

Abstract
We report the enantio- and diastereoselective synthesis of 1,2-difluorides via chiral aryl iodide-catalyzed difluorination of cinnamamides. The method uses HF-pyridine as a fluoride source and mCPBA as a stoichiometric oxidant to turn over catalyst, and affords compounds containing vicinal, fluoride-bearing stereocenters. Selectivity for 1,2-difluorination versus a rearrangement pathway resulting in 1,1-difluorination is enforced through anchimeric assistance from a N-tert-butyl amide substituent.
Graphical Abstract
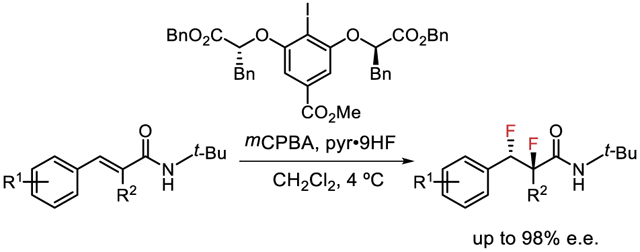
The stereocontrolled introduction of fluorine atoms into organic molecules is a long-standing challenge in synthetic chemistry driven, to a significant extent, by the beneficial properties fluorination can impart to the physical and biological properties of organic molecules.1 Due to their known preference for adopting gauche conformations, vicinal difluorides represent a particularly interesting subset of organofluorine compounds.2 The direct, enantioselective 1,2-difluorination of alkenes represents a most appealing approach to this class of compounds, but no general methods have yet been identified for accomplishing such a transformation.3 Reported examples of enantiocontrolled synthesis of vicinal difluorides most often involve deoxyfluorination of 1,2-fluoroalcohols derived from stereodefined epoxides or diols.4 However, these reactions are prone to competitive elimination pathways and are often low-yielding.5 New methods for direct, enantioselective vicinal difluorination could enable a more thorough exploration of the gauche effect on molecular structure and function.
There has been remarkable progress over the past decade in the development of enantioselective alkene difunctionalization reactions using hypervalent iodine reagents and catalysts.6 In that context, the Gilmour lab and our group recently developed catalytic variants of the alkene 1,2-difluorination first reported by Hara.3n,6g-h Our system engaged HF-pyridine as a nucleophilic fluoride source and meta-chloroperbenzoic acid (mCPBA) as the stoichiometric oxidant,7 and included a single example of an enantioselective variant in the 1,2-difluorination of trisubstituted cinnamamide 2q catalyzed by chiral aryl iodide 1b (Scheme 1).8 However, in subsequent work, we found that the scope of that reaction was severely limited due to competing rearrangement pathways. Here we address that selectivity challenge through a systematic study of the factors influencing product distribution, leading to the development of a protocol for the highly chemo- and enantioselective 1,2-difluorination of trisubstituted cinnamamide substrates. These reactions provide versatile synthetic building blocks bearing contiguous secondary and tertiary fluorine-bearing stereocenters. Concurrent with our efforts, Gilmour and coworkers reported a complementary method for the enantioselective 1,2-difluorination of simples, electron-deficient styrenes.9
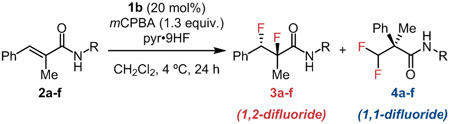
Scheme 1.

Product Selectivity in Aryl Iodide-Catalyzed Difluorination of Cinnamamides
Styrenyl substrates are susceptible to rearrangement pathways under electrophilic fluorination conditions, thereby affording 1,1-difluorinated products.10,11 For example, in the attempted difluorination of trisubstituted cinnamamide 2a promoted by aryl iodide 1b, a mixture of 1,2- and 1,1-difluoride products was obtained unselectively (Scheme 1A). Product partitioning is proposed to arise from the initial fluoroiodination adduct A, which can undergo aryl iodide displacement either by the amide carbonyl oxygen or by the aryl group (Scheme 1B).6g,6j The basis for enantioinduction is likely common to both pathways, and was explored computationally in a recent collaborative study.11e We hypothesized that the amide anchimeric assistance pathway leading to the 1,2-product might be enhanced through judicious introduction of N-substituents, since substitution has been demonstrated to lower the strain energy in small rings in specific cases.12
We evaluated a series of N-substituted amides as model substrates for the enantioselective 1,2-difluorination reaction with catalyst 1b (Table 1). While tertiary amide derivatives of 2 displayed poor reactivity, secondary amides underwent reaction more efficiently than the primary amide 2a. Thus, the difluorination of N-methyl amide 2b (entry 2) proceeded with improved yield and enantioselectivity, although without any change in product ratio. Decreasing the HF-pyridine concentration led to a modest improvement in selectivity for the 1,2-product 3b, with optimal yields obtained using 5.6 equivalents (entry 3). The dependence of product ratio on HF-pyridine loading might be attributable to attenuation of amide nucleophilicity by hydrogen bonding between the amide and HF.13 Increasing the size of the secondary amide N-substituent resulted in increased selectivity for formation of 1,2-difluoride products (entries 3–6), with the N-tert-butyl amide 2f affording the desired 1,2-difluoride 3f almost exclusively (entry 7). Notably, the reaction of 2f proceeded with significantly diminished chemoselectivity when 11 equiv. of HF-pyridine were used (entry 8).14
Table 1.
