

Abstract
Lymphatic vessels (LVs) have been suggested as a preferential conduit for metastatic progression in breast cancer, where a correlation between the occurrence of lymph node metastasis and an increased extracellular matrix (ECM) density has been reported. However, the effect of ECM density on LV function is largely unknown. To better understand these effects, we used a microfluidic device to recreate tubular LVs in a collagen type I matrix. The density of the matrix was tailored to mimic normal breast tissue using a low-density collagen (LD-3 mg/mL) and cancerous breast tissue using a high-density collagen (HD-6 mg/mL). We investigated the effect of ECM density on LV morphology, growth, cytokine secretion, and barrier function. LVs cultured in HD matrices showed morphological changes as compared to LVs cultured in a LD matrix. Specifically, LVs cultured in HD matrices had a 3-fold higher secretion of the pro-inflammatory cytokine, IL-6, and a leakier phenotype, suggesting LVs acquired characteristics of activated vessels. Interestingly, LV leakiness was mitigated by blocking the IL-6 receptor on the lymphatic ECs, maintaining endothelium permeability at similar levels of LV cultured in a LD matrix. To recreate a more in vivo microenvironment, we incorporated metastatic breast cancer cells (MDA-MB-231) into the LD and HD matrices. For HD matrices, co-culture with MDA-MB-231 cells exacerbated vessel leakiness and secretion of IL-6. In summary, our data suggest that (1) ECM density is an important microenvironmental cue that affects LV function in the breast tumor microenvironment (TME), (2) dense matrices condition LVs towards an activated phenotype and (3) blockade of IL-6 signaling may be a potential therapeutic target to mitigate LV dysfunction. Overall, modeling LVs and their interactions with the TME can help identify novel therapeutic targets and, in turn, advance therapeutic discovery.
Graphical Abstract
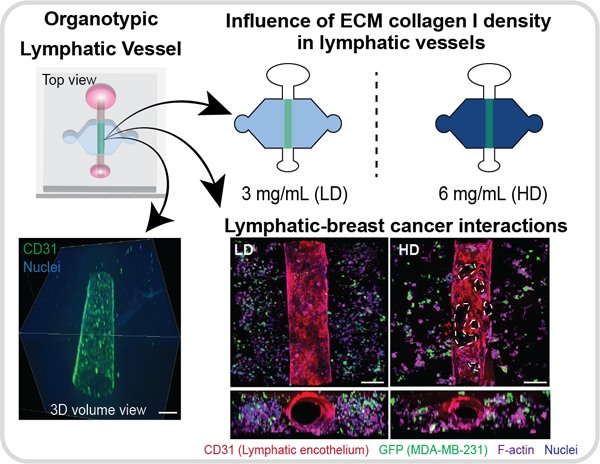
1. Introduction
The tumor microenvironment (TME) is the niche where tumors develop, and comprises many cellular components such as cancer cells, immune cells, fibroblasts, blood and lymphatic vessels, as well as non-cellular components such as the extracellular matrix (ECM)1. The ECM consists of a complex meshwork of fibrillar collagens, glycoproteins, and proteoglycans that shape the biochemical and biophysical properties of tissues2–4, regulating cell behavior in normal physiology5. In the TME, ECM remodeling contributes to tumor development and progression by altering cell behavior and, importantly, the presence of tumor-associated ECM architecture is a predictive biomarker of patient outcome 6–9. Another aspect of the tumor-associated ECM is the increase in deposition of type I collagen by fibroblasts. The increase in collagen deposition forms a dense fibrous tissue surrounding the tumor, which has been implicated in promoting cancer progression and metastasis in numerous solid tumors10–13, including breast cancer14.
ECM remodeling is critical for regulating tumor escape (i.e. metastasis), the leading cause of mortality in cancer patients15. For example, it has been demonstrated that increased ECM density enhances cancer progression by promoting cancer cell migration16, proliferation17, and altering cellular metabolism18. Additionally, cancer cells can interact with other components of the TME, such as blood and lymphatic vasculature, that are also exposed to and could be modified by the remodeled matrix. The vasculature is of interest as it is an essential component that facilitates metastasis, providing a route for cancer cells to intravasate and disseminate to distant organs. Numerous studies have investigated how ECM density influences the physiology of vasculature in the TME and how these changes might contribute to metastasis. In this context, dense ECM has been found to reduce capillary morphogenesis19,20 and angiogenesis21, but also increases the duration of endothelial cell-cell adhesion, cell proliferation, and sprout width22. To date, much less is known about the effect of ECM density on conditioning lymphatic vessel morphology and function.
Lymphatic endothelial cells (LECs) have unique structural and functional characteristics compared to endothelial cells from blood vessels23. LECs are morphologically different as they lack basement membrane, making them leakier than blood vessels and providing a more advantageous route for cancer metastasis24,25. However, it is not known whether a dense ECM alters lymphatic vessel (LV) phenotype. Understanding the influence of a dense ECM on LVs is critical in breast cancer, since increased collagen I deposition has been correlated with increased lymph node metastasis26 and evidence indicates that breast cancer metastasis preferentially occurs through LVs as compared to blood vessels27,28. Therefore, elucidation of the effects of ECM density on lymphatic vasculature is critical to advancing our understanding of breast cancer metastasis. Unfortunately, traditional in vitro and animal models of lymphatic vessels present challenges in recapitulating 3D vessel structure and physiology or have low tractability 29. We have previously reported the development of microfluidic organotypic in vitro models for (1) generating endothelial vessels30–32 and (2) demonstrating the importance of tissue structure on tissue behavior33.
Microfluidic organotypic in vitro models are becoming more widely used due to their potential for recapitulating in vivo tissue structure and function33–35. We and others have previously demonstrated the capability of microfluidic devices to recreate luminal geometries in collagen hydrogel33,36,37, and we recently reported a LV model in these luminal geometries32. In the current work, we have used the microfluidic LV model to investigate the effects of ECM density (i.e., low- vs high-collagen density) on LV physiology. We found that LVs cultured in a dense collagen I matrix exhibited a leakier, more proliferative phenotype and a pro-inflammatory secretion profile, suggesting that dense ECM conditions the LVs toward an activated endothelial phenotype. The inflammatory cytokine, IL-6, was identified as a potential mediator of LV barrier dysfunction given its significantly higher secretion in the dense collagen matrix. Therapeutic targeting of the IL-6/IL-6R pathway, using an anti-IL-6R antibody, decreased LV leakiness. Moreover, LVs were co-cultured with MDA-MB-231 tumor cells in both LD and HD matrices, where vessel dysfunction was heightened in the HD case. Collectively, our findings demonstrate for the first time that ECM density is an important signaling factor that affects LV physiology within the breast TME.
2. Materials and Methods
2.1. Cell culture
Human lymphatic endothelial cells (HLECs, ScienCell, 2500) were cultured in standard cell culture flasks coated with fibronectin (5 µg/cm2, Sigma Aldrich, F1141–5MG) at a starting cell concentration of 5·105. Cultures were maintained with endothelial basal medium-2 (Lonza, CC-3156) supplemented with EGM-2 MV SingleQuot Kit (Lonza, CC-4147). HLECs were cultured to 90–95% confluency at passages 3 to 5 for all experiments and 3 different lots of lymphatic endothelial cells were used for the experiments. We used human mammary adenocarcinoma cells, MDA-MB-231, transfected to stably expressing green fluorescent protein (GFP), a kind gift from Dr. Suzanne Ponik (University of Wisconsin, Madison). MDA-MB-231s were routinely cultured in high glucose DMEM (Gibco, 11965092) supplemented with 10% fetal bovine serum (FBS, VWR, 97068–085) and 1% penicillin/streptomycin (ThermoFisher, 15140–122). For all experiments, a one to one mixture of lymphatic endothelial cell and MDA-MB-231 cell media was used (i.e., EGM-2 MV to 10% FBS, 1% P/S high glucose DMEM), called experimental media through the text. All cultures were kept in a humidified incubator at 37°C with 5% CO2.
2.2. Device Fabrication
Fabrication of the organotypic lumen structure was performed as previously described36. The microdevice consists of two PDMS layers, which define the microchamber; and a suspended PDMS rod, which is removed after polymerization of a hydrogel in the main chamber to create a tubular lumen structure. In order to fabricate the top and bottom layers of the microdevice, a traditional soft lithography technique was used, in which the layers were spun using SU-8 (MicroChem, Y13273) to create the silicon master molds. Polydimethylsiloxane (PDMS, Dow Corning, Sylgard 184) was mixed at a 10:1 base to curing agent ratio and poured over the SU-8 silicon master molds. Using the same PDMS mixture, PDMS rods were fabricated by filling up a 25 gauge (Fisher Scientific, 14–840-84) hypodermic needle with PDMS. PDMS components were then baked at 80°C for 4 h. After baking, the PDMS rods were extracted from the needles, yielding PDMS rods of 280 μm in diameter. The two layers were aligned, ethanol bonded together and the PDMS rods were placed into the microdevice chamber. Finally, the microdevice was oxygen plasma bonded to a glass-bottom MatTek dish (MatTek Corporation, P50G-1.5–30-F), following a general protocol. The microdevices were sterilized using UV irradiation for 15–20 min for further use.
2.3. Organotypic Culture Preparation
2.3.1. Device preparation
To achieve maximum hydrogel adhesion to the PDMS chamber, a two-step coating of 2% poly(ethyleneimine) (PEI, Sigma-Aldrich, 03880) diluted in deionized DI water for 10 minutes was loaded into the side ports. The PEI solution was aspirated and 0.4% glutaraldehyde (GA, Sigma-Aldrich, G6257) diluted in deionized DI water was loaded into the side ports and incubated at room temperature for 30 minutes. During the GA incubation, the collagen solution was prepared on ice (refer to section 2.3.2). After the 30-minute of GA incubation, the microdevices were washed three times with sterile DI water to remove any GA excess. At this point, devices are ready to be loaded with the collagen solution. To minimize evaporation, sacrificial phosphate buffered saline (PBS) was added around the side of the MatTek dish.
