

Abstract

New palladium (Pd)II and platinum (Pt)II complexes (C1–C5) from the Schiff base ligands, R-(phenyl)methanamine (L1), R-(pyridin-2-yl)methanamine (L2), and R-(furan-2-yl)methanamine (L3) (R-(E)-N-((1H-pyrrol-2-yl) methylene)) are herein reported. The complexes (C1–C5) were characterized by FTIR, 1H and 13C NMR, UV–vis, and microanalyses. Single-crystal X-ray crystallographic analysis was performed for the two ligands (L1–L2) and a Pt complex. Both L1 and L2 belong to P21/n monoclinic and P-1 triclinic space systems, respectively. The complex C5 belongs to the P21/c monoclinic space group. The investigated molar conductivity of the complexes in DMSO gave the range 4.0–8.8 μS/cm, suggesting neutrality, with log P values ≥ 1.2692 ± 0.004, suggesting lipophilicity. The anticancer activity and mechanism of the complexes were investigated against various human cancerous (Caco-2, HeLa, HepG2, MCF-7, and PC-3) and noncancerous (MCF-12A) cell lines using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Apopercentage assays, respectively. C5 demonstrated strong DNA-binding affinity for calf thymus DNA (CT-DNA) with a binding constant of 8.049 × 104 M–1. C3 reduced cell viability of all the six cell lines, which included five cancerous cell lines, by more than 80%. The C5 complex also demonstrated remarkably high selectivity with no cytotoxic activity toward the noncancerous breast cell line but reduced the viability of the five cancerous cell lines, which included one breast cancer cell line, by more than 60%. Further studies are required to evaluate the selective toxicity of these two complexes and to fully understand their mechanism of action.
Introduction
According to WHO, Cancer is the second leading cause of death globally and is responsible for an estimated 9.6 million deaths in 2018.1 Globally, about 1 in 6 deaths is due to cancer.2 New anticancer agents, which can effectively treat this disease with minimal side effects, remain of primary importance to the world research community.3−5
The development of drug resistance and disease relapse are major challenges in anticancer drug development.6 This factor is of crucial significance in the world where this disease continues to burden global health care systems and escalate the mortality rates.7 Metals have long been useful in treatment of various ailments known to man.8 Because of their unique properties, metal complexes, especially those containing transition metals have gained tremendous attention as anticancer agents.9 Most recently, the metal complexes derived from pyrrole moieties have shown good antiproliferative activity against lymphocytic, cervical, and breast cancer cells.10−12 A series of pyrrole-based ferrocene complexes were also reported to be cytotoxic against breast cancer (MCF-7),13 colon cancer (HT-29) cell lines, and noncancerous breast cells (MCF-10A).14 Equally, the known cancer drug to date, cisplatin has undergone various derivatization. The analogues containing bulky groups have displayed enhanced cytotoxicity profiles against cisplatin-resistant cells. This led to a conclusion that the steric shielding of the metal center can regulate ligand substitution and consequently increase an efficiency to DNA binding.15 The binding interactions is best followed by spectroscopy studies, where an observation of the alterations in the λmax absorption values appear as a result of π–π* intraligand transitions, ligand → metal/metal → ligand charge transfers, or on the d–d transitions.16 During any of these mentioned processes, the DNA concentration is serially increased to the metal complex. Various modes of binding are then deduced from the spectral response, whereby an intercalative manner of interaction is specified by a hypochromic shift, bathochromic shift, or hypsochromism.17 Intercalation of a complex with the base pairs of DNAs is as a result of overlaps of π*-antibonding orbitals of the ligands bound to the complex with π-bonding orbitals of the DNA base pairs.18 This decreases the probable transitions, and the effect is observed as a hypochromic shift.19 On the other hand, a hyperchromic shift suggests electrostatic mode of binding. The spectral shift in the metal complex absorption is correlated to the binding strength from which the kinetics can then be determined.20 The investigation of drug–DNA interaction is important in understanding the molecular mechanism of drug action and for the design of the specific DNA-targeted drug.
Although the Schiff base ligands are reported in literature for C–C coupling reactions,21 their use in Pt or Pd complexes in anticancer studies has been limitedly studied, particularly, the pyrrole based Schiff bases. In this regard, the ligands and their corresponding Pd(II) and Pt(II) complexes were synthesized and characterized followed by investigation of their DNA binding and anticancer activities. The motivation behind this study was two-fold. First, these complexes have square planar geometry analogous to the common anticancer drug, cisplatin, but bears sterically bulky substituents around the metal center. This is a strategy reported in literature for prevention of an axial approach to the metal atom from the z2 direction, thus hindering the formation of a trigonal bipyramid intermediate, which leads to ligand substitution.22 In this way, the sterically bulky groups shield the labile ligands, therefore reducing the rate of hydrolysis and substitution long enough for the complexes to bind to the desired target. Second, the trans-geometry complexes have not been widely explored as anticancer agents despite literature reports that they could exhibit cytotoxicity equivalent to or greater than that of cisplatin.23−25
Results and Discussion
Synthesis of Organometallic Complexes
A series of pyrrole-based ligands and their corresponding Pd(II) and Pt(II) complexes are synthesized, as shown in Scheme 1. The ligands (L1–L3) were synthesized by standard Schiff base condensation reaction of pyrrole-2-carboxaldehyde either with benzyl amine (L1), 2-furanmethyl amine (L2), or 2-picolyamine (L3), under reflux overnight. To the best of our knowledge, this is the first time that the crystal structures of L1 and L2 are reported. The metal complexes (C1–C5) were obtained through the reaction of Pd(cod)Cl2, Pd(cod)ClMe, or Pt(DMSO)2Cl2 precursors with a solution of ligand in DCM at room temperature. The complexes (C1 and C2) were formed through a reaction of 1:2 (metal/ligand) molar ratios using ligands L1 and L2, while C3 and C4 were formed from a 1:1 M ratio using L3 and L1, respectively, as shown in Error! Reference source not found.. Because of steric factors, the Pd complexes binding at 1:2 ratio (metal/ligand) are believed to be of trans-geometry. Similar work was reported on the bis-ferrocenyl Schiff base complexed with Pd which confirmed a monodentate binding of the Schiff bases to the metal center through the imine-N without involving the heteroatoms on the rings.26 This resulted in a square planar geometry at the Pd center. These ratios were confirmed by elemental analysis.
Scheme 1. Synthetic Routes for the Ligands and Their Corresponding Complexes; (i) Pd(cod)ClMe, DCM, Room Temperature, 12 h; (ii) Pt(DMSO)2Cl2, DCM, Room Temperature; (iii) and (iv) Pd(cod)Cl2, DCM, Room Temperature, 12 h; (v) Crystals Grown by Slow Evaporation of a DCM Solution of C4.

The elemental analysis showed close agreement with the theoretical calculations confirming the proposed structures of the compounds. An interesting observation was made for the Pt complex which was shown to exist either as a monomer or a chlorobridged dimer. A chlorobridged dimer (C4) was confirmed by elemental analysis but the structure obtained after growing single crystals corresponded to a monomer (C5). This suggests that the existence of the compound in the amorphous form differed from the crystalline state, whereby the crystal lattice constraints affect a major conformational rearrangement of the individual molecules favoring the free monomer.27
Characterization of the Complexes
Fourier Transform Infrared Spectra
The Fourier transform infrared (FTIR) analysis for the compounds was performed using potassium bromide (KBr) pellets. Comparisons were drawn between the transmission frequencies for the azomethine functional groups and those of free ligands. The IR spectrum for the ligands showed clear imine C=N (stretch) vibration band which occurred between 1636 and 1644 cm–1. Similar azomethine bands for the complexes occurred between 1592 and 1628 cm–1. The shift was an indication of chelation of Pd or Pt through the imine nitrogen, and the magnitude could be attributed to the different ligands attached to the metal center and to the groups attached to the azomethine region. In general, the shifting to lower frequencies is ascribed to the pi-back donation from metal d-orbitals to empty pi-antibonding orbitals of CH=N.28 A characteristic peak at 696 cm–1 for platinum complexes and at 598 cm–1 for Pd complexes appeared and was ascribed to the ν[M – N] coordination. The FTIR spectra are shown in Figures S1–S7 in the Supporting Information.
