

Abstract
PaaA is a RiPP enzyme which catalyzes the transformation of two glutamic acid residues within a substrate peptide into the bicyclic core of Pantocin A. Here for the first time, we use mRNA display techniques to understand RiPP enzyme-substrate interactions to illuminate PaaA substrate recognition. Additionally, our data revealed insight into the enzymatic timing of glutamic acid modification. The technique developed is quite sensitive and a significant advancement over current RiPP studies and opens the door to enzyme modified mRNA display libraries for natural product-like inhibitor pans.
Graphical Abstract
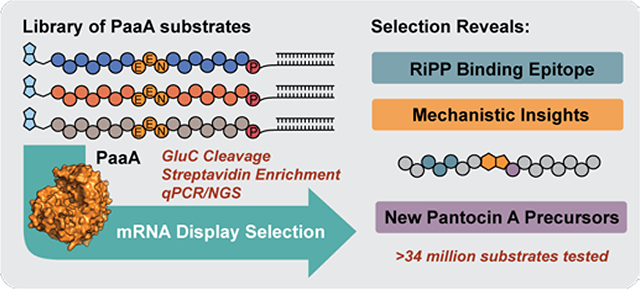
Ribosomally-synthesized and post-translationally modified peptide natural products (RiPPs) are an exciting family of natural products that have seen a surge in research due to exceptional versatility of their biosynthetic enzymes.1 Recent studies have shown that, in many cases, RiPP chemistry is guided by specific interaction between a RiPP recognition element (RRE) and the peptide substrate.2–6 The RRE binds one region of the substrate and feeds the remainder into the catalytic domain for modification. This modular strategy allows remarkable substrate promiscuity and, as a result, RiPP enzymes are increasingly being exploited to make complex libraries of peptide derivatives.7–16 Most recently, RiPPs have been paired with phage and yeast display to identify high affinity, RiPP-based peptide binders.17–19 While this work hints at the extraordinary promiscuity and generality of RiPP enzymes, verifying and measuring enzymatic modification of library members remains a significant challenge. Indeed, in many display libraries where chemical modification is used, including phage,20,21 mRNA display,22 and even DNA-encoded libraries (DELs),23,24 panning hits wind up being the most significant evidence that chemistry worked. In the case of RiPP enzymes, quantitative assessment and comparison of enzymatic modifications on large display libraries could inform broadly on substrate promiscuity and mechanistic features of these enzymes, leading the way to their more effective implementation in benchtop campaigns.
Inspired by several reports of high-throughput assays for proteases and ligases,25–28 we envisioned a coupled mRNA display assay that might allow expedient study of RiPP enzymology and promiscuity. mRNA display is a powerful peptide display technology where peptides are linked to their own encoding RNA during in vitro ribosomal translation.29 mRNA display allows the easy incorporation of non-natural amino acids by Flexizyme reprogramming,30 which could aid in our assay design and significantly expand library diversity one day. To substantiate mRNA display as a biochemical tool to study RiPPs, we first chose PaaA, an RRE-containing RiPP enzyme from the biosynthesis of the antibiotic Pantocin A (1).31 PaaA is a ThiF/E1-like activating enzyme that catalyzes the double dehydration/decarboxylation of two glutamic acid residues (E16 and E17) in substrate peptide, PaaP, to form the fused-bicyclic core of the active tripeptide natural product. Interestingly, the enzymatic mechanism may proceed by either of two imide intermediates to achieve the same product (Fig 1b).32 Since PaaA chemically modifies two glutamic acids, we could use the indiscriminate Glu-protease GluC to cleave unmodified substrate analogs in an mRNA display library and report on PaaA activity. This approach would confirm and measure PaaA modification of substrate peptide mutants and constitute a first application of RiPP enzymes to mRNA display libraries.
Figure 1. Pantocin A biosynthesis and mRNA display activity assay to study PaaA.

