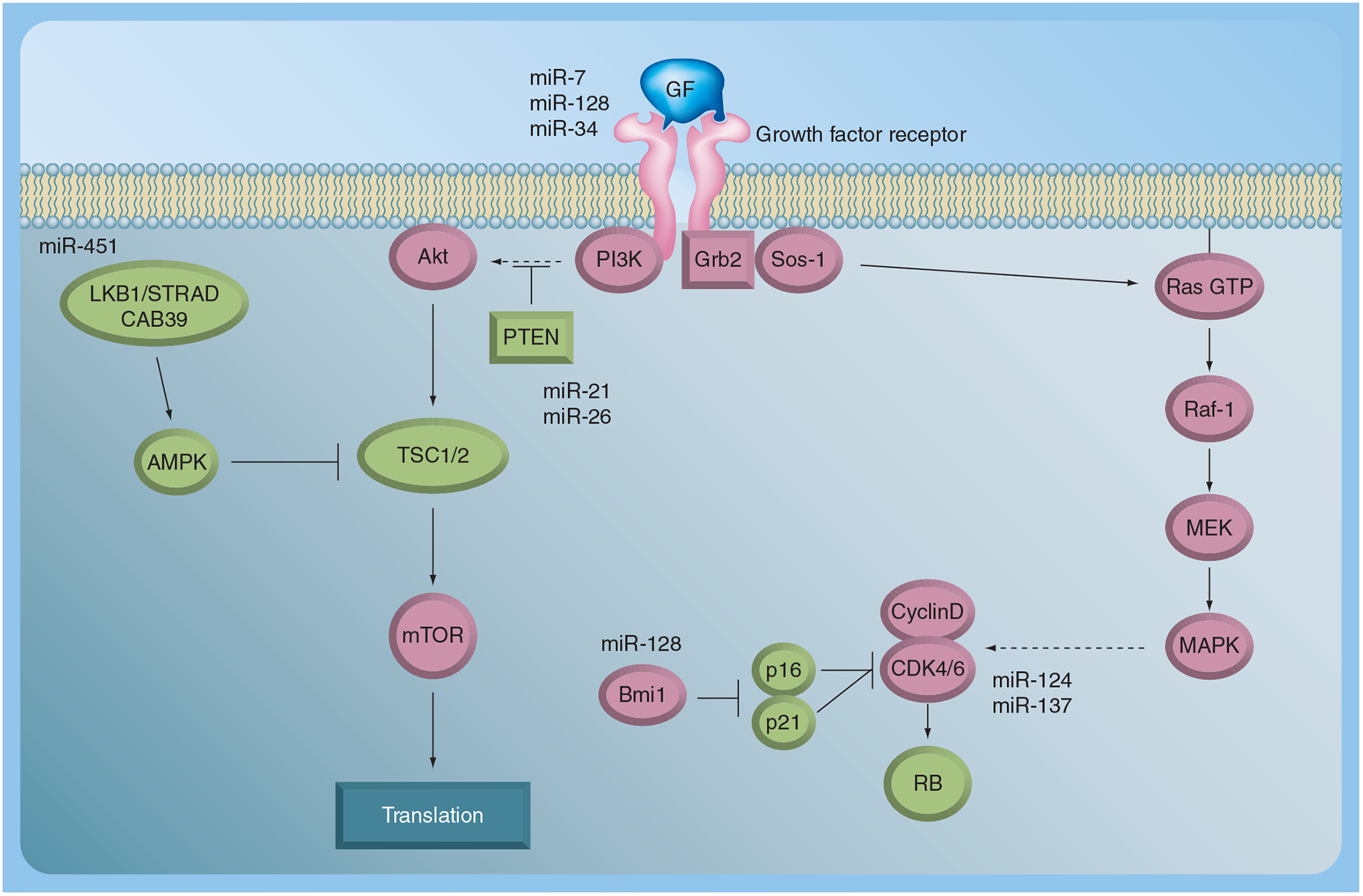
Growth factor tyrosine kinase-mediated signaling is shown. The potential interactions of miRNAs implicated in glioblastoma with these pathways are shown. Tumor suppressors are in green, oncogenes in pink.
Table 1.
miRNA alterations in glioblastoma multiforme with potential targets and functional implications.
| miRNA | Expression in glioblastoma multiforme | Phenotype | Targets |
|---|---|---|---|
| miR-7 | Decreased | Increases apoptosis Decreases invasion | EGFR |
| miR-15 | Increased | Regulates cell cycle progression | CCNE1 |
| miR-21 | Increased | Oncomir, antiapoptosis | RECK, PDCD4, PTEN |
| miR-26 | Increased | Induces tumor growth, part of oncomir/oncogene cluster with CDK4 and CENTG1 | PTEN PI3K/Akt pathway |
| miR-34 | Decreased | Inhibits proliferation, survival, migration and invasion | TP53, c-Met Notch-1/2 |
| miR-124 | Decreased | Inhibits proliferation, cell differentiation | CDK6, PTBP, SCP1 |
| miR-125 | Increased | Induces proliferation, inhibited apoptosis | ERBB2, ERBB3, TP53 |
| miR-128 | Decreased | Inhibits proliferation | BMI1, E2F3a, EGFR |
| miR-137 | Decreased | Inhibits proliferation, cell differentiation | CDK6 |
| miR-146 | Decreased | Inhibits migration | MMP16 |
| miR-181 | Decreased | Reduced colony formation and migration. | Tcl1 |
| miR-221 | Increased | Cell proliferation | p27Kip1, p57Kip2 |
| miR-222 | Increased | Cell proliferation | p27Kip1, p57Kip2 |
| miR-296 | Increased | Induces neovascularization | HGS |
| miR-326 | Decreased | Decreases cell viability and invasion | Notch-1/2 |
| miR-451 | Increased | Inhibits migration, induces proliferation | CAB39 |
EGFR: EGF receptor; MMP: Matrix metalloproteinase.