1H-NMR Spectra
The 1H NMR spectra for the complexes also showed a similar trend in the shifts in frequency of the methylene protons compared to the free ligands. The peak because of CH=N– in C1 and C4 occurred at 7.41 and 7.81 ppm, respectively, compared to 8.15 ppm in the parent ligand L1. In complex C2, the peak occurred at 7.69 ppm compared to 8.12 ppm in the parent ligand, L2. In complex C3, the methylene peak appeared at 7.69 ppm compared to 8.57 ppm in the parent ligand L3. This up-field shifts of 0.74, 0.34, and 1.04 ppm in C1, C3, and C4, respectively, to lower frequencies is because of pi-electrons of the azomethine group shifting toward the d-orbitals of metal leading to deshielding of the azomethine proton29,30 as a result of coordination to the azomethine nitrogen. The 1H-NMR spectra are shown in Figures S8–S13 in the Supporting Information.
13C-NMR Spectra
Similar trends in shifts to higher frequencies were observed in the 13C-NMR peaks for the compounds. The 13C-NMR spectra are shown in Figures S14–S18 in the Supporting Information.
Electronic Spectra
The UV–vis spectra of the ligands and the Pd complexes in dimethyl sulfoxide (DMSO) were measured at the 200–800 nm range. The molar absorptivity for all the compounds was determined using UV titrations. Ligand spectra showed peaks corresponding to intraligand, spin-allowed pi → π* centered on the aromatic units in addition to n → π*. These transitions had molar extinction coefficients ranging from 103 to 104 L mol–1 cm–1. On the other hand, the UV–vis spectra for all the complexes showed two peaks corresponding to a combination of the spin allowed 1A1g → 1B1g transitions and dπ–π*ligand transitions with molar extinction coefficients ranging from 103 to 104 L mol–1 cm–1. This transition is characteristic of square planar geometry. The UV–vis spectra are shown in Figures S26–S32 in the Supporting Information. Representative spectra for complex C1 are shown in Figure 1.
Figure 1.

Representative spectra; (i) 1H NMR for C1, (ii), 13C-NMR for C1, and (iii) LC–MS for Complex C1.
Molar Conductance
The molar conductance was measured for the complexes in DMSO and showed values ranging from 4.0 to 8.2 S cm2 mol L–1. This is in agreement with other reports in the literature, which indicates a less than 30 S cm2 mol L–1 as an indication of nonconductance. This confirms that these types of complexes are nonelectrolytic in solution.31
Partition Coefficient
The partition coefficient was determined as log P values by the UV–vis method. The values were 1.3953 ± 0.0065 for C1, 1.3816 ± 0.0032 for C2, 1.2692 ± 0.004 for C3, and 1.3072 ± 0.0082 for C5, indicating a more than 20-times partitioning of the compounds in the oil layer compared to the water layer. This implies that the compounds can be suitable for oral administration.32
Electrochemical Properties of the Pd and Pt Complexes
Electrochemical studies were performed on C2, C3, and C5. The cyclic voltammograms are shown in Figure 2 below.
Figure 2.

Cyclic voltammograms for C2 and C3 showing irreversible reduction peaks.
Complex C2 exhibits two irreversible reduction peaks at −0.57 V and at −1.7 V, while C3 showed reduction peaks with no anodic peaks present at −0.57 V and at −1.47 V (Figure 2). The observed processes most likely correspond to a metal-centered one-electron reduction process for the two peaks as shown in eqs 1 and 2.
| 1 |
| 2 |
The cyclic voltammetry for the Pt complexes was performed at a scan rate of 200 mV/s at pH 7.0 versus Ag/AgCl, glassy carbon working electrode, and Pt wire counter electrode. As shown in Figure 3, one peak was observed at −0.5 V which was credited to the diffusion-controlled H+ reduction to H2 which occurs around −0.41 V at pH 7 versus Ag/AgCl.33
Figure 3.

Cyclic voltammograms for C5 showing diffusion-controlled H-reduction.
In physiological systems, the redox stability appears within an accessible window of potential ranging between −0.4 and +0.8 V versus normal hydrogen electrode (NHE).34 These values correspond to the potentials for the main reducing agents in biological systems; NADPH with approximately −0.38 V and oxygen at around +0.815 V, at pH 7.0 versus NHE.23 Typically, all redox processes with a bigger or lesser value of potential compared to this physiological range cannot happen in the biological setting. However, there are other routes by which metal complexes interfere with cellular redox reactions through interaction via their redox centers either on the metal or ligand or through coordinating to biomolecules which facilitate cellular redox mechanisms.35
Stability Studies
Stability tests were performed for C1 and C5 in DMSO-d6 over a 72 h period in the presence of 200 μL D2O. 1HNMR scans were conducted at 6 h intervals to monitor any changes in proton environments. There were no shifts in peaks or any other change in spectra over the 72 h period. This indicated that the compounds were stable in solution as shown in the 1H-NMR plots and could be biocompatible.26 The spectra are shown in Figure S33a–c in the Supporting Information.
X-ray Crystal Structure Analysis for Ligands L1 and L2 and C5 Complex
Crystals suitable for X-ray crystallography were grown for L1 and L2 and for the C5 Pt complex. The compounds crystallize in the monoclinic P21/n space system for L1, P-1 for L3, and P21/c for C5.
The crystal data for the four compounds are shown in Table 1.
Table 1. Single-Crystal Data and Structure Refinement Parameters for L1, L2, and C5.
| compound | L1 | L2 | C5 |
|---|---|---|---|
| formula | C12H12N2 | C10H10N2O | C14H18Cl2N2OPtS |
| space group | P21/n | P-1 | P21/c |
| cell lengths | a = 9.8330(3) b = 17.3251(7) c = 11.7851(4) | a = 4.8147(1) b = 9.1561(3) c = 10.0726(3) | a = 8.0734(1) b = 18.0661(3) c = 11.5367(2) |
| cell angles | α 90 | α 96.269(1) | α = 90 |
| β 94.219(2) | β 97.570(2) | β = 94.229(1) | |
| γ 90 | γ 94.171(1) | γ = 90 | |
| cell volume | 2002.24 | 435.82(2) | 1678.10(5) |
| Z | 8 | 2 | 4 |
| temperature | 100 K | 100 K | 100 K |
The structure and ORTEP ellipsoid diagram for L1 is shown in Figure 4. The ligand exists as a molecular pair. Stabilization within the molecule is through intermolecular hydrogen bonding, between the imine nitrogen of one molecule and the hydrogen attached to the pyrrole nitrogen of the second molecule. The intermolecular bond distances are 2.136 and 2.193 Å. The intermolecular bond angles subtended by the H-bonded hydrogen are 161.41° for N2–H1–N3 and 162.04° for N1–H2–N4. The pyrrole and the benzyl rings lie at a torsion angle of 123.57° for one molecule and 118.97° for the second molecule.
Figure 4.

ORTEP Ellipsoids for L1 at 50% probability showing the intramolecular stabilization through H-bonding.
The ORTEP plot for L2 is shown in Figure 5a,b. The crystal is stabilized by intermolecular hydrogen bonding of the hydrogens attached to imine and pyrrole nitrogen of adjacent molecules but facing away from the center of the molecule to the adjacent crystal units in opposite directions. The pyrrole and furan moieties making up one molecule exist at an almost perpendicular planes to each other, at an angle of 109.98°.
Figure 5.

Unit cell for L2 viewed along the crystallographic a-axis showing hydrogen bonding in the unit cell (a) and the near perpendicular (109.98°) molecular planes (b).
The ORTEP plot for the Pt complex (C5) is shown in Figure 6a,b with the unit cell consisting of four molecules. The angles subtended around the Pt center are approximately right angles confirming the square planar geometry of a Pt(II) metal oxidation state.
Figure 6.

Unit cell packing showing hydrogen bonding, (a), and bond angles around the Pt center (b) for C5. Hydrogen atoms are omitted for clarity.
The crystal is stabilized through intermolecular hydrogen bonding (3.198 Å) between the pyrrole hydrogen and chloride atom para to the imine nitrogen of adjacent molecules. A notable property of this complex is the unequal bond distances between the Pt metal center and the two chloride atoms attached to it. The bond length between the Pt and chloride para to the imine nitrogen is 2.302 Å, while the distance between the Pt center and chloride atom para to the sulfur atom of DMSO is 2.317 Å. This is credited to the stronger ground state trans-influence of sulfur compared to nitrogen, leading to a lengthening (and weakening) of interatomic bonds.
These observations are similar to what was reported by Murray and Hartley on N- and S-bonded PdCl2 on amino acid backbones. In their work, a bond length of 2.305 Å for Pd–Cl trans-to N-atom and 2.324 Å for Pd–Cl trans to S was observed.36 It is observed that coordination occurs preferentially between Pt and sulfur of DMSO rather than with the oxygen atom despite the latter being less sterically hindered. This is expected because Pt, being a soft acid, prefers to coordinate with soft donors such as sulfur as opposed to nitrogen.