2.3.2. Extracellular matrix preparation and loading into the device
High-density rat-tail collagen type 1 (Corning, 354249, referred as collagen through the text) was diluted with 5X PBS and neutralized with 0.5 M NaOH (Fisher Scientific, S318) achieving a final concentration of 1X PBS, and a pH of 7.4. To achieve a final concentration of 3 mg/mL (low collagen density-LD) or 6 mg/mL (high collagen density-HD) dilutions with fibrinogen (Sigma-Aldrich, F8630), fibronectin (Sigma-Aldrich, F1141) and media were performed. For experiments with cancer cells in the matrix, MDA-MB-231s, a final concentration of 250 cells/µL was added to the respective collagen solution (recipes for different collagen gel densities and cultures conditions can be found in Table S1). Right after the washes with sterile DI water, 6 µl of collagen solution was loaded through the side ports and polymerized at room temperature for 10 min. Finally, a small droplet of media (5 μL) was placed on top of the side ports to prevent evaporation, and devices were transferred to 37°C for 1 hour to allow collagen to fully polymerize.
2.3.3. Lymphatic endothelial cell seeding in lumens
After incubation, a small droplet of media (5 μL) was added to the input port under sterile conditions. To remove the PDMS rod, the rod was pulled through the output port using a sterilized tweezers, leaving a hollow lumen filled up with media within the collagen matrix. All fluid handling procedures were conducted with standard pipettes, uniquely enabled by passive pumping38 to transport media through the channel. With passive pumping, a droplet of media is transported from the small port to the large port due to the difference in Laplace pressures of fluid droplets at the ports. This procedure was performed 2–3 times a day on each lumen for maintenance. Human lymphatic endothelial cells (HLECs) were trypsinized with 0.05% Trypsin-EDTA (ThermoFisher Scientific, 25300062), counted, resuspended in experimental media at 20,000 cells/μL and seeded into the lumens (4 µL per lumen). HLEC-filled lumens were incubated at 37°C for 2 h to allow for cell attachment, flipping devices every 25 min to ensure homogeneous cell coverage of the lumen wall. After 2 hours, lumens were supplemented with 10 μL of experimental media and cultured overnight at 37°C. Cultured vessel media was refreshed twice a day by flowing experimental media 2–3 times through the lumen to remove dead cells and for vessel maintenance.
2.4. Immunofluorescence staining and imaging
During immunofluorescence staining, cells were washed with PBS for 30 minutes between each step. Unless specified otherwise, steps took place at room temperature. Washing buffer (0.1% PBS-Tween 80 (Sigma-Aldrich, P1754) and blocking buffer (3% Bovine Serum Albumin (BSA, Sigma-Aldrich, A9056) in 0.1% PBS-Tween 80) were made in advance and stored at 4° C until use. Cells were fixed with 4% paraformaldehyde (PFA) (EMScience, 15700) for 15 min, then incubated with 0.2% Triton® X-100 (MP Biomedicals, 807426) for 30 min for permeabilization. Finally, vessels were incubated with 10 µL of blocking buffer at 4°C overnight.
Primary antibodies were diluted to desired concentrations with staining buffer (blocking buffer with 1% PBS-Tween 80 at 10:1 v/v). Vessels were incubated with primary antibodies at 4°C overnight (Table S2). Then, vessels were incubated with the secondary antibodies diluted using staining buffer supplemented with 10% goat serum to reduce unspecific binding for 2 hours. Stained vessels were washed over two days with the washing buffer and stored in sterile PBS until imaging. Texas Red-X Phalloidin (ThermoFisher Scientific, T7471) and DAPI (ThermoFisher Scientific, D3571) were used to stain actin cytoskeleton and nuclei, respectively. Fluorescent images were acquired at 10X using a Nikon TI® Eclipse inverted microscope (Melville, New York) and processed using the National Institutes of Health ImageJ software. Confocal images were acquired using a Leica SP8 3X STED Super-resolution microscope (Wetzlar, Germany) in the UW-Madison Optical Imaging Core.
2.5. Matrix visualization by SHG imaging
SHG images were taken on a custom-built inverted multiphoton microscope (Bruker Fluorescence Microscopy, Middleton, WI), as described previously39. Briefly, the system consists of a titanium:sapphire laser (Spectra Physics, Insight DS-Dual), an inverted microscope (Nikon, Eclipse Ti), and a 40x water immersion (1.15NA, Nikon) objective. SHG images were taken using an excitation wavelength of 890 nm, an emission bandpass filter of 440/80 nm, and a GaAsP photomultiplier tube (H7422P-40, Hamamatsu).
2.6. Collagen fiber quantification
SHG images of low- and high-density collagen matrices were analyzed using CT-FIRE V1.3 Beta2, an open source image processing program developed by the Laboratory for Optical and Computational Instrumentation (https://loci.wisc.edu/software/ctfirev1.3, University of Wisconsin-Madison). As per instructions in the CT-FIRE manual, the count and width of collagen fibers were measured for three separate field of views per device (i.e., top, middle, and bottom planes of the device). The average fiber parameter per device was calculated as the average value of the three planes.
2.7. Fluorescent image quantification
For all images, we conducted a rolling ball background subtraction and a region of interests (ROIs) was drawn over the lumens in one Z-plane. The ROI dimension and background subtraction was kept constant throughout a dataset. To quantify actin stress fibers, we measured the percentage of F-actin covered area within the ROI. To count total Ki67 positive nuclei, we counted all maxima within the ROI defined by a set threshold. To evaluate the percentage of Ki67 positive cells, the number of Ki67 positive nuclei was divided by the total number of nuclei for each lumen.
2.8. Dextran diffusion assay
The permeability of the lymphatic vessels was measured by dextran diffusion assays using Texas Red dextran (70kDa, ThermoFisher Scientific, D1830) prepared in PBS to 1 µM. For each replicate, 3 µL of dextran solution was added to the vessel such that fluid was flush with the lip of the ports to minimize flow from a pressure head. Diffusion was measured over 15 minutes per vessel. Permeability coefficients were calculated using equation 140:
| Eq. 1 |
where Io is the total initial intensity outside the vessel, If is the total intensity outside the vessel at 15 minutes, to is the initial time point, tf is the final time point of 15 minutes, and D is vessel diameter. All vessels were imaged with the Nikon TI® Eclipse inverted microscope (Melville, New York), and maintained at 37 °C and 5% CO2 by a stage-top incubator (Okolab, Italy).
2.9. Targeted blocking and exogenous IL-6 treatment
Anti-IL-6R antibody (Abcam, ab47215) was used to inhibit IL-6/IL-6R signaling. Mouse IgG1 antibody (BioLegend, 400102) was used as an isotype control for the IL-6/IL-6R inhibition experiments. To block vessels cultured in the high-density matrix, vessels were treated with anti-IL-6R antibody (25 µg/mL) from day 2 to day 5 and used for dextran diffusion analysis on day 5. To stimulate vessels cultured in the low-density matrix, IL-6 solutions were prepared to either 5 ng/mL or 30 ng/mL in EGM-2 MV media. Vessels were supplemented with the IL-6 solutions on day 2 and refreshed daily until day 5 for dextran diffusion analysis.
2.10. Cytokine secretion assay
Multiplexed protein secretion analysis was performed on HLEC cultured vessels, HLEC vessels co-cultured with MDA-MB-231s and MDA-MB-231s monocultures for both types of matrix densities. The analysis was performed using the Magnetic Bead-Based Multiplex ELISA system MAGPIX (Luminex Corporation) with the Milliplex human cytokine panel bead kit (R&D Systems, LXSAHM-10) as described elsewhere30. Collected media (20 µL per lumen) was combined to increase the sample volume in each cultured condition. Briefly, media collection was performed on days 3 and 4 from six cultured vessels pooled per cultured condition, yielding 240 µL in total. Sample preparation and detection was performed following the manufacturer’s protocol. Data were collected with xPonent software (Luminex), and soluble factor concentrations in media were calculated using mean fluorescence intensities (MFI) by creating a standard curve for each analyte using a five-parameter logistic (5-PL) curve fit.
2.11. Statistical analysis
All the experiments were repeated at least three times as independent biological repeats. All results are presented as the mean ± one standard deviation of the mean. Data were analyzed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA) and statistical significance was set at p < 0.05. One-to-one comparisons were performed with an unpaired Student t-test with Welch’s correction (if SD were not the same) after the normal distribution was proved via Shapiro-Wilk test. If the normality test was not passed, a non-parametric test was performed (Mann-Whitney test) (Fig. 2,3, 4 and 5). Multiple comparisons by One-way ANOVA were corrected using the Dunnett test and multiple comparisons by 2-way ANOVA were corrected with a Sidak’s test (Fig. 6D).
figure 2.
