

Abstract
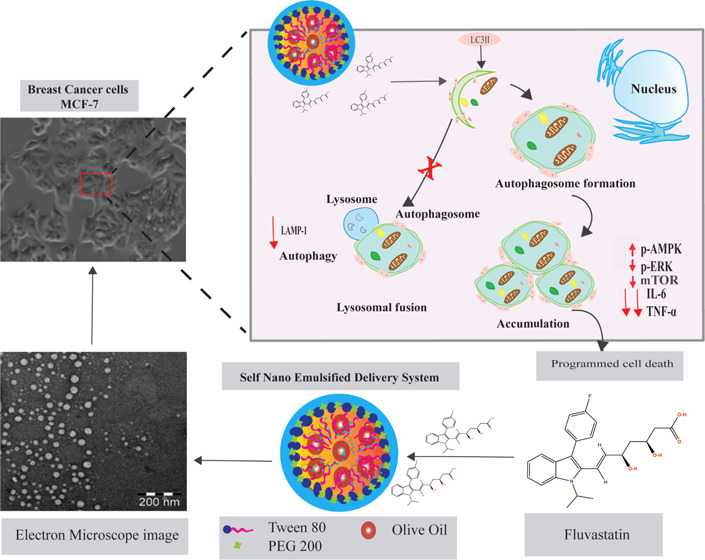
Fluvastatin (FLV) is a statin family member that may play a role in modulating a variety of medical disorders such as atherosclerosis and breast cancer. The present study addresses the ability of FLV to modulate the cellular immune response and provides a new nanosized FLV formula (self-nanoemulsifying delivery system, SNED) potentially more effective for suppression of breast cancer development. We monitored autophagic machinery through the expression of microtubule-associated protein 1A/1B-light chain 3 (LC3I/II). Lysosomal activity upon treatment was evaluated by mRNA and protein expression of lysosomal-associated membrane protein 1 (LAMP-1). Mitogen-activated protein kinase (MAPK) signaling and its association with proinflammatory cytokine secretion were assessed in treated cells. Autophagosome formation was significantly increased in cells that were pretreated with FLV-SNED in comparison to FLV-treated cells. Activation of autophagy was accompanied with arrest of LAMP-1 expression, which correlates with lysosomal activity. Simultaneously, both FLV and FLV-SNED activated MAPK signaling and modified interleukin-6 and tumor necrosis factor-α levels in treated cells. These findings indicate that FLV reduces cell viability via depletion of lysosomal activities along with accumulation of autophagosomes leading to disturbance of autophagosome-lysosomal fusion in treated cells. Furthermore, our data reveal the effectiveness of both FLV agents in the modulation of proinflammatory cytokine secretion from treated cells via regulation of MAPK signaling cascades and indicate that FLV-SNED is more efficient than FLV. This study provides new insights into how FLV regulates breast cancer cell viability via modulation of AMPK-mTOR and ERK-mTOR signaling, and through autophagosome formation accompanied by lysosomal degradation.
