

Abstract
We report inducible dimerization strategies to control protein positioning, enzymatic activity, and organelle assembly inside synthetic cell-like compartments upon photostimulation. Using a photocaged TMP-Haloligand compound, we demonstrate small molecule and light-induced dimerization of DHFR and Haloenzyme to localize proteins to a compartment boundary and reconstitute tripartite sfGFP assembly. Using caged rapamycin and fragments of split TEV protease fused to FRB and FKBP, we establish optical triggering of protease activity inside cell-size compartments. We apply light-inducible protease activation to initiate assembly of membraneless organelles, demonstrating the applicability of these tools for characterizing cell biological processes in vitro. This modular toolkit, which affords spatial and temporal control of protein function in a minimal cell-like system, represents a critical step toward the reconstitution of a tunable synthetic cell, built from the bottom up.
Keywords: Optochemical, Optogenetic, Light-Induced Dimerization, cTMP, CTH, Halo, DHFR, FRB-FKBP, dRap, Split Enzymes, sfGFP, TEV protease, Synthetic Cell, Compartmentalization, Cell-like
Introduction
Attaining conditional control of biological organization and activity provides a unique strategy to interrogate the processes that govern cell behavior. Approaches based on optical or chemical control of dimerization exemplify this strategy, enabling regulation of protein localization and function in space and time1. Optogenetics is one such approach; it utilizes proteins whose oligomeric states are naturally light sensitive, such as plant phytochromes (Pif/PhyB)2,3, phototropins (Lov domains)4–6, engineered fluorescent proteins such as Dronpa7, fungal photoreceptors8, and cryptochromes (Cry2)5,9. However, these systems often require continuous illumination to maintain protein dimerization and have only moderate affinity in the illuminated state.
Optochemical approaches overcome limitations of optogenetic systems by using modular protein switches that dimerize in response to a high-affinity ligand, and by photocaging the ligand through synthetic means in order to block binding in the absence of light1. For example, a bivalent ligand containing trimethoprim (TMP) linked to HaloTag ligand (Haloligand), referred to as TMP-Haloligand (TH), binds tightly to E. Coli dihydrofolate reductase (DHFR) and HaloTag protein (Haloenzyme). However, a coumarin-photocaged version, called caged TMP-Haloligand (CTH), sterically hinders TMP, which blocks its binding to DHFR. In the dark, DHFR and Haloenzyme do not interact, whereas in the presence of violet light, CTH uncages and induces dimerization of these protein domains12. This strategy has been successfully implemented in vivo to optically control protein localization to organelles and kinetochores10–12. Similarly, rapamycin induces dimerization of the canonical FK506-binding protein (FKBP) and FKBP rapamycin binding domain (FRB)13, and light-activated analogs have been generated. In particular, a light-cleavable rapamycin dimer called dRap allows FKBP to bind but sterically blocks association with FRB in the absence of light. Photo-induced association of FRB and FKBP is achieved by cleaving dRap with light to remove the steric constraints, allowing each FKBP:rapamycin to bind FRB14. By fusing these optochemically sensitive domains to a protein of interest, it is feasible to modulate enzymatic localization and function15,16.
The complexity of the cellular milieu has motivated efforts to characterize biological processes in a minimal and carefully controlled context, such as through biochemical reconstitution of purified components17. Recently, it has become possible to carry out more complex ‘cellular reconstitutions’ to study chemical reactions in a cell-like context18,19. For example, cell-like compartments, such as liposomes and emulsions, have been used to encapsulate proteins and cytoplasm20–22. The geometries and picoliter volumes of these compartments mimic those of a natural cell. However, several limitations have hampered further development of these cell-like systems: only a handful of tools are available to pattern protein localization23, and the compartment boundary, whether a lipid monolayer or bilayer, is impermeable to hydrophilic molecules. The latter makes it challenging to chemically regulate enzymatic processes housed within these cell-like compartments. Light-inducible control of protein localization and activity promises to be a critical advance for cell biological studies in synthetic cell systems. Optical triggering is ideal because light can be readily controlled with temporal precision, localized irradiation can convey spatial control, and light easily penetrates into cell-like compartments, regardless of their permeability to small molecules.
By fusing split proteins to inducible dimerization domains, optochemical inputs can be converted into biological outputs. Split proteins, such as split GFP24, have been traditionally used to identify native interacting partners in protein complementation assays (PCA) and to characterize in-vivo protein-protein interactions (PPI) and their inhibitors25. When fragments of split GFP are brought into close proximity via interaction of binding partners, they reconstitute fluorescence26. Independent of their uses to identify constitutive binding interactions, split proteins have also been used to create triggerable switches. For example, a split version of Tobacco Etch Virus (TEV) protease has been fused to FRB and FBKP domains to reconstitute TEV activity in the presence of rapamycin27,28. Additionally, split enzymes have been optically reconstituted by illumination of caged compounds14.