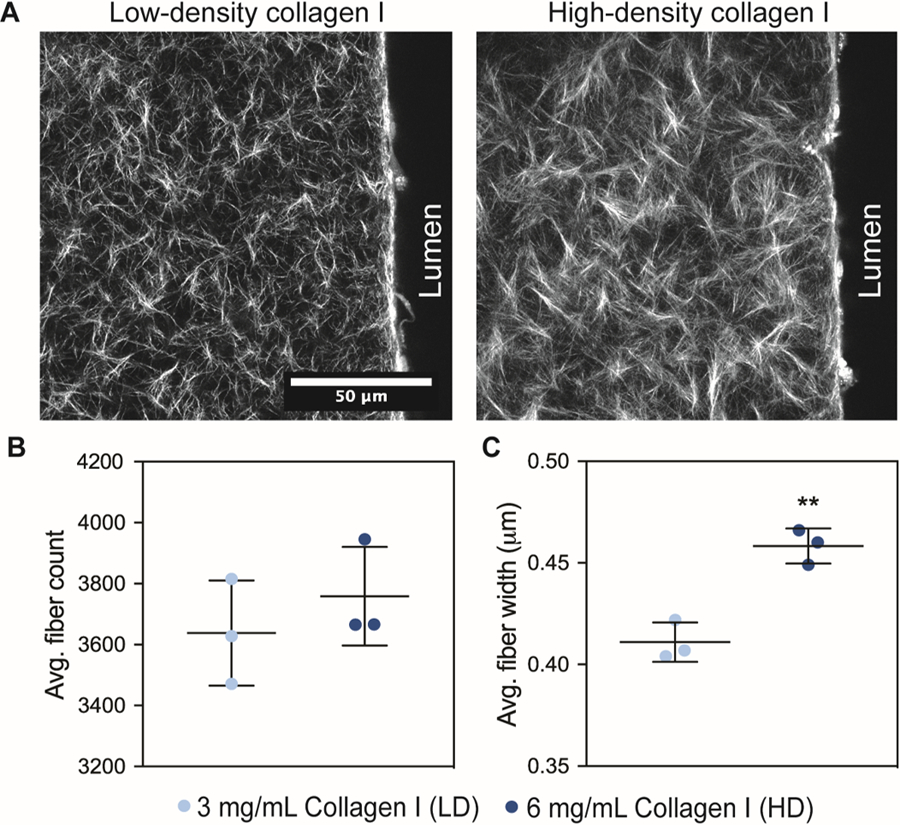
Extracellular matrix characterization of a low-density (LD) and high-density (HD) collagen hydrogel. a) Second Harmonic Generation images of the collagen type I fibers for the low-density (LD) and high-density (HD) matrices in the microdevice, next to the lumen structure. b) average fiber count for LD and HD matrices. c) average fiber width for LD and HD matrices. fiber quantification was performed in one optical plane. bars represent average ± SD of n=3 independent replicates. **p ≤ 0.01
figure 3.
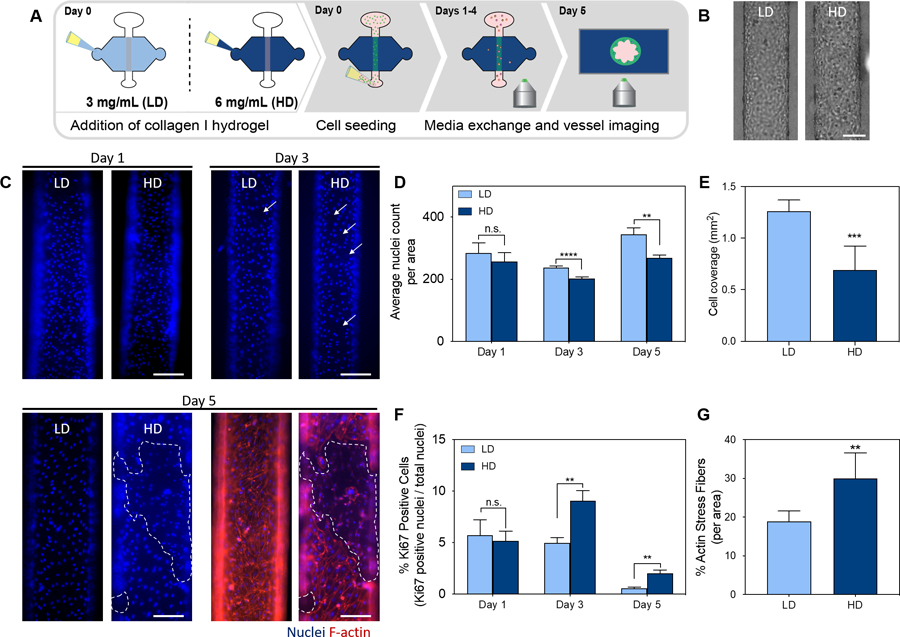
Influence of ecm density in lymphatic vessel morphology, cell coverage, growth, and F-actin stress fibers. a) schematic of the culture conditions and experiment timeline. b) brightfield images of a lymphatic vessel cultured in LD and HD collagen matrices on day 1. scale bar = 200 μm c) images of lymphatic endothelial cells nuclei cultured in LD and HD matrices at day 1(top left), day 3 (top right) and day 5 (bottom left) with f-actin in red and nuclei in blue (bottom right). the dashed outline indicates endothelial cell detachment. d) nuclei count of cells conforming the lymphatic vessels per area at days 1, 3 and 5. e) lymphatic endothelial cell coverage area for each lumen cultured in LD and HD collagen matrices on day 5. f) % Ki67 positive cells (proliferation) per lumen area in lymphatic vessels cultured in LD and HD matrices at days 1, 3 and 5. g) % f-actin stress fibers per lumen area for vessels cultured in LD and HD collagen matrices on day 5. bars represent average ± SEM, n at least 4 individual vessels. scale bars = 140 μm. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
figure 4.
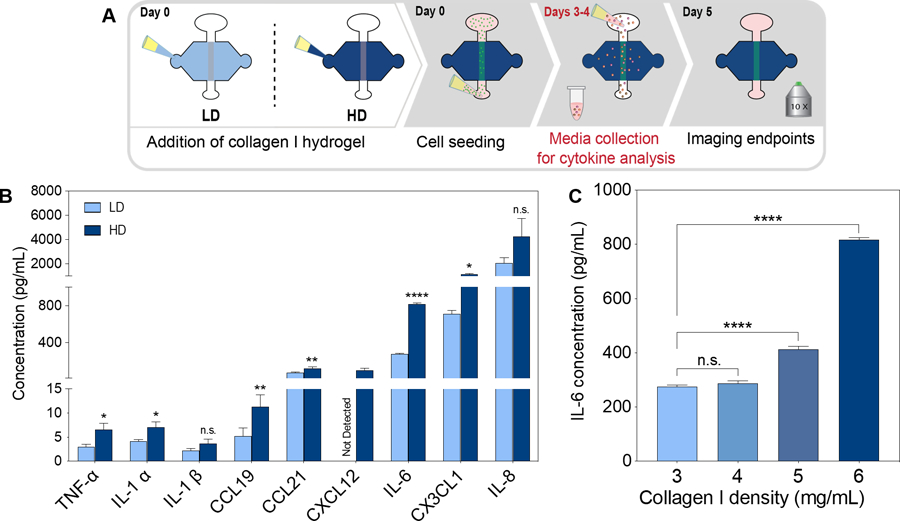
Effect of ECM density in lymphatic vessel cytokine secretion. a) schematic of the culture conditions and experimental timeline. b) pro-inflammatory cytokine and chemokine concentrations for vessels cultured in ld and hd collagen matrix. c) IL-6 cytokine concentration in a range of collagen I density matrices (3, 4, 5 and 6 mg/ml). (n =4, pooled samples over 2 days from at least 6 lumens). *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001.
figure 5.
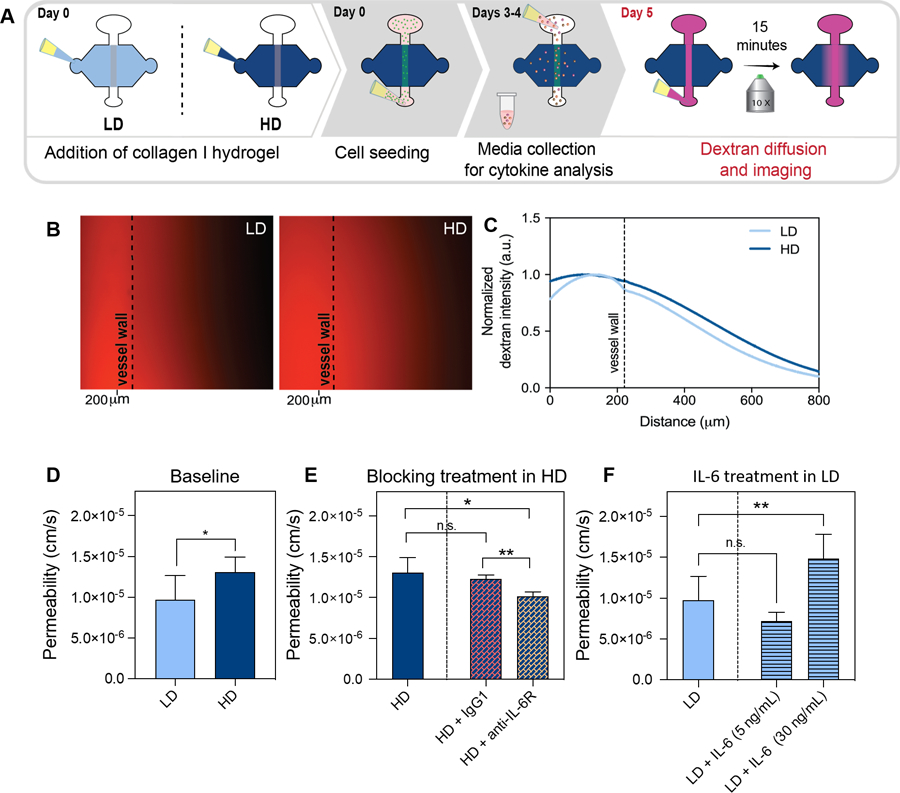
Effect of ECM density in lymphatic vessel barrier function a) schematic of the culture conditions and experimental timeline. b) representative image of diffusion assay in vessels cultured in LD and HD matrices at t=15 min. dashed lines indicate lymphatic vessel wall c) normalized dextran intensity profile for vessels cultured in LD and HD matrices. d-f) quantification of solute permeation for vessels cultured in LD and HD matrices. d) vessel permeability in LD and HD matrices (baseline levels). e) vessel permeability in HD matrices treated with Igg1 (control) or anti-IL-6R (blocking treatment) f) vessel permeability in LD matrices treated with IL-6 (5 mg/ml and 30 ng/ml). bars represent average ± sd, n at least 3 individual vessels. *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001.
figure 6.