Introduction
Cancer is a disease in which a group of abnormal cells grows rapidly and randomly due to dysregulation of normal cell division or apoptosis. Typically, a variety of cellular signals constantly regulate cell division, differentiation, and cell death, including mitogen-activated protein kinase (MAPK) signaling, autophagosome formation, and apoptotic signaling cascades.1 Several protein kinase cascades are activated in response to extracellular growth factors, including the small GTP-binding protein (Ras), which in turn activates the core protein Raf and MEK, resulting in the stimulation of extracellular signal-regulated kinase (ERK) activity. Activated ERK1/2 dimers translocate to the nucleus and phosphorylate a variety of transcription factors that regulate gene expression.1−3 Moreover, AMP-dependent protein kinase (AMPK) could be a target of ERK in growth factor-induced proliferation.4−6
Formation of autophagosomes requires recruitment of autophagy-related proteins (Atgs) in three different steps: initiation, elongation, and maturation.7 Importantly, fusion of autophagosomes with lysosomes is critical for cell survival through degradation and recycling of cargo contents. Several studies reported a possible connection between autophagy and apoptotic signaling via inhibition of the binding of lysosomes and autophagy vacuoles.8
Statins are a drug class used clinically for the treatment of hypercholesterolemia to reduce the incidence of cardiovascular and cerebrovascular diseases. These drugs competitively inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in the mevalonate synthesis pathway.9−11 HMG-CoA reductase is also involved in various cellular functions, including cell differentiation and proliferation.12,13 Many cancer cell lines display less cholesterol than normal cells, which may occur due to excess usage of cholesterol by cancer cells, during cell division. A meta-analysis published in 2006 found that statins have no protective effect against breast cancer.14 However, preclinical and clinical data developed in the past decade support a beneficial role for the use of statins in breast cancer control.15 Various breast cancer cell lines have been used to elucidate the mechanism by which statins may exert their anticancer effects. Statins increase apoptosis and radiosensitivity and inhibit proliferation and invasion of tumor cells.16,17 This anticancer effect could be due to their lipid-lowering function; however, the complete molecular interaction is still poorly understood.18,19
Several strategies can be used to develop drugs in nanostructured systems. These strategies focus on several drug-delivery issues and aim to attain numerous advantages, including protection of drugs from degradation, extension of biological half-lives, control of drug release, and maximizing drug efficacy, while reducing adverse effects, frequency of drug administration, and drug dosage.20 Nanostructured drug-delivery systems significantly influence autophagic pathways.21 Recent studies reveal that the nanosized drug can alter such autophagic pathways by initiation of signaling induced by oxidative stress,22,23 amplifying Akt-mammalian target of rapamycin (mTOR) suppression,24 and modulating autophagy associated with gene/protein expression,25
The aim of this study is to characterize the molecular mechanisms for the anticancer effects of fluvastatin (FLV) and its nanocarrier formulation (self-nanoemulsifying delivery system, SNED). The latter was previously developed to improve the solubility and bioavailability26 in human breast adenocarcinoma cells (MCF-7).
Results
Evaluation of the FLV-SNED Formulation
The globule size of FLV-SNED shows a unimodal distribution in the developed NEs (Figure 1A). FLV-SNED particles were small, 129.9 ± 17 nm, with a low polydispersity index (PDI) of 0.33, indicating a narrow size distribution. Small size is a key factor that influences in vitro drug release, in vivo absorption across membranes, and uptake into biological systems.27,28 The ζ-potential was −24.9 mV, which indicates reasonable stability upon dilution (Figure 1B). Moreover, optical and transmission electron microscopy (TEM) images (Figure 1C,D) show spherical particle morphology in the colloidal dispersion in an aqueous medium, indicating that FLV-SNED forms homogeneous nanoscale structures. Images confirm uniformity of size distribution, in agreement with the distribution obtained using dynamic light scattering. Finally, NEs obtained by dilution of FLV-SNED with distilled water were robust to all dilutions and did not show any separation or drug precipitation after 24 h of storage.
Figure 1.

Evaluation of FLV-SNED: Photomicrographs of the particle size distribution curve (A), ζ-potential distribution curve (B), optical microscope image (40 000×) (C) (bar = 0.5 μm), and transmission electron microscope image (D) (bar = 200 nm).
FLV and FLV-SNED Affect Viability of MCF-7 Cells
The possible toxic effects of both original FLV and FLV-SNED were monitored in living MCF-7 cells, and representative images of the morphology of treated cells were collected at different time points. Furthermore, lactate dehydrogenase (LDH) release into the medium was measured to evaluate cell viability. Notably, LDH is a key enzyme in living cells and is responsible for converting lactate and NAD to pyruvic acid and NADH in mitochondria. In cultured cells, release of LDH indicates disruption of the plasma membranes due to necrotic or late-apoptotic cell death.29 MCF-7 cells were plated in a six-well plate at a density of 2 × 105 cells/well and were incubated for 24 h. Cells were then treated with 10 μM concentration of either original FLV or FLV-SNED and were incubated for the indicated time points. FLV and FLV-SNED significantly affected the number of viable cells while maintaining cellular shape during the incubation period (Figure 2A,B). Furthermore, the LDH release from cells treated with FLV was increased compared to dimethyl sulfoxide (DMSO)-treated cells (P < 0.05) (Figure 2C). FLV-SNED treatment induced greater release of LDH compared to control cells treated with SNED without FLV (P < 0.01). These findings indicate that FLV treatment has a limited cytotoxic effect on MCF-7 cells, while FLV-SNED induces cell death in cancer cells.