(a) Pantocin A gene cluster, including paaA, an ATP-dependent ThiF/E1-like enzyme; paaB, α-ketogluterate-dependent iron oxidase; paaC, efflux pump; and paaP, precursor peptide. (b) Potential mechanisms for PaaA catalyzed modification of PaaP. (c) Overview of mRNA display assay with N-terminal biotinylated (blue) PaaP linked to mRNA via puromycin “P”. Modified substrates are enriched by streptavidin (green) pulldown and analyzed by qPCR or next generation sequencing (NGS). “x” is any given peptide, “wt” is wild type. (d) E scores for each single mutant PaaP peptide analyzed after a single round of PaaA activity selection. Blue indicates sequence enrichment worse than WT and orange indicates sequence enrichment better than WT. Each square represents an average (n=3).
To implement this approach, we first sought to confirm that PaaA could modify an mRNA displayed PaaP substrate: we established a gel shift assay, wherein treatment of 35S-Met-labeled and mRNA displayed-PaaP with PaaA rendered the peptide resistant to GluC cleavage (Supplementary Fig. 1a). To further optimize reaction conditions with mRNA display substrates and allow for enrichment of modified peptides from larger libraries, we adapted this assay to include affinity purification and qPCR-based quantitation (Fig. 1c). This new iteration of the assay involves four key steps: 1) display of an N-terminal biotinylated PaaP substrate via Flexizyme codon reprogramming, 2) treatment with PaaA, 3) protease treatment with GluC to remove RNA tags from unmodified substrates, and finally 4) streptavidin enrichment of only modified substrates. The assay was optimized for GluC cleavage conditions, PaaA concentration, and reaction time (Supplementary Fig. 1b–d). With a functioning activity assay in hand we next designed a saturation mutagenesis single variant library (smSVL) to explore how point mutations along PaaP would affect PaaA activity. The library was treated with 1 μM PaaA for 5, 22.5, and 60 mins before GluC treatment and streptavidin purification. Recovered sequences were PCR amplified and submitted for next generation sequencing (NGS). The sequencing data was processed by calculating E scores (Supplementary Fig. 2),33 which are normalized values for success of each mutant compared to wild type (WT) (Fig.1d and Supplementary Fig 3). In this analysis a score > 1 is better than WT and a score < 1 is worse than WT. As an important assay control, we examined Glu mutants within the leader and follower. Because these mutations lie outside the core, we anticipate that they will go unmodified and thus be readily cleaved by GluC irrespective of PaaA activity at E16 and E17. Satisfyingly, these Glu mutants have consistently low E scores, demonstrating that the assay selection conditions are stringent (Fig 1d, grey arrow). Beyond this internal control four key trends are readily apparent. 1) First, PaaA is broadly tolerant of point mutations. Of the 26 positions within the leader and follower probed by saturation mutagenesis, 22 appear numb to mutation. This is especially evident in the follower sequence as no single mutations strongly inhibited PaaA processing. 2) While the leader sequence also shows high tolerance to mutations, four positions (F4, L7, R10, and I11) consistently enriched poorly when mutated (fig 1d, black arrows). With increasing PaaA incubation time this result lessened suggesting that these mutations slow the reaction but do not prevent it. However, aspartic acid replacements at these positions appear to severely inhibit PaaA processing (Fig 1d. and Supplementary Fig. 3ab). Given that PaaA exhibits an RRE domain, we speculate that this FXXLXXRI motif may be involved in substrate recognition at the RRE/peptide interface where backbone hydrogen bonding typically drives β-strand interactions and hydrophobic residues fill key hotspots.2–6 3) The smSVL data also shows that while E16 mutations were highly susceptible to GluC cleavage, E17 mutants were strongly protected implying modification of E16 (Fig 1d, blue arrows). These results suggest that PaaA may exhibit a preference for activation of E16 over E17 which may further play a role in the mechanism of modification (Fig 1b). 4) Finally, core residue N18 also displays a high tolerance to mutation (Fig 1d, red arrow), suggesting that PaaA can be used to make new Pantocin A analogs.