The experimental studies described in this section often follow a similar approach: identification of candidate miRNAs, and identification of targets and cellular phenotypes induced by altering the expression of that miRNA, using either miRNA restoration for miRNAs that are weakly expressed, or miRNA antagonists - antagomirs - to block the expression of highly expressed miRNAs. The most commonly used screening tools involve microarray chips (e.g., [11]), of which many are commercially available, or quantitative TaqMan®-based RT-PCR (Roche, Basel, Switzerland). Several studies have used these approaches in order to observe differential expression between tumors, and between tumors and normal brain tissue. Another approach that has been employed is to identify miRNAs present in commonly altered regions in tumors. A final approach to identify miRNAs for further study is to start at a gene of interest and work backwards. This involves the use of target prediction algorithms [101,102], which allow the rapid identification of potential miRNA target sites in the 3′-UTRs of specific genes. This was carried out in the miR-7 study, in which the EGFR 3′-UTR was analyzed for potential miRNA binding sites [12].
Once a candidate miRNA has been identified, it is important to assess the phenotype that arises when that miRNA is altered in an experimental model. The majority of studies have examined common tumor-associated phenotypes such as proliferation, apoptosis and migration in transfected cell lines. Some more specific studies have analyzed stem cell-like behavior and angiogenesis. If a phenotype and a particular miRNA/target pair make biological sense, then most studies have gone on to show that the predicted target site is active using 3′-UTR reporter constructs, in which the miRNA is examined in cell culture for its ability to alter luciferase expression using wild-type and mutant constructs where the predicted target site is ablated. The main limitation of the studies carried out so far is that they have focused on very specific target and miRNA pairs. It is clear that a single miRNA can have effects on multiple (hundreds and perhaps thousands of) genes, so the phenotypes are likely to be the result of combined effects on multiple pathways. This makes pathway-specific markers such as phospho-specific antibodies useful tools to assess more general effects. Many of the miRNA/target pairs identified to date may be valid, but there are occasions when hundreds of more stable predicted interacting pairs score higher than those reported, and there are some cases of ‘cherry picking’ in the literature. It is recommended that all studies should include a detailed and rigorous assessment of the importance of any miRNA/target pairs identified. This can be through rescue experiments, the use of multiple downstream readouts, or correlation of expression in human patient samples. Ultimately high-throughput proteomics and gene expression microarray studies could provide a more comprehensive picture, although as yet these have been seldom reported.
miR‑7
miR-7 was shown to regulate expression of EGFR and activity of the Akt pathway [12,13]. Three distinct miR-7 binding sites were detected in the 3′-UTR of EGFR, and subsequently it was confirmed that EGFR is directly targeted by miR-7 through these binding sites, and can be effectively knocked down by transfection of miR-7 [12]. An additional finding of this study was that miR-7 caused downregulation of Akt-activating phosphorylation, but that a specific siRNA against EGFR did not. This suggests that in the cell lines studied, other genes could be responsible for Akt activation rather than EGFR. Further investigation detected multiple miR-7 binding sites in the 3′-UTR of IRS1 and IRS2, which are regulators of the Akt pathway. miR-7 was shown to directly target IRS1 and IRS2 independent of its ability to inhibit EGFR [11]. MiR-7 was also found to indirectly attenuate activation of Akt and ERK1/2 [12]. This demonstrates that miRNAs have the ability to target multiple components of similar pathways, which may make them powerful from a therapeutic standpoint.
Expression of miR-7 is reduced in glioblastoma cell lines [13,14] and primary tissue samples [12]. This may be due at least in part to processing effects at the pri-miRNA to pre-miRNA processing step mediated by Drosha in the nucleus. Levels of the pri-miR-7 isoforms were similar in matched GBM and normal brain tissues, whereas levels of pre-miR-7s were decreased in GBM samples. Transfection of established GBM cells lines with pre-miR-7 leads to an increase in apoptosis. Furthermore, pre-miR-7 transfection decreased in vitro invasion of the GBM cells [12].
miR‑15
Overexpression of miR-15b was identified in glioma tissue samples compared with normal brain. Subsequent transfection studies showed that miR-15b was able to regulate cell cycle progression in glioma cells, which could be explained by its direct targeting of the cell cycle-related molecule CCNE1 (encoding cyclin E1) [15]. More work is needed to further evaluate the significance of these findings.
miR‑21
miR-21 overexpression was first described in GBM where all studied cell lines expressed high levels [16]. Moreover, miR-21 is highly abundant in GBM in terms of differential expression between tumor tissue and normal brain tissue [16,17]. Inhibition of miR-21 with locked nucleic acid (LNA) or 2′-O-methyl-miR antisense oligonucleotides led to an increase in caspase-3-dependent apoptosis, suggesting that miR-21 acts as an oncogene in GBM by suppressing apoptosis [16]. Furthermore, LNA–anti-miR-21-transfected U87 GBM cell lines showed significantly reduced intracranial tumor formation in nude mice [18]. The same group also demonstrated that miR-21 inhibition sensitized GBM cell lines to the short isoform of S-TRAIL. S-TRAIL selectively induces apoptosis in GBM by binding to death domain-linked receptors [19].