Optimization of the 1,2-Difluorination Reaction
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate | R | pyr·9HF (equiv.) |
3 : 4 | yield of 3 (%) |
e.e. of 3 (%) |
| 1a | 2a | H | 11 | 1.1 : 1.0 | 17 | 91 |
| 2a | 2b | Me | 11 | 1.1 : 1.0 | 38 | 95 |
| 3 | 2b | Me | 5.6 | 3.3 : 1.0 | 51 | 94 |
| 4 | 2c | Et | 5.6 | 4.3 : 1.0 | 61 | 95 |
| 5 | 2d | i-Pr | 5.6 | 4.8 : 1.0 | 56 | 95 |
| 6 | 2e | n-Bu | 5.6 | 6.9 : 1.0 | 63 | 94 |
| 7 | 2f | t-Bu | 5.6 | 39 : 1.0 | 81 | 96 |
| 8a | 2f | t-Bu | 11 | 5.9 : 1.0 | 65 | 96 |
Unless noted otherwise, reactions were conducted on a 1.00 mmol scale and isolated yields of 3 are listed. Reported ratios of 1,2-difluoride to 1,1-difluoride were determined by 19F NMR analysis of crude product mixtures.
Reaction conducted on 0.10 mmol scale, with yields of 1,2-difluoride determined by 1H NMR against an internal standard.
Under the optimized conditions, a variety of tert-butyl cinnamamide derivatives were found to undergo highly diastereo- and enantioselective formation of the corresponding 1,2-difluorination products (Figure 1A).15 Substrates bearing electron-withdrawing and mildly electron-donating substituents (2g–i) were particularly effective. The electron-rich cinnamamide 2j underwent reaction with only modest chemoselectivity to generate a 2.2:1.0 ratio of the desired 1,2-difluoride to the 1,1-difluoride, with the 1,2-difluoride isolated in 40% yield and 98% e.e. This result is nonetheless notable because it overturns the overwhelming selectivity for 1,1-difluoride observed for the analogous primary amide substrate (see Supporting Information). A further increase in chemoselectivity for the 1,2-product was obtained by increasing the ratio of pyridine to HF from 1:9 to 1:4.5. Although we have not performed a systematic investigation of the effect of the reaction medium on product distribution, Gilmour and coworkers have demonstrated clearly that 1,2:1,1 product ratios are dependent on amine concentrations in difluorninations of electron deficient styrenes.9 Chemoselectivity for 1,2- vs. 1,1-difluorination was observed to be correlated directly to the nucleophilicity of the arene, as evidenced by the positive linear correlation (ρ+ = 4.34) between the Hammett substituent σ+ constants and log(3:4) for substrates 2f and 2h–j (Figure 1B). The α-alkyl substituent of the cinnamamide could also be varied (Figure 1A). Substrate 2l, which bears an ethyl substituent at the α-position of the cinnamamide, undergoes 1,2-difluorination exclusively. Indene 2m, which is not susceptible to an aryl migration pathway, afforded 3m in moderate yield and enantioselectivity. Non-styrenyl unsaturated amides display poor reactivity under the reaction conditions.
Figure 1.