Bioactivity of the Organometallic Complexes
DNA Binding Studies
Observation on the spectral changes of metal complexes in the UV–vis range after addition of increasing concentrations of DNA is a generally accepted technique to assess the mode of binding and the kinetics of DNA-metal complex interaction.37 In this study, calf thymus DNA (CT-DNA) was used to simulate how the complexes bind to DNA. The metal complex–DNA mixture was allowed to equilibrate for 10 min prior to taking UV–vis measurements. With continued addition of DNA, the peaks of the complexes decreased up to a point where they disappeared, and a peak at 260 nm corresponding to DNA appeared. The appearance of the DNA peak at λ260 nm was used as an indication of the interaction with the complexes. DNA concentration was calculated using the molar absorptivity of the CT-DNA, ε = 6600 M–1, as shown in Figure S34 in the Supporting Information.
The binding kinetics for the complex–NA interaction was then determined from the intrinsic binding constant, Kb, by plots of the Benesi–Hildebrand host–guest equation,38 (eq 3), where the strength of the interaction is reflected by the value of the binding constant.
| 3 |
Ao and A are the absorbance of the blank sample without DNA, and absorbance of complex–DNA after addition of DNA, respectively, εG and εH–G are the molar extinction coefficients of complex only and complex–DNA, respectively. The binding constant is determined by plotting Ao/(A – Ao) versus 1/[DNA] to give a straight–line graph. The equation of the straight line gives the slope and intercept, where the Kb value was calculated from the ratio of the intercept to the slope. From the Kb values, Gibbs free energy changes, ΔG, for the complex–DNA interactions were calculated using eq 4.
| 4 |
where R is the gas constant and T is the absolute temperature.
The absorption spectra of the interaction of CT-DNA with the complexes together with the Benesi-Hildebrand plots are shown in Figures 7–10. The arrows show the intensity changes upon increasing concentration of CT DNA. Inset is the Benesi–Hildebrand plots for the determination of the binding constant.
Figure 7.

UV–vis absorption spectra for the CT-DNA Interactions with C1.
Figure 10.

UV–vis absorption spectra for the CT-DNA Interactions with C5.
The spectra for C1 (Figure 6) showed a 5% hyperchromism at 294 nm. The observed hyperchromism is attributed to external associations by electrostatic interactions of the complex with the groove bases in the DNA possibly through the planar aromatic phenyl and pyrrole moieties and from hydrogen bonding with the negatively charged phosphate backbone.39 This is further supported by the hypsochromic shift of 6 nm because a decrease in electron density in pi-orbitals stabilizes the orbitals, which causes an increase in the energy gap between pi and pi* orbitals, shifting the absorption to a lower wavelength.
On the other hand, the spectra for C2 (Figure 7), C3 (Figure 8), and C5 (Figure 9) showed hypochromic shifts with no shift in wavelength. The hypochromism is explained by the probability of intercalative interactions of the chromophores in the complexes with the DNA base pairs.40 Furthermore, distinct isosbestic points appeared at 300 nm in the spectrum of C1 and 285 nm in the spectra of C2, C3, and C5. This is a clear indication of equilibrium between bound DNA and the free form of the compounds and that no other species apart from the free and the intercalated forms were present in the mixture.41 The mode of binding, Kb values, and Gibbs free energy for the complexes are shown in Table 2.
Figure 8.

UV–vis absorption spectra for the CT-DNA interactions with C2.
Figure 9.

UV–vis absorption spectra for the CT-DNA interactions with C3.
Table 2. Binding Constants and Gibbs Free Energy Values for the Complex–DNA Interactions.
| complex | mode of binding | binding constant Kb (M–1) | gibbs free energy (ΔG) (kJ mol–1) |
|---|---|---|---|
| C1 | electrostatic | 1.6969 × 104 | –24.13 |
| C2 | intercalative | 1.5327 × 104 | –23.88 |
| C3 | intercalative | 1.0091 × 104 | –22.84 |
| C5 | intercalative | 8.049 × 104 | –27.99 |
These values indicate a comparatively strong interaction between the complexes and CT-DNA. Complexes C1, C2, and C3 had lower binding constants which are comparable to the values of the known DNA intercalators: Lumazine, Epirubicin, and Proflavine (2.32 × 104 M–1).42 The Pt complex C5 showed higher Kb values than those of Lumazine, Epirubicin (3.4 × 104 M–1),42 and Proflavine (1.74 × 104 M–1)43 but lower than that of ethidium bromide (1.4 × 106 M–1).44 This strong affinity to bind to the DNA indicates its significance as a possible candidate in the line of potential anticancer drug candidates. The high binding constant for C5 suggests that it can also be a viable candidate for new intercalator compounds. The ΔG values for all the complexes were negative which specified the spontaneity of interaction during the complex–DNA formation process.42
Anticancer Activity of the Organometallic Complexes
The main objective of the study was to demonstrate that stabilizing Pt and Pd by complexing with sterically bulky substituents around the metal center can block the attack from the axial z2-direction of the square planar complexes, thereby reducing ligand substitution reactions when in solution8 and improving their activities. Cytotoxicity of the compounds was assessed by MTT assay on selected cancerous (Caco-2, HeLa, Hep-G2, MCF-7, and PC-3), and noncancerous (MCF-12A) cell lines. The cells were initially treated with a single dose (100 μg/mL) of the ligands (L1–3) and their respective complexes (C1–5) to evaluate their potential anticancer activity. The ligands did not show any substantial activity at this dose (data not shown) compared to their corresponding complexes, and therefore, the reduced cell proliferation shown in Figure 11 could be accredited to the complexed compounds.
Figure 11.
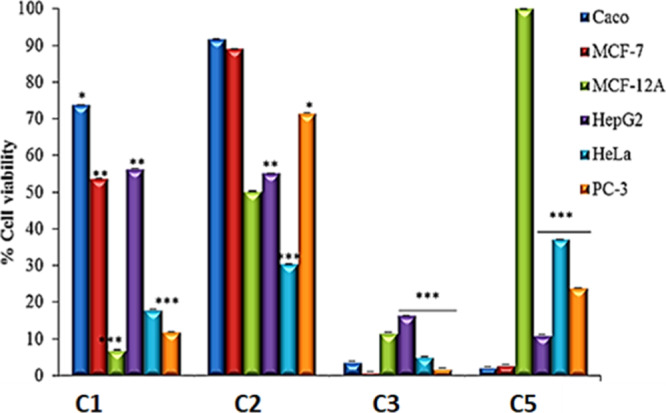
Screening of anticancer activity of Pd and Pt complexes (C1, C2, C3, and C5) against selected human cell lines. The cells were exposed to 100 μg/mL of compounds for 24 h; cell viability was assessed by MTT assay. *indicates statistical significance at p < 0.05 when compared to untreated cells, while ** indicates p < 0.01 and ***p < 0.001.
Two complexes (C3 and C5) were highly toxic to the cancer cells. C3 reduced cell viability by more than 80% in the six cell lines evaluated in this study and was the most cytotoxic complex. C5 demonstrated remarkable selective toxicity. It reduced viability of all the cancer cell lines by 60% or more but did not affect the viability of the noncancerous KMST6MCF-12A cells. In general, C1 and C2 was less toxic but also demonstrated some level of selective toxicity. C2, for example, did not significantly reduce the viability of Caco-2 and MCF-7 cells but was highly toxic to the other four cell lines (HeLa, Hep-G2, PC-3, and MCF-12A). The different responses observed for the different cell lines may be attributed to genetic and transcriptional heterogeneity between the cell lines. Specific genetic mutations in these cells may have resulted increased susceptibility or conversely may result in increased resistance to the complexes. Further testing using whole proteome or genome analysis is needed to fully understand why the different cell lines responded in the way they did.