To validate selection results, we chose several mutants for confirmation by in vitro enzymatic assay and mass spectrometric characterization. Each substrate was translated with NEB PURExpress, then treated with 1 μM PaaA, and reaction products analyzed by MALDI-TOF. As seen in the smSVL data, PaaA proved promiscuous to point mutations within the leader and follower peptides (Fig 2a, Supplementary Fig 10, Supplementary Table 1). Notably, N18D was processed to the final bicycle demonstrating that PaaA can be used for preparation of novel Pantocin A analogs. Also, aligned with the smSVL data, qualitative analysis shows that PaaA struggles to process F4D, L7D, R10D, and I11D mutants, and the quadruple alanine mutant went completely unmodified. In contrast a quadruple mutant outside these four residues was accepted (K3A, T6A, S8A, T15A). These data further support that PaaA has a wide tolerance for single mutations and strengthens the importance of F4, L7, R10, and I11 for PaaA reactivity. To investigate whether these four residues are involved in PaaA recognition, we prepared synthetic variants of the N-terminal 12 residues of PaaP and a TAMRA-labeled fluorescent probe that could be used for competitive fluorescence polarization (FP) assays. In these FP assays, only WT and T6D peptides competed effectively with TAMRA-WT for PaaA binding (Fig. 2b and Supplementary Fig. 4). This was further substantiated by isothermal titration calorimetry (ITC, Fig. 2b and Supplementary Fig. 5). Together, the MALDI-TOF data and binding experiments confirm that PaaA is a promiscuous RiPP enzyme and that these four residues are essential for PaaP binding and subsequent processing. A BLAST of available PaaP homologs shows significant conservation across the central third of the precursor peptide (L7-Y21, Supplementary Fig. 6). F4, L7, R10, and I11 are heavily conserved, as are many others, thus highlighting the power of this approach in effectively discriminating between essential residues where bioinformatics could not.
Figure 2. PaaA substrate promiscuity, binding, and processing.

(a) MALDI-TOF analysis of PaaA activity on PaaP mutants. Total extracted ion integration areas for substrate remaining “S” and observed products “P” were summated and used to calculate percent substrate or products for display in a heatmap. (b) PaaA binding to PaaP measured by competitive fluorescence polarization (FP, mean and error for n=3) and isothermal calorimetry (ITC, mean and error for n=2). (c, d) MALDI-TOF analysis of PaaA catalyzed dehydration (−18 Da) of PaaP core E mutants. (e) PaaP A13C was prepared by native chemical ligation with 13C-labeled E17. Orange dots indicate labeled carbons. Reaction of E17 labeled PaaP A13C with PaaA leads to a mass shift of 81 Da indicating loss of a single 13C atom due to E17 decarboxylation. (f) Weblogo analysis of 996 most enriched sequences with PaaP NNK 6.
We next turned to the difference in enrichment between E16 and E17 mutants (Fig 1d, blue arrows). To better understand this result, we first validated several core E mutants in MALDI-TOF assays. Notably, as exemplified by E17A and E17Q (Fig 2c), E17 mutants were readily modified to a single dehydration product, suggesting partial processing to putative imide intermediates (e.g. Fig. 1b). In good agreement with the selection data, only hydrophobic mutations to E16 are converted to this intermediate (Fig. 2d and Supplementary Table 2). The mechanism of PaaA can be reasonably written from a first step condensation of either E16 or E17 and both routes could yield the same bicyclic Pantocin A core (Fig. 1b and Supplementary Fig. 7). If E16 is condensed first, then E17 must undergo decarboxylation and vice versa. The general rejection of E16 mutations suggests that the E16 side chain is the preferred initial substrate. To confirm this biosynthetic timing, we prepared a PaaA substrate selectively 13C-enriched at E17 (Supplementary Fig. 8). Treatment with PaaA transformed the peptide into the product with a mass shift consistent with loss of a single 13C label (Fig 2e, E17 Decarboxylation, Supplementary Table 3). The loss of a 13C label agrees with a mechanism in which E16 is modified first followed by E17 cyclization and decarboxylation.
The smSVL experiment proved informative for studying point mutations, but the power of mRNA display is its capacity to screen much larger libraries, containing multiple simultaneous mutations. To show that RiPPs and mRNA display might be compatible with larger and more diverse libraries, we prepared a ~34 million-member library where the 6 positions from T6-I11 were simultaneously randomized with NNK codons. This library was treated with 1 μM PaaA before purification, GluC treatment, and streptavidin pull-down. After enrichment, the recovered DNA was PCR amplified, submitted for NGS and analyzed by Weblogo. Significantly, positions 7, 10, and 11 showed strong enrichment to a generalized version of the natural epitope evinced by smSVL experiments: FXXBXXRB (B = V, L, or I, Fig. 2f). Additionally, this data shows that other residues are significantly less important for substrate recognition and processing. Such broad permissiveness has previously been hypothesized in RRE-substrate interactions but not validated to this extent.