(A) Scope of the enantioselective 1,2-difluorination of N-tert-butyl cinnamamides. Reactions were conducted on 1.00 mmol scale with 5.6 equiv. HF. Ratios of 1,2-difluoride to 1,1-difluoride were determined by 19F NMR analysis of crude product mixtures. Isolated yields of diastereomerically pure 1,2-difluoride are reported. The relative and absolute configurations of all 1,2-difluorination products were assigned by analogy to those of 3q (ref 16) a Reaction conducted with 2.8 equiv. HF. b Reaction conducted with 2.8 equiv. HF and added pyridine (pyr:HF = 1:4.5). (B) Hammett plot of σ+ values of the aryl substituents in 2f and 2h–j versus the product ratio (log(1,2 : 1,1)) obtained for each substrate.
We sought to elucidate the basis for the significant impact of amide N-substituent on product ratio. A strong linear free-energy correlation was observed between the 1,2- vs. 1,1-product ratios and the Charton values (ν) of the amide N-substituents for 2a–f (Figure 2A), indicating that the effect is primarily steric in nature.17 Larger substituents thus appear to enhance amide anchimeric assistance relative to aryl migration, thereby favoring the 1,2-difluorination pathway.
Figure 2.

(A) Plot of Charton values (ν) for amide N-substituents of 2a–f versus the product ratio (ln(1,2 : 1,1)) obtained for each substrate. (B) Plot of amide carbonyl stretching absorptions for 2c and 2n–p versus the product ratio (ln(1,2 : 1,1)) obtained for each substrate. Reactions were conducted on a 1.00 mmol scale. Reported ratios of 1,2-difluoride to 1,1-difluoride were determined by 19F NMR analysis of the crude mixture.
The electronic effect of the amide N-substituent on the competition between the aryl migration and amide trapping pathways was probed by examining substrates bearing fluorinated N-substituents (2n–p). Substrates bearing electron-withdrawing N-alkyl substituents underwent difluorination with lower product selectivity for the 1,2-difluoride (Figure 2B, top). The experimentally measured infrared stretching frequencies of the amide carbonyls of 2c and 2n–p correlate to ln(3:4) (Figure 2B, bottom). As might be anticipated, decreased nucleophilicity of the amide oxygen disfavors anchimeric assistance relative to phenonium ion formation.
The products of the difluorination reaction can be derivatized to access versatile, enantioenriched vicinal difluoride building blocks (Figure 4). Treatment of 3f with a solution of hydrogen bromide in acetic acid resulted in efficient cleavage of the tert-butyl group to afford primary amide 3a. The arene of 3f can be degraded oxidatively to give carboxylic acid 5, thereby providing a 1,4-dicarbonyl bearing a second functional handle off the stereodefined difluoride framework.
In conclusion, we have developed a catalytic, enantioselective 1,2-difluorination of cinnamamides. The competing 1,1-difluorination resulting from phenonium rearrangement was suppressed through enhancement of anchimeric assistance by a proximal tert-butyl amide. The resulting products and their derivatives may serve as versatile building blocks for the preparation of 1,2-difluoride-containing compounds, enabling further study of this interesting motif. Efforts are underway to extend the scope of this methodology to other enantioselective fluorofunctionalization reactions.
Supplementary Material
Scheme 2.

Product Derivatization
ACKNOWLEDGMENT
This work was supported by the NIH (GM043214) and by an NSF predoctoral fellowship to S.M.B. We thank Dr. Adam Trotta for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and characterization data (PDF)
The authors declare no competing financial interests.