Figure 2.

Cytotoxicity of FLV and FLV-SNED: Representative photomicrographs of MCF-7 cells treated with either FLV or FLV-SNED (10 μM) compared to DMSO- or SNED-treated cells (A). The number of living cells was evaluated by visual counting at different time points. *P < 0.05 FLV vs DMSO at 8, 16, and 24 h, **P < 0.01 FLV-SNED vs SNED at 8, 16, and 24 h. Two experiments performed in duplicate (B). MCF-7 cells were treated with FLV- or FLV-SNED (1 μM) for 24 h. LDH release from these cells was compared to Triton X-100 (10 μL), DMSO, or SNED-treated cells. *P < 0.05 FLV vs DMSO, **P < 0.01 FLV-SNED vs SNED. Two experiments performed in duplicate (C). Dose–response curves for each treatment (1, 3, 10, 30, and 60 μM); 24 h incubation. LDH release from the treated cells was used to monitor cytotoxicity. FLV-SNED induced significantly greater LDH release compared to FLV. ***P < 0.001 FLV-SNED vs FLV at dose 30 μM, ****P < 0.0001 FLV-SNED vs FLV at 60 μM, three experiments performed in duplicate (D). Representative fluorescence micrographs of nuclear Hoechst H33342 staining in MCF-7 cells treated with FLV or FLV-SNED (10 μM) (E). Apoptotic cells contain condensed/bright nuclei (arrows).
We then examined the cytotoxicity of FLV and FLV-SNED at different doses by monitoring LDH release from MCF-7 cells after 24 h incubation. As shown in the dose–response curves (Figure 2D), both treatments induced LDH release in a dose-dependent manner. Notably, a significantly greater cytotoxic effect of FLV-SNED was found at doses of 30 and 60 μM (P < 0.001 and P < 0.0001, respectively) compared to FLV; however, 10 μM FLV-SNED induced a comparable release of LDH to FLV (the means were not significantly different; Figure 2D). To exclude overwhelming toxicity, we selected 10 μM as a therapeutic dose of FLV and FLV-SNED. Finally, we evaluated apoptosis by visual counting of cell nuclei stained with Hoechst H33342 (Figure 2E). In this assay, apoptotic cells (showing condensed/bright nuclei) constituted <1% of total in the FLV group and 6.9% in FLV-SNED (total of 216–245 cells per group).
Fluvastatin Induces Autophagosome Formation in MCF-7 Cells
Since FLV was reported to play a role in the suppression of breast cancer adenocarcinoma,15,17 we addressed potentially relevant mechanisms. We found that FLV induces autophagy in MCF-7 cells. After incubation of cells with FLV, SNED, or FLV-SNED for different time points, quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed to assess the expression levels of the LC3 gene. Treatment with FLV-SNED significantly increased the relative expression of LC3 up to 10-fold, in a time-dependent manner, compared to cells treated with SNED without FLV or with DMSO. Treatment with FLV showed limited stimulation of LC3 expression compared to the more robust upregulation by FLV-SNED (Figure 3A). The extent of LC3 lipidation (i.e., conversion of LC3-I to LC3-II) is the most commonly used method for assessing autophagy. Typically, an increase in LC3-II correlates with the number of autophagosomes formed.30,31 Thus, cell lysates were immunoblotted with anti-LC3 antibody. Under basal conditions (4 h incubation), FLV-treated MCF-7 did not show a detectable difference in LC3-II, compared to DMSO-treated cells (data not shown). After 24 h, in the presence of chloroquine (CQ, 25 μM for 6 h), the ratio of LC3-II/actin showed a greater increase in both FLV and FLV-SNED-treated cells, compared to DMSO at the corresponding time points (Figure 3B). Densitometric quantification showed that LC3-II was markedly greater in FLV- or FLV-SNED-treated cells at 24 h, compared to DMSO-treated cells (Figure 3C). In the presence of CQ, FLV and FLV-SNED drastically increased LC3-II (P < 0.0001) compared to DMSO + CQ-treated cells. FLV-SNED significantly augmented LC3-II compared to FLV-treated cells, with or without CQ (P < 0.05 and <0.01, respectively), implying that FLV-SNED induces autophagosome formation more efficiently than FLV over the course of 24 h.
