

Abstract
Mitral valve prolapse (MVP) affects 1 in 40 people and is the most common indication for mitral valve surgery. MVP can cause arrhythmias, heart failure, and sudden cardiac death, and to date, the causes of this disease are poorly understood. We now demonstrate that defects in primary cilia genes and their regulated pathways can cause MVP in familial and sporadic nonsyndromic MVP cases. Our expression studies and genetic ablation experiments confirmed a role for primary cilia in regulating ECM deposition during cardiac development. Loss of primary cilia during development resulted in progressive myxomatous degeneration and profound mitral valve pathology in the adult setting. Analysis of a large family with inherited, autosomal dominant nonsyndromic MVP identified a deleterious missense mutation in a cilia gene, DZIP1. A mouse model harboring this variant confirmed the pathogenicity of this mutation and revealed impaired ciliogenesis during development, which progressed to adult myxomatous valve disease and functional MVP. Relevance of primary cilia in common forms of MVP was tested using pathway enrichment in a large population of patients with MVP and controls from previously generated genome-wide association studies (GWAS), which confirmed the involvement of primary cilia genes in MVP. Together, our studies establish a developmental basis for MVP through altered cilia-dependent regulation of ECM and suggest that defects in primary cilia genes can be causative to disease phenotype in some patients with MVP.
INTRODUCTION
Cilia are microtubule-containing structures (axonemes) that project from the cell. There are two main types of cilia: motile and immotile. Whereas motile cilia (>100 per cell) are largely used to move fluid or propel gametes, immotile cilia (primary cilia) are solitary (typically one per cell), extending from the centriole/basal body, and were previously thought to be vestigial evolutionary remnants with no persisting function (1). However, this traditional belief was challenged by recent genetic discoveries that linked mutations in cilia genes to a spectrum of rare syndromic diseases, now known as ciliopathies (2, 3). These findings spurred concerted efforts to understand ciliogenesis and downstream pathways, as well as additional genetic conditions that stem from defects in cilia structure and/or function. The emerging data suggest that primary cilia are essential structures that function to transduce mechanical, electrical, and chemical signals in a tissue-specific and time-dependent context. In doing so, they relay growth factor [transforming growth factor–β (TGFb) (4), platelet-derived growth factor (PDGF) (5), WNT (6, 7), and Hedgehog (8)] and extracellular matrix (ECM) (9) information from their surrounding microenvironment to influence cell survival, differentiation, and tissue organization.
Recent murine and human genetic discoveries have linked primary cilia to a spectrum of disorders involving cardiac, renal, skeletal, and neurologic tissues (10–12). A classic example is the ciliopathy, autosomal dominant polycystic kidney disease (ADPKD), in which ~25% of patients have mitral valve prolapse (MVP), a 10-fold increase above background (13, 14). Both MVP and ADPKD have a congenital etiology and are characterized by excessive deposition of proteoglycans, as well as disorganization and fragmentation of the collagen and elastin in the ECM (15, 16). These molecular changes, which culminate in abnormal tissue hypertrophy, impair normal tissue architecture and function in both mitral valves and kidneys. The comorbidity of MVP and ADPKD, in concert with the pathological similarities of disease phenotypes, raises the possibility that MVP can be caused by cilia gene defects in some individuals.
RESULTS
Primary cilia are spatially and temporally regulated during cardiac valve morphogenesis
Because clinical data support conservation of cilia structure and function in the kidney and heart valves (13, 14), we initiated experiments to determine whether primary cilia contribute to valvulogenesis and whether they are involved in the etiology of MVP by first performing immunohistochemistry (IHC) on murine and human mitral valve leaflets. Primary cilia were infrequently observed on valve endocardium at all time points analyzed (Fig. 1, A to F, and fig. S1A). Cilia were, however, detected on most interstitial (mesenchymal) cells within the murine and human embryonic, fetal, and neonatal valve primordia (Fig. 1, A to E, figs. S1 and S2, A and B, and movies S1 to S3) and were confirmed by three-dimensional (3D) electron tomography as immotile primary cilia with the typical 9 + 0 microtubule configuration (Fig. 1, G and H, and movies S4 and S5). During embryonic/fetal gestation and early neonatal life, primary cilia grow in length with a maximum length observed shortly after birth (Fig. 1I and data file S1). The presence of cilia correlated with the type of ECM produced within the mitral valves. For example, the early valve (E11.5) expresses abundant proteoglycans and very little collagen, coinciding with the expression of cilia on nearly every interstitial cell (fig. S2, A and B). However, by postnatal day 0 (P0), increased collagen within the valve had an inverse correlation with both the presence and length of primary cilia (fig. S2, A, C, and D), and although cilia were abundant at this stage, their presence was primarily confined to proteoglycan-rich regions (fig. S2, B to D, movies S6 and S7, and data file S2). Valve cells expressing cilia in the adult were rarely observed (Fig. 1, F and I). Thus, there is a correlation between the type of ECM produced and the presence of cilia, which supports their putative function in regulating, or responding to, the extracellular environment during mitral valve development.
Fig. 1. The numbers of primary cilia on valve interstitial cells vary spatially and temporally during mitral development.

IHC of axonemes (green, acetylated a-tubulin) and basal bodies (red, g-tubulin). (A) E11.5, (B) E13.5, (C) E15.5, (D) E17.5, (E) P0, and (F) adult (Ad) at 3 months of age. Primary cilia are abundant on valve interstitial cells (VICs). Arrows designate the lack of axonemes on valve endocardial cells. Blue, nuclei (Hoechst); AL, anterior leaflet; PL, posterior leaflet. Three levels of magnification are shown from left to right. Scale bars, 100, 10, and 2 mm [(A) to (E)]; 200, 50, and 5 mm (F). (G) Tomographic electron microscopy of VICs at P0 showing a primary cilium (axoneme), basal body, and mother centriole. Scale bar, 100 nm. (H) Higher magnification of the mother centriole showing a classic triplet microtubule 9 + 0 organization indicative of a primary cilium. Scale bar, 20 nm. (I) Box plots of primary cilia lengths in the mitral valves throughout the murine life cycle. The blue boxes show the main distribution of the cilia length values. The bottom of the box is the 25th percentile, the middle line is the median value, and the top of the box is the 75th percentile. Red dot denotes the mean cilia length. Error bars represent 95% confidence intervals. *P < 0.01; **P < 0.001; ***P < 0.0001.
Primary cilia orchestrate valve development through an ECM mechanism
To determine whether primary cilia are relevant to matrix synthesis and/or its organization during valve development, we conditionally removed the ciliogenic gene, intraflagellar transport protein 88 (Ift88) (17) from endothelial-derived mesenchyme by using the NfatC1Cre driver (18). Loss of Ift88 in valve mesenchyme resulted in failure of ciliogenesis, as indicated by the lack of axonemes (Fig. 2A), a finding that was concomitant with valve leaflet enlargement at P0 (Fig. 2, B to D, and data file S3). In vivo cell analyses demonstrated that loss of primary cilia does not affect the proliferation or the total number of valve progenitor cells (fig. S3 and data file S4) but does result in a significant decrease in VIC density, P < 0.001 (fig. S4 and data file S5). Whole transcriptome analyses [RNA sequencing (RNA-seq)] and subsequent IHC confirmed that loss of primary cilia results in robust activation of ECM gene pathways in the anterior mitral leaflets (GEO accession no. GSE125092; figs. S5 and S6 and data file S6) that are consistent with early stages of myxomatous degeneration. We next sought to determine whether developmental loss of primary cilia results in progressive tissue changes that culminate in adult mitral valve disease. As shown in Fig. 2 (E to G), genetic disruption of primary cilia via Ift88 deletion results in adult myxomatous mitral valve disease as evidenced by increased proteoglycans (such as versican) and loss of the normal ECM distribution within the valve. Thus, loss of primary cilia causes developmental defects that are characterized by expansion of ECM and disrupted histological architecture, which progress to an adult myxomatous valve pathology, similar to that observed in patients with MVP.
Fig. 2. Loss of Ift88 impairs ciliogenesis during development and results in myxomatous mitral valve disease.

(A) IHC for acetylated tubulin (green) and basal bodies (red) at neonatal day 0 in conditional knockout for Ift88 [NfatC1Cre(+);Ift88f/f] and control mice shows axoneme structures (loss indicated by arrowheads). (B) Hematoxylin and eosin (H&E) staining for Ift88 conditional knockout (cKO) and control mice at P0. (C) 3D reconstructions of the mitral valve anterior and posterior leaflets (valve thickening indicated by arrowheads). (D) Quantification of valve 3D reconstructions of control [ NfatC1Cre(+);Ift88+/+] and Ift88 conditional knockout [ NfatC1Cre(+);Ift88f/f] mitral leaflets. *P < 0.03; n = 6 for conditional knockout and n = 4 for control. Dots represent means, and error bars are SD of the mean. (E) IHC for collagen (red), myocardium (green), and nuclei (blue) on adult Ift88-deficient mitral leaflets and controls. (F) IHC for versican (red), myocardium (green), and nuclei (blue) on adult Ift88-deficient mitral leaflets and controls. (G) Movats histological stain on adult Ift88-deficient mitral leaflets and controls [arrows indicate increased proteoglycans (blue) and enlarged valve leaflets] compared to controls. Proteoglycan, blue; collagen, yellow; myocardium, red. n = 4 per genotype.
Genome-wide association studies and familial genetics identify cilia gene variants in patients with MVP
On the basis of the clinical observation of ADPKD and MVP co-morbidity, combined with cilia expression studies and genetic ablation experiments, we hypothesized that cilia may play a role in human mitral valve disease. To test our hypothesis, we took three approaches. First, we took advantage of data from a previous genome-wide association study (GWAS) of 1412 MVP cases and 2439 controls (19) and performed a gene set enrichment analysis of the 278 genes implicated in primary cilia biology (20), mainly as harboring rare mutations in a diverse panel of ciliopathies (data file S7). We found a modest but significant enrichment of MVP-associated variants in this cilia gene set [P = 0.009, false discovery rate (FDR) = 0.024; data file S8]. Second, we then evaluated the two known nonsyndromic MVP genes [DCHS1 (16) and FLNA (21–24)] to determine whether they play a role in ciliogenesis. Immunohistological examination of murine knockout models of Dchs1 and Flna showed a significant reduction in mitral valve primary cilia length, P < 0.001 (fig. S7 and data file S9), consistent with a role for these organelles in the molecular etiology of MVP. Third, we obtained additional evidence for an involvement of primary cilia in causing MVP, which came from clinical and genetic analyses of a multigenerational family with inherited autosomal dominant MVP that we previously linked to chromosome 13 (Fig. 3A) (25). Of 43 family members enrolled in the original study, 6 were coded with minimal MVP and 11 patients met the full clinical diagnostic criteria for MVP, with 2 individuals having moderate to severe mitral regurgitation and 1 with ruptured chordae that required surgical intervention. None of the individuals in the family exhibited extracardiac manifestations, and all MVP-affected individuals were deemed “nonsyndromic.” The proband (III-12) of this family displayed bileaflet prolapse coincident with mitral regurgitation (fig. S8). In the original study, MVP was linked to an 8.2-Mb region of chromosome 13. Recent examination of the genes in the linked region showed that DZIP1, a gene known to regulate ciliogenesis (26–28) and/or cilia signaling (29–32), is located within the linked interval (Fig. 3B). RNA in situ hybridization and IHC experiments revealed a robust expression of Dzip1 in wild-type murine developing anterior and posterior mitral leaflets, with protein expression being localized to basal bodies and nuclear speckles within mitral VICs in vivo (fig. S9, A to D, and movie S8). Whole-exome sequencing (WES) of four affected family members (II-3, III-4, III-10, and IV-12) revealed a single heterozygous missense variant resulting in a serine-to-arginine change in both known DZIP1 isoforms (p.S70R and p.S24R), which was confirmed by Sanger sequencing (Fig. 3C). This DZIP1 variant was the only coding change identified within the linkage interval that segregated with disease phenotype. Ontology database comparisons indicate high evolutionary conservation of this amino acid. Moreover, this variant is not currently found in Single-Nucleotide Polymorphism Database (dbSNP) and observed in only 1 of 31,166 genomes and 0 of 61,336 exomes in gnomAD with a minor allele frequency of 1.081 × 10−5 (https://gnomad.broadinstitute.org/variant/13-96294074-G-T). Furthermore, this particular S70R/S24R variant has a CADD (combined annotation-dependent depletion) score of 14.27, which places it in the top 3.7% of deleterious single-base changes possible in the entire genome (33) and within the 95% confidence interval of gene-specific CADD scores corresponding to high-confidence pathogenic mutations for DZIP1 (Fig. 3D) (34). Sequencing on an additional 15 sporadic patients with MVP by WES revealed rare, potentially pathogenic DZIP1 variants in two individuals (table S1). On the basis of the strong association of the segregating DZIP1S24R variant in a multigenerational family with MVP phenotype, follow-up studies were performed to examine mutation pathogenicity.