Here we characterize the CTH photocaged dimerizer system as a tool to modulate protein localization and assembly in emulsion-based, synthetic cell-like compartments. We further test a second, complementary photochemical system, composed of a light-cleavable rapamycin dimer, dRap, paired with FRB and FKBP, as a strategy for transducing optical inputs into enzymatic function based on split protease reconstitution. We demonstrate the utility of this optochemically-regulated protease through light-induced phase separation and formation of membraneless organelles within our synthetic cells.
Results
To engineer spatial control of protein positioning in vitro within synthetic cells, we tested whether proteins could be relocalized from the lumen to compartment boundary within a water-in-oil emulsion. The proteins included His 10-RFP-Haloenyzme, whose His-tag enables anchoring to DGS-NTA(Ni) lipid, and GFP-DHFR (Fig. 1A, Fig. S1A,B). The dimerizer, non-caged TH, and the caged dimerizer, CTH, were prebound to Haloenyzme through a covalent interaction with Haloligand. To determine whether the components were biochemically active, we tested whether Haloenzyme and DHFR could be chemically and optically dimerized in vitro. First, we conducted pulldown binding assays using MBP-GFP-DHFR immobilized on amylose beads as bait. We found that in the presence of non-caged TH, His10-RFP-Haloenzyme prey bound to MBP-GFP-DHFR (Fig. 1B). In the absence of dimerizer an interaction could not be detected via pulldown assay (Fig. 1B). Using the photocaged compound, we found undetectable interaction in the absence of light, and significant binding after exposure to 405 nm light (Fig. 1B, C). Prey binding was consistently much higher in the light than in the dark and within a factor of two of binding observed in positive control, non-caged TH dimerizer (Fig 1C, Fig. S1C). These results demonstrated that the core components were functional and bind specifically to one another, only in the presence of small molecule dimerizer or in response photouncaging of the caged dimerizer.
Fig. 1. Optochemical control of protein spatial localization in cell-like compartments.
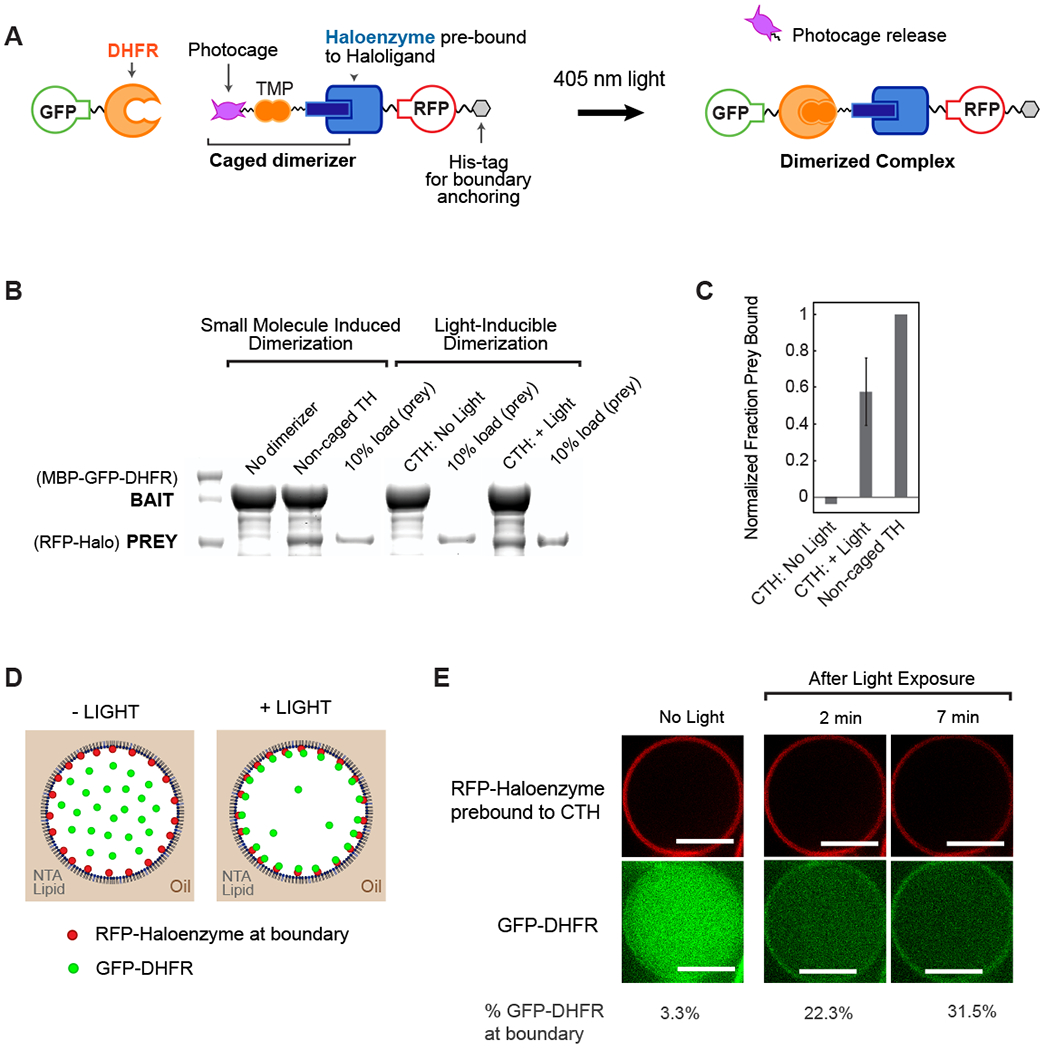
(A) Schematic overview of photocaged TMP-Halo inducible dimerization system. (B) Haloenzyme and DHFR bind to one another in the presence of non-caged TMP-Halo dimerizer, but not in absence of dimerizer, in a pulldown assay. Photouncaging of caged TMP-Halo (CTH) leads to comparable binding to TH; there is undetectable binding in the dark. Caged and non-caged compounds were prebound to Haloenzyme protein. (C) Quantification of CTH and non-caged TH pulldown binding, normalized to positive control, non-caged TH binding. Error bar shows standard deviation from the mean, n = 3 (D) Schematic of light-inducible protein recruitment to the boundary inside synthetic cell-like compartments. 