Figure 3.

Alteration of autophagy in FLV- and FLV-SNED-treated cells: Relative expression of autophagy-related LC3B mRNA in MCF-7 cells at indicated time points after treatment with DMSO, SNED, FLV, or FLV-SNED (10 μM). *P < 0.05 FLV vs DMSO at 16 and 24 h, **P < 0.01 FLV-SNED vs SNED at 16 and 24 h. Two experiments performed in duplicate (A). MCF-7 cells were treated with FLV, FLV-SNED, or DMSO and were incubated with or without chloroquine (CQ, 25 μM). Lysates were immunoblotted with anti-LC3 antibody. Representative immunoblot of LC3 proteins, LC31 (18 kDa) and LC3II (16 kDa). Actin served as loading control (B). Densitometric quantification of LC3II. ****P < 0.0001 FLV + CQ vs DMSO + CQ, **P < 0.01 FLV vs FLV-SNED, *P < 0.05 FLV-SNED + CQ vs FLV + CQ. Four experiments performed in duplicate (C).
In parallel to the above experiment, conversion of LC3-I to LC3-II in MCF-7 cells was addressed via the alternative method of monitoring the formation of LC3-II puncta (using fluorescence microscopy). Cultured MCF-7 cells on coverslips were transfected with RFP-LC3. The cells were treated with FLV or FLV-SNED for 18 h and then with CQ (25 μM) for 6 h. At low levels of autophagy, RFP-LC3 is distributed diffusely in the cytoplasm, presumably in the form of LC3-I. An increase in autophagy is reflected by the formation of puncta, indicating the conversion of LC3-I to LC3-II and accumulation of LC3-II in autophagosomes. As shown in Figure 4A, control (DMSO-treated) MCF-7 cells contained relatively few puncta. RFP-LC3-II puncta in FLV- or FLV-SNED-treated cells were markedly enhanced, consistent with the formation of autophagosomes. Quantification of the puncta demonstrated that in the presence of CQ, FLV or FLV-SNED treatment significantly increased the number of puncta in MCF-7 cells compared to control (P < 0.01). Interestingly, in the presence of CQ, the number of puncta significantly increased in FLV-SNED-treated cells compared to FLV-treated cells (P < 0.05) (Figure 4B), thus confirming enhanced autophagy.
Figure 4.

FLV and FLV-SNED induce formation of LC3-II puncta: MCF-7 cells were transfected with plasmid RFP-LC3 and treated with FLV or FLV-SNED (10 μM) for 24 h in the presence or absence of chloroquine (CQ, 25 μM) in the last 6 h. Representative photomicrographs; bar = 25 μm (A). Quantification of LC3-II puncta is presented as puncta count (B). **P < 0.01 FLV + CQ vs DMSO + CQ, *P < 0.05 FLV-SNED + CQ vs FLV + CQ; 14–30 cells per group in three experiments. Quantification of steady-state mRNA expression in MCF-7 cells transfected with Atg7- or luciferase (Luci)-directed siRNAs (qRT-PCR). Atg7 siRNA reduced Atg7 and increased P53 and Casp3 expression compared to control and luciferase siRNA (C). **P < 0.01 Atg7, ***P < 0.001 P53 *P < 0.05 Casp3 in Atg7 siRNA vs control. Two experiments performed in duplicate.
The connection between autophagosome formation and cell viability was investigated in MCF-7 cells independently of drug treatment. Cells were transfected with siRNA that targets either Atg7 or luciferase, as a control. The relative gene expression of the tumor suppressor gene, P53, which promotes apoptosis, and the apoptotic effector, caspase-3 (Casp3), was assessed in transfected cells. Knockdown of Atg7 stimulated expression of P53 and Casp3, indicating that the interruption of autophagic machinery is associated with induction of factors that can lead to programmed cell death (Figure 4C).