C1 and C2 only differ in structure by a CH3- in C1 and a Cl- in C2 around the metal center. Because the CH3 group is a strong σ-bonding ligand with a stronger trans-influence compared to Cl-, it causes the σ-bonding character of the Pd–Cl bond in C1 to decrease. This means a better lability of the Cl-trans to CH3- in C1 compared to C2, which led to the observed differences in their cytotoxicity profile. The IC50 values for the complexes (C1, C2, C3, and C5) are shown in Table 3. The ligands (L1–3) were not cytotoxic at the highest concentration (100 μg/mL) tested. The complexes with cis geometry, C3 and C5, showed enhanced cytotoxicity compared to the trans-geometry complexes C1 and C2. The IC50 values varied significantly depending on the cell line and complex used. The IC50 value for C5 (0.3 ± 0.0054 μg/mL) on the Hep-G2 cell line was the lowest IC50 value obtained. The IC50 values also demonstrate the selective toxicity of some of the complexes. While the IC50 values for C1 and C2 varied between 22 and 86 μg/mL for MCF-12A, HeLa, and PC-3 cells, the IC50 values for C1 and C2 were more than 100 μg/mL for Caco-2, MCF-7, and Hep-G2 cells. Interestingly, C5 was highly toxic to MCF-7 cells with an IC50 value of 23.3 ± 0.0031 μg/mL, while the IC50 value for MCF-12A cells was more than 100 μg/mL. This is significant because MCF-7 and MCF-12A are, respectively, cancerous and noncancerous cell lines from breast tissue. This demonstrates the selective toxicity of C5 toward MCF-7 breast cancer cells compared to the noncancerous breast cells.
Table 3. IC50 Values for the Ligands and Their Respective Complexes.
| [complex] (μg/mL) |
|||||
|---|---|---|---|---|---|
| L1–3 Ligands | C1 | C2 | C3 | C5 | |
| Caco-2 | >100 | >100 | >100 | 15.81 ± 0.039 | 16.63 ± 0.006 |
| MCF-7 | >100 | >100 | >100 | 64.5 ± 0.0171 | 23.3 ± 0.0031 |
| MCF-12A | >100 | 22.95 ± 0.014 | 53.5 ± 0.0753 | 24.9 ± 0.0199 | >100 |
| HeLa | >100 | 66.62 ± 0.051 | 86.4 ± 0.0286 | 45.5 ± 0.1665 | 84.86 ± 0.087 |
| Hep-G2 | >100 | >100 | >100 | 13.0 ± 0.0294 | 0.3 ± 0.0054 |
| PC-3 | >100 | 25.1 ± 0.0161 | >100 | 20.9 ± 0.0179 | 29.5 ± 0.0961 |
The cells treated with these complexes demonstrated morphological changes, as shown in Figure S35, such as cell shrinking, loss of cell contact (detachment), membrane blebbing, and formation of apoptotic bodies, which are features that can be associated with apoptosis.45,46 To further confirm if the cytotoxic effects of the complexes are exerted through apoptosis, phosphatidyl serine (PS) externalization as one of the molecular markers of apoptosis was investigated using APOPercentage dye. PS is localized in the inner membrane of viable intact cells. Transmembrane lipid translocation during apoptosis results in the translocation of PS to the outside of the membrane. At this stage of apoptosis, apoptotic cells can be detected with the APOPercentage dye, which become entrapped within apoptotic cells.47
APOPercentage dye uptake was studied in selected cells (Hep-G2, PC-3, and Caco-2) treated with the complexes for 24 h, and the number of apoptotic cells was quantified using a flow cytometer as described by Meyer et al. (2008);48Figure 12 shows that the cell lines differed in their response to the complexes. Significantly higher levels of apoptosis were observed for the Caco-2 cell line when the cells were treated with C2, C3, and C5. Interestingly, C1 did not induce significant levels of apoptosis in Caco-2 cells. The Caco-2 cell line was the only cell line that showed a significant increase in the percentage of apoptotic cells when treated with C5. Only C1 and C3 were able to induce significant levels of apoptosis in Hep-G2 cells, while C1, C2, and C3 induced significant levels of apoptosis in Hep-G2 cells PC3 cells. Cisplatin, which was used as a positive control, induced significant levels of apoptosis in PC3 cells but not Hep-G2 cells. The Caco-2 cell line was not treated with cisplatin.
Figure 12.

Apoptotic effects of organometallic complexes on human cancer cells.
The cells were exposed to IC50 of complexes (100 μg/mL) for 24 h; apoptosis was assessed by the APOPercentage assay. Representative histograms (A) depict untreated (in black) and C3-treated (in red) Caco-2 cells. The cell population that is stained with the APOPercentage is labeled as +Apo Dye, and the cell population that are not stained is labeled as -Apo Dye. The bar graph in (B) shows the percentage apoptotic cells when Hep-G2, PC3, and Caco-2 cells are treated with the complexes. Treatment with Cisplatin (Cis) was used as a positive control. *indicates statistical significance at p < 0.05 when compared to untreated cells, while ** indicates p < 0.01 and ***p < 0.001
Conclusions
New complexes based on pyrrole were synthesized and fully characterized. The structures were confirmed by elemental analysis, and the molecular structure and geometry of the Pt complex were confirmed by single-crystal X-ray crystallography.
Despite literature reports that the use of bulky ligands in trans-Pd complexes may confer increased cytotoxic activity, this was not observed in this study. This shows that bulky ligands, which shield the metal center to a point where it is not accessible for ligand substitution, may in fact reduce the efficacy of these compounds. The challenge is to balance the extent of shielding and the extent of accessibility of the metal center to bioligands. This conclusion is based on the bioactivity of C2 (trans-geometry with bulky ligands) compared to C3 (cis-geometry with unshielded metal centre). C2 showed selective cytotoxicity, while C3 displayed high cytotoxicity to all six cell lines tested in this study.
The Pt complex (C5), also of cis-conformation, showed enhanced and selective cytotoxicity and selectivity to cancer cells. This compound also demonstrated strong DNA intercalation activity. This suggests that C5 can be a viable candidate for cancer treatment and requires further in vivo tests.
Experimental Section
Standard inert atmosphere Schlenk line techniques under nitrogen or argon in the Laminar flow were used for all the synthetic procedures employed in this study. Reaction progress was monitored on a silica gel thin layer chromatography (Fluka) plates of 15 μm particle size. Solvent was removed under vacuum via a liquid nitrogen trap, and samples are stored and dried as recommended and distilled prior to use. All reagents utilized in synthesis were of analytical grade; they were purchased from Sigma-Aldrich or Merck and used without further modifications.
Determination of melting points was performed on open glass capillaries using the SMP-10 instrument. FTIR spectra were run using a PerkinElmer Spectrum 100 Series FTIR instrument on the range 4000–400cm–1using KBr pellets. Proton nuclear magnetic resonance (1H NMR) spectra were run on a Bruker 400 MHz spectrometer available at the University of Western Cape, South Africa. The chemical shifts were measured in ppm relative to TMS standard. Elemental analysis was performed on Server 1112 series Elemental Analyzer at Stellenbosch University. UV–vis spectra were recorded in DMSO at a range of 200–800 nm using Nicolet evolution 100 spectrophotometer from Thermo Electron Corporation, and the data were analyzed using VisionPro software. Cyclic voltammetry studies were performed using the PalmerScan3 instrument equipped with glassy carbon electrodes as working electrodes, Ag/AgCl as the reference electrode, and platinum wire as the counter electrode, scan range of −1.8 to +1.8, and a scan rate of 200 mV/s. The supporting electrolyte was 5 mL of 10 mMol phosphate-buffered saline (PBS) buffer at pH 7. Single-crystal X-ray crystallography was performed on Bruker SMART CCD APEXII area-detector diffractometer available at Rutgers University, USA.
Synthesis of the Ligands and Complexes
(E)-N-((1H-Pyrrol-2-yl)methylene) (Phenyl) Methanamine (L1)
The ligand was synthesized using 0.3882 g (4 mmol) Pyrrole-2-carbaldehyde and 440 μL (4 mmol) benzylamine in dry methanol and refluxed overnight. The solvent was rotavaped to dryness, and the product was obtained as orange powder. It was recrystallized in DCM/Et2O and kept in fridge overnight for complete precipitation. The mother liquor was filtered off, and the product dried under vacuum. Crystals suitable for X-ray crystallography were grown by slow diffusion of hexane into DCM, yielding light brown crystals. Yield: 95% mp 110–112 °C. FTIR (KBr cm–1); 1636, (C=N); 2829 (sp3 −C–H); 2888 (−C–H benzene), 3369, (−N–H). 1H NMR (400 MHz, CDCL3, δ-ppm): δ 8.15 (s, 1H), 7.24–7.37 (m, 5H), 6.90 (s, 1H), 6.53 (dd, J = 1.22, 3.55 Hz, 1H), 6.22–6.27 (m, 1H), 4.71 (s, 2H). 13C NMR (400 MHz, CDCL3): δ 152.35 (imine C=N), 137.04 (C2C–N pyrrole), 128.55, 128.19, 127.98, 127.56, 127.07, 120.23 (CC–N pyrrole), 109.88, 88.93, 55.29 (CH2). Molecular weight (Calcd); 184.2371, ESI–MS (m/z): [M + H]+ (obtained): 184.2. Molecular formula; C12H12N2, elemental analysis; Calcd (obtained) N, 15.21 (15.63); C, 74.2 (74.46); H, 6.8 (7.0)
(E)-N-((1H-Pyrrol-2-yl)methylene) (Furan-2-yl) Methanamine (L2)
The ligand was synthesized as for the previous ligand using 0.4853 g (5 mmol) pyrrole-2-carbaldehyde and 450 μL (5 mmol) furfurylamine with MgSO4 and stirred in room temperature for 18 h. Orange powder turned to brown. It was recrystallized from DCM with ether. Yield: 0.8166 g (94%). mp 86–89 °C. Soluble in methanol, chloroform, DCM, Et2O.