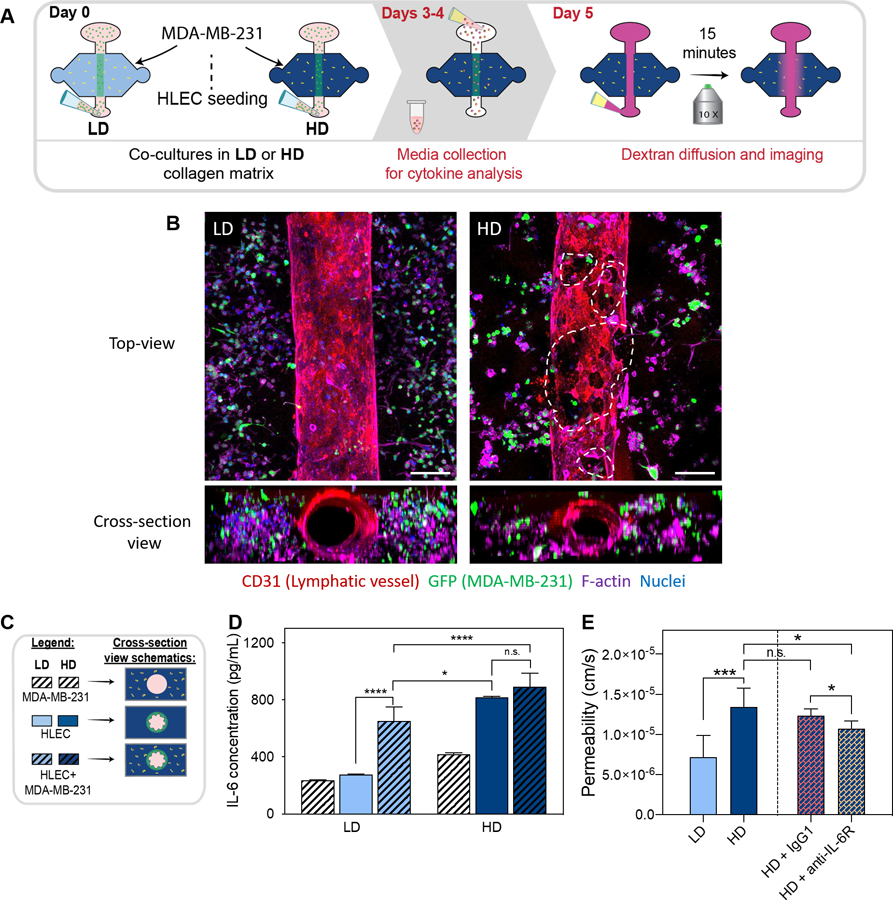
Lymphatic vessel co-culture with metastatic breast cancer cells (MDA-MB-231) in LD and HD matrices. a) schematic of the experimental conditions and timeline. b) top-view and cross-section view of immunofluorescent images of lymphatic vessels co-cultured with metastatic breast cancer cells in LD (left) and HD (right) matrices (f-actin in purple, cd31 in red, MDA-MB-231-GFP in green and nuclei in blue. dashed outlines indicate endothelial cell detachment in the vessel wall. c) legend and cross-section view schematic of conditions. d) co-cultures IL-6 protein secretion levels in LD and HD matrices. e) quantification of solute permeation for lymphatic lumens in LD and HD matrices and for lymphatic lumens in LD and HD matrices treated with anti-Igg1 (control) and anti-il-6r (blocking treatment). bars represent average ± SD, n at least 3 individual vessels. (n =4, pooled samples over 2 days from at least 6 lumens). scale bar = 140 μm. *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001.
3. Results
3.1. 3D organotypic lymphatic vessel model generation.
To investigate how ECM density affects LV function, we adapted a recently published and validated in vitro model that recreates a physiological lymphatic microenvironment, such as lumen structure and matrix composition32, as illustrated in Fig. 1A and Fig 1B. LVs were generated within a 3 mg/mL collagen hydrogel by lining the lumen structure with primary human lymphatic endothelial cells (HLECs) (Fig. 1C). After 5 days, LVs developed a confluent endothelial monolayer visualized via CD31 staining (Fig. 1D) with evidence of evident tubular structure in cross-section (Fig. 1E), demonstrating generation of a 3D tubular lymphatic vessel (Fig. 1F and Supplemental video 1). To characterize LV phenotype, the expression of prospero homeobox protein 1 (PROX-1), a protein that co-localizes with the nucleus of lymphatic endothelial cells41, was assessed and confirmed by immunofluorescent staining (Fig. 1G). We previously demonstrated that HLECs also express lymphatic endothelial hyaluronan receptor 1 and podoplanin32, which are markers specific to lymphatic ECs.
figure 1.
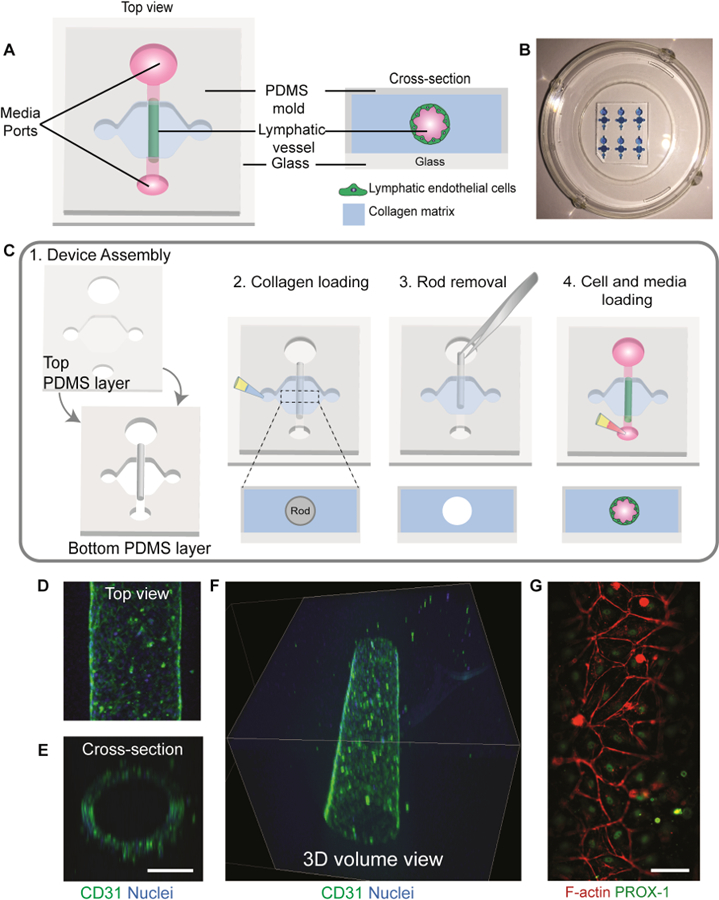
Organotypic lymphatic vessel model. a) Top view of an assembled microdevice (left) with a cross-sectional view of the device showing the lymphatic endothelial cells lining the lumen structure within a collagen matrix (right). b) Representative image of microdevice array. Microdevices were filled with a blue dye for visualization purposes. c) Microdevice design and fabrication scheme. 1) The device consists of two PDMS layers bonded together with a suspended PDMS rod. The PDMS layers defined the microchamber, while the rod allows for the generation of the lumen structure. The top layer of the microdevice contains ports for fluid handling and a cover for the microchamber. For device operation, after plasma bonding to a glass-bottom dish: 2) the microchamber is filled with a hydrogel solution and left to polymerize, 3) lumen rod is removed exposing an empty lumen within the hydrogel, 4) cells are seeded into the lumen with media and cultured at 37ºC. d) Top view of a lymphatic vessel stained with a classical endothelial cell junction marker, cluster of differentiation 31 (CD31), and nuclei. e) Orthogonal view of the vessel. Scale bar= 140 μm f) confocal image of the lymphatic vessel showing a 3d tubular structure. g) Top view of cultured vessels stained with a lymphatic-specific marker, prospero homeobox protein 1 (PROX-1), and F-actin. Scale bar = 70 μm.
3.2. Formation and characterization of low-density and high-density collagen matrices.
Using the LV model, we sought to investigate how LVs are affected by ECM density. To do so, we used a low-density (LD) and a high-density (HD) collagen I matrix. The selected collagen concentration of 3 mg/mL (LD) is representative of healthy normal tissue such as the mammary gland, whereas the 6 mg/mL (HD) collagen gel mimics the tissue stiffening occurring in solid tumors 16,42,43. Thus, we first aimed to elucidate the differences in matrix architecture in the LD and HD matrices using Second Harmonic Generation (SHG) to visualize and analyze collagen fibers (Fig. 2A). CT-FIRE was used to compare the average fiber count and average fiber width in the LD and HD matrices44. The average fiber count was 3638 ± 172.2 for LD and 3759 ± 161.4 for HD, revealing that the number of collagen fibers per field of view is similar for both matrices (Fig. 2B). However, the average fiber width was significantly different for HD at 0.4583 ± 0.008 µm as compared to 0.411 ± 0.009 µm for LD (Fig. 2C). Overall, we found that changes in ECM collagen density alters matrix architecture resulting in higher fiber width.