5% DGS-NTA(Ni) lipid and 95% POPC in decane phase. 1 μM His10-RFP-Halo, 0.1 μM GST-GFP-DHFR in aqueous phase. (E) His10-RFP-Halo binds to DGS-NTA(Ni) lipid in the droplet boundary. Recruitment of GST-GfP-DHFR to the boundary is triggered by 405 nM illumination, which uncages CTH. Scalebar 10 μm.
To implement control of protein localization in synthetic cells, we tethered His10-RFP-Haloenzyme to DGS-NTA(Ni) lipid present in the lipid monolayer boundary and tested light-induced recruitment of GST-GFP-DHFR to the boundary. The components were recombinantly expressed and then encapsulated in water-in-oil emulsions with a lipid monolayer composed of 95% POPC and 5% DGS-NTA(Ni) lipid (Fig. 1D). The DGS-NTA(Ni) lipid recruited His10-RFP-Haloenzyme bait to the boundary (Fig. 1E). In the dark, only 3% of GST-GFP-DHFR prey is localized to the compartment boundary. After illumination using 405 nm light nearly one-third of the protein relocalizes to the compartment boundary, via light-inducible dimerization to the tethered bait (Fig. 1E, Fig. S2). These results demonstrate successful light-inducible protein localization in water-in-oil emulsions for patterning cell-like organization, such as cortical protein recruitment, with temporal control over this spatial organization.
Next, we wanted to test whether a small molecule or light stimulus could be used for temporal control of protein assembly and reconstitution within synthetic cells. Towards this goal, we used a tripartite split superfolder GFP (sfGFP) (Fig. 2A), which only fluoresces when strand 10 and strand 11 are dimerized in the presence of strands 1-9. This variant of split GFP was developed to have reduced auto-reconstitution29 compared to an older bipartite split sfGFP30. We fused sfGFP.Strand10 to Haloenzyme and DHFR to sfGFP.Strand11, and generated GST-sfGFP.Strands1-9 (Fig. S3A,B). Using these components, we tested whether assembly of the trimeric complex and ultimate generation sfGFP fluorescence could be triggered by the addition of non-caged TH or by uncaging CTH with 405 nm light. First, we tested whether we could stimulate tripartite sfGFP assembly and fluorescence in bulk solution using the non-caged TH dimerizer. We found that non-caged TH led to significant reconstitution of sfGFP fluorescence, comparable to 21% of the intact sfGFP control (Fig. S3C). Only minimal auto-reconstitution in the absence of dimerizers was observed, yielding a greater than a 65-fold increase in fluorescence of the tripartite split sfGFP in the presence of the dimerizer (Fig. 2B).
Fig. 2. Inducible reconstitution of tripartite sfGFP assembly and activity.