Since its first description, multiple studies have been carried out with the aim of identifying miR-21 target transcripts. One such study demonstrated that miR-21 targets multiple important components of the p53, TGF-β and mitochondrial apoptosis tumor-suppressive pathways [20]. These findings indicate that miR-21 negatively regulates multiple tumor-suppressive networks in GBM, thereby promoting oncogenic transformation in glioma precursor cells [20].
In addition to apoptosis- and cell cycle-related genes, a number of important extracellular matrix remodeling factors that were predicted miR-21 targets were identified. RECK, a major inhibitor of matrix metalloproteinases, was identified as a direct target of miR-21 [21]. PDCD4 has also been identified as a direct target of miR-21 in GBM cells [22]. Thus, high miR-21 expression in glioma cells simultaneously suppresses the expression of multiple tumor-suppressor genes, thereby promoting tumor growth, survival and invasion. It is of interest that in other tumor types, miR-21 has also been shown to target the major tumor suppressor PTEN, although this has yet to be confirmed in glioma cells.
It is apparent from the studies highlighted earlier that overexpression of miR-21 in high-grade gliomas can result in the activation of multiple oncogenic pathways. This increasing evidence supports targeting of miR-21 as a candidate approach for GBM treatment.
miR‑26
Recently, miR-26a was found to directly target the PTEN tumor suppressor in GBM samples [23]. It was also found that miR-26a is only overexpressed in a subset of GBMs, and further analyses demonstrated that miR-26a overexpression is mediated by an increase in copy number at the miR-26a-2 genomic region and is associated with monoallelic PTEN loss, suggesting that amplification of miR-26a serves to silence residual PTEN transcripts. miR-26a-mediated PTEN depression also precluded loss of heterozygosity at the PTEN locus in a murine glioma model. The repression of PTEN by miR-26a provides an intriguing link to the oncogenic Akt pathway, and strengthens the case for therapeutic inhibition of Akt signaling in glioma treatment.
A second group was also able to provide evidence that miR-26a amplification provides a significant growth advantage in gliomas [24]. They studied one of the most commonly amplified genomic regions in GBM on chromosome 12q13.3–14.1. Remarkably, this genomic region encodes for miR-26a as well as for the oncogenes CDK4 and CENTG1. They demonstrated that overexpression of miR-26a alone induces tumor growth. However, more importantly, they were able to show that over-expression of the combination of miR-26a with these genes collaboratively enhances cell growth. Taken together, these observations strongly suggest the presence of a functionally integrated oncomir/oncogene cluster that is frequently amplified in GBM.
Functional analysis of miR-26a demonstrated a coordinated antagonism of the JNK and RB1 pathways, and the activation of the PI3K/Akt pathway. These latter two pathways are among the most frequently altered in GBM. Importantly, amplification of this oncomir/oncogene cluster is associated with a particularly poor prognosis in GBM patients.
miR‑34
The miRNA-34 family has been shown to directly interact with TP53, which is one of the most commonly mutated tumor-suppressor genes in cancers. p53 directly activates the transcription of the miR-34 family, which themselves suppress cell proliferation by direct interaction with the p53 pathway [25].
More recent studies showed that miR-34a is downregulated in high-grade gliomas [26]. The same group screened multiple potential miR-34a targets and demonstrated that miR-34a inhibits GBM cell proliferation, survival, migration and invasion in vitro and in vivo. Their research also suggests that miR-34a may directly target c-Met, Notch-1 and Notch-2, making miR-34a a potential tumor suppressor in malignant gliomas. Further studies on miR-34a effects on glioma stem cells showed that miR-34a induces glioma stem cell differentiation [27]. All these findings make miR-34a an attractive candidate for glioma treatment.
miR‑124 & miR‑137
miR-124 and miR-137 alterations were discovered through screening of glioma samples, which showed that these miRNAs were downregulated in high-grade gliomas compared with normal brain tissues. Further tissue screening revealed that the same miRNAs are upregulated during adult neural stem cell neurogenesis induced by growth factor deprivation in vitro, and that overexpression of miR-124 and miR-137 can induce neuron-like differentiation of glioma stem cells in vitro [28].
CDK6, which plays an important role in the G1/S-phase transition, was identified as one of the targets for miR-137 and miR-124 [28,29]. Other genes involved in neurogenesis were also found to be inhibited by miR-124, including PTBP1, which regulates pre-mRNA splicing, resulting in a neuronal-specific alternative splicing pattern [30]. miR-124 also inhibits the expression of SCP1, an antineural factor expressed in non-neuronal cells [31]. miR-124a expression itself is partially regulated by REST. REST inhibits miR-124a expression in non-neuronal cells and neural progenitors, enabling their differentiation into non-neuronal cells. However, as neuronal progenitors mature into neurons, REST leaves the miR-124a promoter regions, and non-neuronal transcripts are degraded [32]. Further studies are needed to evaluate the therapeutic potential of miR124/137. These studies highlight the potential link between GBM and the suppression of differentiation programs that would normally lead to the formation of fully differentiated nonproliferating and nontumorigenic cells.
miR‑125
miR-125 was found at high levels in differentiated cells of the adult CNS, including neurons and astrocytes [33]. miR-125 was also found to be upregulated during neuronal differentiation of P19 embryonal carcinomas [34], and is highly expressed in oligodendroglial tumors [35]. These findings prompted investigations of miR-125 function in high-grade gliomas, which showed that overexpression of miR-125b promoted GBM cell-line proliferation and inhibited apoptosis [36].
miR‑128
Recent data draws close parallels between GBM and the biology of stem and progenitor cells, suggesting novel avenues for research and therapeutic targets [37]. A striking theme in recent literature is the increasing number of reports showing that miRNAs play an important role in the regulation of stem cell biology, differentiation and cell ‘identity’ (see discussion of miR-124) [38]. Another study showed that miR-128 was significantly downregulated in GBM samples compared with normal brain tissue [17]. Overexpression of miR-128 in GBM cells resulted in reduced cell proliferation in vitro and in vivo. The TargetScan miRNA target prediction algorithm for miR-128 [101] identified multiple potential direct targets including Bmi-1, an oncogene that has been linked to various cancers including GBM [39].