FTIR (KBr ν–1); 1644, C=N (stretch); 2868 sp3 −C–H stretch; 2975 −C–H (aromatic) stretch, 3177; N–H stretch. 1H NMR (400 MHz, CDCl3): δ 8.12 (s, 1H), 7.38 (s, 1H), 6.89 (s, 1H), 6.53 (br s, 1H), 6.34 (d, J = 3.06 Hz, 1H), 6.24 (s, 2H), 4.67 (s, 2H). 13C NMR (400 MHz, Chloroform-d): δ 153.19 (C=N imine), 153.09 (C2C–O furan), 152.68 (C–C–O furan), 149.24 (C2C=N pyrrole), 142.21 (CC–N pyrrole), 124.79, 122.09, 120.07, 114.97, 56.47 (CH2)
Molecular weight (Calcd); 174.1992, ESI–MS (m/z): [M + H]+ (obtained); 174. Molecular formula; C10H10N2O, elemental analysis; Calcd (obtained) C, 68.95(68.61); H, 5.79(6.2); N, 16.08(16.47).
(E)-N-((1H-Pyrrol-2-yl)methylene) (Pyridin-2-yl) Methanamine (L3)
The ligand was synthesized as for the previous ligands using 0.3882 g (4 mmol) pyrrole-2-carbaldehyde and 4 mmol (420 μL) 2-methylamino pyridine (picolyamine). The reaction mixture turned yellow during reflux. The solvent was evaporated to dryness under rotavap, and the product was isolated as a paste. It was recrystallized in DCM/Et2O and kept in fridge overnight to complete precipitation; mother liquor was filtered off, and the product dried under vacuum. Yield: 94%. mp 96–99 °C. Soluble in methanol, chloroform, DCM, Et2O.
FTIR (KBr ν–1); 1632, C=N (stretch); 2962 sp3 −C–H stretch; 2847 −C–H (aromatic) stretch, 3080; N–H stretch. 1H NMR (400 MHz, CDCL3): δ 8.56 (d, J = 4.16 Hz, 1H), 8.24 (s, 1H), 7.61 (dt, J = 1.71, 7.64 Hz, 1H), 7.31 (d, J = 7.82 Hz, 1H), 7.15 (dd, J = 5.32, 6.91 Hz, 1H), 6.83 (s, 1H), 6.54 (dd, J = 1.10, 3.55 Hz, 2H), 6.23 (dd, J = 2.81, 3.42 Hz, 1H), 4.86 (s, 2H). 13C NMR (400 MHz, CDCL3): δ 160.03 (C2C=N pyridine)), 153.67 (C=N imine), 150.50, 149.30, 142.99, 136.62 (C2C–N pyrrole), 131.59, 130.08, 119.80 (CC–N pyrrole), 114.87, 111.11, 66.18 (CH2).
Molecular weight (Calcd); 185.2251, ESI–MS (m/z) [M – H]+ 183.1. Molecular formula; C11H11N3. Elemental analysis Calcd (obtained); N, 22.69(22.45); C, 70.33(69.38); H, 5.99(5.6).
Trans-Chloridobis[(pyrrolymethylidene) (Benz-2-yl-methyl) Amine-κN] Methylpalladium(II) (C1)
To a stirring solution of 92 mg (0.5 mmol) L1 in CH2Cl2 under nitrogen was added dropwise a solution of 0.25 mmol (66 mg) Pd(COD)ClMe in CH2Cl2. The solution turned green, and stirring was sustained overnight at room temperature conditions. The solution was reduced under vacuum to about 1 mL, and hexane was added in excess to precipitate out the product. The mixture was kept in fridge overnight to allow complete precipitation; liquor was filtered off, and the precipitate was washed with more hexane, filtered, and dried in inert conditions under vacuum. Yield: 52%, mp 216 °C (decompose). FTIR (KBr cm–1); 3025, (aromatic C–H); 2937, (aliphatic C–H); 1602, (C=N). 1H NMR (400 MHz, CDCL3): δ 7.78 (s, 2H), 7.41 (m, 5H), 7.29 (m, 5H), 6.92 (s, 2H), 6.64 (s, 4H), 6.09 (br. s., 2H), 4.93 (s, 4H), 1.58 (br. s., 3H). 13C NMR (101 MHz, CDCL3): δ = 162.8, 139.5, 137.5, 136.7, 129.0, 127.8, 117.7, 111.0, 61.3. Molecular weight (Calcd); 525.38, molecular formula; liquid chromatography–mass spectrometry (LC–MS) (m/z): [M + H]+ (obtained) 525.30; C25H27ClN4Pd, elemental analysis; Calcd (obtained); N, 110.7 (11.3); C, 55.2 (55.79); H, 5.2 (5.1).
Trans-Dichloridobis[(pyrrolymethylidene) (furan-2-yl-methyl) Amine-κN] Methylpalladium(II) (C2)
The complex was synthesized for the previous one using 174 mg (1 mmol) of L3 and 144 mg (0.5 mmol) Pd(COD)Cl2. The mixture formed an orange solution which was reduced to about 1 mL by vacuum, and the product was recrystallized with diethyl ether. It was filtered, washed with 5 mL Et2O and further with 2 × 5 mL hexane, and dried under vacuum to afford an orange product. Yield: 60 mg, 23%. The complex was analyzed by FTIR, UV–vis, and elemental analysis. FTIR (KBr cm–1) 3117, (aromatic C–H); 3015, (aliphatic C–H); 1662, (C=N). 1H NMR (400 MHz, CDCl3): δ 7.69 (s, 1H), 7.54 (br s, 2H), 7.13 (br s, 1H), 6.46 (br s, 3H), 4.81 (br s, 2H), 3.49 (s, 2H), 1.59 (s, 3H), 1.21 (s, 3H). Molecular formula; C20H20Cl2N4O2Pd, molecular weight (Calcd); 525.72, LC–MS (m/z): [M + H]+ (obtained) 525.29. Elemental analysis; Calcd (obtained); C, 45.69(45.84); H, 2.84(2.79); N, 5.30(5.65).
cis-Dichlorido[(pyrrolymethylidene) (Pyridyl-2-yl-methyl) Amine N,N] Pd(II) (C3)
The complex was synthesized as for the previous one using 93 mg (0.5 mmol) L3 and 0.5 mmol (144 mg) Pd(COD)Cl2. The product precipitated immediately, and the mixture was stirred overnight after which the precipitation was deemed complete. It was filtered, washed with 5 mL Et2O and further with 2 × 5 mL hexane, and dried under vacuum to afford a gray product. The compound was analyzed by FTIR, UV–vis, and elemental analysis. Yield: 90%. mp 250 °C (decompose). FTIR (KBr cm–1) 1610, C=N (stretch); 1576, C=N (aromatic). 1H NMR (400 MHz, CDCl3): δ 7.44 (d, 1H), 7.26 (s, 1H), 6.44 (m, 1H), 6.31 (m, 2H), 5.30 (m, 1H), 4.81 (m, 1H), 3.49 (m, 1H), 1.57 (d, 1H), 1.21 (s, 2H). Molecular formula; C11H11Cl2N3Pd, Molecular weight (Calcd); 362.55; LC–MS (m/z): [M + H]+ (obtained) 362.04. Elemental analysis Calcd (obtained); C, 36.4(36.5); H, 3.1(3.5); N, 11.6(11.2).