REFERENCES
- 1. (a).Zhou Y; Wang J Zhanni G; Wang S; Zhu W; Aceña JL Soloshonok VA; Izawa K; Liu H Review pubs.acs.org/CR Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II−III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev 2016, 116, 422. [DOI] [PubMed] [Google Scholar]; (b) Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315. [DOI] [PubMed] [Google Scholar]; (c) Wang J; Sanchez- Rosello M; Acena JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001−2011). Chem. Rev 2014, 114, 2432. [DOI] [PubMed] [Google Scholar]; (d) Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in medicinal chemistry. Chem. Soc. Rev 2008, 37, 320. [DOI] [PubMed] [Google Scholar]; (e) Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51, 4359. [DOI] [PubMed] [Google Scholar]
- 2. (a).Thiehoff C; Rey YP; Gilmour R The Fluorine Gauche Effect: A Brief History. Isr. J. Chem 2017, 57, 92. [Google Scholar]; (b) Schüler M; O’Hagan D; Slawin AMZ The vicinal F–C–C–F moiety as a tool for influencing peptide conformation. Chem. Commun 2005, 4324. [DOI] [PubMed] [Google Scholar]; (c) Yamamoto I; Jordan MJT; Gavande N; Doddareddy MR; Chebib M; Hunter L The enantiomers of syn-2,3-difluoro-4-aminobutyric acid elicit opposite responses at the GABAC receptor. Chem. Commun 2012, 48, 829. [DOI] [PubMed] [Google Scholar]; (d) Hu X-G; Thomas DS; Griffith R; Hunter L Stereoselective Fluorination Alters the Geometry of a Cyclic Peptide: Exploration of Backbone-Fluorinated Analogues of Unguisin A. Angew. Chem. Int. Ed 2014, 53, 6176. [DOI] [PubMed] [Google Scholar]; (e) Huchet QA; Kuhn B; Wagner B; Kratochwil NA; Fischer H; Kansy M; Zimmerli D; Carreira EM; Müller K Fluorination Patterning: A Study of Structural Motifs That Impact Physicochemical Properties of Relevance to Drug Discovery. J. Med. Chem 2015, 58, 9041. [DOI] [PubMed] [Google Scholar]
- 3.For racemic alkene fluorination reactions employing F2, see: (a) Merritt RF; Johnson FA Direct Fluorination. Addition of Fluorine to Indenes and Acenapthylenes. J. Org. Chem 1966, 31, 1859. [Google Scholar]; (b) Barton DHR; Lister-James J; Hesse RH; Pechet MM; Rozen S Electrophilic fluorination of some steroidal α,β-unsaturated ketones. J. Chem. Soc., Perkin Trans. 1 1982, 1105. [Google Scholar]; (c) Purrington ST; Kagen BS; Patrick TB The Application of Elemental Fluorine in Organic Synthesis. Chem. Rev 1986, 86, 997. For reactions using XeF2: [Google Scholar]; (d) Šket B; Zupan M Fluorination with xenon difluoride. Part 15. Stereochemistry of fluorine addition to acenaphthylene and dihydronaphthalenes. J. Chem. Soc., Perkin Trans. 1 1977, 2169. [Google Scholar]; (e) Shellhamer DF; Conner RJ; Richardson RE; Heasley VL; Heasley GE Fluorination of 1,3-dienes with xenon difluoride and (difluoroiodo)benzene. J. Org. Chem 1984, 49, 5015. [Google Scholar]; (f) Stavber S; Sotler T; Zupan M; Popovic A Fluorination with XeF2 Part 40 The Important Role of pi-Bond Disruption in Fluorine Addition to Phenyl-Substituted Alkenes. J. Org. Chem 1994, 59, 5891. [Google Scholar]; (g) Shellhamer DF; Chiaco MC; Gallego KM; Low WSC; Carter B; Heasley VL; Chapman RD The fluorination of cyclopentadiene and 3,4-epoxycyclopentene. J. Fluorine Chem 1995, 72, 83. [Google Scholar]; (h) Tius MA Xenon difluoride in synthesis. Tetrahedron 1995, 51, 6605. [Google Scholar]; (i) Tamura M; Takagi T; Quan H -d.; Sekiya, A. Utility of silicon tetrafluoride as a catalyst of reactions with xenon difluoride: fluorinations of phenyl alkenes and benzaldehydes. J. Fluorine Chem 1999, 98, 163. For reactions using Selectfluor: [Google Scholar]; (j) Lal GS Site-Selective Fluorination of Organic Compounds Using 1-Alkyl-4-fluoro-1,4-diazabicyclo[2.2.2]octane Salts (Selectfluor Reagents). J. Org. Chem 1993, 58, 2791 For reports of diastereoselective 1,2-difluorination using