Fluvastatin Inhibits Autolysosome Formation in MCF-7 Cells
Next, we examined the hypothesis that FLV disturbs lysosomal activity by evaluating expression of LAMP-1 (a protein localized on lysosomal membranes) in MCF-7 cells treated with FLV or FLV-SNED.32 Relative expression of the LAMP-1 gene (quantitative RT-PCR) was significantly reduced in FLV-treated cells (P < 0.05) and completely abolished in FLV-SNED-treated cells after 24 h (P < 0.01) (Figure 5A). We also employed immunoblotting to confirm the above result. LAMP-1 is a heavily glycosylated lysosomal membrane protein that protects the lysosomal membranes from intracellular proteolysis.33 Glycosylated LAMP-1 protein expression is relatively high in MCF-7 cells treated with DMSO (Figure 5B). In keeping with the reduced expression level of LAMP-1 mRNA in FLV-treated cells, FLV treatment tended to decrease glycosylated LAMP-1 protein expression, though not significantly (Figure 5B,C). Densitometric quantification of glycosylated LAMP-1 revealed that FLV-SNED treatment for 24 h significantly reduced LAMP-1 expression in comparison to FLV (P < 0.05) and DMSO-treated cells (P < 0.001) (Figure 5C). Thus, autolysosome formation is inhibited by FLV-SNED.
Figure 5.

Lysosomal activity in FLV- and FLV-SNED-treated cells: Relative expression of the lysosomal marker, LAMP-1, at indicated time points following FLV or FLV-SNED treatment of MCF-7 cells, compared to DMSO and SNED *P < 0.05 FLV vs DMSO at 8 and 16 h, **P < 0.01 FLV vs DMSO at 24 h, **P < 0.01 FLV-SNED vs SNED at 8 and 16 h, ***P < 0.001 FLV-SNED vs SNED at 24 h. Two experiments performed in duplicate (A). MCF-7 cells were treated with FLV, FLV-SNED, or DMSO at the indicated time points. Lysates were immunoblotted with anti-LAMP-1 antibody. Representative immunoblot of glycosylated LAMP-1 (90–120 kDa). α-Tubulin served as loading control (B). Densitometric quantification of glycosylated LAMP-1 at different time points. ***P < 0.001 FLV-SNED vs DMSO at 24 h, +P < 0.05 FLV-SNED vs FLV at 24 h. Four experiments performed in duplicate (C). Representative fluorescent photomicrographs of LAMP-1; bar = 25 μm (D).
The LAMP-1 protein was also examined by monitoring LAMP-1-positive organelles using fluorescence microscopy. Consistent with the above results, LAMP-1 expression, visualized as green staining with a dotlike pattern, was higher in MCF-7 cells treated with DMSO compared to FLV and FLV-SNED treatments (Figure 5D).
FLV and FLV-SNED Modulate AMPK and MAPK Signaling in MCF-7 Cells
We next examined the signaling pathways potentially involved in the activation of autophagy by FLV. First, we monitored the phosphorylation of AMPK, which can be stimulated by reduced cellular levels of ATP.30 We found that FLV and FLV-SNED markedly induced phosphorylation of AMPK in MCF-7 cells compared to DMSO (Figure 6A). Densitometry of pAMPK/AMPK levels in FLV-SNED-treated cells, in the presence of CQ, showed a significant upregulation compared to DMSO + CQ (P < 0.0001)- and FLV + CQ-treated cells (P < 0.05). Moreover, FLV-SNED alone significantly amplified the ratio of pAMPK/AMPK compared to FLV (Figure 6B). Second, ERK, the upstream kinase of the tuberous sclerosis complexes 1 and 2 (TSC1 and TSC2),34,35 was less phosphorylated/activated by FLV and FLV-SNED compared to DMSO (Figure 6A). These results suggest that AMPK phosphorylation and inhibition of basal ERK phosphorylation may be potential mediator pathways in FLV-induced autophagy.