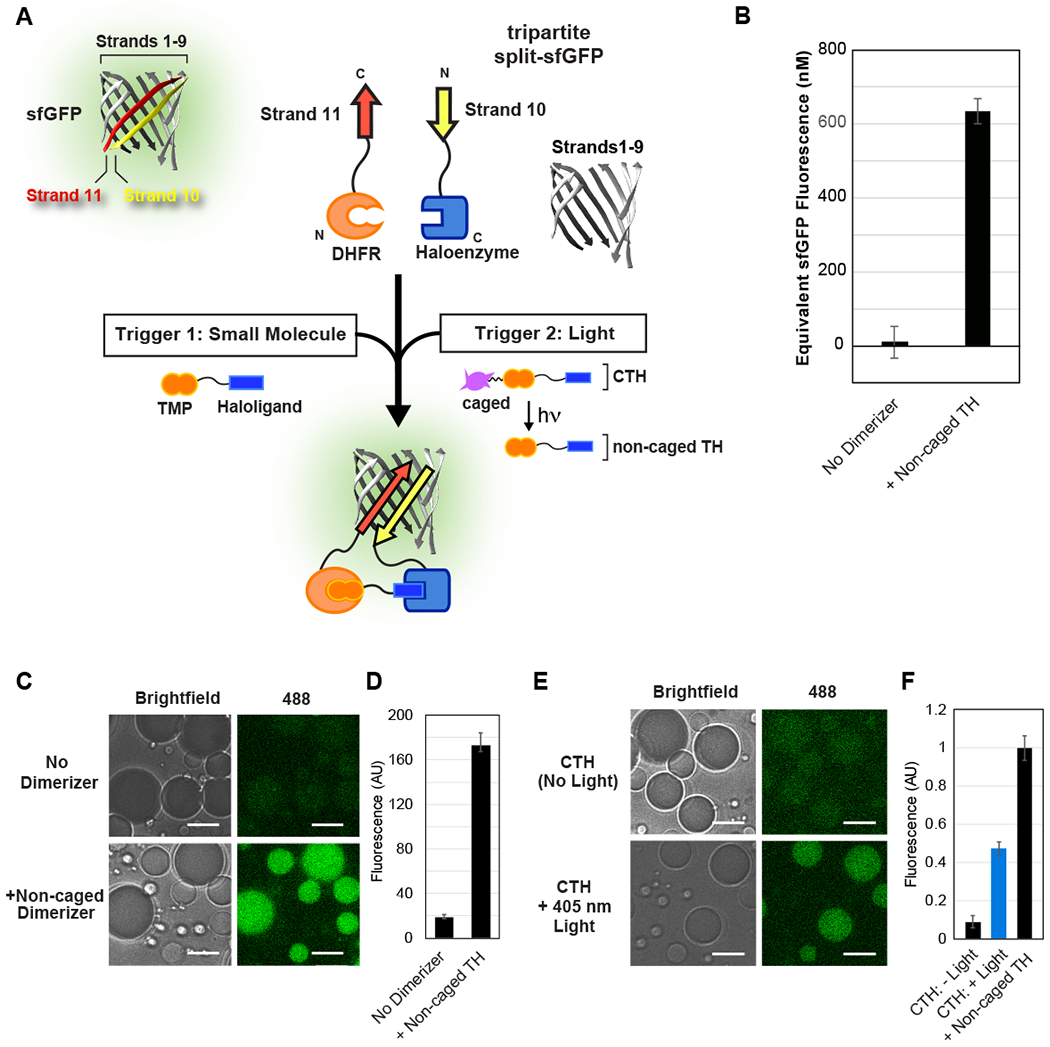
(A) Schematic of tripartite sfGFP system, and fusion to DHFR and Halo domains to enable chemical or optical control of sfGFP reconstitution. Non-caged and caged versions of TMP-Halo compound are prebound to the sfGFP.Strand10-Haloenzyme protein. (B) Non-caged dimerizer promotes reconstitution of sfGFP fluorescence in a plate reader assay. The increase in fluorescence over 12 hours was interpolated to a dilution series of full sfGFP (Fig. S3C), yielding concentrations of sfGFP reconstitution. (C-D) Small molecule induced assembly of tripartite sfGFP inside cell-like compartments depends on the presence of non-caged TH dimerizer. Fluorescence was quantified 18 hours after addition of dimerizer and encapsulation, to allow for sufficient chromophore maturation. (E-F) Light-inducible reconstitution of sfGFP inside water-in-oil emulsions, normalized to non-caged TH positive control. Approximately 50% uncaging achieved using 1 s exposure to 405 nm laser light. All experiments used 3 μM DHFR-sfGFP.Strand11,3 μM sfGFP.Strand10-Haloenzyme, and 24 μM sfGFP.Strands1-9. Scale bar: 10 μm.