dilute F2 at low temperature, see: [Google Scholar]; (k) Rozen S; Brand M Direct Addition of Elemental Fluorine to Double Bonds. J. Org. Chem 1986, 51, 3607. [Google Scholar]; (l) Vints I; Rozen S Fluorination of Flavones and Chromones Using Elemental Fluorine. J. Org. Chem 2014, 79, 7261. [DOI] [PubMed] [Google Scholar]; (m) Vints I; Rozen S Fluorination of α,β-unsaturated carbonyl compounds using elemental fluorine. Tetrahedron 2016, 72, 632 For the seminal report of alkene difluorination reactions using ArIF2/HF, see: [Google Scholar]; (n) Hara S; Nakahigashi J; Ishi-i K; Sawaguchi M; Sakai H; Fukuhara T Yoneda N Difluorination of Alkenes with Iodotoluene Difluoride. Synlett 1998, 495. [Google Scholar]
- 4. (a).Bell HM; Hudlicky M Stereochemistry of α,α′-Difluorosuccinic Acids. J. Fluorine Chem 1980, 15, 191. [Google Scholar]; (b) Burmakov AI; Motnyak LA; Kunshenko BV; Alexeeva LA; Yagupolskii LM Treatment of Dimethyl (+)-L-tartrate with Sulfur Tetrafluoride. J. Fluorine Chem 1981, 19, 151. [Google Scholar]; (c) Hamatani T; Matsubara S; Matsuda H; Schlosser M A stereocontrolled access to vicinal difluoroalkanes. Tetrahedron 1988, 44, 2875. [Google Scholar]; (d) Marson CM; Melling RC The first enantioselective syntheses of vicinal difluoropyrrolidines and the first catalytic asymmetric synthesis mediated by the C2 symmetry of a –CHFCHF– unit. Chem. Commun 1998, 1223. [Google Scholar]; (e) O’Hagan D; Rzepa HS; Schüler M; Slawin AMZ The vicinal difluoro motif: The synthesis and conformation of erythro- and threo-diastereomers of 1,2-difluorodiphenylethanes, 2,3-difluorosuccinic acids and their derivatives. Beilstein J. Org. Chem 2006, 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hunter L; Jolliffe KA; Jordan MJT; Jensen P; Macquart RB Synthesis and Conformational Analysis of α,β-Difluoro-γ-amino Acid Derivatives. Chem. Eur. J 2011, 17, 2340. [DOI] [PubMed] [Google Scholar]
- 5. (a).Clark JL; Hollecker L; Mason JC; Stuyver LJ; Tharnish PM; Lostia S; McBrayer TR; Schinazi RF; Watanabe KA; Otto MJ; Furman PA; Stec WJ; Patterson SE; Pankiewicz KW Design, Synthesis, and Antiviral Activity of 2′-Deoxy-2′-fluoro-2′-C-methylcytidine, a Potent Inhibitor of Hepatitis C Virus Replication. J. Med. Chem 2005, 48, 5504. [DOI] [PubMed] [Google Scholar]; (b) Tavasli M; O’Hagan D; Pearson C; Petty MC The fluorine gauche effect. Langmuir isotherms report the relative conformational stability of (±)-erythro- and (±)-threo-9,10-difluorostearic acids. Chem. Commun 2002, 1226. [DOI] [PubMed] [Google Scholar]
- 6.For selected recent reviews: (a) Romero RM; ste TH; Muñiz K Vicinal Difunctionalization of Alkenes with Iodine(III) Reagents and Catalysts. Chem. - Asian J 2014, 9, 972. [DOI] [PubMed] [Google Scholar]; (b) Yoshimura A; Zhdankin VV Advances in Synthetic Applications of Hypervalent Iodine Compounds Chem. Rev, 2016, 116, 3328. [DOI] [PubMed] [Google Scholar]; (c) Kohlhepp SV; Gulder T Hypervalent iodine(III) fluorinations of alkenes and diazo compounds: new opportunities in fluorination chemistry. Chem. Soc. Rev 2016, 45, 6270. [DOI] [PubMed] [Google Scholar]; (d) Claraz A; Masson G Asymmetric iodine catalysis-mediated enantioselective oxidative transformations. Org. Biomol. Chem 2018, 16, 5386. [DOI] [PubMed] [Google Scholar]; (e) Flores A; Cots E; Berg J; Muñiz K Enantioselective Iodine(I/III) Catalysis in Organic Synthesis Adv. Synth. Catal 2019, 361, 2. For examples of enantioselective alkene fluorofunctionalizations: [Google Scholar]; (f) Kong W; Feige P; de Haro T; Nevado C Regio‐ and Enantioselective Aminofluorination of Alkenes. Angew. Chem. Int. Ed 2013, 52, 2469. [DOI] [PubMed] [Google Scholar]; (g) Banik SM; Medley JW; Jacobsen EN Catalytic, Diastereoselective 1,2-Difluorination of Alkenes. J. Am. Chem. Soc 2016, 138, 5000. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Molnár IG; Gilmour R Catalytic Difluorination of Olefins. J. Am. Chem. Soc 2016, 138, 5004. [DOI] [PubMed] [Google Scholar]; (i) Woerly EM; Banik SM; Jacobsen EN Enantioselective, Catalytic Fluorolactonization Reactions with a Nucleophilic Fluoride Source. J. Am. Chem. Soc. 2016, 138, 13858. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Banik SM; Medley JW; Jacobsen EN Catalytic, asymmetric difluorination of alkenes to generate difluoromethylated stereocenters. Science 2016, 353, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Mennie KM; Banik SM; Reichert EC; Jacobsen EN Catalytic Diastereo- and Enantioselective Fluoroamination of Alkenes. J. Am. Chem. Soc 2018, 140, 4797. For other enantioselective alkene difunctionalizations: [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Fujita M; Yoshida Y; Miyata K; Wakisaka A; Sugimura T Enantiodifferentiating endo-Selective Oxylactonization of ortho-Alk-1-enylbenzoate with a Lactate-Derived Aryl-λ3-Iodane. Angew. Chem. Int. Ed 2010, 49, 7068. [DOI] [PubMed] [Google Scholar]; (m) Romero RM; Wöste TH; Muñiz K Vicinal Difunctionalization of Alkenes with Iodine(III) Reagents and Catalysts. Chem. - Asian J 2014, 9, 972. [DOI] [PubMed] [Google Scholar]; (n) Mizar P; Laverny A; El-Sherbini M; Farid U; Brown M; Malmedy F; Wirth T Enantioselective Diamination with Novel Chiral Hypervalent Iodine Catalysts. Chem. - Eur. J 2014, 20, 9910. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Haubenreisser S; Wöste T; Martínez C; Ishihara K; Muñiz K Structurally Defined Molecular Hypervalent Iodine Catalysts for Intermolecular Enantioselective Reactions. Angew. Chem. Int. Ed 2016, 55, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Wöste TH; Muñiz K Enantioselective Vicinal Diacetoxylation of Alkenes under Chiral Iodine(III) Catalysis. Synthesis 2016, 48, 816. [Google Scholar]; (q) Muñiz K; Barreiro L; Romero RM; Martínez C Catalytic Asymmetric Diamination of Styrenes. J. Am. Chem. Soc 2017, 139, 4354. [DOI] [PubMed] [Google Scholar]; (r) Gelis C; Dumoulin A; Bekkaye M; Neuville L; Masson G Chiral Hypervalent Iodine(III) Catalyst Promotes Highly Enantioselective Sulfonyl- and Phosphoryl-oxylactonizations. Org. Lett 2017, 19, 278. [DOI] [PubMed] [Google Scholar]
- 7.The HF/mCPBA protocol for catalytic fluorination reactions was first described by Kita, Shibata, and co-workers: Suzuki S; Kamo T; Fukushi K; Hiramatsu T; Tokunaga E; Dohi T; Kita Y; Shibata N Iodoarene-catalyzed fluorination and aminofluorination by an Ar–I/HF-pyridine/mCPBA system. Chem. Sci 2014, 5, 2754. [Google Scholar]
- 8.The initial Gilmour report (ref. 6h) also included a single example of an enantioselective 1,2-difluorination.
- 9.Scheidt F; Schäfer M; Sarie J; Daniliuc C; Molloy J; Gilmour R Enantioselective, Catalytic Vicinal Difluorination of Alkenes. Angew. Chem. Int. Ed 2018, 57, 16431. [DOI] [PubMed] [Google Scholar]
- 10. (a).Dimroth O; Bockemüller W Versuche zur Fluorierung organischer Verbindungen, I. Die Einwirkung von Blei(IV)-fluorid auf einige organische Verbindungen. Ber. Dtsch. Chem. Ges 1931, 64, 516. [Google Scholar]; (b) Bockemüller W Versuche zur Fluorierung organischer Verbindungen, II. Die Einwirkung von Aryljodidfluoriden auf einige organische Verbindungen. Ber. Dtsch. Chem. Ges 1931, 64, 522. Both preceding references originally reported products resulting from 1,2-difluorination, but were later shown to produce 1,1-difluorinated products: [Google Scholar]; (c) Bornstein J; Borden MR; Nunes F; Tarlin HI Rearrangement Accompanying the Addition of Fluorine to 1,1-Diarylethylenes. J. Am. Chem. Soc 1963, 85, 1609. For examples of racemic 1,1-difluorination of alkenes with hypervalent iodine reagents and catalysts, see: [Google Scholar]; (d) Hara S; Nakahigashi J; Ishi-i K; Fukuhara T; Yoneda N Fluorinative Ring-Contraction of Cyclic Alkenes with p-Iodotoluene Difluoride. Tetrahedron Lett. 1998, 39, 2589. [Google Scholar]; (e) Ilchenko NO; Tasch BOA; Szabó KJ Mild Silver-Mediated Geminal Difluorination of Styrenes Using an Air- and Moisture-Stable Fluoroiodane Reagent. Angew. Chem. Int. Ed 2014, 53, 12897. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kitamura T; Muta K; Oyamada J Hypervalent Iodine-Mediated Fluorination of Styrene Derivatives: Stoichiometric and Catalytic Transformation to 2,2-Difluoroethylarenes. J. Org. Chem 2015, 80, 10431. [DOI] [PubMed] [Google Scholar]; (g) Scheidt F; Neufeld J; Schäfer M; Thiehoff C; Gilmour R Catalytic Geminal Difluorination of Styrenes for the Construction of Fluorine-rich Bioisosteres. Org. Lett 2018, 20, 8073. [DOI] [PubMed] [Google Scholar]
- 11.For additional examples of fluorinative hypervalent iodine-reactions involving aryl migration, and for related computational studies, see: (a) Zhou B; Yan T; Xue X-S; Cheng J-P Mechanism of Silver-Mediated Geminal Difluorination of Styrenes with a Fluoroiodane Reagent: Insights into Lewis-Acid-Activation Model. Org. Lett 2016, 18, 6128. [DOI] [PubMed] [Google Scholar]; (b) Ulmer A; Brunner C; Arnold AM; Pöthig A; Gulder T A Fluorination/Aryl Migration/Cyclization Cascade for the Metal-Free Synthesis of Fluoro-Benzoxazepines. Chem. Eur. J 2016, 22, 3660. [DOI] [PubMed] [Google Scholar]; (c) Yan T; Zhou B; Xue X-S; Cheng J-P Mechanism and Origin of the Unexpected Chemoselectivity in Fluorocyclization of o-Styryl Benzamides with a Hypervalent Fluoroiodane Reagent. J. Org. Chem 2016, 81, 9006. [DOI] [PubMed] [Google Scholar]; (d) Zhou B; Xue X-S; Cheng J-P Theoretical study of Lewis acid activation models for hypervalent fluoroiodane reagent: The generality of “F-coordination” model. Tetrahedron Lett. 2017, 58, 1287. [Google Scholar]; (e) Zhou B; Haj MK; Jacobsen EN; Houk KN; Xue X-S Mechanism and Origins of Chemo- and Stereoselectivities of Aryl Iodide-Catalyzed Asymmetric Difluorinations of β-Substituted Styrenes. J. Am. Chem. Soc 2018, 140, 15206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For studies on the effect of substitution on the strain energy of small rings, see: Bach RD; Dmitrenko O The Effect of Substitutents on the Strain Energies of Small Ring Compounds. J. Org. Chem 2002, 67, 2588. [DOI] [PubMed] [Google Scholar]
- 13.However, primary amide 2a did not display the same sensitivity to HF-pyridine loading (see Supporting Information).
- 14.The 1,2- to 1,1-selectivity in the difluorination of secondary amides 2b–2e also displayed significant sensitivity to HF-pyridine loading (see Supporting Information).
- 15.The reactions of 2j and 2m yielded the 1,2-difluorides as 6.1:1.0 and 5.1:1.0 mixtures of diastereomers, respectively (diastereomeric ratios were determined by 19F NMR analysis of the crude reaction mixtures). For all other substrates, difluorination reactions proceeded with greater than 25:1.0 d.r.
- 16.The N-tert-butyl derivative of cinnamamide 2q (Scheme 1B) was prepared and subjected to the difluorination reaction conditions to afford the corresponding 1,2-difluoride as the exclusive product in 97% ee. This product was treated with 33 wt.% hydrobromic acid in acetic acid to afford the primary amide product 3q (see Supporting Information for details). The sign of the measured optical rotation of 3q prepared by this method matched that previously reported for 3q prepared directly by difluorination of 2q. The relative and absolute configurations of 3q have been previously assigned by X-ray diffraction of a single crystal (see ref 6g).
- 17.Charton M Steric Effects. I. Esterification and Acid-Catalyzed Hydrolysis of Esters. J. Am. Chem. Soc 1975, 97, 1552. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.