Fig. 3. DZIP1 is identified as an MVP gene.

(A) Multigenerational family with inherited, autosomal dominant, nonsyndromic MVP. Black circles and squares are affected individuals, green circles and squares are individuals who exhibit minimal MVP, and white circles and squares are unaffected. Circles, female; squares, male. ID designations for family members are denoted under the circles or squares. “?,” unknown phenotype. Proband is identified with the black arrow. (B) Human transcript and marker maps of the linkage interval on chromosome 13 (Chr 13). Candidate region is between 13q31.3 and 13q32.1, and all RefSeq genes and their orientations are shown within the 8.2-Mb interval. DZIP1, the only cilia gene within the locus, is highlighted in pink. (C) Sanger sequencing identified a single missense mutation within exon 5 of DZIP1, resulting in a serine-to-arginine change. The mutation segregates with the affected patients and is designated by “+/−” in the pedigree. (D) Population frequency showing the rarity of the identified DZIP1 variant in the population.
Validation of DZIP1 variant reveals a developmental etiology for MVP
To assess pathogenicity of this variant, point mutation knock-in (KI) mice were generated through CRISPR-Cas9 (fig. S10). On the basis of GenBank accession numbers (human: NP_945319.1 and mouse: NP_080219.2), the murine DZIP1 lacks 10 N-terminal amino acids compared to the human, and thus, the murine mutant will be designated as DzipS14R/+. Adult mice harboring this single missense mutation (Dzip1S14R/+) develop myxomatous mitral valves (Fig. 4, A to C) and functional MVP (Fig. 4D and movie S9). MVP was never observed in control animals, and although end-diastolic volume was moderately increased in the Dzip1S14R/+ hearts, there were no apparent differences in ventricular contractile function between the Dzip1S14R/+ and control animals (table S2). This genetically accurate model for nonsyndromic MVP has allowed us to test whether the disease could be traced back to errors during development. Histological and 3D quantification at P0 revealed that 100% of Dzip1S14R/+ mice exhibited a mitral valve phenotype with variable severity compared to littermate Dzip1+/+ controls (Fig. 5, A to C), which correlated with a reduction in cilia length in the mutant animals (Fig. 5, D and E, and data files S10 and S11). To gain insight into the function of DZIP1 at early stages of morphogenesis, RNA-seq was conducted on E13.5 hearts (GEO accession no. GSE125092) and compared to the expression data that we obtained for the Ift88 RNA-seq (fig. S5, described above). Gene ontology (GO) analyses at E13.5 demonstrated that the most significant changes (P < 0.02) observed in the Dzip1S14R/+ hearts compared to controls were those associated with ECM pathways (fig. S11), consistent with our findings in the NfatC1Cre(+);Ift88f/f study. The changes in ECM composition at E13.5 likely represent early changes in the molecular profile of the developing heart. Many of the differentially expressed genes in the Dzip1S14R/+ mice are also altered in the context of various syndromic diseases that include valve disease as a comorbidity (data file S12).
Fig. 4. Dzip1S14R/+ mice have MVP and myxomatous valves.

(A) Movats staining of control hearts at 6 months of age. (B) Movats staining of Dzip1S14R/+ hearts at 6 months of age showing dysmorphic posterior leaflet (arrowhead). (C) Movats staining of Dzip1S14R/+ hearts at 6 months of age showing dysmorphic anterior leaflet (arrowhead). n = 4 per genotype. (D) Echocardiography of adult (6-month-old) Dzip1S14R/+ mice. Arrows indicate a prolapsing posterior leaflet (PL) and excess tissue on the anterior leaflet (AL). Ao, aorta. n = 4 per genotype.
Fig. 5. Dzip1S14R/+ mice have dysmorphic valves concomitant with altered ciliogenesis during development.

(A) H&E staining of Dzip1S14R/+ and control mitral valves at P0. Arrowheads indicate regions of abnormal thickening. Scale bars, 200 μm. (B) 3D reconstructions of Dzip1S14R/+ and control mitral valves at P0. White arrowhead indicates valve thickening. (C) Quantification of valve dimensions. *P < 0.05. (D) IHC for primary cilia in Dzip1S14R/+ and control P0 mitral valves. Axonemes, green; basal bodies, red; nuclei, blue. (E) Box plots of primary cilia lengths in the mitral valves of Dzip1S14R/+ and control mitral valves at P0. The blue boxes show the main distribution of the cilia length values. The bottom of the box is the 25th percentile, the middle line is the median value, and the top of the box is the 75th percentile. Red dots denote the mean cilia length. Error bars represent 95% confidence intervals. Each gray circle is a single cilia length measurement.
To test potential pathogenicity of the mutation, we quantified the protein half-lives of wild-type and mutant human DZIP1 proteins by using the protein translation inhibitor cycloheximide. These experiments revealed that the DZIP1S24R mutation results in a significant (P < 0.05) reduction in protein half-life (14.75 hours versus 40.77 hours for control) and likely loss of function (fig. S12, A and B, and data file S13). To further evaluate the possibility that the loss of DZIP1 function can cause developmental defects and myxomatous degeneration in the adult, we genetically removed Dzip1 from valve progenitor cells [NfatC1Cre(+);Dzip1f/f]. As shown in fig. S13 (A to C) (data file S14), the loss of Dzip1 in valve mesenchyme progenitor cells results in reduction in primary cilia length during development and concomitant anatomical changes in the mitral valves, similar to phenotypic observations made in the Ift88 and Dzip1S14R/+ mutants (Figs. 2 and 5). Additional analyses of adult Dzip1 conditional knockout mice revealed a myxomatous phenotype with increased proteoglycans and collagen and loss of the normal ECM zonal boundaries when compared to control mitral leaflets (fig. S13D). Functional echocardiographic assessment revealed that these structurally altered mitral valves exhibited prolapse and leaflet elongation in Dzip1-deficient animals (fig. S14 and movie S10). MVP was never observed in control animals (n = 4), and there were no significant changes in cardiac function in adult Dzip1 conditional knockout (table S3). Together, these data demonstrate that rare, damaging DZIP1 mutations can cause MVP by altering ciliogenic programs during development.
DISCUSSION
Our understanding of the role of primary cilia in the heart is still in its infancy, but there is an increasing recognition of cilia as central biomechanical and molecular regulators of cardiac development. Large datasets generated from clinical data or mutagenic screens have indicated fundamental roles for these structures in early cardiac development and congenital heart disease (10, 11, 35–42), yet mechanisms driving cilia-dependent morphogenic events remain poorly understood. Understanding how cilia function as either mechanosensors or signaling hubs during embryonic, fetal, and postnatal growth will be important if we are to identify the mechanical, molecular, and cellular pathways that are altered when cilia are perturbed in the context of human cardiovascular diseases.
This study demonstrates that cilia defects can cause MVP in humans. This was shown through analyses of our GWAS datasets, defects in cilia in various MVP murine models, demonstration of MVP in a pure ciliopathy model (Ift88), and identification of a specific, rare, and damaging variant in the cilia gene DZIP1 in a large family with inherited, autosomal dominant nonsyndromic MVP. These studies led to the generation of a genetically accurate model for nonsyndromic MVP (Dzip1S14R/+), which showed that altered developmental processes involving primary cilia can lead to MVP and provided evidence that MVP is not always an aging disease. Although the mechanisms that contribute to the deterioration of the valve leaflets as seen in patients with degenerative mitral valve disease and MVP are not well understood, we posit that understanding the function of primary cilia will shed light on MVP disease pathogenesis. Related to this, our studies suggest that primary cilia constrain ECM production during early valve morphogenesis and that the premature loss of cilia (in either Ift88 or Dzip1S14R/+ models) results in dysregulation of ECM synthesis. Thus, defining the mechanisms by which cilia regulate ECM synthesis and/or its organization through various pathways such as hedgeghog (43, 44), Wnt (45–47), Pdgf (48), Tgfβ (4, 49), Notch1 (40, 50, 51), and Tor (52) will be an important step in understanding the cause of a diverse set of disease states. Of note, recent clinical studies have identified pronounced fibrosis in the left ventricle (LV) of patients with MVP (53, 54), further supporting a model whereby some patients with cilia defects may be more prone to left ventricular dysfunction and heart failure due to dysregulated ECM synthesis. In addition, fibrosis is commonly observed in the setting of ciliopathies, especially in patients with ADPKD, strongly supporting conservation of cilia-driven molecular and cellular mechanisms across organ systems. This “molecular economy,” whereby organ systems use the same pathway for tissue growth and organization, is further supported by data showing that mutations in the DZIP1-Like (DZIP1L) gene cause polycystic kidney disease (55). Although the affected individuals within our DZIP1 MVP family were not diagnosed with cystic kidneys at the time of evaluation, our studies highlight the molecular conservation that occurs between organ systems in the regulation of ECM production.
Our data have demonstrated that primary cilia are developmentally regulated and disappear after birth in the mitral valves. This finding establishes an interesting paradox between the etiology of disease and its clinical presentation much later in life. This brings about the question of how a developmental defect can give rise to a disease recognized in the adult setting. We posit that degenerative mitral valve disease in patients with MVP can initiate through altered developmental processes, which result in changes in valve geometry and biomechanics. The altered genetic, anatomical, and biomechanical information may result in an aberrant induction of secondary factors such as inflammation, which can contribute to the tissue destruction over time. How secondary factors respond to changes in valve geometry and/or biomechanics to exacerbate a developmental defect is currently unknown. By using the model systems generated in this project, pathogenic mechanisms and disease pathways can be uncovered, which may provide keys to developing effective treatment options beneficial to patients with MVP.