We further wanted to test whether such a strategy for inducing protein reconstitution would work inside cell-size compartments, so we encapsulated the reactions in water-in-oil emulsions. Because we were not interested in protein recruitment to the compartment boundary we selected a passivating surfactant, Cithrol DPHS, to prevent non-specific interactions. We found that water-in-oil emulsions containing the three proteins plus non-caged TH displayed a 9fold higher sfGFP fluorescence compared to those that lacked the dimerizer (Fig. 2C,D). This result demonstrated that small molecule induced split protein reconstitution was feasible within cell-like compartments. However, these experiments are limited since dimerizer must be added prior to encapsulation; the boundary and continuous oil layer prevents non-caged TH from diffusing from the exterior environment into the lumen of the compartment. Therefore, we decided to test light as a trigger, because photons are able to transduce the compartment.
To determine whether an optical trigger was capable of reconstituting tripartite sfGFP in synthetic cells, we loaded sfGFP.Strand10-Haloenyzme with CTH and encapsulated it along with DHFR-sfGFP.Strand11 and sfGFP.Strands1-9 in water-in-oil emulsions. In the absence of light, minimal fluorescence was observed. However, upon stimulation with light, we observed sfGFP reconstitution consistent with approximately one-half the fluorescence signal in the non-caged TH control sample (Fig. 2E,F). This outcome demonstrates that we can achieve robust light-triggered protein assembly within our cell-like compartments using the CTH system.
Following our initial demonstration of light-inducible protein assembly, we wanted to expand our optochemical toolkit and add-in triggerable enzymatic activity. We decided to use a split version of Tobacco Etch Virus (TEV) protease, a member of the cysteine family of proteases that recognizes and efficiently cleaves the recognition site ENLYFQG31. N and C terminal domains of TEV were fused to FRB and FKBP (Fig. S4A,B), respectively to enable small molecule-based dimerization and activation of split TEV (Fig. 3A), as shown previously27. Because split TEV had not been previously characterized outside cells, we were unsure whether the proteins would be functional and if the protease activity could be successfully triggered by rapamycin. Using an in vitro fluorimetric TEV protease assay (Fig. 3B, Fig. S4C), we tested the activity of a mixture of FRB-TEV-N and FKBP-TEV-C with varying concentrations of dimerizer (Fig. 3C). We observed TEV protease activity dependent on rapamycin dosage, with little background activity in its absence. Next, we tested whether we could introduce a similar strategy to chemically induce TEV protease activity in cell-size compartments. We found that water-in-oil emulsions containing split TEV constructs in the presence of rapamycin displayed 9-fold higher activity than emulsions without rapamycin (Fig. 3D,E). To the best of our knowledge, these studies represent the first demonstration of a small molecule-activated protease in a cell-free system.
Fig. 3. Activation of split TEV protease in cell-like compartments using small molecule and light-inducible dimerization.
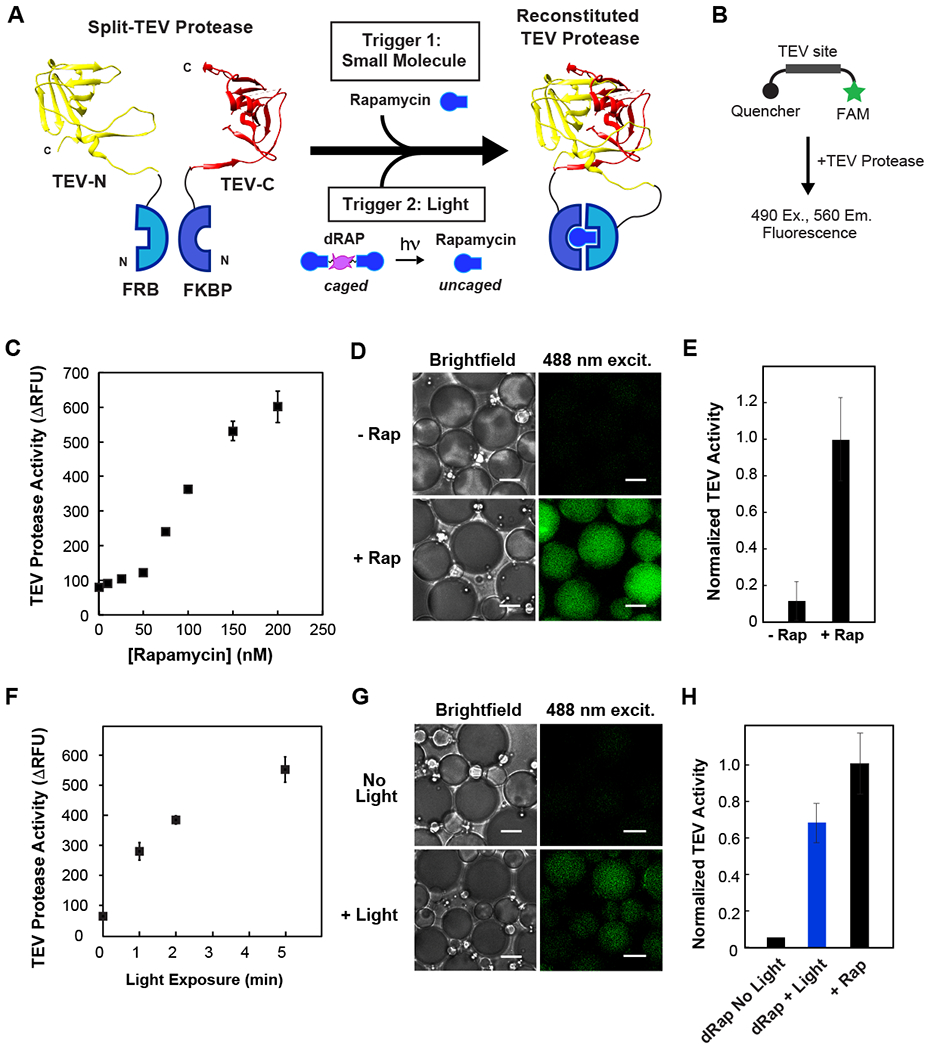