Bmi-1 is well established as a stem cell-renewal factor [40]. Like other proteins involved in stem cell renewal in embryonic and adult stem cells, Bmi-1 also plays a role in carcinogenesis. This is probably due to its ability to confer self-renewal capacity to tumor stem cells [41,42]. Bmi-1 is a member of a family of genes involved in transcriptional silencing known as polycomb genes. These have an essential role in embryogenesis, regulation of the cell cycle, and lymphopoiesis by acting as transcriptional repressors that are essential for the silencing of other families of genes. Bmi-1 acts as part of the polycomb repressor complex (PRC1), which plays a role in epigenetic gene silencing by modifying chromatin structure [42]. It has recently been shown that Bmi-1 plays an important oncogenic role in glioma formation in a mouse model [39], and also that it has a key role in stem cell renewal, including neuronal stem cells, through the repression of genes associated with differentiation and senescence such as p16INK4A and p21WAF1 [40,43].
Analysis of miR-128-transduced GBM cells showed significant Bmi-1 knockdown, and GBM-derived stem cell cultures demonstrated a marked reduction in neurosphere formation [17]. These findings were highly suggestive for a block in glioma stem cell self-renewal by inhibition of the Bmi-1 pathway. These data also suggest that repressed miR-128 expression may contribute to an increased expression of Bmi-1, promoting glioma cell proliferation. However, the effects of miR-128 are likely the sum of effects on multiple genes such as EGFR [44] and E2F3a, which has also been shown to be targeted by miR-128 in GBM [45]. In addition, there are further very high-scoring TargetScan targets that have not yet been studied, and whose function in glioma is not yet known.
miR‑146
Recent findings show that miR-146b expression is downregulated in GBM samples. In vitro studies using miR-146b overexpression or knockdown in glioma cell lines had no effect on cell proliferation. However, overexpression demonstrated reduced cell migration, knockdown of miR-146b promoted cell migration in vitro and matrix metalloproteinase-16 was identified as a direct target of miR-146b [46]. Further studies are needed to evaluate the significance of miR-146 on cell migration in vivo.
miR‑181
Among the downregulated miRNAs in GBM samples is the group of miR-181a–c [11,47]. GBM cell lines transduced to overexpress miR-181a or miR-181b showed a reduction in cell numbers, as well as reduced colony formation and in vitro migration. Furthermore, a recent study found a significant correlation between downregulation of miR-181b/c and temozolomide chemo therapy responders [47]. However, the molecular basis for these findings are unknown. Interestingly, miR-181 was identified as a tumor suppressor in glioma by bioinformatics, using PCR-based miRNA expression profiling along with gene-expression profiling in 12 patient samples [48]. Correlation of miRNA and gene expression led to the identification and validation of a tumor-suppressive function for miR-181c.
miR‑221 & miR‑222
miR-221 and miR-222 belong to a group of upregulated miRNAs in GBM and target the expression of the tumor suppressor p27Kip1 [49]. The p27Kip1 protein is a CDK inhibitor, and is frequently inactivated in gliomas where expression is inversely correlated with patient prognosis [50]. Inhibition of miR-221 and miR-222 induced a growth reduction of GBM cells, and expression of miR-221/222 in U87 GBM cells was required to maintain cell proliferation [49]. These findings were complemented by a study showing that miR-221 and miR-222 not only target p27Kip1 but also the related p57Kip2. Furthermore, functional studies demonstrated that miR-221/222 overexpression is not sufficient for uncontrolled cell proliferation [51].
miR‑296
A recently described group of miRNAs modulating angiogenesis known as angiomirs have also shown a strong correlation with high-grade gliomas [52]; specifically, miR-296, which seems to play a significant role in neovascularization, possibly by inducing overexpression of growth factor receptors on endothelial cells. The same group also showed that inhibition of miR-296 resulted in reduced tumor angiogenesis in vitro and in vivo. Furthermore, it was shown that glioma cells and angiogenic growth factors (VEGF and EGF) induce miR-296 expression in human brain microvascular endothelial cells, as well as in glioma endo thelial cells isolated from surgical specimens [52]. Finally, miR-296 knockdown resulted in inhibition of angiogenesis and endothelial cell migration in vitro and in vivo. The results of this study, with observations that miR-296 is expressed at higher levels in blood vessels derived from GBMs relative to blood vessels from normal brain tissue, indicate that miR-296 is an important regulator of glioma angiogenesis.
miR‑326
miR-326 was first identified among a set of miRNAs with high expression in neurons [53]. However, miR-326 was found to be downregulated in GBM samples, and only recently it was shown that miR-326 inhibits the Notch pathway and is in turn inhibited by Notch [54]. Notch is critical in stem cell maintenance and cell survival, and it is instrumental for cell fate diversification during neural development [55]. The connection of miR-326 to Notch was made after the observation that miR-326 decreased cell viability and invasion in glioma cell lines. Furthermore, transfection of glioma cells with miR-326 reduced their tumorigenicity. Functional target ana lysis demonstrated that miR-326 down-regulates both Notch-1 and Notch-2. Notch inhibition seems to be an essential component in the cytotoxic effects of miR-326 on glioma cells, since restoration of Notch expression partially but significantly ‘rescues’ the phenotypic effects of miR-326. Inhibition of Notch by miR-326 may also help explain its cytotoxicity to stem-like glioma cells.