MVP is a heterogeneous disease with diverse genetic causes. Although our data demonstrate that cilia defects can lead to myxomatous mitral valve disease, it remains unclear what proportion of MVP results from ciliary defects. Our limited data from exome sequencing of probands with MVP suggest that DZIP1 mutations are not a frequent cause of MVP, but larger scale sequencing studies will be needed to fully answer this question. However, we do note that all of the known genetic causes of nonsyndromic MVP result in ciliary defects, leaving open the possibility that MVP may turn out to be a disease of valvular cilia defects.
MATERIALS AND METHODS
Study design
The rationale and objective of this study was to determine the contribution of primary cilia to the etiology of MVP. Familial genetic studies and analysis of GWAS data were performed to determine gene mutation and cilia variant burden and its association with MVP. We generated a genetically accurate model of nonsyndromic MVP based on the particular DZIP1 familial gene mutation. This model and additional MVP models were developed and used for a variety of in vivo and in vitro assays during developmental and adult time points (including volumetric analysis of 3D reconstruction, IHC 3D confocal microscopy, 3D electron microscopy, and echocardiography). Power analyses were conducted to determine sample size assuming α = 0.05 with a power of 0.80. For analyses, two independent study groups were tested, one being control and the other being genetically modified animals with a primary endpoint of valve defects (proliferation, ECM production, geometry changes, functional prolapse of the mitral valve, cell density, and protein half-life). For all measurements, animals/samples were put into the two study groups based on genotypes. The researchers were blinded to all the animals’ genotypes for purpose of the analyses. Genotyping code was held by one individual not associated with measurement calculations. Analyses were conducted by at least two independent investigators who were blinded to genotype. After procurement of all measurements, the code was broken and genotype/phenotype correlations were graphically presented. The total numbers of replicates are represented in the figure legends.
Gene-targeted animals used in the study
Ift88 conditional mice were genotyped as previously described (56). Histology was performed on neonatal (P0) and adult wild-type (NfatC1Cre–;Ift88f/f), conditional heterozygous (NfatC1Cre–;Ift88f/+), and conditional knockout (NfatC1Cre+;Ift88f/f) hearts on mixed background sv129:C57Bl/6. Conditional Dzip1 knockout mice were generated by a targeted homologous recombination approach using a Dzip1-targeting construct (PG00125_Z_2_E04) purchased from the Knockout Mouse Project (KOMP) repository. The conditional knockout deletes exons 8 and 9 (Dzip1–202 transcript) and causes a reading frame shift and premature translational termination. The conditional mutant allele was genotyped using forward primer: 5′-GCCAAAGT-GGTTTGCCTGACA-3′ and reverse primer: 5′-GCAGGTTAAA-CACTCATATAGC-3′ [210 base pairs (bp) for wild type and 290 bp for mutant]. The Dzip1 conditional allele was generated on a C57Bl/6 J background. When bred with NfatC1Cre lines, the resulting background was a mixed sv129:C57Bl/6 J. Tie2Cre(+); Flna xfy and Dchs1−/− mice were used for cilia analyses, and generation of these models was previously described (15, 16, 23, 24, 57).
Generation of the Dzip1S14R KI mouse model
A p.Ser24Arg (S24R) substitution observed in a conserved region near the N terminus of human DZIP1 [National Center for Biotechnology Information (NCBI) RefSeq: NP_055749] was identified in members of a large family with autosomal dominant nonsyndromic MVP (Fig. 3). To examine the functional relevance of this polymorphism, we made a mouse model mirroring this substitution. To achieve this, the serine at amino acid position 14 in mouse DZIP1 in a region homologous to its human ortholog was targeted for substitution with arginine (S14R). This was achieved through a c.42C > G single-nucleotide exchange in mouse Dzip1 (NCBI RefSeq: NM_025943.3) using CRISPR-Cas9–mediated genome editing in zygotes from C57BL/6 J mice. Through a simultaneous silent c.45C > T substitution, we created an Aat I restriction site to facilitate genotyping. Single-guide RNA (sgRNA; ctctggccaacagccccgagngg) and single-stranded oligonucleotide (ssODN; cgcgctgtctcttcttccgcagcccttccagaagcacgtc-tactaccctctggccaacaggcctgaggggcctgacgcctctgccataggcgcggctcccatg-gcctttgtccccccagcgcg) were designed and synthesized at the Genome Engineering and iPSC Center (GEiC), Washington University, St. Louis, MO; for design strategy and sequences, see fig. S10. Pronuclear injection of single-cell embryos was performed as described previously (58). Microinjection cocktails included sgRNA (MS721. DZIP1.sp4) (2.5 ng/μl), Alt-R s.P. Cas9 nuclease 3NLS (5.0 ng/μl) (Integrated DNA Technologies Inc.), and ssODN (10 ng/μl) (MS721. DZIP.ssODN.S14R.Sense) in MI buffer [10 mM tris-HCl (pH 7.4); 0.25 mM EDTA]. We also performed injections with Cas9 mRNA supplied by the GEiC at a concentration of 5 ng/μl. In both cases, successful targeting and faithful editing were achieved as determined by polymerase chain reaction and sequence analysis of DNA extracted from tail biopsies of pups derived from injected embryos.
Injections with cocktails containing Cas9 mRNA resulted in 16 pups. Of these 16 pups (32 alleles), sequence confirmed two homozygous KI (Dzip1S14R/S14R) and one heterozygous KI (Dzip1S14R/+). Injection cocktails containing Cas9 protein resulted in 18 pups. Of these 18 pups (36 alleles), sequence analysis confirmed one homozygous KI (Dzip1S14R/S14R) and two heterozygous KI ( Dzip1S14R/+). CRISPR-mediated targeting events were also identified in other pups but occurred in concert with frameshift mutations that precluded their use in the study. In 6 of 32 targeted alleles derived from injections using Cas9 mRNA, a frameshift was identified, whereas injections using Cas9 protein resulted in a frameshift rate of 10 of 36 alleles. In total, four male heterozygous KI animals (Dzip1S14R/+) generated from this study were used for echocardiography and breeding as detailed below.
Visualization of murine mitral valve cilia by IHC
IHC was performed to identify primary cilia during the life span of mice. Embryonic and adult tissue were harvested, processed, and sectioned for IHC as previously described (41). Cilia stains to assess expression and measure cilia length by IHC were done on 15-μm-thick sections. Antigen retrieval was performed for 1 min using antigen unmasking solution (Vector Laboratories, catalog no. H-3300) in a pressure cooker (Cuisinart). After antigen retrieval, two antibodies and their dilutions were used to identify the primary cilia: acetylated tubulin (Sigma-Aldrich, catalog no. T6793, 1:500) and g-tubulin (Abcam, catalog no. ab11317, 1:1000). Primary antibodies were detected using fluorescent secondary antibodies, goat anti-mouse immunoglobulin G (IgG) (Thermo Fisher Scientific, Alexa Fluor 488; catalog no. A-11029, 1:100) or goat anti-rabbit IgG (Thermo Fisher Scientific, Cyanine5; catalog no. A-10523, 1:100). Nuclei were counterstained in all IHC experiments with Hoechst (Life Technologies, catalog no. H3569, 1:10,000) for 10 min, and slides were cover-slipped with SlowFade mounting medium (Life Technologies, catalog no. S36937). To define the spatial distribution of primary cilia with ECM, we costained for axonemes with previously validated antibodies against either versican or collagen I (1: 250 dilution) (16, 23). We used a previously validated DZIP1-specific antibody (31) with acetylated tubulin (or g-tubulin) to determine colocalization of DZIP1 with axonemes and/or basal bodies. Secondary antibodies used for this staining are listed above. IHC was also performed for collagen I and versican as described above. Mf20 (Developmental Studies Hybridoma Bank, dilution 1:50) was used to stain myocardial tissue, with n = 4 for each of these IHC experiments. Fluorescence imaging was performed using the Leica TCS SP5 AOBS Confocal Microscope System (Leica Microsystems Inc.).
3D electron tomography
Samples were prepared as previously described (59). Briefly, neonatal mouse hearts (P0) were excised after cervical dislocation, the LV was opened by an apico-basal incision under stereomicroscopic control, and the mitral valve was exposed by slight spreading of the cut LV tissue. Tissue was fixed by gently dripping iso-osmotic Karnovsky’s fixative [0.45% paraformaldehyde, 0.57% glutaraldehyde, and 0.97% sodium cacodylate; 300 mOsm (60)] onto the opened LV. The anterior mitral valve leaflet was carefully excised and kept in fixative for 24 hours. Tissue was then washed in 0.1 M sodium cacodylate, post-fixed in 1% OsO4 for 1 hour, dehydrated in graded acetone, and embedded in Epon-Araldite resin (Electron Microscopy Sciences). Semithick (280 nm) sections were placed on formvar-coated slot grids and post-stained with 2% aqueous uranyl acetate and Reynold’s lead citrate (Electron Microscopy Sciences). Colloidal gold particles (15 nm) were added to both surfaces of the sections to serve as fiducial markers for tilt series alignment. Images were acquired using a 300-kV Tecnai TF30 (FEI Company, now Thermo Fisher Scientific) and a 4 × 4–K charge-coupled device camera (UltraScan; Gatan) at the EMBL Heidelberg Electron Microscopy Core facility. The specimen holder was tilted from +60° to −60° at 1° intervals. For dual-axis tilt series, the specimen was then rotated by 90° in the X-Y plane, and another +60° to −60° tilt series was taken. The images from each tilt series were aligned by fiducial marker tracking and back-projected to generate two single full-thickness reconstructed volumes (tomograms), which were then combined to generate a single high-resolution 3D reconstruction of the original partial cell volume. Isotropic voxel size was 1.01 nm. All data were processed and analyzed using IMOD software.
Quantification of cilia
Quantification of primary cilia number and length was conducted in a blinded fashion using images acquired with the Leica TCS SP5 AOBS Confocal Microscope System (Leica Microsystems Inc.). Z-stacks were set by finding the highest and lowest depth (with 10× and 40× objectives) with visible fluorescence and using the system optimized setting to determine steps. Z-stacks were then compiled to form maximum projection images. 3D reconstructions of these images were performed by importing Z-stack confocal images into Imaris 9.0 (Bitplane Inc.) and creating surface renderings based on stain intensities. 3D reconstructions were used to generate movie representations of data or quantifiable metrics of cilia length. Cilia length was measured from the base of the axoneme (acetylated tubulin–positive stain) to the tip. For measuring the cilia length in the Dzip1 control (Dzip1+/+) versus Dzip1S14R/+, values were plotted every 0.5 mm to assess cilia length distribution differences between the two genotypes at P0. For control mitral leaflets, three animals were analyzed with a total of n = 466 cilia lengths quantified. For Dzip1S14R/+ mitral leaflets, six animals were analyzed with a total of n = 1178 cilia lengths quantified. For quantifying the spatial localization of cilia relative to collagen or versican expression, three independent replicates were performed with a total of n = 422 cells analyzed. For comparison between genotypes of MVP models Flna, Dchs1, and control, a total of five independent P0 anterior leaflets were analyzed with the following total number of cells quantified for cilia length: control, n = 224; Flna conditional knockout [Tie2Cre(+);Flna xfy], n = 185; Dchs1 knockout (Dchs1−/−), n = 154. For comparison between genotypes of the Dzip1 conditional knockout model [NfatC1Cre(+);Dzip1+/+; f/+ or f/f], a total of five independent P0 anterior leaflets were analyzed for each strain with the following total number of cells quantified for cilia length: control [NfatC1Cre(+);Dzip1+/+], n = 163; conditional heterozygote [NfatC1Cre(+);Dzip1f/+], n = 330; conditional knockout [NfatC1Cre(+); Dzip1f/f], n = 185.