3.3. Influence of low-density and high-density collagen matrices on lymphatic vessel phenotype.
To determine the effect of LD and HD collagen on LV phenotype, we seeded HLEC lumens within LD and HD collagen matrices (Fig. 3A), and assessed their phenotype at days 1, 3 and 5. Bright-field images of LVs cultured after one day revealed that HLECs attached to the lumen wall equally in both matrices (Fig. 3B), which was also confirmed via nuclei count (Fig. 3C-top left), revealing no significant differences in the number of cells attached to the lumen on day one as shown by the average nuclei count per area on day 1 (Fig. 3D). However, the average nuclei count significantly decreased in the HD matrix as compared to the LD matrix for day 3 and day 5 (Fig. 3D). In addition, LVs were stained for F-actin at day 5. Interestingly, there were noticeable morphological changes to the lymphatic endothelium in the HD matrix, such as cell detachment, as compared to the lymphatic endothelium in the LD matrix (Fig. 3C- bottom right). As previously described by us and others, cell detachment was quantified by measuring the cell coverage area of the lumen, in addition to the average nuclei count per area45,46. Cell coverage area significantly decreased in HD matrices as compared to LD matrices (Fig 3E). These observations are consistent with previous reports that point to the capacities of the ECM to modulate capillary network formation and structural integrity of endothelial vessels47.
In addition, previous studies have demonstrated that endothelial cell proliferation increases in stiffer matrices as compared to more compliant matrices48. Therefore, we hypothesized that LVs cultured in HD matrices would be more proliferative than LVs cultured in LD matrices. To evaluate cell proliferation, LVs were stained for the cell proliferation marker Ki67 at days 1, 3 and 5. The percentage of Ki67 positive over total nuclei (DAPI) was quantified using ImageJ49 and found to be similar at day 1, revealing no differences in LEC proliferation (Fig. 3F). However, the proliferation rate significantly increased in the HD matrix as compared to the LD matrix for day 3 and day 5 (Fig. 3F).
Similarly, previous studies have suggested that increases in matrix stiffness, which are associated with increases in matrix density, alters EC-EC adhesion and EC-ECM adhesion by increasing cell contractility and actin stress fiber formation 50,51. Interestingly, in our model, we observed more actin stress fibers in HD matrices (Fig. 3G). The percentage of actin stress fibers was quantified as 18.9% ± 2.7% for LD matrices and 30% ± 6.6% for HD matrices, indicating a significant increase in actin stress fiber formation per vessel area in the HD matrices.
3.4. Dense collagen matrix promotes chemokine and pro-inflammatory cytokine secretion and disrupts barrier function in LVs
After studying the morphological changes of LVs cultured in LD and HD matrices, we focused on studying the secretory profiles of cultured lumens within the different matrices (as shown in Fig. 4A) using a pre-made bead-based ELISA panel (Luminex MAGPIX), from which all factors were within detectable ranges. LVs cultured in HD matrices showed an increase in most of the chemokines and pro-inflammatory cytokines (Fig. 4B) such as IL-1β (1.4-fold) and IL-8 (1.7-fold), as compared to vessels cultured in LD matrices. The chemokine CXCL12 was not detectable in LD matrices but it was detectable in HD matrices, showing an increase in secretion in HD matrices. In addition, the specific inflammatory cytokines that significantly increased in HD matrices were TNF-α (2-fold), IL-1α (1.8-fold), CCL19 (2.1-fold), CCL21 (1.6-fold), CX3CL1 (1.6-fold) and the most strongly upregulated cytokine was IL-6, which increased 3-fold in HD matrices. IL-6 is of interest given that accumulating evidence establishes IL-6 as a key player of the tumor microenvironment which critically regulates endothelial cell dysfunction and tumor progression52–54.
To further evaluate the change in IL-6 concentration observed in LD and HD matrices, we sought to investigate if there is a relationship between ECM density and IL-6 secretion. For this, we cultured the LVs in two additional matrix densities, covering a range of collagen concentrations between the LD and HD matrices (i.e., 4 mg/mL and 5 mg/mL matrix). Interestingly, we observed an increase of IL-6 secretion in an ECM density-dependent manner (Fig. 4C). Specifically, similar levels of IL-6 were observed in the LD (3 mg/mL) and 4 mg/mL matrices, whereas there was a significant increase in IL-6 secretion in the 5 mg/mL and HD (6mg/mL) matrices as compared to a LD matrix. These results confirm that IL-6 secretion increases in an ECM dependent manner. In the literature, IL-6 is known to be released from endothelial cells in inflammatory states, which can then alter endothelial permeability via autocrine and paracrine interactions55,56. Therefore, we hypothesized that LV culture in HD matrices have an increase in vessel permeability as a result of the increase in IL-6 secretion.
To test our hypothesis, we assessed the barrier function of cultured vessels in LD and HD matrices by diffusion assays using 70 kDa-Texas Red dextran (as shown by the schematic in Figure 5A), which represents the regulation of diffusion for biomolecules in the size range of serum albumin, ~67 kDa. Specifically, the solution of dextran was perfused through the lumen and tracked using time-lapse fluorescent microscopy for 15 minutes. In the HD matrix, localized leakage was observed at time 0 (Fig. S1). Representative image of the LD matrix (left image) and HD matrix (right image) after 15 min of dextran perfusion shows more dextran outside of the vessel wall for the HD matrix (Fig. 5B). As shown by the representative curve in the normalized dextran intensity graph, LVs cultured in HD collagen matrices have higher intensity values outside of the vessel wall through the matrix as compared to the LD collagen matrices (Fig. 5C). These results indicate that LVs cultured in HD matrices are leakier than those cultured in LD matrices. We also calculated the permeability coefficient of the vessels which revealed that in HD density matrices LVs were 1.4-fold leakier than LVs cultured in LD matrices (Fig. 5D). However, the increase in permeability in HD vessels was significantly reduced to the level of LD vessels, by treatment with an IL-6 receptor blocking antibody, IL-6R, (25 μg/mL) (Fig. 5E). In addition, blocking with an IL-6R antibody significantly reduced vessel permeability compared to the control (IgG blocking), while the permeability values for the control and HD matrix were not significantly different. To confirm that IL-6 was responsible for the decrease in barrier function, we added exogenous IL-6 (5 ng/mL and 30 ng/mL) to the vessels cultured in the LD matrix. The addition of exogenous IL-6 led to a significant increase in permeability at 30 ng/mL, increasing permeability to a level equivalent to vessels cultured in HD matrices, 1.5·10−5 ± 0.3·10−5 cm/s (Fig. 5F) and leading to a significant increase in vessel permeability as compared to the LD condition. However, treatment with 5 ng/mL of IL-6 did not result in increased vessel permeability, 0.7·10−5 ± 0.1·10−5cm/s (Fig. 5F). Our results show that HD matrices promote an activated vessel phenotype in LVs by inducing the secretion of pro-inflammatory cytokines such as IL-6. Overall, IL-6 drives the decrease in LV barrier function in response to HD conditions. Additionally, our results demonstrate the potential of therapeutic treatment using anti-IL-6R to rescue LV barrier function. Taken together, the increase in secretion of pro-inflammatory cytokines and concurrent increase in vessel leakiness in dense matrices suggests that LVs are conditioned by the increased ECM density, which promotes the activation of endothelial cells and the development of a leaky vessel phenotype.
3.5. Lymphatic vessel co-culture with cancer cells in dense matrix enhances vessel dysfunction.
Next, we investigated the interactions of breast cancer cells and LVs in the LD and HD matrices. Specifically, triple negative MDA-MB-231 cancer cells expressing GFP were embedded in the LD or HD matrix on day zero and co-cultured with LVs for five days, referred to as 231-lymphatic co-cultures (schematic in Figure 6A). After five days of culture, phenotype assessment of co-cultured vessels in LD and HD collagen matrices was performed by staining the vessels for CD31, F-actin, and nuclei to identify all cells whereas cancer cells were identified by GFP. In HD matrix conditions, there was an increase in endothelial cell detachment compared to LVs cultured in LD matrices, similar to our LV monocultures (Fig. 6B). Our previous permeability data (Fig. 5 D–F) suggested that LV barrier dysfunction is caused by IL-6. Therefore, to evaluate the levels of IL-6 in co-culture conditions, conditioned media from the 231-lymphatic co-cultures and monocultures in LD and HD was collected on days 3 and 4 for the conditions shown in the schematic and legend (Fig. 6C). Then, the concentration of IL-6 was determined by MAGPIX analysis (Figure 6D). In the LD matrix, IL-6 concentration significantly increased in the LV monoculture as compared to the 231-lymphatic co-culture. However, in the HD matrix, similar concentrations of IL-6 were found in LV monoculture as compared to 231-lymphatic co-cultures. Interestingly, IL-6 concentration was significantly higher in 231-lymphatic co-cultures in HD matrices as compared to the LV monocultures and 231-lymphatic co-cultures in LD matrices.