Figure 6.

Effect of FLV and FLV-SNED on AMPK, MAPK, and mTOR signaling: MCF-7 cells were treated with FLV, FLV-SNED (10 μM), or DMSO for 24 h in the presence or absence of chloroquine (CQ, 25 μM; final, 6 h). Lysates were immunoblotted with antibodies to AMPK, pAMPK, ERK 42/44, pERK 42/44, and LC3. Representative immunoblots of corresponding proteins. Actin served as loading control (A). Densitometric quantification of pAMPK/AMPK. *P < 0.05 FLV + CQ vs DMSO + CQ, **P < 0.01 FLV-SNED vs FLV, ***P < 0.001 FLV-SNED + CQ vs DMSO + CQ, +P < 0.05 FLV-SNED + CQ vs FLV + CQ. Three experiments performed in duplicate (B). The same lysates were immunoblotted with antibodies to S6 kinase, phospho-S6 kinase, and LC3. Representative immunoblots of corresponding proteins. Actin served as loading control. Phosphorylated S6 kinase, 85 kDa isoform (pp85), is reduced in FLV-SNED (C).
FLV and FLV-SNED Inhibit mTOR Activity
The above results led us to examine whether FLV-mediated autophagy activation is dependent on the mechanistic target of rapamycin (mTOR). We undertook to examine the activation of the mTOR pathway. mTOR activity was monitored by phosphorylation of the mTOR substrate, S6 kinase, at Thr389.36 There are two isoforms of S6 kinase (70 and 85 kDa), and both isoforms are expressed in MCF-7 cells. Phosphorylated S6 kinase, 85 kDa isoform (pp85), was reduced in FLV-SNED-treated cells compared to DMSO- and FLV-treated cells (Figure 6C), indicating that mTOR is likely inactive in SNED-treated cells and supporting a role for mTOR in autophagy.
FLV and FLV-SNED Prevent Inflammation in MCF-7 Cells
Proinflammatory cytokines, IL-6 and TNF-α, play a critical role in cancer development and associated inflammation. Therefore, levels of IL-6 and TNF-α were assessed in treated cells at distinct time points (4, 8, 16, and 24 h) using ELISA. As expected, the highest concentrations of both IL-6 and TNF-α were detected in SNED- and DMSO-treated cells (more than 180 and 300 pM, respectively). Importantly, the lowest concentrations were found in cells treated with FLV-SNED in a time-dependent manner (less than 100 pM) (Figure 7A,B). Accordingly, our findings support a role of FLV-SNED and original FLV in the regulation of proinflammatory cytokine secretion, autophagy, and MAPK signaling in breast cancer MCF-7 cells.
Figure 7.

Cytokine secretion in FLV- and FLV-SNED-treated cells: MCF-7 cells were treated with FLV, FLV-SNED, DMSO, or SNED at the indicated time points. Concentrations of IL-6 and TNF-α were measured in the culture media. Basal IL-6 levels were detected between 150 and 180 pM, in DMSO and SNED cells. Both FLV and FLV-SNED reduced IL-6 levels. ++P < 0.01 FLV vs DMSO at 16 h, +++P < 0.001 FLV vs DMSO at 24 h, *P < 0.05 FLV-SNED vs SNED at 4 h, **P < 0.01 FLV-SNED vs SNED at 8 h, ***P < 0.001 FLV-SNED vs SNED at 16 h, ****P < 0.0001 FLV-SNED vs SNED at 24 h. Two experiments performed in duplicate (A). Basal TNF-α levels were detected between 250 and 350 pM in DMSO- and SNED-treated cells. Both FLV and FLV-SNED reduced TNF-α levels. ++P < 0.01 FLV vs DMSO at 8 h, +++P < 0.001 FLV vs DMSO at 16 h, ++++P < 0.0001 FLV vs DMSO at 24 h, **P < 0.01 FLV-SNED vs SNED at 8 h, ***P < 0.001 FLV-SNED vs SNED at 16 h, ****P < 0.0001 FLV-SNED vs SNED at 24 h. Two experiments performed in duplicate (B).