Visualization of human mitral valve cilia by IHC
One fetal heart (12 weeks) was obtained under Institutional Review Board approval (2010P001333) from an elective termination that was lawfully performed at Brigham and Women’s Hospital, Boston, MA. Samples were stored in RNAlater (Qiagen Inc.) and maintained at the Harvard BioBank. Tissues were washed and refixed in formalin, followed by tissue processing, embedding, and sectioning as we have previously described (16). Staining for primary cilia and 3D reconstructions of the stains were performed as described above for the mouse IHC experiments.
Histology and 3D reconstructions
3D reconstructions of H&E images were performed to generate volumetric measurements of P0 anterior and posterior mitral leaflets, as described previously (16). Briefly, 5-mm sections throughout the entirety of the mitral valve were H&E-stained and imaged using an Olympus BX40 bright-field microscope. Images were then aligned using ImageJ FIJI and imported into Imaris 9.0. Manual reconstruction was performed by tracing each individual leaflet on every section and combining all traces to create a 3D structure. 3D reconstructions were performed on P0 mice from these genotypes: NfatC1Cre(+);Ift88f/f, Dzip1S14R/+, NfatC1Cre(+);Dzip1f/+, and NfatC1Cre(+);Dzip1f/f. For all analyses, littermates were used to ensure that background strains were similar. The numbers of mice for each genotype were as follows: NfatC1Cre(+);Ift88f/f, n = 6; NfatC1Cre(+);Ift88+/+ (control), n = 4; Dzip1S14R/+, n = 5; Dzip1+/+(control), n = 5; NfatC1Cre(+);Dzip1f/+, n = 5; NfatC1Cre(+);Dzip1f/f, n = 5; NfatC1Cre(+);Dzip+/+, n = 5. Quantification was performed using Imaris 9.0 software, and data were generated for volume, surface area, and width. Width measurements were taken at three locations along the longitudinal axis of the mitral leaflets at the base, mid region, and tip. For these measurements, five sections throughout the leaflet were used while cognizant of keeping anatomical positions between the experimental and controls animals comparable. P values for each dataset are provided in representative figures or their figure legends.
Movats pentachrome histological stain
Movats stain was performed as previously described (16, 23) on adult NfatC1Cre(+);Ift88f/f, Dzip1S14R/+, and NfatC1Cre(+);Dzip1f/+ and f/f mice and compared to control littermates with n = 4 per genotype.
Section in situ
RNA in situ hybridization for Dzip1 at E14.5 was performed through Genepaint (61). A 2900-bp riboprobe was used to analyze Dzip1 RNA expression at E14.5. This probe was generated against region 84–2983 of accession no. NM_0025943.3. This probe spans all known isoforms for Dzip1.
Proliferation studies
Ki67-positive cells and total cell numbers were counted throughout eight sections, with each section separated by 20 mm (to ensure counts were not duplicated) per heart. For NfatC1Cre(+);Ift88+/+ (control), a total of five individual anterior and posterior leaflets were analyzed. For NfatC1Cre(+);Ift88f/+ (conditional heterozygote), a total of eight individual anterior and posterior leaflets were analyzed. For NfatC1Cre(+);Ift88f/f (conditional knockout), a total of seven individual anterior and posterior leaflets were analyzed. The average number of Ki67-positive cells and total number of cells counted throughout the eight sections were averaged among the five leaflets analyzed per genotype. Measurements were compared to control data to obtain fold change values. To detect statistically significant differences between test groups with two-sided α = 0.05, Student’s t test was used. Error bars represent SD.
Cell density studies
Cell density was quantified by counting all nuclei in a specified area (15 mm2). Measurements were taken at three points (base, middle region, and tip) along the craino-caudal (longitudinal) axis of each section of the anterior mitral leaflet. A total of five sections were analyzed in each heart, thus generating a total of 15 measurements per animal. For the NfatC1Cre(+);Ift88+/+ (control), and NfatC1Cre(+);Ift88f/f (conditional knockout), a total of five hearts were analyzed per genotype. Measurements were compared to control data to obtain fold change values. P values for each dataset are provided in representative figures or their figure legends. Error bars represent SD.
Mouse echocardiography
Mice were anesthetized with 3 to 5% isoflurane vapor in an anesthesia chamber (Vetequip Inc.) and then placed on a biofeedback warming station (Indus Electronics) with nose cone anesthesia of 1.5 to 2.5% isoflurane (Piramal Critical Care), which was regulated to maintain a heart rate between 500 to 600 beats/min while providing anesthesia (abolition of the toe pinch reflex). The hair over the chest was removed using a commercially available depilatory cream (Nair). Ultrasound gel was placed on the chest, and echocardiography measurements were performed using a 40-MHz probe with a spatial resolution of 30 mm (Vevo2100; Visualsonics). 2D and M-mode echo images were obtained in the parasternal short- and long-axis views. LV volumes and ejection fractions were computed from the parasternal long-axis recordings, and LV mass was computed from the short-axis measurements (62–64). For terminal studies, heart harvest was performed after this procedure. The entire echocardiography procedure took ~20 to 30 min per mouse. The following 6-month-old Dzip1 conditional knockout animals were used: NfatC1Cre(+);Dzip1+/+ (n = 4), NfatC1Cre(+);Dzip1f/+ (n = 5), and NfatC1Cre(+);Dzip1f/f (n = 3). Total 5-month-old Dzip1 KI animals used were Dzip1+/+ (n = 4) and Dzip1S14R/+ (n = 4). The Dzip1S14R/+ mice used for echocardiography were independent founders that resulted from the CRISPR-Cas9 targeting experiment.
RNA-seq analyses
Mitral leaflets were dissected from P0 NfatC1Cre(+);Ift88+/+ (n = 2), NfatC1Cre(+);Ift88f/+(n = 2), and NfatC1Cre(+);Ift88f/f (n = 3) mice. A second RNA-seq experiment was performed using RNA isolated from E13.5 Dzip1+/+ (n = 3) and Dzip1S14R/+ (n = 3) whole hearts. Total RNA was isolated using MicroRNeasy (Qiagen). Purity and quantification were determined by Bioanalyzer. The library preparation was done using the SMART-Seq v4 RNA-seq kit (Clontech Laboratories), following the manufacturer’s instructions. The analysis was carried out on an OnRamp Bioinformatics Genomics research platform (OnRamp Bioinformatics). OnRamp’s advanced Genomics Analysis Engine used an automated RNA-seq workflow to process the data, including (i) data validation and quality control, (ii) read alignment to the mouse genome (mm10) using STAR RNA-seq aligner, (iii) generation of gene-level count data with HTSeq, and (iv) differential expression (DE) analysis with DEseq2, which en-abled the inference of differential signals with robust statistical power (65–67). Transcript count data from DESeq2 analysis of the samples were sorted according to their adjusted P value or Q value, which is the smallest FDR at which a transcript is called significant. FDR is the expected fraction of false-positive tests among significant tests and was calculated using the Benjamini-Hochberg multiple testing adjustment procedure (68). All RNA-seq datasets were uploaded to the GEO accession viewer through the National Institutes of Health (NIH) with accession number GSE125092 and web link www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125092. The DE list was then submitted to the iPathway Guide tool from Advaita Bioinformatics; this tool uses a systems biology approach to identify pathways that are significantly affected in any condition from high-throughput gene expression data. The impact analysis incorporates the classical probabilistic component of the magnitude of the expression changes of each gene, the position of the differentially expressed genes in the given pathways, the topology of the pathway that describes how these genes interact, and the types of signaling interactions between them (69). GO analyses and heat map output derived from the Advaita software with FDR correction is presented for RNA-seq datasets.
Collagen I quantification
IHC for collagen I (red) and MF20 (green) on neonatal (P0) NfatC1Cre(+);Ift88+/+ (controls) was compared to NfatC1Cre(+);Ift88f/f littermates. Quantification of IHC was performed in Adobe Photoshop (Adobe Photoshop CS5 Extended) by conversion of the immuno-fluorescent channel to inverted gray scale. Integrated density of identical sized acquisition boxes was measured at the base, middle, and tip of the anterior leaflet. Collagen I expression in the epicardium was used as a normalization control for the staining between genotypes because the Cre is not active in this tissue. n = 3 per genotype. Measurements were compared to control data to obtain fold change data. P values for each dataset are provided in representative figures or their legends. Errors bars represent SD.
DZIP1 protein half-life calculations
Cycloheximide experiments were performed on transfected immortalized mouse atrioventricular valve cells (mAVCs). The mAVCs were a gift from J. Barnett (Vanderbilt University) and were previously published (70). Control human DZIP1 plasmid was purchased from Origene (clone ID: 198968). The DZIP1 mutation was incor-porated through a QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) per the manufacturer’s recommendations, and both control and DZIP1S24R constructs were transfected into the mAVC cell line (1 × 105 on 35-mm dishes). Both constructs had a V5-epitope tag at the C terminus of the construct. For the cycloheximide experiments, medium containing cycloheximide (100 ng/ml) was added at 48 hours after transfection, and each well was harvested as above at the indicated time points. Western blots were probed with a mouse anti-V5 primary antibody (1:4000 dilution, Invitrogen) and a horseradish peroxidase–linked secondary at the same dilution (Thermo Fisher Scientific). Blots were also probed with a mouse anti-tubulin primary (Millipore) at a 1:4000 dilution and the same secondary antibody as above. Blots were treated with West Femto (Thermo Fisher Scientific) and visualized on film. For quantitation, blot pixel intensity was measured by Photoshop CS and normalized to tubulin. Each experiment was repeated a minimum of three times and all samples were run in triplicate, and a single exponential decay regression curve was fit and half-lives calculated using Excel (Microsoft). Measurements at each time point were compared to control data.
Human echocardiography
MVP was diagnosed in 2D long-axis echocardiographic views by ≥2-mm leaflet displacement, superior to the annulus hinge points (71, 72). Prodromal/minimal morphology was noted on the basis of leaflet coaptation abnormally displaced >40% anterior relative to the mitral annulus, and minimal superior displacement was based on <2-mm displacement (25, 73). These morphologies associate with progressive prolapse and link to the haplotype of affected individuals with fully diagnostic displacement (25).
Familial genetics and WES
Complete details on the family in Fig. 3A and the original linkage analysis can be found in (25). Exome sequencing was performed on four individuals (II-3, III-4, III-10, and IV-12) carrying the linked haplotype, and all genes in the candidate interval were analyzed for coding sequence mutations. Exome capture was carried out using the SureSelect Human All Exon System using the manufacturer’s protocol version 1.0 (Agilent Inc.) that is compatible with Illumina paired-end sequencing. Exome-enriched genomes were multiplexed by flow cell for 101-bp paired-end read sequencing according to the protocol for the HiSeq 2000 sequencer (version 1.7.0; Illumina) to allow a minimum coverage of 30×. Reads were aligned to the human reference genome (UCSC NCBI36/hg19) using the Burrows-Wheeler Aligner (version 0.5.9). Quality control to determine sample and genotyping quality and to potentially remove poor SNPs and/or samples was performed in PLINK, a whole-genome association analysis toolset (74).