Then, we sought to compare vessel permeability in 231-lymphatic co-cultures with LV monocultures. Therefore, we analyzed changes in LV barrier function by measuring the permeability coefficient as described previously. The permeability coefficient of the 231-lymphatic co-cultures in LD was 0.7 ·10−5 ± 0.2 ·10−5 cm/s and 1.3 ·10−5 ± 0.2·10−5 cm/s for HD, revealing that in HD matrices LVs were 1.9-fold leakier (Fig. 6E). Interestingly, the permeability values in the co-cultures are lower for both matrices as compared to the permeability values for the LVs monocultures (Fig. 5D). Based on the results described in the previous figure, we hypothesized that IL-6 is responsible for decreasing LVs barrier capacity in the 231-lymphatic co-cultures within dense matrices. To test this hypothesis, co-cultures in dense matrices were treated with anti-IL-6R (25 μg/mL) antibody that was previously described for the LV monocultures. Compared to the 231-lymphatic co-cultured in HD matrices, the increase in vessel permeability was significantly reduced to 1.0·10−5 ± 0.09 ·10−5 cm/s by supplementing the anti-IL6R to the media (Fig. 6E) while the control (IgG blocking) did not change (1.2 ·10−5 ± 0.08·10−5 cm/s). Overall, these results indicate that IL-6 signaling through IL-6R was likely a primary mediator of LV barrier function.
4. Discussion
Despite the crucial connection between ECM density and lymphatic metastasis in breast cancer, little is known about how ECM density affects LV function. Elucidating the mechanisms by which a dense ECM mediates lymphatic metastasis can help identify new therapeutic targets and, in turn, improve patient outcome. To this end, we applied a LV model recently developed and characterized by our lab32. This microfluidic model is capable of reproducing LV hallmarks, such as the expression of lymphatic specific markers, characteristic cytokine profiles, leakier barrier function than blood vessels, and increased drainage capacity57. These characteristics make our model representative of in vivo LVs with the additional capability to model different microenvironment conditions32. We adapted the LV model to study the effect of the density of the ECM in conditions resembling normal and cancerous breast tissue. To this end, we included cancer cells in the model to study the effects of high/low matrix density on LV and cancer cell crosstalk. To the authors’ knowledge, this is the first LV model that incorporates the lumen structure, matrix proteins, and cancer cells into a single system.
It is known that collagen deposition is increased in the early stages of invasive breast cancer. The resulting increased collagen density is known to promote tumorigenesis, local invasion, and metastasis14. In this article, we have decided to recapitulate this increased collagen deposition by modulating the ECM density. Specifically, we chose to study the effect of ECM density on LV function by using two collagen densities: low density (3 mg/mL, abbreviated LD) and high density (6 mg/mL, abbreviated HD) since in the literature, a range of 1 mg/mL to 6 mg/mL collagen matrices have been used and correlated to tumor cell behavior in vitro (i.e., low density produced a normal phenotype, whereas high density correlated with a tumor-like phenotype)17,43,58. We found that a higher collagen density matrix produces significantly thicker collagen fibers, which is observed in breast cancerous tissues and contributes to enhanced mechanical rigidity, leading to tumor progression59,60. Some reports also indicate that fiber thickness increases in vivo in mammary gland environments highly prone to breast cancer, which is consistent with our results. Overall, the effect of fiber thickness and breast cancer is currently an area of high interest in the field59, and our model presents a high potential to help unravel the underlying mechanisms through which the ECM can promote breast cancer migration, invasion and metastasis. Another important characteristic of the matrix implicated in promoting breast cancer is the stiffness. In this regard, previous reports of stiffness from similar hydrogels measured via rheometry determined that stiffness for 3 mg/ml (LD) is around the few hundreds of Pa, whereas in 5 mg/ml hydrogels (HD are 6 mg/ml) is around 1 kPa61. Comparably, the stiffness of normal breast tissue is around 0.5 kPa in vivo, whereas tumors are in the range of the few kPa62. In this sense, our model captures representative stiffnesses of the states we mimic. However, due to complexities of the system (i.e., small matrix) and the challenges to measure the stiffness within the device, we did not focus on this parameter in the present study. Taking everything together, the selected matrices recapitulate several aspects of the microenvironment found in vivo. With these matrices we sought to investigate the effects of collagen density in LV morphology, changes in actin stress fibers and cell proliferation. After one day lymphatic cells had similar nuclei counts and proliferation levels on both matrices. However, after 3 days of culture, we observed a significant decrease in nuclei count in the HD matrices and a significant increase in cell proliferation, suggesting that dense matrices can condition vessel behavior. Although the similar trend was observed on day 5, compared to day 3, there was an overall increase in nuclei count in both matrices but a decrease in cell proliferation. These results suggest the vessel is stabilizing at day 5, hence, the decrease in cell proliferation. In addition, we observe a significant decrease in cell coverage area and a significant increase in F-actin stress fibers at day 5, revealing that LVs respond to dense collagen matrices by going through vascular damage which results in poor endothelial integrity. Therefore, our results are consistent with the vascular damage observed in vivo. Specifically, the increase in cell proliferation that is observed in our model in the HD matrices is described by one of the repair mechanisms of vascular damage where adjacent mature endothelial cells can replicate locally and replace the lost and damaged cells63. This local repair mechanism is usually sufficient to maintain vascular integrity in healthy conditions. However, in disease states, loss of endothelial integrity develops as observed in our HD matrices. Although we did not explore this mechanism in more detail, it is important to note that in vivo there is another repair mechanism that relies in circulating endothelial progenitor cells for the maintenance and repair of the endothelium64,65. Consequently, the phenotype that we observe in vitro could potentially be mitigated by the addition of endothelial progenitor cells.
Another characteristic of vascular damage described in the literature is the activation and dysfunction of endothelial cells. The activation of endothelial cells represents the switch from a quiescent phenotype toward one that involves a response from the endothelium, resulting in the expression of chemokines, cytokines and adhesion molecules. Then, the activation of endothelial cells progress to endothelial cell detachment, leading to loss of endothelial integrity63,66,67. Therefore, to examine the activation of LV, we assessed the secretion of inflammatory cytokines and chemokines of cultured lumens within the different matrices by measuring the secreted factors in the culture media. Interestingly, we found a significant increase in most of the cytokines and chemokines analyzed. We also identified a significant increase in IL-6 secretion in LVs cultured in HD matrices as compared to LD matrices and we found that IL-6 secretion increases in a density dependent manner as we observed the increase in the 5mg/mL and 6 mg/mL (HD) matrices but not the 3mg/mL (LD) or 4 mg/mL matrices. Hence, these results indicate IL-6 increases in response of the increase in ECM density and demonstrate that LV respond to changes in the surrounding ECM density, suggesting that in dense ECM conditions endothelial cells are being activated.
Previous studies have shown that the increase in pro-inflammatory cytokine secretion results in endothelial barrier dysfunction68–70. In particular, endothelial cells have been shown to secrete IL-6 in inflammatory states (e.g., vascular damage), thereby altering vessel permeability55,56. For this reason, we next examined the permeability and barrier function of the LV in LD and HD matrices through diffusion assays using 70 kDa-Texas Red dextran. We determined that LV cultured in HD matrices were significantly more permeable than LV cultured in LD matrices, which has implications in the context of cancer metastasis. Based on the literature and our results, we hypothesized that the increase in permeability was a result of the increase in IL-6 secretion which in turn led to an increase in cell actin stress fibers and cell detachment, resulting in gaps in the LV.
IL-6 is of importance as it has been implicated with the regulation of LV barrier function in vitro70,71 and in promoting breast cancer72–76. In our model, blocking the IL-6 receptor mitigated vessel leakiness in HD, whereas leakiness was induced in LV cultured in LD matrices after treatment with IL-6. Thus, these results confirm that IL-6 is responsible for increasing LV permeability. Interestingly, treatment with 5 ng/mL of IL-6 did not induce leakiness in the LV cultured in LD matrices. This could indicate that a constant and localized source of IL-6 might be necessary to induce vessel permeability in an autocrine manner. The mitigation of HD-induced LV leakiness may have implications in breast cancer metastasis by reducing the potential of intravasation events through the stabilization of the endothelium. In addition to IL-6, the overexpression of chemokines that serve as chemotactic signals for cancer cells (e.g. CX3CL1, CXCL12) have previously been found to be secreted by lymphatic vessels, having an important contribution to cancer metastasis77–81. In our system we observed that an increased ECM density results in an increase in cytokine secretions, which in turn disrupted the endothelial barrier.
As for cancer cells, breast cancer cells have been shown to play a role in the conditioning of the lymphatic vasculature82,83. For example, a study reported that LECs support tumor growth in breast cancer84, demonstrating important interplay between lymphatic cells and breast cancer cells. To our knowledge, no study has investigated the effect of matrix density in a microenvironment that incorporates both LVs and cancer cells, despite the known correlation between dense matrices and lymph node metastasis in breast cancer26. Therefore, we leveraged our model to investigate the effect of the ECM density in breast cancer cell crosstalk with LVs, where we also found a significant increase in IL-6 secretion in HD matrices. Although IL-6 secretion increased in both matrices, we observed disruption of the LV in dense matrices only, which illustrates the importance of the matrix and cellular composition in organotypic models of cancer. Despite the apparent presence of gaps in the LVs co-cultured with breast cancer cells in dense matrices, we observed lower permeability coefficient levels as compared to monocultures. This discrepancy could be due to matrix remodeling exerted by the breast cancer cells. For example, the density of the matrix increases due to cell proliferation and reorganization of the collagen fibers by the cells, which may limit the diffusion of the dextran molecule through the matrix85. Another explanation for this discrepancy could be due to the crosstalk between the LVs and cancer cells. It is also important to note that the increase in IL-6 secretion in the co-culture within a LD matrix did not result in a permeability increase. Interestingly, the increase in IL-6 in LD matrix co-cultures did not reach the levels of LV monocultures cultured in HD matrices, suggesting that there might be a threshold of IL-6 that produces LV barrier destabilization.