Notch and miR-326 suppress each other, and in GBM this axis is shifted toward high Notch and low miR-326 activity. Reversing this axis through miR-326 delivery provides a potential new therapeutic rationale.
miR‑451
In order to identify miRNAs involved in glioma cell migration, a 3D glioma spheroid migration model was used. Alterations of previously described miRNAs with cell migration were observed, including upregulation of miR-21 [21] and downregulation of miR-126 [56]. However, the most striking change observed was the downregulation of miR-451 in migrating cells. Statistical analysis looking at the correlation between miR-451 and patient survival demonstrated a significant reduction in survival in patients expressing high levels of miR-451 [57].
To further examine the effects of miR-451 on cell migration, glioma cells were stably transfected with pre-miR-451. This reduced glioma cell migration significantly, and at the same time a change in cell morphology was noted with extensive retraction of the long, slender protrusions that characterize wild-type tumor cells. Remarkably, further examination of miR-451-transfected glioma cells demonstrated an increase in cell proliferation [57].
The search for potential miR-451 targets revealed 14 potential candidates. The putative target CAB39 was of particular interest owing to its known function in LKB1/AMPK regulation. CAB39 mRNA and protein levels were reduced in cells overexpressing miR-451 and inhibition of miR-451 resulted in increased CAB39 levels. Functional assays also demonstrated a direct interaction of miR-451 with the CAB39 3′-UTR site. Further investigations analyzing the effects of miR-451 on the LKB1/AMPK pathway showed that miR-451 regulates LKB1 activity through direct targeting of CAB39, resulting in disruption of LKB1 signaling, which leads to reduced AMPK phosphorylation and to impaired cell survival in response to glucose withdrawal. Thus, when glucose is sufficient, miR-451 seems to act as an oncogene by suppressing the LKB1/AMPK pathway. All these findings indicate that miR-451 may act as a switch that would regulate the balance of migration and proliferation in response to extracellular stimuli. However, it is not known how glucose acts to reduce miR-451 levels.
Expert commentary
It is becoming increasingly clear that miRNAs are important regulators of many of the key pathways implicated in tumor pathogenesis, and the increasing evidence that miRNA expression is altered in high-grade gliomas and its involvement in tumorigenesis provides the rationale for using miRNAs as potential therapeutic agents as well as diagnostic and prognostic markers.
In order to utilize miRNA for therapeutic purposes, one needs to fully understand their functional involvement in tumor pathology and whether they act in the manner of an oncomir or a tumor suppressor. This approach not only requires detailed understanding of the molecular pathways affected by miRNAs, but also the development of therapeutic agents that specifically alter these genes and pathways.
At present, not all altered miRNAs in GBM have been studied from a functional perspective. Moreover, studies performed thus far are selective in the kinds of assays performed. The field would benefit from comprehensive comparative studies in which multiple miRNAs or antagonists were examined in parallel for phenotypic effects in relevant assays. These would include proliferation, tumorigenicity, cell cycle, cell survival/apoptosis, cell migration/invasion, radio- and chemosensitization, angiogenesis, and effects under conditions of low oxygen and low glucose. This would allow the most promising therapeutic candidates to be taken forward for further screening in preclinical models.
Five-year view
miRNA-based gene therapy offers the theoretical appeal of targeting multiple gene networks that are controlled by a single miRNA [58]. Inhibition of miRNA expression can be achieved using antisense oligonucleotides (‘antagomirs’), or their chemically modified versions, such as LNAs, which allow more stable and specific interactions with targets [59]. Restoring miRNA expression can be achieved by introducing pre-miRs through various vectors, which could be either viral or nanoparticle based. The issue of delivery of small RNA-based oligonucleotides to humans remains a challenge, particularly in the CNS. There has been much interest in this area, which is largely driven by an interest in RNAi as a therapeutic tool. The kinds of approaches and challenges of RNAi delivery apply equally to miRNAs due to their similar chemical nature. These challenges include the develop ment of tumor-specific delivery systems, the selection of optimal miRNAs and combinations, as well as optimization of treatment dose and administration schedules, and evaluation of the efficacy of combination treatment strategies. The safety of miRNA-based therapies and potential for miRNA-specific resistance mechanisms (e.g., altered processing) has not yet been addressed. However, the preclinical studies mentioned later show promise.
A recent study showed that a therapeutic miRNA delivered by adeno-associated virus could have dramatic effects on tumor growth in an animal model [60]. In this case, the model was hepato-cellular carcinoma, and the miRNA expressed by the virus was miR-26a. Therapeutic miRNA delivery has also been shown to suppress prostate cancer metastasis to bone using systemic injection of miR-16 in a nonviral delivery system [61]. Thus, proof of principle of therapeutic miRNA delivery now exists. There is no reason similar studies cannot be performed in glioblastoma models, using injection of a tumor-targeting viral vector carrying a candidate therapeutic miRNA. A wide variety of approaches are in development for glioblastoma [62], and a TGF-β2 antisense approach is being tested in a clinical trial using convection-enhanced delivery [63]. For brain tumors, the most obvious candidates for therapeutic miRNA delivery based on current data would include restoration of miR-124, miR-7 and miR-128, all of which have potent effects on glioma tumorigenicity. These could be administered both singly or in combination. The potential therapeutic applications are shown in Figure 3. A great advantage of miRNA-based therapies are that multiple genes and pathways are targeted by a single molecule, and also that the small amount of sequence required to express a miRNA (~100 bp) would allow multiple miRNAs to be expressed on the same vector, or be coadministered with another type of gene or shRNA. Based on fold-change of expression and effects on cell survival and migration, miR-21 would be an excellent candidate for miRNA knockdown therapy using antagomirs.