Genome-wide association studies
We used an enrichment analysis method called improved gene set enrichment analysis for GWAS (i-GSEA4GWAS) (75). This method uses all tested SNPs in the GWAS (6.6 million) that it maps to genes if they are exonic/intronic or to the closest genes if they are within 20 kb upstream/downstream of genes. Of the 6.6 million SNPs that we included, this analysis mapped 4,349,539 variants to 21,167 genes and tested 130 different gene sets from the preexisting catalog Kyoto Encyclopedia of Genes and Genomes (KEGG), to which we added our list of 278 known ciliopathy genes that we predefined as a cilia gene set (table S1). Enrichment was considered as highly confident and statistically significant at FDR < 0.05, as recommended in (75) (table S2).
Animal studies
All animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committees at the Medical University of South Carolina and University Heart Center Freiburg. Before cardiac resection, mice were euthanized by isoflurane (Piramal) induction, followed by cervical dislocation in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85–23, revised 1996). Comparisons of the data generated for both male and female sexes showed no appreciable differences. As such, combined data for both sexes are shown.
Human studies
All studies involving human research were approved by the Institutional Review Board of Partners Healthcare, Boston, MA, and all participants provided written informed consent.
Overall statistical analyses
All data are shown as means ± SD. To detect statistically significant differences between test groups with two -sided α = 0.05, Student’s t test was used. P values for each dataset are provided in representative figures or their figure legends. For all box plots, a generalized linear mixed model was used to compare the likelihood of cilia presence across genotypes using a logit link function and litter, mouse, and genotype by litter interaction as random effects in the model. For mice with cilia present, mixed model analysis of variance was used to compare mouse average cilia length across genotypes, with litter serving as a random effect. Cilia values were analyzed using SPSS v25. P values for each dataset are provided in representative figures or their figure legends. Error bars designate the 95% confidence intervals.
Supplementary Material
Fig. S3. Loss of primary cilia has no effect on cell proliferation.
{kind=link}
Movie S1. 3D reconstruction of primary cilia in E13.5 murine anterior mitral leaflets.
Table S3. Echocardiographic analyses of Dzip1 conditional heterozygote and homozygote mice.
Table S1. WES to identify additional DZIP1 variants.
{kind=link}
Table S2. Echocardiographic analyses of Dzip1S14R/+ and control mice.
Movie S3. 3D reconstruction of primary cilia in 10-week-old human fetal anterior mitral leaflets.
Movie S2. 3D reconstruction of primary cilia in E15.5 murine anterior mitral leaflets.
Movie S5. EM tomography showing a compilation of transmission electron microscopy slices and renderings of the microtubule triplets that comprise the mother centriole/basal body.
Movie S6. 3D reconstruction of primary cilia and versican in P0 anterior mitral leaflets.
Movie S4. EM tomography showing the presence of the axoneme and basal body.
Movie S7. 3D reconstruction of primary cilia and collagen IaI in P0 anterior mitral leaflets.
Movie S9. Echocardiography: Parasternal long-axis view of Dzip1S14R/+ mitral leaflet.
Movie S8. 3D reconstruction of DZIP1 protein expression in an E13.5 AV cushion mesenchymal cell.
Movie S10. Echocardiography: Parasternal long-axis view of Dzip1 conditional knockout mice.
Fig. S1. Primary cilia are detected in human mitral valves during development.
{kind=link}
Fig. S2. Primary cilia are located in areas rich in versican.
{kind=link}
Fig. S5. RNA-seq analyses correlate loss of cilia with ECM gene activation.
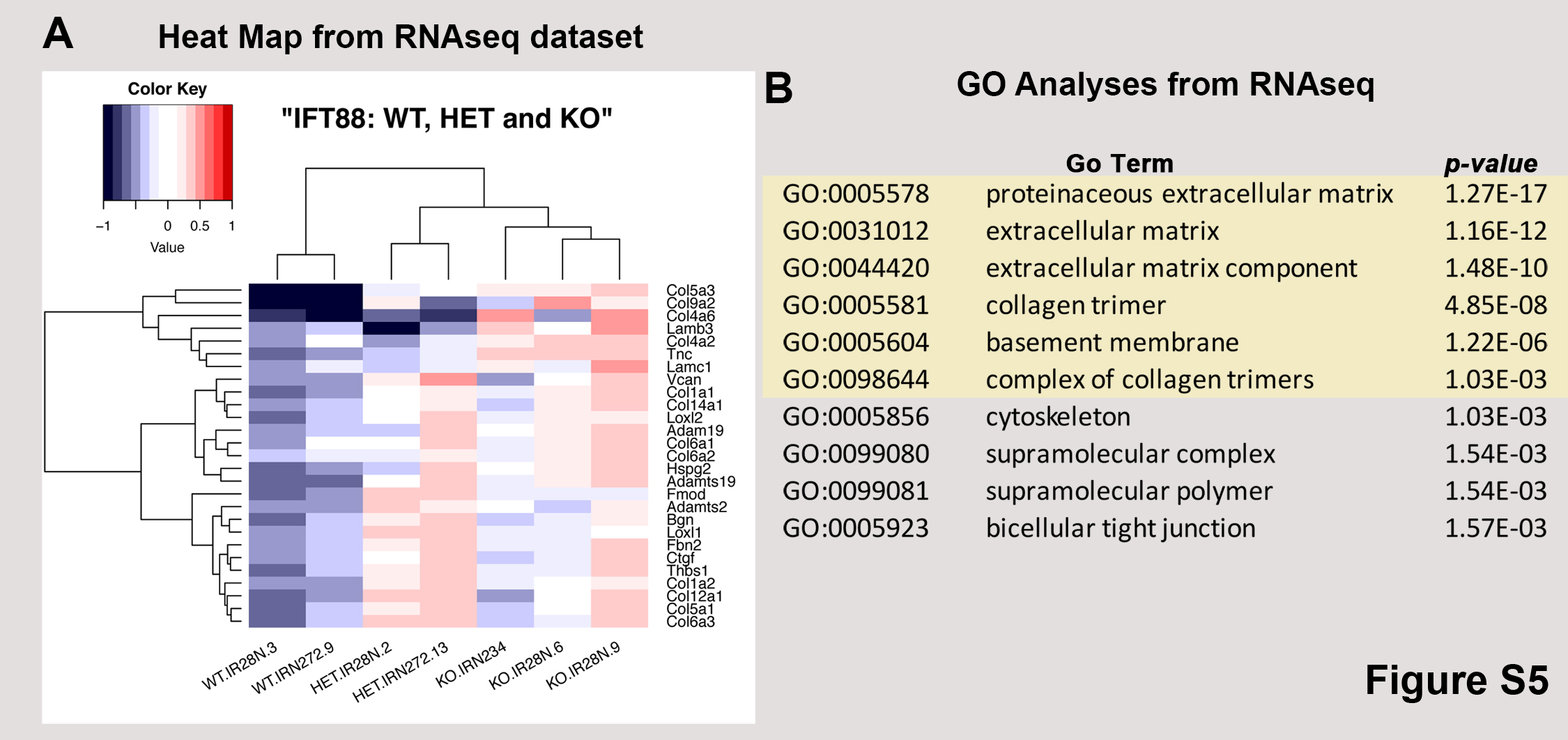{kind=link}
Fig. S6. Loss of primary cilia results in increased collagen I expression in mitral valves.
{kind=link}
Fig. S7. Cilia length is decreased in mouse models associated with MVP.
{kind=link}
Fig. S4. Loss of primary cilia results in decreased cell density.
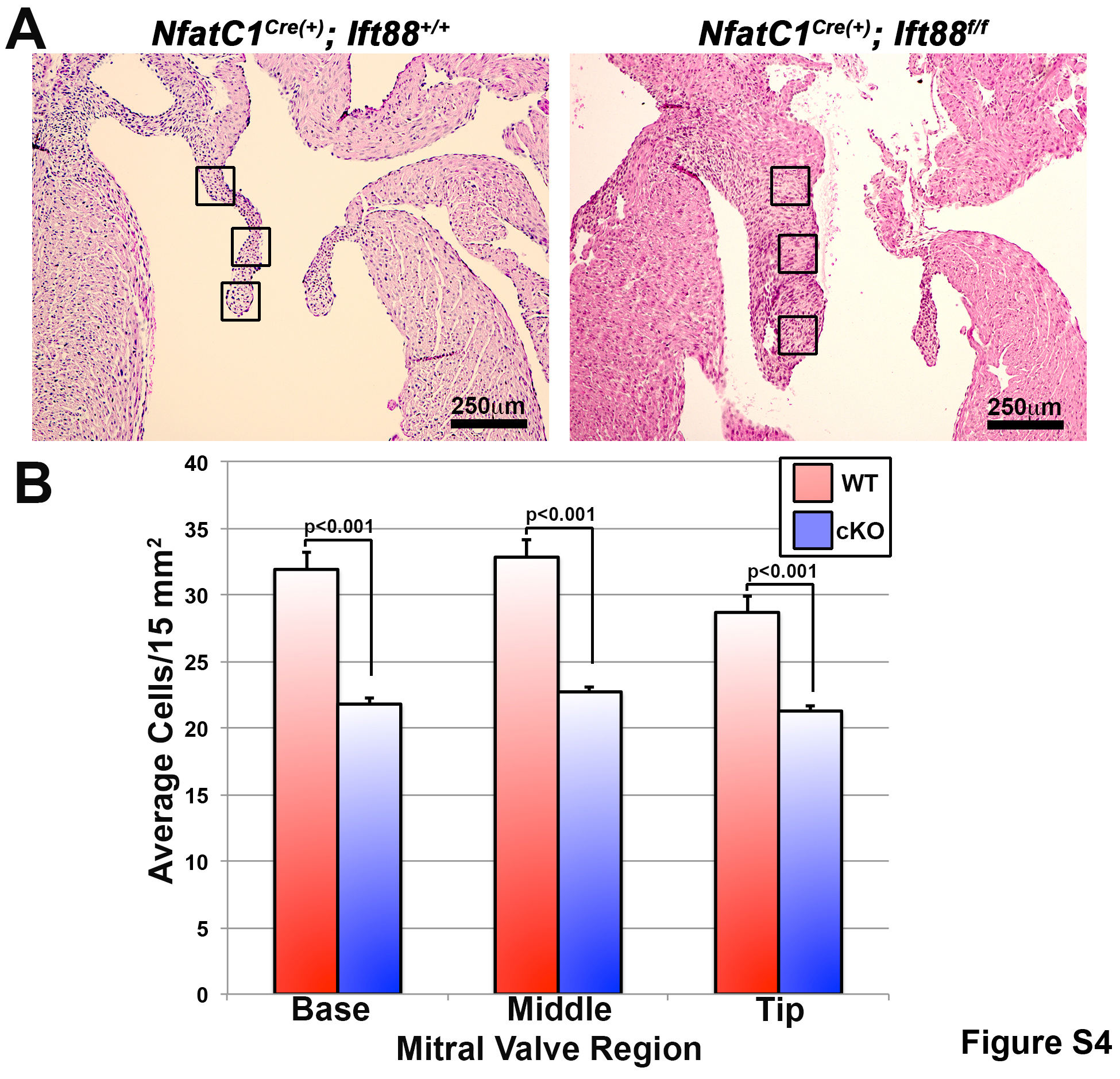{kind=link}
Fig. S8. MVP of proband from family with chromosome 13 mutation is observed through echocardiographic assessment.