There is ample literature demonstrating that IL-6 also has a role in in breast cancer cell migration28,86, growth and metastasis87. Specifically, a study demonstrated that breast cancer cells secrete IL-6 and educate LECs in pre-metastatic organs, facilitating breast cancer metastasis88 and revealing the significance of IL-6 as a therapeutic target in cancer therapy. Therefore, the 231-lymphatic co-culture model allowed us to test the effect of IL-6 receptor blocking, a potential therapeutic target, to mitigate LVs leakiness in the HD matrix. Although the increase in permeability was significantly reduced by supplementing the anti-IL-6R to the media, the effect was lower than in the LV monocultures and the permeability levels did not return to baseline levels. These results suggest that there may be other factors besides cancer cells and IL-6 that contribute to the permeability of the vessels, which is an important factor that allows metastasis.
Altogether, our findings demonstrate the usefulness of our model to dissect the contribution of different microenvironment components (e.g., ECM density and cancer cells) on LV biology. Therefore, the model allows the incorporation of different cancer cell types that represent distinctive types of cancers, which is a powerful tool to advance cancer research. For example, triple negative breast cancer (TNBC) is a highly aggressive and migratory cancer type and, in this study, the cell line MDA-MB-231 was used to represent TNBC. However, tumor progression, treatment and patient outcome depends on the specific breast cancer subtype. In fact, the heterogeneity of this disease is highlighted in a recently published study using an adaptation of this microfluidic lymphatic model. The study used the lymphatic model for co-cultures with MCF7 or MDA-MB-231 cells and revealed differential alteration of genes and functional changes (e.g., MCF-7 conditioning induced a leakier endothelium) in lymphatic vessels89. Therefore, future studies will focus on the interaction of different breast cancer subtypes with lymphatic vessels by incorporating cell lines representative of the different molecular subtypes of breast cancer. Our results demonstrate the utility of our model for investigating tumor-vessel crosstalk within different cells and microenvironments, and indicate that this model could be used to study differential features and mechanisms for distinct breast cancer subtypes as well as other cancers known to traffic through the lymph system.
Finally, our microfluidic in vitro model allowed us to mimic important aspects of the breast tumor microenvironment by including only a few key players in the tumor microenvironment. However, to increase the relevance of the model, future experiments should incorporate different components of the microenvironment to better recapitulate what is found in vivo. For example, we previously demonstrated that the incorporation of stromal cells such as fibroblasts (i.e., normal and cancer-associated fibroblasts) differentially regulate lymphatic vessels32. Interestingly, we revealed that CAFs induce the secretion of inflammatory cytokines and induce lymphatic vessel leakiness32. In terms of the matrix, collagens are the major components of the ECM90. However, most tissues have additional ECM proteins. The use of only collagen to form the matrices is a caveat of this study, although collagen was selected as a starting point due to the ease of use and high reproducibility among batches. Therefore, future experiments could increase the ECM relevance by incorporating other matrix proteins that are found in vivo9. In addition, due to the small number of cells required in our model, we have the potential to use primary patient cells (e.g., cancer cells and fibroblasts) to test translational relevance of our system. In the future, it would be interesting to decouple the effects of matrix density and stiffness on LVs. Taken together, these results demonstrated the capability of our model to investigate the effects of ECM density on LV physiology and tumor-lymphatic crosstalk, which can contribute to the understanding of breast cancer metastasis in the context of ECM mechanics and demonstrates the potential of the model for therapeutic assessment.
5. Conclusions
In this paper, we elucidated how a dense ECM matrix conditions the lymphatic vasculature toward an activated phenotype through the increase in secretion of chemokines and pro-inflammatory cytokines such as IL-6. Furthermore, we demonstrated how IL-6 secretion exacerbated vessel leakiness in LV monoculture and co-culture with breast cancer cells, both of which were mitigated by blocking the IL-6R. Therefore, our results provide a possible therapeutic target to inhibit breast cancer metastasis by modulating the lymphatic vasculature.
Supplementary Material
8. Acknowledgements
We acknowledge a fellowship from the UW-Madison Graduate Engineering Research Scholars program to KML. We also thank the NIH (R01EB010039 BRG) and the Wisconsin Head & Neck SPORE (P50 DE026787) for funding support. The authors would like to acknowledge the help of Sheena Kerr reviewing the manuscript.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/d0lc00099j
Conflicts of interest
David J. Beebe holds equity in Bellbrook Labs LLC, Tasso Inc., Salus Discovery LLC, Lynx Biosciences Inc., Stacks to the Future LLC, Turba LLC, and Onexio Biosystems LLC. David J. Beebe is a consultant for Abbott Laboratories.
9 References
- 1.Balkwill FR, Capasso M and Hagemann T, J. Cell Sci, 2012, 125, 5591–5596. [DOI] [PubMed] [Google Scholar]
- 2.Christian Frantz, Kathleen S and Weaver VM, J. Cell Sci, 2010, 14, 4195–4200. [Google Scholar]
- 3.Theocharis AD, Skandalis SS, Gialeli C and Karamanos NK, Adv. Drug Deliv. Rev, 2016, 97, 4–27. [DOI] [PubMed] [Google Scholar]
- 4.Balasubramanian PG, Chiquet-ehrismann R, Tucker RP and Adams JC, 2010, 21, 4300–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teti A, J. Am. Soc. Nephrol, 1992, 2, S83–7. [DOI] [PubMed] [Google Scholar]
- 6.Cox TR and Erler JT, Dis. Model. Mech, 2011, 4, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu P, Weaver VM and Werb Z, 2012, 196, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conklin MW and Keely PJ, Cell Adhes. Migr, 2012, 6, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomko LA, Hill RC, Barrett A, Szulczewski JM, Conklin MW, Eliceiri KW, Keely PJ, Hansen KC and Ponik SM, Sci. Rep, 2018, 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang M, Yuan J, Peng C and Li Y, 2014, 2871–2882. [DOI] [PMC free article] [PubMed]
- 11.Zhu G-G, Kauppila A, Risteli L, Mäkinen M, Stenbäck F and Risteli J, Cancer, 1995, 75, 1010–1017. [DOI] [PubMed] [Google Scholar]
- 12.Drifka CR, Tod J, Loeffler AG, Liu Y, Thomas GJ, Eliceiri KW and Kao WJ, Mod. Pathol, 2015, 28, 1470–1480. [DOI] [PubMed] [Google Scholar]
- 13.Huijbers IJ, Iravani M, Popov S, Robertson D, Al-Sarraj S, Jones C and Isacke CM, PLoS One, 2010, 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG and Keely PJ, BMC Med, 2008, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.R. a W. Chaffer Christine L, Nat. Med, 2013, 19, 179–92.23389618 [Google Scholar]
- 16.Velez DO, Tsui B, Goshia T, Chute CL, Han A, Carter H and Fraley SI, Nat. Commun, , 10.1038/s41467-017-01556-7. [DOI] [PMC free article] [PubMed]
- 17.Provenzano PP, Inman DR, Eliceiri KW and Patricia J, Matrix, 2009, 28, 4326–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris BA, Burkel B, Ponik SM, Fan J, Condeelis JS, Aguire-Ghiso JA, Castracane J, Denu JM and Keely PJ, EBioMedicine, 2016, 13, 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghajar CM, Chen X, Harris JW, Suresh V, Hughes CCW, Jeon L, Putnam AJ and George SC, 2008, 94, 1930–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kniazeva E and Putnam AJ, 2009, 179–187.