(A) Schematic of photocaged rapamycin, dRap, and split TEV fragments fused to FRB and FKBP. (B) Schematic of TEV activity assay: upon substrate cleavage, a FAM fluorophore is released from quencher. (C) Dose-dependence of rapamycin mediated TEV reconstitution. Assay uses 125 nM of split TEV proteins and varying concentrations of rapamycin. (D-E) Chemically induced reconstitution of TEV activity inside celllike compartments. Equimolar concentration of rapamycin promotes TEV protease activity; there is low background activity in the absence of rapamycin. (F) Optical uncaging of dRap at various exposure times, promotes TEV reconstitution in a plate reader assay. 125 nM split TEV and 73 nM dRap. (G-H) Temporal triggering of TEV activation within cell-like compartments using light. 10 min exposure to 365 nm UV light to uncage dRap within emulsions. Minimal background activity in non-illuminated samples. For 3D-E and 3G-H, 500 nM split TEV proteins with equimolar rapamycin or equivalent dRap. For 3G-H, activity from a control without dimerizer was subtracted from conditions with dimerizer present. Scale bar: 5 μm.
We also set out to implement a light-inducible version of TEV, because chemical triggers cannot permeate the emulsion compartment. As our optochemical tool, we selected a light-cleavable rapamycin dimer (dRap), which, upon illumination with 365 nm light, uncages and dimerizes FRB and FKBP proteins. The light-cleavable rapamycin dimer binds two FKBP proteins in the dark state, and only allows FRB to bind each FKBP:Rapamycin complex after irradiation to cleave the dRap linker14. Using the FRB-TEV-N and FKBP-TEV-C proteins described above, we added dRap and illuminated the samples for varying periods of time. TEV protease activity displayed a dose-dependence to light exposure, with minimal signal in the absence of light (Fig. 3F).
Next, we tested this strategy for light-induced TEV protease activation in cell-like compartments. FRB-TEV-N, FKBP-TEV-C and dRap were encapsulated inside cell-size water-in-oil emulsions. Following illumination with UV light, TEV protease activity achieved a level corresponding to approximately 70% of activity in the positive control containing rapamycin (Fig. 3G,H). Minimal activity was observed in samples that were not exposed to light, illustrating that enzymatic activity can be optically triggered within a synthetic cell system.
To demonstrate that our optochemical protease platform could be used to interrogate cell biological phenomena, and in the presence of cell cytoplasm, we chose to test the formation of membraneless organelles from an intrinsically disordered protein (IDP) inside cell-like compartments (Fig. 4A). We used a domain containing RGG repeats, which self-assembles and phase separates in vitro32. IDP domains, such as this one, demix from solution under physiological conditions but can be made soluble by tagging with a large globular domain33. We fluorescently-tagged the IDP and tested whether phase separation and formation of organelles could be triggered by TEV-mediated removal of an MBP solubilization tag (Fig. S5A,B). In cell-like compartments, the IDP readily phase separates into protein droplets when encapsulated along with split TEV and rapamycin, but remains well-mixed in the absence of rapamycin (Fig. 4B). Next we tested whether formation of membraneless organelles could be triggered using light by encapsulating the components along with dRap. In the absence of exposure to light, cell-like compartments displayed no protein droplets. However, by exposing to 365 nm light, membraneless organelles were formed inside these compartments (Fig. 4C). This finding demonstrates the successful use of our optochemical system for inducible control of cell biological processes such as membraneless organelle formation.
Fig. 4. Triggering formation of membraneless organelles in cell-like compartments using small molecule and light-inducible protease activity.