In summary, we successfully deployed an mRNA display-based enzyme activity assay to study the RiPP enzyme PaaA. This assay provides rapid insight into the broad promiscuity, sequence dependent substrate recognition, and residue specific processing of the PaaP core all in one set of experiments. The results suggest that PaaA is broadly promiscuous outside of its core and binding epitope and might be readily adapted to synthesize new Pantocin analogs or incorporate the indolizidinone core into peptide libraries.34,35 The present version of this assay is limited to reporting on glutamate modification because of reliance on GluC, but future iterations might probe other amino acids by exploiting alternative proteases or biorthogonal chemistries.36 mRNA display is particularly well-suited to broadly probe peptide post-translational enzymatic chemistry in this manner because of ready introduction of non-natural functionality through Flexizyme codon reprogramming. Perhaps most importantly, this work demonstrates compatibility of a RiPP enzyme, here PaaA, with a C-terminal mRNA display tag, suggesting that others might also be used in this manner. These results ultimately pave the way for using RiPP enzymes to transform mRNA display libraries into more natural product-like molecules for inhibitor discovery.
Supplementary Material
ACKNOWLEDGMENT
We thank C. Neumann and B. Li for informative discussions. SRF is recipient of a joint NSF/JSPS EAPSI fellowship. This work was supported by NIH Grant GM125005 (AAB).
Footnotes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Supporting Information
Experimental details, synthetic schemes, figures available at http://pubs.acs.org
REFERENCES
- (1).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; Cotter PD; Craik DJ; Dawson M; Dittmann E; Donadio S; Dorrestein PC; Entian K-D; Fischbach MA; Garavelli JS; Göransson U; Gruber CW; Haft DH; Hemscheidt TK; Hertweck C; Hill C; Horswill AR; Jaspars M; Kelly WL; Klinman JP; Kuipers OP; Link AJ; Liu W; Marahiel MA; Mitchell DA; Moll GN; Moore BS; Müller R; Nair SK; Nes IF; Norris GE; Olivera BM; Onaka H; Patchett ML; Piel J; Reaney MJT; Rebuffat S; Ross RP; Sahl H-G; Schmidt EW; Selsted ME; Severinov K; Shen B; Sivonen K; Smith L; Stein T; Süssmuth RD; Tagg JR; Tang G-L; Truman AW; Vederas JC; Walsh CT; Walton JD; Wenzel SC; Willey JM; van der Donk WA Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 2012, 30 (1), 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Koehnke J; Mann G; Bent AF; Ludewig H; Shirran S; Botting C; Lebl T; Houssen WE; Jaspars M; Naismith JH Structural Analysis of Leader Peptide Binding Enables Leader-Free Cyanobactin Processing. Nat. Chem. Biol. 2015, 11 (8), 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Grove TL; Himes P; Hwang S; Yumerefendi H; Bonanno JB; Kuhlman B; Almo SC; Bowers AA Structural Insights into Thioether Bond Formation in the Biosynthesis of Sactipeptides. J. Am. Chem. Soc. 2017, 139 (34), 11734–11744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ortega MA; Hao Y; Zhang Q; Walker MC; van der Donk WA; Nair SK Structure and Mechanism of the tRNA-Dependent Lantibiotic Dehydratase NisB. Nature 2015, 517 (7535), 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Burkhart BJ; Hudson GA; Dunbar KL; Mitchell DA A Prevalent Peptide-Binding Domain Guides Ribosomal Natural Product Biosynthesis. Nat. Chem. Biol. 2015, 11 (8), 564–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Regni CA; Roush RF; Miller DJ; Nourse A; Walsh CT; Schulman BA How the MccB Bacterial