Figure 3. miRNAs and the hallmarks of glioblastoma.
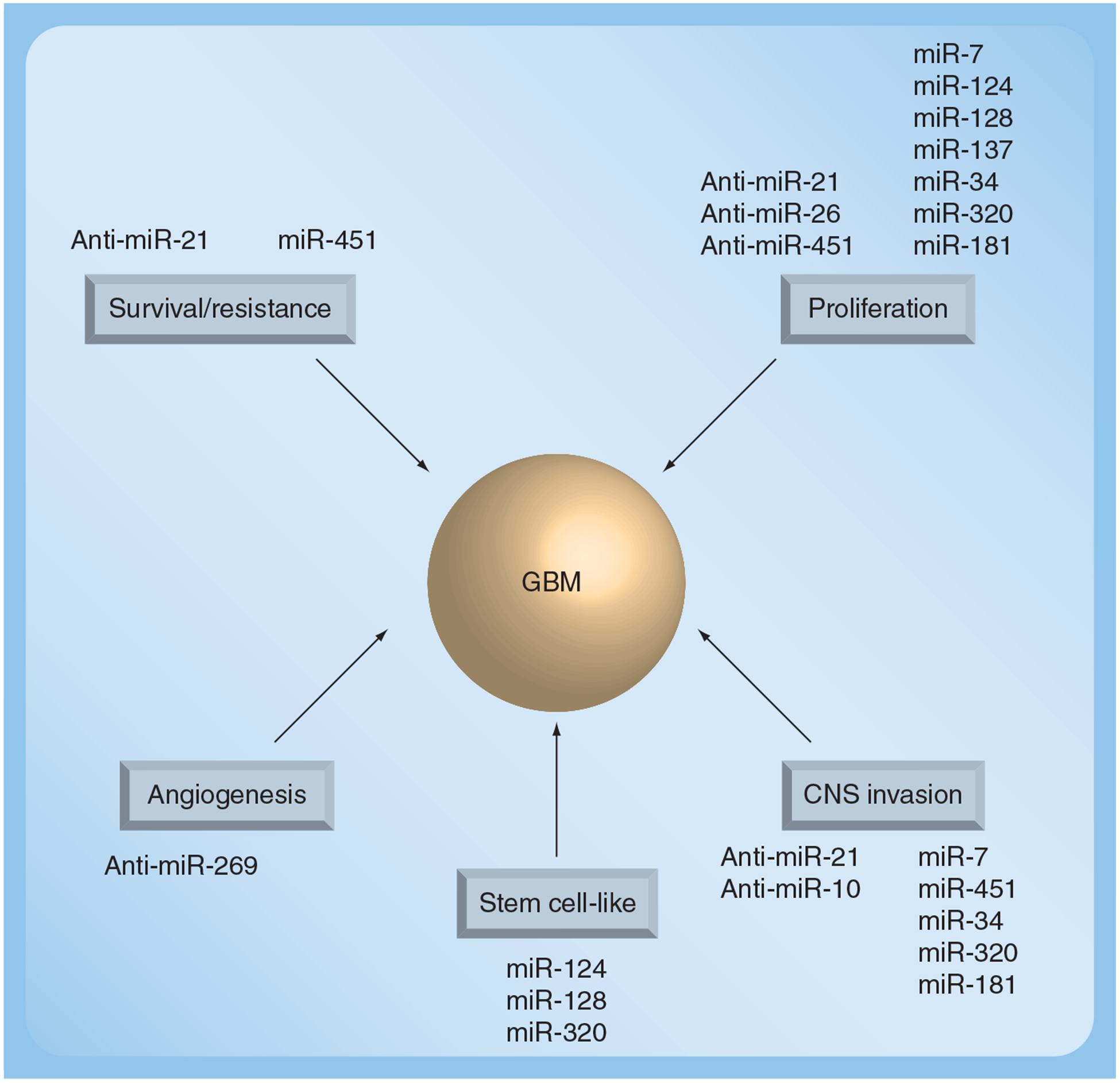
This figure shows which miRNAs have been shown to affect various biological processes linked to glioblastoma. The potential therapeutic approach is shown in terms of whether miRNA restoration or anti-miR treatment would be the appropriate approach to target each process.
GBM: Glioblastoma multiforme.
There are also some examples in which specific miRNA binding sites have been incorporated into gene therapy vectors, and oncolytic viruses, in order to improve tumor targeting. For example, incorporating a miRNA binding site in the 3′-UTR of an essential viral gene for a miRNA expressed in normal tissue surrounding the tumor, but not expressed by the tumor, will increase the tumor tropism of the virus [64].
The completed research on miRNA in gliomas has offered some promising results. However, the results need to prove safe and efficacious in clinical trials in order to live up to the expectations created by experimental data. At present, no miRNA-based human trial for brain tumors is on its way. However, the time of miRNA therapeutics is approaching, and we are looking forward to clinical trials elucidating their potential and benefits in the clinic.
Key issues.
Characteristic miRNA alterations have been characterized in patient glioblastomas. However, at present, knowledge regarding miRNA expression profiles as they relate to survival and treatment response is lacking. In addition, the functional effects of altered miRNAs have not yet been comprehensively analyzed. This information would be useful in selecting appropriate miRNA-based therapeutic approaches that may be tailored to specific tumor genetic backgrounds.
miRNAs play key roles in glioblastoma biology. All the major hallmarks of the disease can be alleviated - at least in vitro - by modification of cellular miRNA levels.