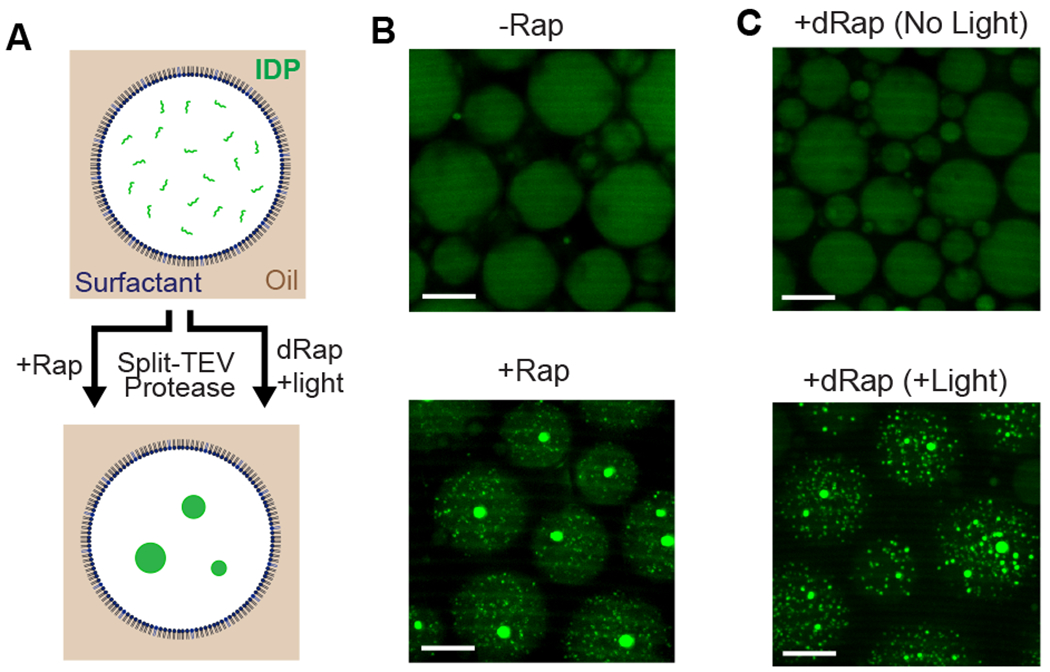
(A) Schematic of fluorescently-tagged IDP formation of membraneless organelles. MBP solubilization domain is cleaved from the IDP, resulting in phase separation and formation of membraneless organelles. (B) Rapamycin-dependent formation of protein droplets within emulsions. In absence of rapamycin, IDP remains soluble and well mixed. 1 μM split TEV was +/− equimolar dimerizer and 30 μM IDP, in the presence of 25% Xenopus egg extract. (C) Light-induced TEV activation and formation of membraneless organelles in cell-like compartments. Compartments encapsulated with 1 μM split TEV, 500 nM dRap, 30 μM IDP, 25% egg extract; either kept in dark or exposed to 365 nm UV light for 10 minutes. For 4B-C, emulsions were imaged 12 hours post-induction. Scalebar: 20 μm.
In summary, our study demonstrates the utility of optochemical approaches for inducing protein dimerization and identifies a robust set of tools for conditional control of protein localization and activity in a minimal cell system. Triggering protein patterning and function using light is particularly critical to the study of biochemical pathways in water-in-oil emulsions because these compartments, once formed, are impermeable to small molecules. Although these proteins were validated in water-in-oil emulsions, they will retain functionality within liposomes or polymersomes and thus represent a robust toolkit for studies within a broad variety of synthetic cell systems. By demonstrating optical triggering of protein-based membraneless organelle assembly, our work paves the way toward spatial and temporal perturbations of additional cell biological processes inside cell-like compartments.
Supplementary Material
Acknowledgements
We thank M. Lampson (U.Penn) for DHFR and Haloenzyme plasmids, M. Rosen (UTSW) for FRB and FKBP plasmids, and members of the Good, Lampson and Chenoweth labs for helpful discussion. JGB is supported by a Cell and Molecular Biology NIH Training Grant (5-T32-GM-007229-39). BSS is supported by an NIH postdoctoral fellowship (F32GM119430). MCG is supported by a CASI grant from Burroughs Wellcome Fund, and an award from the Charles E. Kaufman Foundation. TC is supported by a National Science Foundation Graduate Research Fellowship. AD acknowledges funding from the NSF (CHE-1404836). DMC acknowledges support from NIH (GM118510). DAH acknowledges support from DOE (DE-SC0007063).