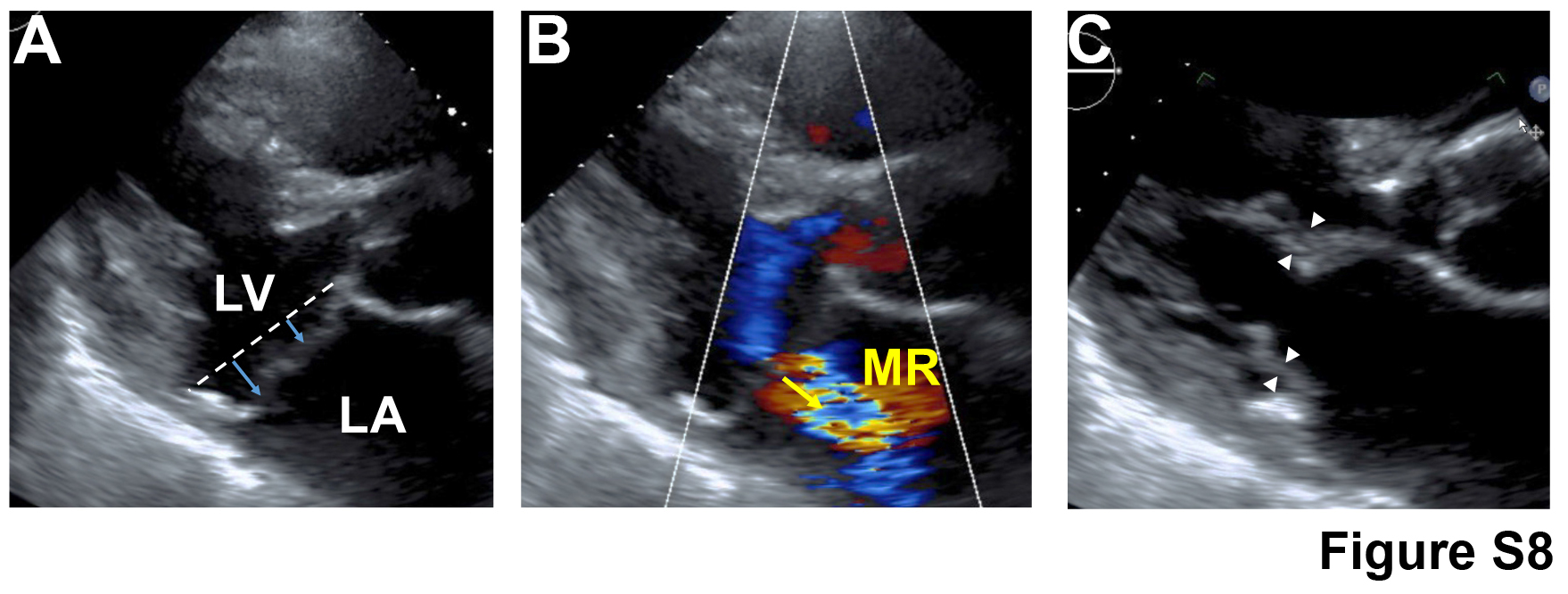{kind=link}
Fig. S9. DZIP1 is expressed at the base of primary cilia in the murine mitral valve.
{kind=link}
Fig. S10. Dzip1S14R/+ KI MVP mouse models were generated through CRISPR-Cas9.
{kind=link}
Fig. S11. GO analyses of RNA-seq datasets reveal changes in ECM synthesis.
{kind=link}
Fig. S12. Mutated DZIP1 is unstable.
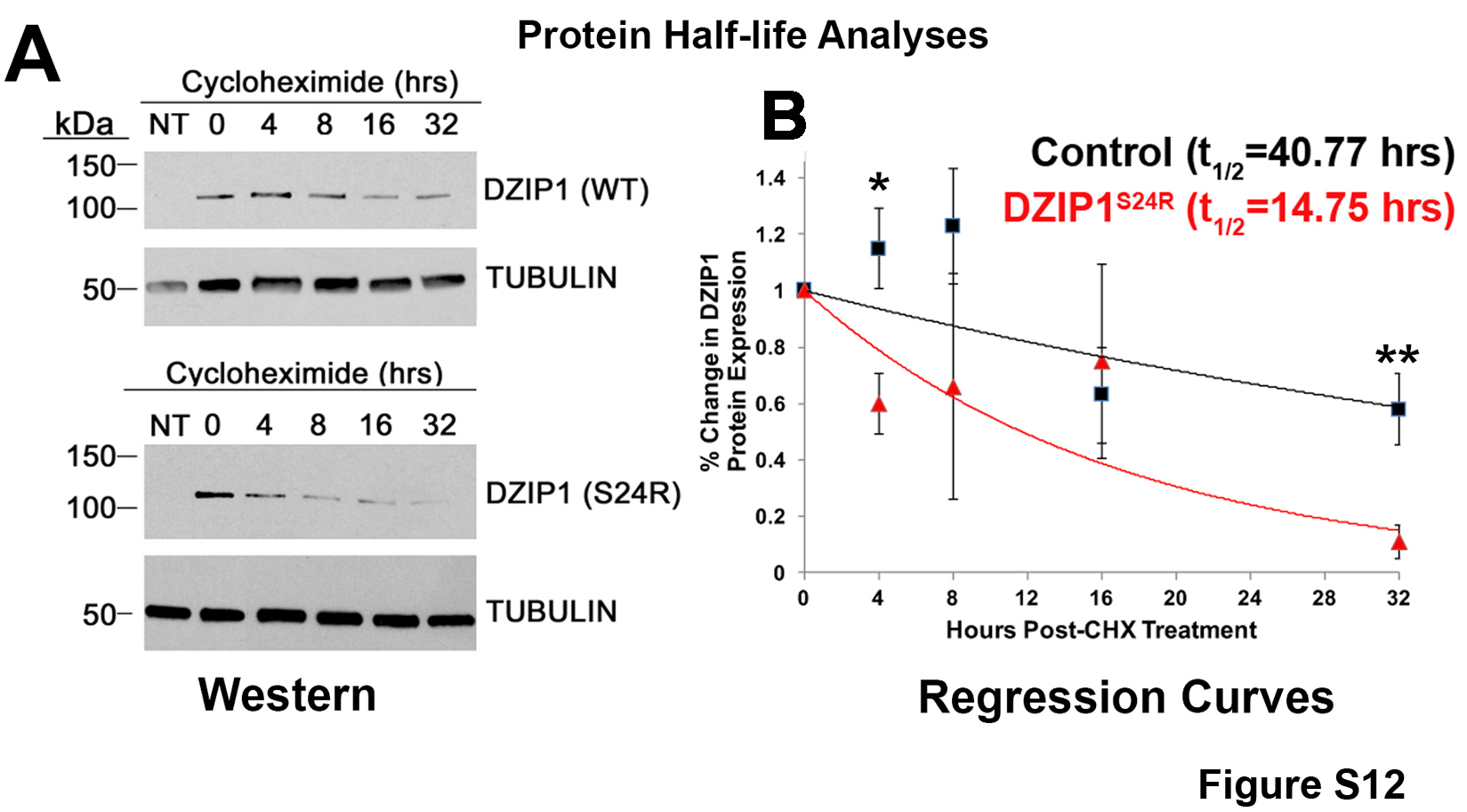{kind=link}
Fig. S14. Echocardiography of Dzip1 conditional knockout mitral leaflets shows MVP.
{kind=link}
Fig. S13. Loss of Dzip1 causes developmental defects and subsequent MVP in the adult.
{kind=link}
Data file S1. Cilia length during development.
Data file S2. Correlation of cilia with the type of ECM produced in the valve.
Data file S3. Morphometrics of valve geometry in Ift88-deficient mitral valves compared to controls at P0.
Data file S4. Cell proliferation in Ift88-deficient mitral valves compared to controls at P0.
Data file S5. Decreased cell density Ift88-deficient valves.
Data file S6. Increased collagen production in Ift88-deficient valves.
Data file S7. List of cilia genes, gene function, and disease association used in GWAS analyses.
Data file S8. Details about cilia gene set enrichment results using i-GSEA4GWAS.
Data file S9. Reduced cilia lengths in Dchs1- and Flna-deficient mitral valves.
Data file S10. Morphometrics of Dzip1S14R/+ mitral valves at P0 compared to controls.
Data file S11. Shortened cilia length in Dzip1S14R/+ mitral valves at P0 compared to controls.
Data file S12. Differentially expressed gene comparison.
Data file S13. Reduced protein half-life of DZIP1S24R variant.
Data file S14. Reduced cilia length in Dzip1 conditional knockout mice.
Acknowledgments:
Ift88 conditional mice were a gift from C. Haycraft (Mississippi College). Versican and collagen I antibodies were gifts from S. Hoffman (Medical University of South Carolina). We thank E. Chleilat (IEKM, University of Freiburg, Germany) for assistance with the transmission electron microscopy (TEM) preparation and P. Machado (EMBL Heidelberg, Germany) for assistance with TEM data acquisition.
Funding: This work was supported by the Fondation Leducq (Paris, France) Mitral Transatlantic Network of Excellence grant 07CVD04. MVP patient studies were supported by the Innovation in Clinical Research award of the Doris Duke Charitable Foundation (to S.S. and R.A.L.). The work at MUSC was performed in a facility constructed with support from the National Institutes of Health grant number C06 RR018823 from the Extramural Research Facilities Program of the National Center for Research Resources. Other funding sources include the following: National Heart Lung and Blood Institute: HL131546 (to R.A.N.), HL122906 (to A.W.), HL33756 (to RRM), COBRE GM103342 (to RRM, R.A.N., and A.W.), HL142159 (to D.F.), GM103444 (to RRM and R.A.N.), HL127692 (to D.J.M., S.A.S., R.A.N., and R.A.L.), HL116652 (to F.N.D.), HL007260 (to K.S.M.), and HL128099 and HL141917 (to R.A.L.); National Institute of Diabetes, Digestive, and Kidney Diseases: P30DK074038 (to J.H.L.); U.S. Department of Veterans Affairs merit award: I01 BX000820 (to J.H.L.); National Institute of Mental Health: R00-MH095867 (to M.E.T.); National Institute of General Medical Sciences: R01GM114429 (to B.W.); National Institute of Drug Abuse: U01DA045300 (to G.H.); the Hassenfeld Scholar Program (to D.J.M.); the March of Dimes (to M.E.T.); MGH Scholars Program (to S.A.S.); American Heart Association: 17CSA33590067 (to R.A.N., S.B., and J.H.L.), 2261354 (to D.M.), 16PRE30970048 (to K.A.T.), and 18PRE34080172 (to L.G.); National Science Foundation: EPS-0903795 (to RRM); the European Research Council: Stg-ROSALING-716628 (to N.B.-N.), Advanced grant CardioNECT (to P.K.); a German Research Foundation Emmy Noether Fellowship (to E.R.-Z.); Chinese Scientific Council Scholarship (to M.Y.); French Ministry of Research: ANR grant ANR-16-CE17–0015-01 (to J.-J.S. and N.B.-N.); The French Society of Cardiology (to A.H.); Fédération Française de Cardiologie, Fondation Coeur et Recherche, French Ministry of Health “PHRC National 2007” and “PHRC-I 2012,” and INSERM Translational Research Grant (T.L.T.); Ellison Foundation, Boston, MA (to R.A.L.) and a gift from M. Zak (to D.M.).