Their small size and important biological functions suggest that a new era of miRNA-based therapeutics is on the way. These therapeutics may involve miRNA re-expression, or the use of antagomir oligonucleotides, to prevent the action of oncogenic miRNAs in cancer cells. In addition, the use of multiple miRNAs, or antagomirs, miRNA/antagomir combinations, or combinations with other therapeutic approaches may be the most effective.
Footnotes
Financial & competing interests disclosure
The authors would like to thank the Esther L Dardinger Endowment for Neuro-oncology and Neurosciences, the Jeffrey Thomas Hayden Foundation, the Neurosurgery Research and Education Foundation (NREF), and the NIH (RO1 CA136803) for their support. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Websites
101 TargetScanHuman www.targetscan.org
102 miRBase www.mirbase.org
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Stupp R, Mason W, van den Bent M et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med 352(10), 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Fulda S, Wick W, Weller M, Debatin K. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med 8(8), 808–815 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Zhu Y, Parada L. The molecular and genetic basis of neurological tumours. Nat. Rev. Cancer 2(8), 616–626 (2002). [DOI] [PubMed] [Google Scholar]
- 4.•Esquela-Kerscher A, Slack F. Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer 6(4), 259–269 (2006). [DOI] [PubMed] [Google Scholar]; Nice review of the roles of miRNAs in cancer.
- 5.Bartel D MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2), 281–297 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Hobert O Gene regulation by transcription factors and microRNAs. Science 319(5871), 1785–1786 (2008). [DOI] [PubMed] [Google Scholar]
- 7.••Bartel D MicroRNAs: targeting and regulatory functions. Cell 136(2), 215–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; Review of more advanced concepts in the miRNA field. Examines more detailed aspects of targeting mechanisms.
- 8.Tay Y, Zhang J, Thomson A, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 455(7216), 1124–1128 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Lytle J, Yario T, Steitz J. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl Acad. Sci. USA 104(23), 9667–9672 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature 455(7209), 58–63 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Ciafre S, Galardi S, Mangiola A et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem. Biophys. Res. Commun 334(4), 1351–1358 (2005). [DOI] [PubMed] [Google Scholar]
- 12.•Kefas B, Godlewski J, Comeau L et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 68(10), 3566–3572 (2008). [DOI] [PubMed] [Google Scholar]; One of the first functional studies of miRNA involvement in glioblastoma multiforme. Nice story develops from identification of EGF receptor as a target, to downregulation in tumor cells, and then further targets based on functional observations.
- 13.Webster RJ, Giles KM, Price KJ et al. Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J. Biol. Chem 284(9), 5731–5741 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Gaur A, Jewell DA, Liang Y et al. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 67(6), 2456–2468 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Xia H, Qi Y, Ng SS et al. MicroRNA-15b regulates cell cycle progression by targeting cyclins in glioma cells. Biochem. Biophys. Res. Commun 380(2), 205–210 (2009). [DOI] [PubMed] [Google Scholar]
- 16.•Chan J, Krichevsky A, Kosik K. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 65(14), 6029–6033 (2005). [DOI] [PubMed] [Google Scholar]; Earliest functional study of miRNA involvement in glioblastoma multiforme, and strong evidence that miR‑21 is a major oncomir.
- 17.•Godlewski J, Nowicki M, Bronisz A et al. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 68(22), 9125–9130 (2008). [DOI] [PubMed] [Google Scholar]; Identification of a miRNA/target pair, followed by further phenotypic studies to show that miR‑128 affects cancer cell self‑renewal as predicted based on its targeting of Bmi‑1.
- 18.Corsten MF, Miranda R, Kasmieh R et al. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 67(19), 8994–9000 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Panner A, James CD, Berger MS, Pieper RO. mTOR controls FLIPS translation and TRAIL sensitivity in glioblastoma multiforme cells. Mol. Cell Biol 25(20), 8809–8823 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.••Papagiannakopoulos T, Shapiro A, Kosik KS. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 68(19), 8164–8172 (2008). [DOI] [PubMed] [Google Scholar]; A more systems‑based approach to looking at miRNA function. There may be more such studies in the future.
- 21.••Gabriely G, Wurdinger T, Kesari S et al. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol. Cell Biol 28(17), 5369–5380 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive analysis of miR‑21 targets and phenotypes. A very good study.
- 22.Chen Y, Liu W, Chao T et al. MicroRNA-21 down-regulates the expression of tumor suppressor PDCD4 in human glioblastoma cell T98G. Cancer Lett. 272(2), 197–205 (2008). [DOI] [PubMed] [Google Scholar]
- 23.•Huse JT, Brennan C, Hambardzumyan D et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 23(11), 1327–1337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; Identifies a miR‑26 involvement in glioblastoma multiforme on the basis of known genetic alterations.
- 24.Kim H, Huang W, Jiang X et al. Integrative genome analysis reveals an oncomir/ oncogene cluster regulating glioblastoma survivorship. Proc. Natl Acad. Sci. USA 107(5), 2183–2188 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He L, He X, Lim LP et al. A microRNA component of the p53 tumour suppressor network. Nature 447(7148), 1130–1134 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Guessous F, Zhang Y et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 69(19), 7569–7576 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guessous F, Zhang Y, Kofman A et al. microRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 9(6) 1031–1036 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.•Silber J, Lim D, Petritsch C et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 6, 14 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrates the role of miR‑124 and miR‑137 using glioma stem cell‑like cultures.