References
- 1.Ankenbruck N, Courtney T, Naro Y, Deiters A Angew Chem Int Ed Engl 57, 2768–2798.(2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levskaya A, Weiner OD, Lim WA, Voigt CA Nature 461, 997–1001. (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckley CE, Moore RE, Reade A, Goldberg AR, Weiner OD, Clarke JDW Dev Cell 36, 117–126. (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strickland D, Lin Y, Wagner E, Hope CM, Zayner J, Antoniou C, Sosnick TR, Weiss EL, Glotzer M Nat Methods 9, 379–384. (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hallett RA, Zimmerman SP, Yumerefendi H, Bear JE, Kuhlman B ACS Synth Biol 5, 53–64. (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guntas G, Hallett RA, Zimmerman SP, Williams T, Yumerefendi H, Bear JE, Kuhlman B Proc Natl Acad Sci U S A 112, 112–117. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou XX, Chung HK, Lam AJ, Lin MZ Science 338, 810–814. (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawano F, Suzuki H, Furuya A, Sato M Nat Commun 6, 6256 (2015) [DOI] [PubMed] [Google Scholar]
- 9.Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL Nat Methods 7, 973–975. (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ballister ER, Aonbangkhen C, Mayo AM, Lampson MA, Chenoweth DM Nat Commun 5, 5475 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ballister ER, Ayloo S, Chenoweth DM, Lampson MA, Holzbaur ELF Curr Biol 25, R407–R408. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Aonbangkhen C, Tarasovetc EV, Ballister ER, Chenoweth DM, Lampson MA Nat Chem Biol 13, 1096–1101. (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banaszynski LA, Liu CW, Wandless TJ Journal of the American Chemical Society 127, 4715–4721. (2005) [DOI] [PubMed] [Google Scholar]
- 14.Brown KA, Zou Y, Shirvanyants D, Zhang J, Samanta S, Mantravadi PK, Dokholyan NV, Deiters A Chem Commun (Camb) 51, 5702–5705. (2015) [DOI] [PubMed] [Google Scholar]
- 15.Karginov AV, Ding F, Kota P, Dokholyan NV, Hahn KM Nat Biotechnol 28, 743–747. (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenstein AE, Echols N, Lombana TN, King DS, Alber T J Biol Chem 282, 11427–11435. (2007) [DOI] [PubMed] [Google Scholar]
- 17.Liu AP, Fletcher DA Nat Rev Mol Cell Biol 10, 644–650. (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bermudez JG, Chen H, Einstein LC, Good MC Genesis 55 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Good MC Methods Mol Biol 1413, 87–108. (2016) [DOI] [PubMed] [Google Scholar]
- 20.Stachowiak JC, Richmond DL, Li TH, Liu AP, Parekh SH, Fletcher DA Proc Natl Acad Sci U S A 105, 4697–4702. (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyazaki M, Chiba M, Eguchi H, Ohki T, Ishiwata S Nat Cell Biol 17, 480–489. (2015) [DOI] [PubMed] [Google Scholar]
- 22.Good MC, Vahey MD, Skandarajah A, Fletcher DA, Heald R Science 342, 856–860. (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudd AK, Valls Cuevas JM, Devaraj NK J Am Chem Soc 137, 4884–4887. (2015) [DOI] [PubMed] [Google Scholar]
- 24.Magliery TJ, Wilson CGM, Weilan Pan DM, Ghosh I, Hamilton AD, Regan L J Am Chem Soc 127, 146–157 (2005) [DOI] [PubMed] [Google Scholar]
- 25.Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E Nat Rev Drug Discov 6, 569–582. (2007) [DOI] [PubMed] [Google Scholar]
- 26.Shekhawat SS, Ghosh I Curr Opin Chem Biol 15, 789–797. (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gray DC, Mahrus S, Wells JA Cell 142, 637–646. (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wehr MC, Laage R, Bolz U, Fischer TM, Grunewald S, Scheek S, Bach A, Nave KA, Rossner MJ Nat Methods 3, 985–993. (2006) [DOI] [PubMed] [Google Scholar]
- 29.Cabantous S, Nguyen HB, Pedelacq JD, Koraichi F, Chaudhary A, Ganguly K, Lockard MA, Favre G, Terwilliger TC, Waldo GS Sci Rep 3, 2854 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cabantous S, Terwilliger TC, Waldo GS Nat Biotechnol 23, 102–107. (2005) [DOI] [PubMed] [Google Scholar]
- 31.Phan J, Zdanov A, Evdokimov AG, Tropea JE, Peters HK 3rd, Kapust RB, Li M, Wlodawer A, Waugh DS J Biol Chem 277, 50564–50572. (2002) [DOI] [PubMed] [Google Scholar]
- 32.Elbaum-Garfinkle S, Kim Y, Szczepaniak K, Chen CC, Eckmann CR, Myong S, Brangwynne CP Proc Natl Acad Sci U S A 112, 7189–7194. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burke KA, Janke AM, Rhine CL, Fawzi NL Mol Cell 60, 231–241. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.