Pt Complex [Pt2(μ-Cl)2(dmso)2(L1)2] (C5) and PtCl2dmsoL1 (C5)
The platinum complex C5 was obtained by reaction of L1 with Pt(DMSO)2Cl2 at room temperature. Into a stirring solution of 42 mg (0.125 mmol) Pt(DMSO)2Cl2 in 15 mL DCM, 0.125 mmol (23 mg) L1 in 5 mL DCM was added dropwise using a nitrogen-filled syringe. Stirring was continued overnight, evaporated under vacuum to yield a yellow precipitate of C5 which was filtered and dried under vacuum. Yield: 65%. FTIR (KBr cm–1) 3300, (2° amine N–H for pyrrole); 3130, (aromatic C–H); 3016, (aliphatic C–H); 1631, (C=N). 1H NMR (400 MHz, CDCl3): δ 7.78–7.85 (m, 2H), 7.26 (s, 10H), 6.90–7.01 (m, 4H), 6.55–6.61 (m, 2H), 3.43–3.52 (m, 2H), 2.17 (s, 4H), 1.56 (s, 12H). Elemental analysis; Calcd (obtained); C, 34.11 (34.15); H, 3.68 (3.70); N, 5.68 (5.69); S, 6.51 (6.50). Crystals suitable for X-ray crystallography were obtained by slow evaporation of the DCM solution and were obtained as light-yellow crystals of C5. 1H NMR (400 MHz, DMSO-d6): δ 8.72 (br. s., 1H), 8.44 (s, 1H), 7.45 (s, 5H), 6.45 (br. s., 1H), 5.11 (s, 1H), 4.92 (s, 1H), 3.35 (s, 2H), 2.54 (s, 6H). Molecular weight (Calcd); 528.3 LC–MS (m/z): [M + H]+ (obtained), 530.07.
Partition Coefficient
The partition coefficient as the log P value was determined for C1–C5 by the shake-flask method. The values were determined by partitioning the complexes between 10 mL of deionized water (the medium inside and outside the cells) and 10 mL of 99% n-octanol (a model of the lipid bilayer of the cell membrane) and stirring for 48 h. The two layers were first presaturated with each other to avoid solubility-induced volume changes during the extraction step.49 Before extraction, the absorbance of the initial stock was determined after appropriate dilutions.
The mixture was then left to settle for 24 h after which 3 mL aliquots were taken from each layer and centrifuged for 2 h at 7000 rpm using the Minispin Epperdorf centrifuge. The UV–vis absorbance for the two layers was taken after appropriately diluting the layers.50 Absorbance was read using a 96-well microplate on a POLARstar Omega microplate reader instrument from BMG Labtech. The partition coefficient was calculated as log (CO/CW), where CO is the concentration in the oil layer, and CW is the concentration in the water layer.
Electrochemical Properties of the Pd Complexes
Electrochemical studies in solution were performed on two of the complexes (C3 and C4) using the PalmerScan3 instrument equipped with glassy carbon electrodes as working electrodes, Ag/AgCl as the reference electrode and platinum wire as the counter electrode. A scan range of −1.8 to +1.8 and a scan rate of 200 mV/s were used. The supporting electrolyte was 5 mL of 10 mMol PBS buffer at pH 7.0.
Single-Crystal X-ray Analysis
Crystals suitable for X-ray crystallography were grown by slow diffusion of hexane into DCM which gave crystal for L1 or by slow evaporation of a DCM solution of the ligand for L2 and the platinum complex, C5. X-ray crystallography was performed on a Bruker SMART CCD APEXII area-detector diffractometer and was kept at 100.03 K during data collection. Using Olex2 software,51 the structure was solved with the ShelXS-199752 structure solution program using Direct Methods and refined with the Olex2 refinement package.53
Evaluation of the Bioactivity of the Organometallic Compounds
DNA Binding Studies
This was done by UV–vis spectroscopy by serial titrations of CT-DNA into a fixed concentration of the complexes in a quartz cuvette. UV–vis readings were taken at increasing amounts of DNA and allowing for 10 min equilibration time after each addition. The binding kinetics for the complex–DNA interaction was then determined from the intrinsic binding constant, Kb, by plots of the Benesi–Hildebrand host–guest equation.54 From the Kb values, Gibbs free energy changes (ΔG) for the complex–DNA interactions were then calculated and compared with known DNA intercalators.
Effect of Compounds on Cancer Cells
Cytotoxicity of the compounds was tested against human cell lines: Caco-2 (colon), HeLa (Cervical), Hep-G2 (Hepatocellular Carcinoma/liver cancer), MCF-7 (breast cancer), PC-3 (prostate), and MCF-12A (non-cancer breast) cells. The cells were cultured in their respective media supplemented with 10% foetal bovine serum (FBS) and 1% pen-strep cocktail comprised of streptomycin (100 μg/mL) and penicillin (100 U/mL). Caco-2, HeLa, Hep-G2, and MCF-7 cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM); MCF-12A in DMEM-F12 containing insulin, EGF, and hydrocortisone, while PC-3 cells were cultured RPMI-1640 media. The cells were incubated at 37 °C in a 5% humidified CO2 incubator (Labotech, South Africa).
The cells were seeded at 1 × 105 cells/mL density in a 96-well plate and incubated at 37 °C for 24 h. The cells were treated with varying concentrations of extracts (0–100 μg/mL) for 24 h. Cytotoxicity of the compounds was assessed by MTT assay following manufacturer’s instructions. After treatment, 10 μL of 5 mg/mL MTT (Sigma, USA) was added to each well and the plates were incubated at 37 °C for 3 h. The insoluble formazan crystals were solubilized by adding 100 μL of DMSO. The reduction of MTT was read at 570 nm using the POLARStar Omega plate reader. Cell viability was calculated using eq 5 given below
| 5 |
The half-maximal cytotoxic concentration (IC50) on each cell line was estimated using GraphPad Prism software version 5 (GraphPad Software, California, USA).
APOPercentage Apoptosis Assay
APOPercentage assay (Biocolor Ltd.) was performed following a protocol described by Alpuche-Aviles et al. (2018).33 Briefly, selected cell lines (Hep-G2, PC-3, and Caco-2) were seeded in 12 well cell culture plates at a density of 1 × 105 cells/mL and treated using concentrations equivalent to the IC50 values obtained using the MTT assay. After treatment, the morphological changes in the cells were observed under inverted contrast microscope (EVOS XL Core microscope; Thermo Fisher Scientific, Germany) at 10× magnification. The cells were then trypsinized and stained with APOPercentage dye. The cells staining positive for the APOPercentage dye were quantified using a Becton Dickinson (BD) Accuri C6 Flow cytometer (BD Biosciences Pharmingen, San Diego, CA, USA). A minimum of 10,000 events per sample was acquired, and the data were analyzed using the BD Accuri software (BD Biosciences).
Statistical Analyses
Statistical analyses for MTT and APOPercentage Apoptosis data were analyzed using Graph pad prism 6.0. The data were presented as means ± S according to the one-way ANOVA test and student t-test. A p value of <0.05 was considered to be of statistical significance and represented by an asterisk (*). *p < 0.05, **p < 0.01, and ***p < 0.001.
Acknowledgments
The authors gratefully acknowledge the International Science Program (ISP) Uppsala, Sweden under the KEN-01 project, and the NRF South Africa (CSUR160323161066) for funding this research; NIC Biolabels Node (Department of Biotechnology, UWC), University of Nairobi, for support and guidance in this work; and the Organometallics and Nanomaterials group (Department of Chemical Sciences, UWC), South Africa, where this work was carried out.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00360.