Competing interests: P.T.E. is supported by a grant from Bayer AG to the Broad Institute focused on the genetics and therapeutics of cardiovascular diseases. P.T.E. has also served on advisory boards or consulted for Quest Diagnostics and Novartis. All other authors declare that they have no competing interests.
Footnotes
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Ishikawa H, Marshall WF, Ciliogenesis: Building the cell’s antenna. Nat. Rev. Mol. Cell Biol 12, 222–234 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Davis EE, Katsanis N, The ciliopathies: A transitional model into systems biology of human genetic disease. Curr. Opin. Genet. Dev 22, 290–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hildebrandt F, Benzing T, Katsanis N, Ciliopathies. N. Engl. J. Med 364, 1533–1543 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gencer S, Oleinik N, Kim J, Panneer Selvam S, De Palma R, Dany M, Nganga R, Thomas RJ, Senkal CE, Howe PH, Ogretmen B, TGF-b receptor I/II trafficking and signaling at primary cilia are inhibited by ceramide to attenuate cell migration and tumor metastasis. Sci. Signal 10, eaam7464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST, PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr. Biol 15, 1861–1866 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Reiter JF, Kif3a constrains b-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol 10, 70–76 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Hulin A, Moore V, James JM, Yutzey KE, Loss of Axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease. Cardiovasc. Res 113, 40–51 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu A, Wang B, Niswander LA, Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 132, 3103–3111 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Seeger-Nukpezah T, Golemis EA, The extracellular matrix and ciliary signaling. Curr. Opin. Cell Biol 24, 652–661 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, Hung W-C, Haider S, Zhang J, Knight J, Bjornson RD, Castaldi C, Tikhonoa IR, Bilguvar K, Mane SM, Sanders SJ, Mital S, Russell MW, Gaynor JW, Deanfield J, Giardini A, Porter GA Jr., D. Srivastava, C. W. Lo, Y. Shen, W. S. Watkins, M. Yandell, H. J. Yost, M. Tristani-Firouzi, J. W. Newburger, A. E. Roberts, R. Kim, H. Zhao, J. R. Kaltman, E. Goldmuntz, W. K. Chung, J. G. Seidman, B. D. Gelb, C. E. Seidman, R. P. Lifton, M. Brueckner, Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet 49, 1593–1601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, Chang C, Yagi H, San Agustin JT, Thahir M, Anderton S, Lawhead C, Vescovi A, Pratt H, Morgan J, Haynes L, Smith CL, Eppig JT, Reinholdt L, Francis R, Leatherbury L, Ganapathiraju MK, Tobita K, Pazour GJ, Lo CW, Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 521, 520–524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaghloul NA, Liu Y, Gerdes JM, Gascue C, Oh EC, Leitch CC, Bromberg Y, Binkley J, Leibel RL, Sidow A, Badano JL, Katsanis N, Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet–Biedl syndrome. Proc. Natl. Acad. Sci. U.S.A 107, 10602–10607 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivy DD, Shaffer EM, Johnson AM, Kimberling WJ, Dobin A, Gabow PA, Cardiovascular abnormalities in children with autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol 5, 2032–2036 (1995). [DOI] [PubMed] [Google Scholar]
- 14.Lumiaho A, Ikäheimo R, Miettinen R, Niemitukia L, Laitinen T, Rantala A, Lampainen E, Laakso M, Hartikainen J, Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am. J. Kidney Dis 38, 1208–1216 (2001). [DOI] [PubMed] [Google Scholar]
- 15.de Vlaming A, Sauls K, Hajdu Z, Visconti RP, Mehesz AN, Levine RA, Slaugenhaupt SA, Hagège A, Chester AH, Markwald RR, Norris RA, Atrioventricular valve development: New perspectives on an old theme. Differentiation 84, 103–116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durst R, Sauls K, Peal DS, deVlaming A, Toomer K, Leyne M, Salani M, Talkowski ME, Brand H, Perrocheau M, Simpson C, Jett C, Stone MR, Charles F, Chiang C, Lynch SN, Bouatia-Naji N, Delling FN, Freed LA, Tribouilloy C, Le Tourneau T, LeMarec H, Fernandez-Friera L, Solis J, Trujillano D, Ossowski S, Estivill X, Dina C, Bruneval P, Chester A, Schott J-J, Irvine KD, Mao Y, Wessels A, Motiwala T, Puceat M, Tsukasaki Y, Menick DR, Kasiganesan H, Nie X, Broome A-M, Williams K, Johnson A, Markwald RR, Jeunemaitre X, Hagege A, Levine RA, Milan DJ, Norris RA, Slaugenhaupt SA, Mutations in DCHS1 cause mitral valve prolapse. Nature 525, 109–113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG, Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J. Cell Biol 151, 709–718 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu B, Zhang Z, Lui W, Chen X, Wang Y, Chamberlain AA, Moreno-Rodriguez RA, Markwald RR, O’Rourke BP, Sharp DJ, Zheng D, Lenz J, Baldwin HS, Chang C-P, Zhou B, Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell 151, 1083–1096 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dina C, Bouatia-Naji N, Tucker N, Delling FN, Toomer K, Durst R, Perrocheau M, Fernandez-Friera L, Solis J; PROMESA investigators, Le Tourneau T, Chen M-H, Probst V, Bosse Y, Pibarot P, Zelenika D, Lathrop M, Hercberg S, Roussel R, Benjamin EJ, Bonnet F, Lo SH, Dolmatova E, Simonet F, Lecointe S, Kyndt F, Redon R, Le Marec H, Froguel P, Ellinor PT, Vasan RS, Bruneval P, Markwald RR, Norris RA, Milan DJ, Slaugenhaupt SA, Levine RA, Schott J-J, Hagege AA; MVP-France Jeunemaitre X; Leducq Transatlantic MITRAL Network, Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nat. Genet 47, 1206–1211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Dam TJP, Wheway G, Slaats GG; SYSCILIA Study Group, Huynen MA, Giles RH, The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia 2, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kyndt F, Gueffet J-P, Probst V, Jaafar P, Legendre A, Le Bouffant F, Toquet C, Roy E, McGregor L, Lynch SA, Newbury-Ecob R, Tran V, Young I, Trochu J-N, Le Marec H, Schott J-J, Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation 115, 40–49 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Le Tourneau T, Le Scouarnec S, Cueff C, Bernstein D, Aalberts JJJ, Lecointe S, Mérot J, Bernstein JA, Oomen T, Dina C, Karakachoff M, Desal H, Al Habash O, Delling FN, Capoulade R, Suurmeijer AJH, Milan D, Norris RA, Markwald R, Aikawa E, Slaugenhaupt SA, Jeunemaitre X, Hagège A, Roussel J-C, Trochu J-N, Levine RA, Kyndt F, Probst V, Le Marec H, Schott J-J, New insights into mitral valve dystrophy: A Filamin-A genotype-phenotype and outcome study. Eur. Heart J 39, 1269–1277 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauls K, de Vlaming A, Harris BS, Williams K, Wessels A, Levine RA, Slaugenhaupt SA, Goodwin RL, Pavone LM, Merot J, Schott J-J, Le Tourneau T, Dix S, Jesinkey S, Feng Y, Walsh C, Zhou B, Baldwin S, Markwald RR, Norris RA, Developmental basis for filamin-A-associated myxomatous mitral valve disease. Cardiovasc. Res 96, 109–119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sauls K, Toomer K, Williams K, Johnson AJ, Markwald RR, Hajdu Z, Norris RA, Increased infiltration of extra-cardiac cells in myxomatous valve disease. J. Cardiovasc. Dev. Dis 2, 200–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nesta F, Leyne M, Yosefy C, Simpson C, Dai D, Marshall JE, Hung J, Slaugenhaupt SA, Levine RA, New locus for autosomal dominant mitral valve prolapse on chromosome 13: Clinical insights from genetic studies. Circulation 112, 2022–2030 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Sekimizu K, Nishioka N, Sasaki H, Takeda H, Karlstrom RO, Kawakami A, The zebrafish iguana locus encodes Dzip1, a novel zinc-finger protein required for proper regulation of Hedgehog signaling. Development 131, 2521–2532 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Wolff C, Roy S, Lewis KE, Schauerte H, Joerg-Rauch G, Kirn A, Weiler C, Geisler R, Haffter P, Ingham PW, iguana encodes a novel zinc-finger protein with coiled-coil domains essential for Hedgehog signal transduction in the zebrafish embryo. Genes Dev 18, 1565–1576 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang B, Zhang T, Wang G, Wang G, Chi W, Jiang Q, Zhang C, GSK3b-Dzip1-Rab8 cascade regulates ciliogenesis after mitosis. PLOS Biol 13, e1002129 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim HR, Richardson J, van Eeden F, Ingham PW, Gli2a protein localization reveals a role for Iguana/DZIP1 in primary ciliogenesis and a dependence of Hedgehog signal transduction on primary cilia in the zebrafish. BMC Biol 8, 65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tay SY, Yu X, Wong KN, Panse P, Ng CP, Roy S, The iguana/DZIP1 protein is a novel component of the ciliogenic pathway essential for axonemal biogenesis. Dev. Dyn 239, 527–534 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Wang C, Low W-C, Liu A, Wang B, Centrosomal protein DZIP1 regulates Hedgehog signaling by promoting cytoplasmic retention of transcription factor GLI3 and affecting ciliogenesis. J. Biol. Chem 288, 29518–29529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arnold CR, Lamont RE, Walker JT, Spice PJ, Chan C-K, Ho C-Y, Childs SJ, Comparative analysis of genes regulated by Dzip1/iguana and hedgehog in zebrafish. Dev. Dyn 244, 211–223 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J, A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, Scott E, Shah I, Stenson PD, Gleeson J, Cooper DN, Quintana-Murci L, Zhang S-Y, Abel L, Casanova J-L, The mutation significance cutoff: Gene-level thresholds for variant predictions. Nat. Methods 13, 109–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Egorova AD, Khedoe PPSJ, Goumans M-JTH, Yoder BK, Nauli SM, ten Dijke P, Poelmann RE, Hierck BP, Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ. Res 108, 1093–1101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedland-Little JM, Hoffmann AD, Ocbina PJR, Peterson MA, Bosman JD, Chen Y, Cheng SY, Anderson KV, Moskowitz IP, A novel murine allele of Intraflagellar Transport Protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum. Mol. Genet 20, 3725–3737 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hierck BP, Van der Heiden K, Alkemade FE, Van de Pas S, Van Thienen JV, Groenendijk BCW, Bax WH, Van der Laarse A, DeRuiter MC, Horrevoets AJG, Poelmann RE, Primary cilia sensitize endothelial cells for fluid shear stress. Dev. Dyn 237, 725–735 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Lopes SS, Lourenço R, Pacheco L, Moreno N, Kreiling J, Saúde L, Notch signalling regulates left-right asymmetry through ciliary length control. Development 137, 3625–3632 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Poelmann RE, Gittenberger-de Groot AC, Hierck BP, The development of the heart and microcirculation: Role of shear stress. Med. Biol. Eng. Comput 46, 479–484 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samsa LA, Givens C, Tzima E, Stainier DYR, Qian L, Liu J, Cardiac contraction activates endocardial Notch signaling to modulate chamber maturation in zebrafish. Development 142, 4080–4091 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toomer KA, Fulmer D, Guo L, Drohan A, Peterson N, Swanson P, Brooks B, Mukherjee R, Body