Ancestor of Ubiquitin E1 Initiates Biosynthesis of the Microcin C7 Antibiotic. EMBO J. 2009, 28 (13), 1953–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Burkhart BJ; Kakkar N; Hudson GA; van der Donk WA; Mitchell DA Chimeric Leader Peptides for the Generation of Non-Natural Hybrid RiPP Products. ACS Cent. Sci. 2017, 3 (6), 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fleming SR; Bartges TE; Vinogradov AA; Kirkpatrick CL; Goto Y; Suga H; Hicks LM; Bowers AA Flexizyme-Enabled Benchtop Biosynthesis of Thiopeptides. J. Am. Chem. Soc. 2019, 141 (2). 758–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Himes PM; Allen SE; Hwang S; Bowers AA Production of Sactipeptides In Escherichia Coli:Probing the Substrate Promiscuity of Subtilosin A Biosynthesis. ACS Chem. Biol. 2016, 11 (6), 1737–1744. [DOI] [PubMed] [Google Scholar]
- (10).Hegemann JD; Bobeica SC; Walker MC; Bothwell IR; van der Donk WA Assessing the Flexibility of the Prochlorosin 2.8 Scaffold for Bioengineering Applications. ACS Synth. Biol. 2019, 8 (5), 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Islam MR; Shioya K; Nagao J; Nishie M; Jikuya H; Zendo T; Nakayama J; Sonomoto K Evaluation of Essential and Variable Residues of Nukacin ISK-1 by NNK Scanning. Mol. Microbiol. 2009, 72 (6), 1438–1447. [DOI] [PubMed] [Google Scholar]
- (12).Young TS; Dorrestein PC; Walsh CT Codon Randomization for Rapid Exploration of Chemical Space in Thiopeptide Antibiotic Variants. Chem. Biol. 2012, 19 (12), 1600–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pan S; Link JA Sequence Diversity in the Lasso Peptide Framework: Discovery of Functional Microcin J25 Variants with Multiple Amino Acid Substitutions. J. Am. Chem. Soc. 2011, 133 (13), 5016–5023. [DOI] [PubMed] [Google Scholar]
- (14).Goto Y; Ito Y; Kato Y; Tsunoda S; Suga H One-Pot Synthesis of Azoline-Containing Peptides in a Cell-Free Translation System Integrated with a Posttranslational Cyclodehydratase. Chem. Biol. 2014, 21 (6), 766–774. [DOI] [PubMed] [Google Scholar]
- (15).Ozaki T; Yamashita K; Goto Y; Shimomura M; Hayashi S; Asamizu S; Sugai Y; Ikeda H; Suga H; Onaka H Dissection of Goadsporin Biosynthesis by in Vitro Reconstitution Leading to Designer Analogues Expressed in Vivo. Nat. Comm. 2017, 8, 14207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wever W; Bogart J; of the American BA Identification of Pyridine Synthase Recognition Sequences Allows a Modular Solid-Phase Route to Thiopeptide Variants. J. Am. Chem. Soc. 2016, 138, (41), 13461–13464. [DOI] [PubMed] [Google Scholar]
- (17).Hetrick KJ; Walker MC; van der Donk WA Development and Application of Yeast and Phage Display of Diverse Lanthipeptides. ACS Cent. Sci. 2018, 4 (4), 458–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Yang X; Lennard KR; He C; Walker MC; Ball AT; Doigneaux C; Tavassoli A; van der Donk WA A Lanthipeptide Library Used to Identify a Protein–Protein Interaction Inhibitor. Nat. Chem. Biol. 2018, 14 (4), 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Urban JH; Moosmeier MA; Aumüller T; Thein M; Bosma T; Rink R; Groth K; Zulley M; Siegers K; Tissot K; Moll GN; Prassler J Phage Display and Selection of Lanthipeptides on the Carboxy-Terminus of the Gene-3 Minor Coat Protein. Nat. Comm. 2017, 8 (1), 1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tjhung KF; Kitov PI; Ng S; Kitova EN; Deng L; Klassen JS; Derda R Silent Encoding of Chemical Post-Translational Modifications in Phage-Displayed Libraries. J. Am. Chem. Soc. 2016, 138 (1), 32–35. [DOI] [PubMed] [Google Scholar]