FTIR spectra (Figures S1-S7); 1HNMR spectra (Figures S8-S13); 13CNMR spectra (Figures S14-S18); mass spectra supplementary data (Figures S19-25); UV/Vis spectra (Figures S26-S32); and check CIF data for L1 (labelled PC-BA), L2 (labelled PC-O), and C5 (labelled PC-BA-Pt) (PDF)
CheckCIF report for Ligand L1 (CIF)
CheckCIF report for Ligand L2: Alert levels A and B explained (CIF)
CheckCIF report for Complex C5 (CIF)
Author Present Address
⊥ The author current address is Department of Chemistry, University of Nairobi. P.O. Box 30197 00100, Nairobi, Kenya.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K.; Jemal A. Cancer Statistics, 2018. Ca—Cancer J. Clin. 2018, 68, 7–30. 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- Ferlay J.; Colombet M.; Soerjomataram I.; Mathers C.; Parkin D. M.; Piñeros M.; Znaor A.; Bray F. : Estimating The Global Cancer Incidence And Mortality In 2018: Globocan Sources And Methods. Int. J. Canc. 2019, 144, 1941–1953. 10.1002/ijc.31937. [DOI] [PubMed] [Google Scholar]
- Nagaraju G. P.; Srivani G.; Dariya B.; Chalikonda G.; Farran B.; Behera S. K.; Alam A.; Kamal M. A. Nanoparticles Guided Drug Delivery and Imaging in Gastric Cancer. Semin. Cancer Biol. 2020, 10.1016/j.semcancer.2020.01.006. [DOI] [PubMed] [Google Scholar]
- Misirlic-Dencic S.; Poljarevic J.; Isakovic A. M.; Sabo T.; Markovic I.; Trajkovic V. Current Development Of Metal Complexes With Diamine Ligands As Potential Anticancer Agents. Curr. Med. Chem. 2020, 27, 380–410. 10.2174/0929867325666181031114306. [DOI] [PubMed] [Google Scholar]
- El-Razek S. E. A.; El-Gamasy S. M.; Hassan M.; Abdel-Aziz M. S.; Nasr S. M. Transition Metal Complexes Of A Multidentate Schiff Base Ligand Containing Guanidine Moiety: Synthesis, Characterization, Anti-Cancer Effect, And Anti-Microbial Activity. J. Mol. Struct. 2020, 1203, 127381. 10.1016/j.molstruc.2019.127381. [DOI] [Google Scholar]
- Hallek M. Chronic Lymphocytic Leukemia: 2020 Update On Diagnosis, Risk Stratification And Treatment. Am. J. Hematol. 2019, 94, 1266–1287. 10.1002/ajh.25595. [DOI] [PubMed] [Google Scholar]
- Ginsburg O.; Bray F.; Coleman M. P.; Vanderpuye V.; Eniu A.; Kotha S. R.; Sarker M.; Huong T. T.; Allemani C.; Dvaladze A.; Gralow J.; Yeates K.; Taylor C.; Oomman N.; Krishnan S.; Sullivan R.; Kombe D.; Blas M. M.; Parham G.; Kassami N.; Conteh L. The Global Burden Of Women’s Cancers: A Grand Challenge In Global Health. Lancet 2017, 389, 847–860. 10.1016/s0140-6736(16)31392-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward R. J.; Crichton R. R. Essential Metals In Medicine: Therapeutic Use And Toxicity Of Metal Ions In The Clinic. Met. Ions Life Sci. 2019, 19, 87–122. 10.1515/9783110527872-004. [DOI] [Google Scholar]
- Sánchez-López E.; Gomes D.; Esteruelas G.; Bonilla L.; Lopez-Machado A. L.; Galindo R.; Cano A.; Espina M.; Ettcheto M.; Camins A.; Silva A. M.; Durazzo A.; Santini A.; Garcia M. L.; Souto E. B. Metal-Based Nanoparticles As Antimicrobial Agents: An Overview. Nanomaterials 2020, 10, 292. 10.3390/nano10020292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali I.; Lone M.; Al-Othman Z.; Al-Warthan A.; Sanagi M. Heterocyclic Scaffolds: Centrality In Anticancer Drug Development. Curr. Drug Targets 2015, 16, 711–734. 10.2174/1389450116666150309115922. [DOI] [PubMed] [Google Scholar]
- Özdemir İ.; Çiftçi O.; Evren E.; Gürbüz N.; Kaloğlu N.; Başak Türkmen N.; Yaşar Ş.; Üstün E.; Hamdi N.; Mansour L.; Özdemir İ. Synthesis, Characterization And Antitumor Properties Of Novel Silver (I) And Gold (I) N-Heterocyclic Carbene Complexes. Inorg. Chim. Acta 2020, 506, 119530. 10.1016/j.ica.2020.119530. [DOI] [Google Scholar]
- Sreenivasulu R.; Tej M. B.; Jadav S. S.; Sujitha P.; Kumar C. G.; Raju R. R. Synthesis, Anticancer Evaluation And Molecular Docking Studies Of 2, 5-Bis (Indolyl)-1, 3, 4-Oxadiazoles, Nortopsentin Analogues. J. Mol. Struct. 2020, 1208, 127875. 10.1016/j.molstruc.2020.127875. [DOI] [Google Scholar]
- Apetrei R.-M.; Camurlu P. Functional Platforms For (Bio) Sensing: Thiophene-Pyrrole Hybrid Polymers. J. Electrochem. Soc. 2020, 167, 037557. 10.1149/1945-7111/ab6e5f. [DOI] [Google Scholar]
- Bavadi M.; Niknam K.; Shahraki O. Novel Pyrrole Derivatives Bearing Sulfonamide Groups: Synthesis In Vitro Cytotoxicity Evaluation, Molecular Docking And Dft Study. J. Mol. Struct. 2017, 1146, 242–253. 10.1016/j.molstruc.2017.06.003. [DOI] [Google Scholar]
- Saeed H.; Sreedharan S.; Thomas JA Photoactive Metal Complexes That Bind Dna And Other Biomolecules As Cell Probes, Therapeutics, And Theranostics. Chem. Commun. 2020, 56, 1464. 10.1039/c9cc09312e. [DOI] [PubMed] [Google Scholar]
- Martinez-Gonzalez A. I.; Díaz-Sánchez Á. G.; De La Rosa L. A.; Bustos-Jaimes I.; Alvarez-Parrilla E. Alvarez-Parrilla, E.: Inhibition Of Α-Amylase By Flavonoids: Structure Activity Relationship (Sar). Spectrochim. Acta, Part A 2019, 206, 437–447. 10.1016/j.saa.2018.08.057. [DOI] [PubMed] [Google Scholar]
- Ren C.; Xiong W.; Li J.; Li B. Comparison Of Binding Interactions Of Cyanidin-3-O-Glucoside To Β-Conglycinin And Glycinin Using Multi-Spectroscopic And Thermodynamic Methods. Food Hydrocolloids 2019, 92, 155–162. 10.1016/j.foodhyd.2019.01.053. [DOI] [Google Scholar]
- Fairbanks S. D.; Robertson C. C.; Keene F. R.; Thomas J. A.; Williamson M. P. Structural Investigation Into The Threading Intercalation Of A Chiral Dinuclear Ruthenium (Ii) Polypyridyl Complex Through A B-Dna Oligonucleotide. J. Am. Chem. Soc. 2019, 141, 4644–4652. 10.1021/jacs.8b12280. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-P.; Li Y.; Xu G.-C.; Li J.-Y.; Luo H.-Y.; Li J.-Y.; Zhang L.; Jia D.-Z. Synthesis, Crystal Structure, Dna/Bovine Serum Albumin Binding And Antitumor Activity Of Two Transition Metal Complexes With 4-Acylpyrazolone Derivative. Appl. Organomet. Chem. 2019, 33, E4668 10.1002/aoc.4668. [DOI] [Google Scholar]
- Morgan S. M.; El-Sonbati A. Z.; Eissa H. R. Geometrical Structures, Thermal Properties And Spectroscopic Studies Of Schiff Base Complexes: Correlation Between Ionic Radius Of Metal Complexes And Dna Binding. J. Mol. Liq. 2017, 240, 752–776. 10.1016/j.molliq.2017.05.114. [DOI] [Google Scholar]
- Layek S.; Anuradha B.; Agrahari B.; Pathak D. D. Synthesis And Characterization Of A New Pd (Ii)-Schiff Base Complex [Pd (Apd) 2]: An Efficient And Recyclable Catalyst For Heck-Mizoroki And Suzuki-Miyaura Reactions. J. Organomet. Chem. 2017, 846, 105–112. 10.1016/j.jorganchem.2017.05.049. [DOI] [Google Scholar]
- Janssen-Müller D.; Schlepphorst C.; Glorius F. Privileged Chiral N-Heterocyclic Carbene Ligands For Asymmetric Transition-Metal Catalysis. Chem. Soc. Rev. 2017, 46, 4845–4854. 10.1039/c7cs00200a. [DOI] [PubMed] [Google Scholar]
- Popova E. A.; Protas A. V.; Trifonov R. E. Tetrazole Derivatives As Promising Anticancer Agents. Anti Canc. Agents Med. Chem. 2017, 17, 1856–1868. 10.2174/1871520617666170327143148. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Luo H.; Xu Q.; Lin L.; Zhang B. Antitumor Activity Of A Trans-Thiosemicarbazone Schiff Base Palladium (Ii) Complex On Human Gastric Adenocarcinoma Cells. Oncotarget 2017, 8, 13620. 10.18632/oncotarget.14620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspari R.; Prota A. E.; Bargsten K.; Cavalli A.; Steinmetz M. O. Structural Basis Of Cis-And Trans-Combretastatin Binding To Tubulin. Chem 2017, 2, 102–113. 10.1016/j.chempr.2016.12.005. [DOI] [Google Scholar]