- 29.Lujambio A, Ropero S, Ballestar E et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 67(4), 1424–1429 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Makeyev E, Zhang J, Carrasco M, Maniatis T. The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27(3), 435–448 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Visvanathan J, Lee S, Lee B, Lee JW, Lee SK. The microRNA miR-124 antagonizes the anti-neural REST/SCP1 pathway during embryonic CNS development. Genes Dev. 21(7), 744–749 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc. Natl Acad. Sci. USA 103(7), 2422–2427 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smirnova L, Grafe A, Seiler A et al. Regulation of miRNA expression during neural cell specification. Eur. J. Neurosci 21(6), 1469–1477 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Wu L, Belasco JG. Micro-RNA regulation of the mammalian lin-28 gene during neuronal differentiation of embryonal carcinoma cells. Mol. Cell Biol 25(21), 9198–9208 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson PT, Baldwin DA, Kloosterman WP et al. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA 12(2), 187–191 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia HF, He TZ, Liu CM et al. MiR-125b expression affects the proliferation and apoptosis of human glioma cells by targeting Bmf. Cell. Physiol. Biochem 23(4–6), 347–358 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Sanai N, Alvarez-Buylla A, Berger M. Neural stem cells and the origin of gliomas. N. Engl. J. Med 353(8), 811–822 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Kloosterman W, Plasterk R. The diverse functions of microRNAs in animal development and disease. Dev. Cell 11(4), 441–450 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Bruggeman S, Hulsman D, Tanger E et al. Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell 12(4), 328–341 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Molofsky A, Pardal R, Iwashita T et al. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 425(6961), 962–967 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pardal R, Molofsky A, He S, Morrison S. Stem cell self-renewal and cancer cell proliferation are regulated by common networks that balance the activation of proto-oncogenes and tumor suppressors. Cold Spring Harb. Symp. Quant. Biol 70, 177–185 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 6(11), 846–856 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Fasano CA, Dimos JT, Ivanova NB, Lowry N, Lemischka IR, Temple S. shRNA knockdown of Bmi-1 reveals a critical role for p21–Rb pathway in NSC self-renewal during development. Cell Stem Cell 1(1), 87–99 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Weiss GJ, Bemis LT, Nakajima E et al. EGFR regulation by microRNA in lung cancer: correlation with clinical response and survival to gefitinib and EGFR expression in cell lines. Ann. Oncol 19(6), 1053–1059 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Chao T, Li R et al. MicroRNA-128 inhibits glioma cells proliferation by targeting transcription factor E2F3a. J. Mol. Med 87(1), 43–51 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Xia H, Qi Y, Ng SS et al. MicroRNA-146b inhibits glioma cell migration and invasion by targeting MMPs. Brain Res. 1269, 158–165 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Slaby O, Lakomy R, Fadrus P et al. MicroRNA-181 family predicts response to concomitant chemoradiotherapy with temozolomide in glioblastoma patients. Neoplasma 57(3), 264–269 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Liu T, Papagiannakopoulos T, Puskar K et al. Detection of a microRNA signal in an in vivo expression set of mRNAs. PLoS One 2(8), e804 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.le Sage C, Nagel R, Egan DA et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 26(15), 3699–3708 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mizumatsu S, Tamiya T, Ono Y et al. Expression of cell cycle regulator p27Kip1 is correlated with survival of patients with astrocytoma. Clin. Cancer Res 5(3), 551–557 (1999). [PubMed] [Google Scholar]
- 51.Medina R, Zaidi SK, Liu CG et al. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 68(8), 2773–2780 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wurdinger T, Tannous BA, Saydam O et al. miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell 14(5), 382–393 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim J, Krichevsky A, Grad Y et al. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc. Natl Acad. Sci. USA 101(1), 360–365 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kefas B, Comeau L, Floyd DH et al. The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J. Neurosci 29(48), 15161–15168 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cau E, Blader P. Notch activity in the nervous system: to switch or not switch? Neural Dev. 4, 36 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crawford M, Brawner E, Batte K et al. MicroRNA-126 inhibits invasion in non-small cell lung carcinoma cell lines. Biochem. Biophys. Res. Commun 373(4), 607–612 (2008). [DOI] [PubMed] [Google Scholar]
- 57.•Godlewski J, Nowicki MO, Bronisz A et al. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 37(5), 620–632 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of complex regulation of glioma growth and invasion by a single miRNA. Shows how detailed mechanistic studies of miRNAs may reveal novel aspects of the disease.
- 58.Tong AW, Nemunaitis J. Modulation of miRNA activity in human cancer: a new paradigm for cancer gene therapy? Cancer Gene Ther. 15(6), 341–355 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Davis S, Lollo B, Freier S, Esau C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 34(8), 2294–2304 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.••Kota J, Chivukula RR, O’Donnell KA et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 137(6), 1005–1017 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; Shows how a miRNA can prevent tumor formation very effectively in vivo.
- 61.Takeshita F, Patrawala L, Osaki M et al. Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol. Ther 18(1), 181–187 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawler SE, Prezzi PP, Chiocca EA. Genetic strategies for brain tumor therapy. Cancer Gene Ther. 13(3), 225–233 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Hau P, Jachimczak P, Bogdahn U. Treatment of malignant gliomas with TGF-β2 antisense oligonucleotides. Expert Rev. Anticancer Ther 9(11), 1663–1674 (2009). [DOI] [PubMed] [Google Scholar]
- 64.•• Brown BD, Naldini L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat. Rev. Genet 10(8), 578–585 (2009). [DOI] [PubMed] [Google Scholar]; Overview of the potential therapeutic impact of and strategies to exploit miRNAs therapeutically.