- 21.Edgar LT, Underwood CJ, Guilkey JE, Hoying JB and Weiss JA, 2014, 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamloo AMIRS, Ohammadaliha NEM, Eilshorn SACH and Auer AMYLB, 2016, 44, 929–941. [Google Scholar]
- 23.Podgrabinska Simona and Pascal Braun MS, Velasco Paula, Kloos Bryan, Pepper Michael S., Jackson David G., 10.1073/pnas.242401399. [DOI] [PMC free article] [PubMed]
- 24.M DM and Baluk P, Cancer Res, 2002, 62, 5381–5385. [PubMed] [Google Scholar]
- 25.Mierke CT, J. Biophys, 2009, 2008, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kakkad SM, Solaiyappan M, Argani P, Sukumar S, Jacobs LK, Leibfritz D, Bhujwalla Z and Glunde K, J. Biomed. Opt, , 10.1117/1.JBO.17.11.116017. [DOI] [PMC free article] [PubMed]
- 27.E. J. L. and Myron Moskowitz JH, Russell Peter, Fidler James, Sutorius Darryl, Cancer, 1975, 36, 2245–2250. [DOI] [PubMed] [Google Scholar]
- 28.Ran S, Volk L, Hall K and Flister MJ, Pathophysiology, 2010, 17, 229–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruyere F and Noel A, FASEB J, 2010, 24, 8–21. [DOI] [PubMed] [Google Scholar]
- 30.Ingram PN, Hind LE, Jiminez-Torres JA, Huttenlocher A and Beebe DJ, Adv. Healthc. Mater, 2018, 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiménez-Torres JA, Virumbrales-Muñoz M, Sung KE, Lee MH, Abel EJ and Beebe DJ, EBioMedicine, 2019, 42, 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gong MM, Lugo-Cintron KM, White BR, Kerr SC, Harari PM and Beebe DJ, Biomaterials, 2019, 214, 119225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bischel LL, Sung KE, Jiménez-Torres JA, Mader B, Keely PJ and Beebe DJ, FASEB J, 2014, 28, 4583–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huh D, Torisawa YS, Hamilton GA, Kim HJ and Ingber DE, Lab Chip, 2012, 12, 2156–2164. [DOI] [PubMed] [Google Scholar]
- 35.Bhatia SN and Ingber DE, Nat. Biotechnol, 2014, 32, 760–72. [DOI] [PubMed] [Google Scholar]
- 36.Jiménez-Torres J BD, Peery S, Sung K, 2016, 165, 255–269. [Google Scholar]
- 37.Bischel S-HLBJD, Lauren L; Lee, J. Lab Autom, 2012, 17, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker GM and Beebe DJ, Lab Chip, 2002, V, 131–134. [Google Scholar]
- 39.Ayuso JM, Gillette A, Lugo-Cintrón K, Acevedo-Acevedo S, Gomez I, Morgan M, Heaster T, Wisinski KB, Palecek SP, Skala MC and Beebe DJ, EBioMedicine, 2018, 37, 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huxley VH, Curry FE and Adamson RH, Am. J. Physiol, 1987, 252, H188–97. [DOI] [PubMed] [Google Scholar]
- 41.Wigle JT and Oliver G, EMBO J, 2002, 21, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanjay Kumar VM, Weaver, Cancer Metastasis Rev, 2009, 28, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fraley SI, Wu P-H, He L, Feng Y, Krisnamurthy R, Longmore GD and Wirtz D, Sci. Rep, 2015, 5, 14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bredfeldt JS, Liu Y, Pehlke CA, Conklin MW, Szulczewski JM, Inman DR, Keely PJ, Nowak RD, Mackie TR and Eliceiri KW, J. Biomed. Opt, 2014, 19, 016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie Z, Ghosh CC, Patel R, Iwaki S, Gaskins D, Nelson C, Jones N, Greipp PR, Parikh SM and Druey KM, Blood, 2012, 119, 4321–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Virumbrales-Muñoz M, Ayuso JM, Olave M, Monge R, De Miguel D, Martínez-Lostao L, Le Gac S, Doblare M, Ochoa I and Fernandez LJ, Sci. Rep, 2017, 7, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lavalley DJ and Reinhart-king CA, Adv. Regen. Biol, , 10.3402/arb.v1.25247. [DOI]
- 48.Yeh YT, Hur SS, Chang J, Wang KC, Chiu JJ, Li YS and Chien S, PLoS One, 2012, 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caroline KWE Schneider A, Rasband Wayne S., Nat Methods, 2012, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krishnan R, Klumpers DD, Park CY, Rajendran K, Trepat X, van Bezu J, van Hinsbergh VWM, Carman CV, Brain JD, Fredberg JJ, Butler JP and van Nieuw Amerongen GP, Am. J. Physiol. Physiol, 2011, 300, C146–C154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.John Huynh CAR-K, Nishimura Nozomi, Rana Kuldeepsinh, Peloquin John M., Califano Joseph P., Montague Christine R., King Michael R., Schaffer Chris B., Sci Transl Med, , 10.1038/mp.2011.182. [DOI] [PMC free article] [PubMed]
- 52.Neiva KG, Warner KA, Campos MS, Zhang Z, Moren J, Danciu TE and Nör JE, BMC Cancer, 2014, 14, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gopinathan G, Milagre C, Pearce OMT, Reynolds LE, Hodivala-Dilke K, Leinster DA, Zhong H, Hollingsworth RE, Thompson R, Whiteford JR and Balkwill F, Cancer Res, 2015, 75, 3098–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sprague AH and Khalil RA, Biochem. Pharmacol, 2009, 78, 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Desai TR, Leeper NJ, Hynes KL and Gewertz BL, J. Surg. Res, 2002, 104, 118–123. [DOI] [PubMed] [Google Scholar]
- 56.Alsaffar H, Martino N, Garrett JP and Adam AP, Am. J. Physiol. Physiol, , 10.1152/ajpcell.00235.2017. [DOI] [PubMed]
- 57.Weber E, Sozio F, Gabbrielli E and Rossi A, Transl. Vasc. Med. Pathog. Diagnosis, Treat, 2013, 5, 137–150. [Google Scholar]
- 58.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA and Weaver VM, Cancer Cell, 2005, 8, 241–254. [DOI] [PubMed] [Google Scholar]
- 59.Seo BR, Bhardwaj P, Choi S, Gonzalez J, Eguiluz RCA, Wang K, Mohanan S, Morris PG, Du B, Zhou XK, Vahdat LT, Verma A, Elemento O, Hudis CA, Williams RM, Gourdon D, Dannenberg AJ and Fischbach C, 2015, 7, 1–12. [Google Scholar]
- 60.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES and Weaver VM, Integr. Biol. (United Kingdom), 2015, 7, 1120–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scott Gehler JLL Baldassarre Massimiliano, Lad Yatish, Wozniak MA, Riching KM, Eliceiri KW, Weaver VM, Calderwood DA and and Keely PJ, Mol. Biol. Cell, 2009, 20, 3224–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bram Piersma VMW, Hayward Mary-Kate, BBA - Rev. Cancer, , 10.1016/j.bbcan.2020.188356. [DOI]
- 63.Deanfield JE, Halcox JP and Rabelink TJ, 2007, 1285–1295. [DOI] [PubMed]
- 64.Den Buijs JO, Musters M, Verrips T, Post JA, Braam B and Van Riel N, Am. J. Physiol. - Hear. Circ. Physiol, 2004, 287, 2651–2658. [DOI] [PubMed] [Google Scholar]
- 65.Asahara T, Murohara T, Sullivan A, Silver M, Van Der Zee R, Li T, Witzenbichler B, Schatteman G and Isner JM, Science (80-.)., 1997, 275, 964–967. [DOI] [PubMed] [Google Scholar]
- 66.Zhao J, Mitrofan CG, Appleby SL, Morrell NW and Lever AML, Stem Cells Int, , 10.1155/2016/1406304. [DOI] [PMC free article] [PubMed]
- 67.Woywodt A, Nephrol. Dial. Transplant, 2002, 17, 1728–1730. [DOI] [PubMed] [Google Scholar]
- 68.Campos SB, Ashworth SL, Wean S, Hosford M, Sandoval RM, Hallett MA, Atkinson SJ and Molitoris BA, Am. J. Physiol. Physiol, 2009, 296, F487–F495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chaitanya GV, Franks SE, Cromer W, Wells SR, Bienkowska M, Jennings MH, Ruddell A, Ando T, Wang Y, Gu Y, Sapp M, Mathis JM, Jordan PA, Minagar A and Alexander JS, Lymphat. Res. Biol, 2010, 8, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walter E Cromer DCZ, Zawieja Scott D., Tharakan Binu, F.A.H.A., Childs Ed W., Newell M. Karen, Angiogenesis, 2014, 17, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zawieja DC and Barrier CÁ, 2014, 395–406. [DOI] [PMC free article] [PubMed]
- 72.Dethlefsen C, Højfeldt G and Hojman P, Breast Cancer Res. Treat, 2013, 138, 657–664. [DOI] [PubMed] [Google Scholar]
- 73.Osuala KO, Sameni M, Shah S, Aggarwal N, Simonait ML, Franco OE, Hong Y, Hayward SW, Behbod F, Mattingly RR and Sloane BF, BMC Cancer, 2015, 15, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gallo M, Frezzetti D, Roma C, Chicchinelli N, Barbieri A, Arra C, Scognamiglio G, Botti G, De Luca A and Normanno N, Oncotarget, 2018, 9, 17543–17553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim HS, Jung M, Choi SK, Woo J, Piao YJ, Hwang EH, Kim H, Kim SJ and Moon WK, 2018, 1–13.
- 76.Hou L, Xie S, Li G, Xiong B, Gao Y, Zhao X, Hu J, Deng S and Jiang J, Basic Clin. Pharmacol. Toxicol, 2018, 123, 549–557. [DOI] [PubMed] [Google Scholar]
- 77.Tsang JYS, Ni YB, Chan SK, Shao MM, Kwok YK, Chan KW, Tan PH and Tse GM, Breast Cancer Res. Treat, 2013, 140, 495–504. [DOI] [PubMed] [Google Scholar]
- 78.Liang Y, Yi L, Liu P, Jiang L, Wang H, Hu A, Sun C and Dong J, J. Cancer, 2018, 9, 3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo F, Wang Y, Liu J, Mok SC, Xue F and Zhang W, Oncogene, 2016, 35, 816–826. [DOI] [PubMed] [Google Scholar]
- 80.Ahirwar DK, Nasser MW, Ouseph MM, Elbaz M, Cuitiño MC, Kladney RD, Varikuti S, Kaul K, Satoskar AR, Ramaswamy B, Zhang X, Ostrowski MC, Leone G and Ganju RK, Oncogene, 2018, 37, 4428–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mirisola V, Zuccarino A, Bachmeier BE, Sormani MP, Falter J, Nerlich A and Pfeffer U, Eur. J. Cancer, 2009, 45, 2579–2587. [DOI] [PubMed] [Google Scholar]
- 82.Harris AR, Perez MJ and Munson JM, BMC Cancer, 2018, 18, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen J, Lai Y, Chu P, Chan S, Wang L and Hung W, Cancers (Basel), , 10.3390/cancers11081120. [DOI] [PMC free article] [PubMed]
- 84.Lee E, Pandey NB and Popel AS, Sci. Rep, 2014, 4, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tilghman RW, Cowan CR, Mih JD, Koryakina Y, Gioeli D, Slack-Davis JK, Blackman BR, Tschumperlin DJ and Parsons JT, PLoS One, 2010, 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim S, Chung M and Jeon NL, Biomaterials, 2016, 78, 115–128. [DOI] [PubMed] [Google Scholar]
- 87.Jin K, Pandey NB and Popel AS, Breast Cancer Res, 2018, 20, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee E, Fertig EJ, Jin K, Sukumar S, Pandey NB and Popel AS, Nat. Commun, 2014, 5, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ayuso JM, Gong MM, Skala MC, Harari PM and Beebe DJ, Adv. Healthc. Mater, 2020, 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nissen NI, Karsdal M and Willumsen N, J. Exp. Clin. Cancer Res, 2019, 38, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.