- Motswainyana W. M.; Onani M. O.; Madiehe A. M. Bis (Ferrocenylimine) Palladium (Ii) And Platinum (Ii) Complexes: Synthesis, Molecular Structures And Evaluation As Antitumor Agents. Polyhedron 2012, 41, 44–51. 10.1016/j.poly.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Ehrmann F. R.; Stojko J.; Metz A.; Debaene F.; Barandun L. J.; Heine A.; Diederich F.; Cianférani S.; Reuter K.; Klebe G. Soaking Suggests ″Alternative Facts″: Only Co-Crystallization Discloses Major Ligand-Induced Interface Rearrangements Of A Homodimeric Trna-Binding Protein Indicating A Novel Mode-Of-Inhibition. PloS One 2017, 12, e0175723 10.1371/journal.pone.0175723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunath M.; Kulkarni A. D.; Bagihalli G. B.; Malladi S.; Patil S. A. Bio-Important Antipyrine Derived Schiff Bases And Their Transition Metal Complexes: Synthesis, Spectroscopic Characterization, Antimicrobial, Anthelmintic And Dna Cleavage Investigation. J. Mol. Struct. 2017, 1127, 314–321. 10.1016/j.molstruc.2016.07.123. [DOI] [Google Scholar]
- Tyagi P.; Tyagi M.; Agrawal S.; Chandra S.; Ojha H.; Pathak M. Synthesis, Characterization Of 1, 2, 4–Triazole Schiff Base Derived 3d-Metal Complexes: Induces Cytotoxicity In Hepg2, Mcf-7 Cell Line, Bsa Binding Fluorescence And Dft Study. Spectrochim. Acta, Part A 2017, 171, 246–257. 10.1016/j.saa.2016.08.008. [DOI] [PubMed] [Google Scholar]
- Ekennia A. C.; Osowole A. A.; Olasunkanmi L. O.; Onwudiwe D. C.; Ebenso E. E. Coordination Behaviours Of New (Bidentate N, O-Chelating) Schiff Bases Towards Copper (Ii) And Nickel (Ii) Metal Ions: Synthesis, Characterization, Antimicrobial, Antioxidant, And Dft Studies. Res. Chem. Intermed. 2017, 43, 3787–3811. 10.1007/s11164-016-2841-z. [DOI] [Google Scholar]
- Al-Fregi A. A. Synthesis, Characterization And Molar Conductivity Study Of Some New Palladium (Ii) And Platinum (Ii) Complexes Containing Heterocyclic Tellurium-Dicarboxylato Ligands. Int. J. 2015, 3, 637–647. [Google Scholar]
- Bhal S. K. Logp—Making Sense Of The Value. Adv. Chem. Dev. 2007, 1–4. [Google Scholar]
- Alpuche-Aviles M. A.; Farina F.; Ercolano G.; Subedi P.; Cavaliere S.; Jones D. J.; Rozière J. Electrodeposition Of Two-Dimensional Pt Nanostructures On Highly Oriented Pyrolytic Graphite (Hopg): The Effect Of Evolved Hydrogen And Chloride Ions. Nanomaterials 2018, 8, 668. 10.20944/preprints201807.0392.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen-Müller D.; Schlepphorst C.; Glorius F. Correction: Privileged Chiral N-Heterocyclic Carbene Ligands For Asymmetric Transition-Metal Catalysis. Chem. Soc. Rev. 2017, 46, 5463. 10.1039/c7cs90067h. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Sadler P. J. Redox-Active Metal Complexes For Anticancer Therapy. Eur. J. Inorg. Chem. 2017, 2017, 1541–1548. 10.1002/ejic.201600908. [DOI] [Google Scholar]
- Murray S. G.; Hartley F. R. Coordination Chemistry Of Thioethers, Selenoethers, And Telluroethers In Transition-Metal Complexes. Chem. Rev. 1981, 81, 365–414. 10.1021/cr00044a003. [DOI] [Google Scholar]
- Olsztynska S.; Komorowska M.. Biomedical Engineering: Trends, Research and Technologies; Bod–Books on Demand, 2011. [Google Scholar]
- Kuntz I. Jr; Gasparro F.; Johnston M. Jr; Taylor R. Molecular Interactions And The Benesi-Hildebrand Equation. J. Am. Chem. Soc. 1968, 90, 4778–4781. 10.1021/ja01020a004. [DOI] [Google Scholar]
- Suntharalingam K.; Mendoza O.; Duarte A. A.; Mann D. J.; Vilar R. A Platinum Complex That Binds Non-Covalently To Dna And Induces Cell Death Via A Different Mechanism Than Cisplatin. Metallomics 2013, 5, 514–523. 10.1039/c3mt20252f. [DOI] [PubMed] [Google Scholar]
- Pyle A. M.; Rehmann J. P.; Meshoyrer R.; Kumar C. V.; Turro N. J.; Barton J. K. Mixed-Ligand Complexes Of Ruthenium (Ii): Factors Governing Binding To Dna. J. Am. Chem. Soc. 1989, 111, 3051–3058. 10.1021/ja00190a046. [DOI] [Google Scholar]
- Arshad N.; Bhatti M. H.; Farooqi S. I.; Saleem S.; Mirza B. Synthesis, Photochemical And Electrochemical Studies On Triphenyltin (Iv) Derivative Of (Z)-4-(4-Cyanophenylamino)-4-Oxobut-2-Enoic Acid For Its Binding With Dna: Biological Interpretation. Arabian J. Chem. 2016, 9, 451–462. 10.1016/j.arabjc.2014.08.018. [DOI] [Google Scholar]
- Aslanoglu M. Electrochemical And Spectroscopic Studies Of The Interaction Of Proflavine With Dna. Anal. Sci. 2006, 22, 439–443. 10.2116/analsci.22.439. [DOI] [PubMed] [Google Scholar]
- Ibrahim M. S.; Shehatta I. S.; Al-Nayeli A. A. Voltammetric Studies Of The Interaction Of Lumazine With Cyclodextrins And Dna. J. Pharmaceut. Biomed. Anal. 2002, 28, 217–225. 10.1016/s0731-7085(01)00624-0. [DOI] [PubMed] [Google Scholar]
- Lepecq J.-B.; Paoletti C. A Fluorescent Complex Between Ethidium Bromide And Nucleic Acids: Physical—Chemical Characterization.. J. Mol. Biol. 1967, 27, 87–106. 10.1016/0022-2836(67)90353-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Chen X.; Gueydan C.; Han J. Plasma Membrane Changes During Programmed Cell Deaths. Cell Res. 2018, 28, 9–21. 10.1038/cr.2017.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M.; Essack M.; Kanyanda S.; Rees J. G. A Low-Cost Flow Cytometric Assay For The Detection And Quantification Of Apoptosis Using An Anionic Halogenated Fluorescein Dye. Biotechniques 2008, 45, 317–320. 10.2144/000112908. [DOI] [PubMed] [Google Scholar]
- Park S.-Y.; Kim I.-S. Engulfment Signals And The Phagocytic Machinery For Apoptotic Cell Clearance. Exp. Mol. Med. 2017, 49, E331 10.1038/emm.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M.; Essack M.; Kanyanda S.; Rees J. G. A low-cost flow cytometric assay for the detection and quantification of apoptosis using an anionic halogenated fluorescein dye. BioTechniques 2008, 45, 317–320. 10.2144/000112908. [DOI] [PubMed] [Google Scholar]
- Sangster J. Octanol–Water Partition Coefficients: Fundamentals And Physical Chemistry. Eur. J. Med. Chem. 1997, 32, 842. 10.1016/s0223-5234(97)82764-x. [DOI] [Google Scholar]
- Minick D. J.; Frenz J. H.; Patrick M. A.; Brent D. A. A Comprehensive Method For Determining Hydrophobicity Constants By Reversed-Phase High-Performance Liquid Chromatography. J. Med. Chem. 1988, 31, 1923–1933. 10.1021/jm00118a010. [DOI] [PubMed] [Google Scholar]
- Liao B.-L.; Yin X.-J. Synthesis, Structure And High Photocatalytic Performance Of A Novel Vanadium (V) Complex Based On Pyridine-2, 4, 6-Tricarboxylic Acid. Inorg. Nano-Met. Chem. 2018, 48, 427–433. 10.1080/24701556.2019.1569684. [DOI] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement With Shelxl. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/s2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourhis L. J.; Dolomanov O. V.; Gildea R. J.; Howard J. A. K.; Puschmann H. The Anatomy Of A Comprehensive Constrained, Restrained Refinement Program For The Modern Computing Environment–Olex2 Dissected. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 59–75. 10.1107/s2053273314022207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Gao L.; Zhao Q.; Zhang Y.; Dong W.-K.; Ding Y.-J. A Highly Selective Fluorescent Chemosensor For Cn– Based On A Novel Bis (Salamo)-Type Tetraoxime Ligand. Spectrochim. Acta, Part A 2018, 190, 111–115. 10.1016/j.saa.2017.09.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.