- (21).He B; Tjhung KF; Bennett NJ; Chou Y; Rau A; Huang J; Derda R Compositional Bias in Naïve and Chemically-Modified Phage-Displayed Libraries Uncovered by Paired-End Deep Sequencing. Sci. Rep. 2018, 8, 1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hofmann FT; Szostak JW; Seebeck FP In Vitro Selection of Functional Lantipeptides. J. Am. Chem. Soc. 2012, 134 (19), 8038–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Malone ML; Paegel BM What Is a “DNA-Compatible” Reaction? ACS Comb. Sci. 2016, 18 (4), 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li Y; Luca R; Cazzamalli S; Pretto F; Bajic D; Scheuermann J; Neri D Versatile Protein Recognition by the Encoded Display of Multiple Chemical Elements on a Constant Macrocyclic Scaffold. Nat. Chem. 2018, 10 (4), 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sugimura Y; Yokoyama K; Nio N; Maki M; Hitomi K Identification of Preferred Substrate Sequences of Microbial Transglutaminase from Streptomyces Mobaraensis Using a Phage-Displayed Peptide Library. Arch. Biochem. Biophys. 2008, 477 (2), 379–383. [DOI] [PubMed] [Google Scholar]
- (26).Kretz CA; Dai M; Soylemez O; Yee A; Desch KC; Siemieniak D; Tomberg K; Kondrashov FA; Meng F; Ginsburg D Massively Parallel Enzyme Kinetics Reveals the Substrate Recognition Landscape of the Metalloprotease ADAMTS13. Proc. Natl. Acad. Sci. 2015, 112 (30), 9328–9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yin J; Straight PD; Hrvatin S; Dorrestein PC; Bumpus SB; Jao C; Kelleher NL; Kolter R; Walsh CT Genome-Wide High-Throughput Mining of Natural-Product Biosynthetic Gene Clusters by Phage Display. Chem. Biol. 2007, 14 (3), 303–312. [DOI] [PubMed] [Google Scholar]
- (28).Matthews D; Wells J Substrate Phage: Selection of Protease Substrates by Monovalent Phage Display. Science 1993, 260 (5111), 1113–1117. [DOI] [PubMed] [Google Scholar]
- (29).Huang Y; Wiedmann M; Suga H RNA Display Methods for the Discovery of Bioactive Macrocycles. Chem. Rev. 2018, 119, 10360–10391 [DOI] [PubMed] [Google Scholar]
- (30).Goto Y; Katoh T; Suga H Flexizymes for Genetic Code Reprogramming. Nat. Protoc. 2011, 6 (6), 779–790. [DOI] [PubMed] [Google Scholar]
- (31).Smits TH; Duffy B; Blom J; Ishimaru CA; Stockwell VO Pantocin A, a Peptide-Derived Antibiotic Involved in Biological Control by Plant-Associated Pantoea Species. Arch. Microbiol. 2019, 201 (6), 713–722. [DOI] [PubMed] [Google Scholar]
- (32).Ghodge SV; Biernat KA; Bassett S; Redinbo MR; Bowers AA Post-Translational Claisen Condensation and Decarboxylation En Route to the Bicyclic Core of Pantocin A. J. Am. Chem. Soc. 2016, 138 (17), 5487–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Rogers JM; Passioura T; Suga H Nonproteinogenic Deep Mutational Scanning of Linear and Cyclic Peptides. Proc. Natl. Acad. Sci. 2018, 115 (43), 10959–10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Atmuri PN; Lubell WD Stereo- and Regiochemical Transannular Cyclization of a Common Hexahydro-1H-Azonine to Afford Three Different Indolizidinone Dipeptide Mimetics. J. Org. Chem. 2020, 85 (3), 1340–1351. [DOI] [PubMed] [Google Scholar]
- (35).Cluzeau J; Lubell WD Design, Synthesis, and Application of Azabicyclo[X.Y.0]Alkanone Amino Acids as Constrained Dipeptide Surrogates and Peptide Mimics. Biopolymers 2005, 80 (2–3), 98–150. [DOI] [PubMed] [Google Scholar]
- (36).Jetson RR; Krusemark CJ Sensing Enzymatic Activity by Exposure and Selection of DNA‐Encoded Probes. Angew. Chem. Int. Ed. 2016, 55 (33), 9562–9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.