S, Lipschutz JH, Wessels A, Norris RA, A role for primary cilia in aortic valve development and disease. Dev. Dyn 246, 625–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willaredt MA, Gorgas K, Gardner HAR, Tucker KL, Multiple essential roles for primary cilia in heart development. Cilia 1, 23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J, Li Q, Kuehn MR, Litingtung Y, Vokes SA, Chiang C, Sonic hedgehog signaling directly targets Hyaluronic Acid Synthase 2, an essential regulator of phalangeal joint patterning. Dev. Biol 375, 160–171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagy N, Barad C, Graham HK, Hotta R, Cheng LS, Fejszak N, Goldstein AM, Sonic hedgehog controls enteric nervous system development by patterning the extracellular matrix. Development 143, 264–275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Christensen ST, Pedersen SF, Satir P, Veland IR, Schneider L, The primary cilium coordinates signaling pathways in cell cycle control and migration during development and tissue repair. Curr. Top. Dev. Biol 85, 261–301 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Gerdes JM, Katsanis N, Ciliary function and Wnt signal modulation. Curr. Top. Dev. Biol 85, 175–195 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Lienkamp S, Ganner A, Walz G, Inversin, Wnt signaling and primary cilia. Differentiation 83, S49–S55 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Ivarsson M, McWhirter A, Borg TK, Rubin K, Type I collagen synthesis in cultured human fibroblasts: Regulation by cell spreading, platelet-derived growth factor and interactions with collagen fibers. Matrix Biol 16, 409–425 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Clement CA, Ajbro KD, Koefoed K, Vestergaard ML, Veland IR, Henriques de Jesus MPR, Pedersen LB, Benmerah A, Andersen CY, Larsen LA, Christensen ST, TGF-b signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep 3, 1806–1814 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Liu Y, Pathak N, Kramer-Zucker A, Drummond IA, Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development 134, 1111–1122 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Grisanti L, Revenkova E, Gordon RE, Iomini C, Primary cilia maintain corneal epithelial homeostasis by regulation of the Notch signaling pathway. Development 143, 2160–2171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan S, Li J, Diener DR, Choma MA, Rosenbaum JL, Sun Z, Target-of-rapamycin complex 1 (Torc1) signaling modulates cilia size and function through protein synthesis regulation. Proc. Natl. Acad. Sci. U.S.A 109, 2021–2026 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kitkungvan D, Nabi F, Kim RJ, Bonow RO, Khan MA, Xu J, Little SH, Quinones MA, Lawrie GM, Zoghbi WA, Shah DJ, Myocardial fibrosis in patients with primary mitral regurgitation with and without prolapse. J. Am. Coll. Cardiol 72, 823–834 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Levine RA, Jerosch-Herold M, Hajjar RJ, Mitral valve prolapse: A disease of valve and ventricle. J. Am. Coll. Cardiol 72, 835–837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu H, Galeano MCR, Ott E, Kaeslin G, Kausalya PJ, Kramer C, Ortiz-Brüchle N, Hilger N, Metzis V, Hiersche M, Tay SY, Tunningley R, Vij S, Courtney AD, Whittle B, Wühl E, Vester U, Hartleben B, Neuber S, Frank V, Little MH, Epting D, Papathanasiou P, Perkins AC, Wright GD, Hunziker W, Gee HY, Otto EA, Zerres K, Hildebrandt F, Roy S, Wicking C, Bergmann C, Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet 49, 1025–1034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK, Intraflagellar transport is essential for endochondral bone formation. Development 134, 307–316 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Wessels A, van den Hoff MJB, Adamo RF, Phelps AL, Lockhart MM, Sauls K, Briggs LE, Norris RA, van Wijk B, Perez-Pomares JM, Dettman RW, Burch JBE, Epicardially-derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol 366, 111–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gordon JW, Ruddle FH, Gene transfer into mouse embryos: Production of transgenic mice by pronuclear injection. Methods Enzymol 101, 411–433 (1983). [DOI] [PubMed] [Google Scholar]
- 59.Rog-Zielinska EA, Johnston CM, O’Toole ET, Morphew M, Hoenger A, Kohl P, Electron tomography of rabbit cardiomyocyte three-dimensional ultrastructure. Prog. Biophys. Mol. Biol 121, 77–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lohezic M, Teh I, Bollensdorff C, Peyronnet R, Hales PW, Grau V, Kohl P, Schneider JE, Interrogation of living myocardium in multiple static deformation states with diffusion tensor and diffusion spectrum imaging. Prog. Biophys. Mol. Biol 115, 213–225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Visel A, Thaller C, Eichele G, GenePaint.org: An atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res 32, D552–D556 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kholmukhamedov A, Logdon C, Hu J, McKinney RA, Spinale FG, Lemasters JJ, Mukherjee R, Cyclosporin A in left ventricular remodeling after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol 306, H53–H59 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mani SK, Balasubramanian S, Zavadzkas JA, Jeffords LB, Rivers WT, Zile MR, Mukherjee R, Spinale FG, Kuppuswamy D, Calpain inhibition preserves myocardial structure and function following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol 297, H1744–H1751 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zavadzkas JA, Stroud RE, Bouges S, Mukherjee R, Jones JR, Patel RK, McDermott PJ, Spinale FG, Targeted overexpression of tissue inhibitor of matrix metalloproteinase-4 modifies post-myocardial infarction remodeling in mice. Circ. Res 114, 1435–1445 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davis-Turak J, Courtney SM, Hazard ES, Glen WB Jr., da Silveira WA, Wesselman T, Harbin LP, Wolf BJ, Chung D, Hardiman G, Genomics pipelines and data integration: Challenges and opportunities in the research setting. Expert Rev. Mol. Diagn 17, 225–237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Benjamini Y, Hochberg Y, Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B. Methodol 57, 289–300 (1995). [Google Scholar]
- 69.Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, Georgescu C, Romero R, systems A biology approach for pathway level analysis. Genome Res 17, 1537–1545 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peng Y, Song L, Li D, Kesterson R, Wang J, Wang L, Rokosh G, Wu B, Wang Q, Jiao K, Sema6D acts downstream of bone morphogenetic protein signalling to promote atrioventricular cushion development in mice. Cardiovasc. Res 112, 532–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levine RA, Stathogiannis E, Newell JB, Harrigan P, Weyman AE, Reconsideration of echocardiographic standards for mitral valve prolapse: Lack of association between leaflet displacement isolated to the apical four chamber view and independent echocardiographic evidence of abnormality. J. Am. Coll. Cardiol 11, 1010–1019 (1988). [DOI] [PubMed] [Google Scholar]
- 72.Levine RA, Handschumacher MD, Sanfilippo AJ, Hagege AA, Harrigan P, Marshall JE, Weyman AE, Three-dimensional echocardiographic reconstruction of the mitral valve, with implications for the diagnosis of mitral valve prolapse. Circulation 80, 589–598 (1989). [DOI] [PubMed] [Google Scholar]
- 73.Delling FN, Gona P, Larson MG, Lehman B, Manning WJ, Levine RA, Benjamin EJ, Vasan RS, Mild expression of mitral valve prolapse in the Framingham offspring: Expanding the phenotypic spectrum. J. Am. Soc. Echocardiogr 27, 17–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC, PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang K, Cui S, Chang S, Zhang L, Wang J, i-GSEA4GWAS: A web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Res 38, W90–W95 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S3. Loss of primary cilia has no effect on cell proliferation.
Movie S1. 3D reconstruction of primary cilia in E13.5 murine anterior mitral leaflets.
Table S3. Echocardiographic analyses of Dzip1 conditional heterozygote and homozygote mice.
Table S1. WES to identify additional DZIP1 variants.
Table S2. Echocardiographic analyses of Dzip1S14R/+ and control mice.
Movie S3. 3D reconstruction of primary cilia in 10-week-old human fetal anterior mitral leaflets.
Movie S2. 3D reconstruction of primary cilia in E15.5 murine anterior mitral leaflets.
Movie S5. EM tomography showing a compilation of transmission electron microscopy slices and renderings of the microtubule triplets that comprise the mother centriole/basal body.
Movie S6. 3D reconstruction of primary cilia and versican in P0 anterior mitral leaflets.
Movie S4. EM tomography showing the presence of the axoneme and basal body.
Movie S7. 3D reconstruction of primary cilia and collagen IaI in P0 anterior mitral leaflets.
Movie S9. Echocardiography: Parasternal long-axis view of Dzip1S14R/+ mitral leaflet.
Movie S8. 3D reconstruction of DZIP1 protein expression in an E13.5 AV cushion mesenchymal cell.
Movie S10. Echocardiography: Parasternal long-axis view of Dzip1 conditional knockout mice.
Fig. S1. Primary cilia are detected in human mitral valves during development.
Fig. S2. Primary cilia are located in areas rich in versican.
Fig. S5. RNA-seq analyses correlate loss of cilia with ECM gene activation.
Fig. S6. Loss of primary cilia results in increased collagen I expression in mitral valves.
Fig. S7. Cilia length is decreased in mouse models associated with MVP.
Fig. S4. Loss of primary cilia results in decreased cell density.
Fig. S8. MVP of proband from family with chromosome 13 mutation is observed through echocardiographic assessment.
Fig. S9. DZIP1 is expressed at the base of primary cilia in the murine mitral valve.
Fig. S10. Dzip1S14R/+ KI MVP mouse models were generated through CRISPR-Cas9.
Fig. S11. GO analyses of RNA-seq datasets reveal changes in ECM synthesis.
Fig. S12. Mutated DZIP1 is unstable.
Fig. S14. Echocardiography of Dzip1 conditional knockout mitral leaflets shows MVP.
Fig. S13. Loss of Dzip1 causes developmental defects and subsequent MVP in the adult.
Data file S1. Cilia length during development.
Data file S2. Correlation of cilia with the type of ECM produced in the valve.
Data file S3. Morphometrics of valve geometry in Ift88-deficient mitral valves compared to controls at P0.
Data file S4. Cell proliferation in Ift88-deficient mitral valves compared to controls at P0.
Data file S5. Decreased cell density Ift88-deficient valves.
Data file S6. Increased collagen production in Ift88-deficient valves.
Data file S7. List of cilia genes, gene function, and disease association used in GWAS analyses.
Data file S8. Details about cilia gene set enrichment results using i-GSEA4GWAS.
Data file S9. Reduced cilia lengths in Dchs1- and Flna-deficient mitral valves.
Data file S10. Morphometrics of Dzip1S14R/+ mitral valves at P0 compared to controls.
Data file S11. Shortened cilia length in Dzip1S14R/+ mitral valves at P0 compared to controls.
Data file S12. Differentially expressed gene comparison.
Data file S13. Reduced protein half-life of DZIP1S24R variant.
Data file S14. Reduced cilia length in Dzip1 conditional knockout mice.