Discussion
FLV is a member of the statin drug family currently recognized for the treatment of various diseases and medical disorders, including cardiovascular disease and cancer.37,38 Recent evidence demonstrates that FLV successfully inhibits the expression of estrogen receptor α (ERα), resulting in the regulation of basal-type and high-grade breast cancer.37,39 However, while the action of FLV as an anticancer agent has been reported, the molecular aspects of statin-regulated innate immune responses and cell proliferation are poorly understood.
Accordingly, we investigated the molecular interactions of FLV treatment on breast cancer cells using the MCF-7 cell line. In addition, we provide a well-established method to increase the efficiency of FLV by creating an FLV-loaded SNED agent. Thus, we treated MCF-7 cells with either original FLV or a formulated version (nanosized structure, FLV-SNED). Following treatment, cytotoxic effects were monitored by the amounts of LDH released from treated cells. Autophagosome formation, lysosomal activities, and MAPK-mTOR signaling cascades were also investigated to underline the molecular aspects of FLV-actions in breast cancer cells. Finally, levels of proinflammatory cytokines, IL-6 and TNF-α, were monitored to assess the anti-inflammatory potential of FLV treatment.
First, our results showed reduced viability of both FLV- and FLV-SNED-treated cells; however, FLV-SNED induced the greatest reduction in viability. Furthermore, the expression profile of autophagy-related genes, including LC3B, as well as autophagosome formation was markedly increased in FLV-SNED-treated cells, more than in FLV-treated cells, as indicated by immunoblotting and immunofluorescent assays. Similarly, activation of MAPK cascades, including ERK1/2 and arrest of lysosomal activity, was more pronounced in FLV-SNED-treated cells. These results reveal an inhibitory effect of unmodified FLV and FLV-SNED on LAMP-1-induced lysosomal activity and suggest crosstalk between autophagic flux and signals that mediate cell viability.
Autophagy is an intracellular digestive system that participates in the degradation of unnecessary or unneeded proteins by delivering cytosolic constituents to lysosomes.40 A variety of cellular signaling pathways are involved in the regulation of autophagy; these signals either inhibit or support autophagosome formation. Besides the phosphatidylinositol 3-kinase/Akt/mTOR pathway, Ras/protein kinase A signaling downregulates autophagy, while the LKB1-AMPK pathway induces autophagy in response to bioenergetic stress during nutrient deprivation.41 Activation of AMPK (e.g., by reduced cellular levels of ATP30,42) redirects cellular metabolism by acting on mTORC1, p53, fatty acid synthase, and other substances that control cell growth.43 An upward trend in pAMPK protein levels was evident in FLV- and FLV-SNED-treated cells (Figure 6A). TSC2 integrates signals from various kinases leading to mTORC1 regulation. For example, phosphorylation by AMPK increases TSC2 activity, leading to inactivation of mTORC1.35 In the present study, the activity of mTORC1 was reduced in FLV-SNED-treated cells. These findings support a cascade, where AMPK phosphorylation inhibits mTORC1 activation via promoting TSC1/2 complex formation and phosphorylating the associated regulatory protein of mTOR (Raptor).44 Alternatively, downregulation of basal ERK1/2 phosphorylation (Figure 6A) may potentially inhibit TSC2 phosphorylation, allowing TSC1/2 complex formation that could also render mTORC1 inactive.34 Inhibition of mTORC1 promotes autophagosome maturation. Notably, autophagosome formation requires conjugation and association of multiple autophagy-related proteins (Atgs) to complete initiation and elongation steps before fusion of autophagosomes and lysosomes. Lysosomes are organelles that contain hydrolyzing enzymes that break down biopolymers at low pH, including proteins, carbohydrates, and lipids. However, while autophagic machinery maintains cell survival, interruption of lysosomal fusion with autophagy vesicles can induce apoptosis.45,46
We investigated autophagy and lysosomal activation in FLV-treated cells, focusing on the relative expression of the autophagy-related LC3 gene and lysosomal marker LAMP-1 gene. Treatment with FLV or FLV-SNED increased the relative expression of LC3 more than threefold compared to control (Figure 3A). Meanwhile, the relative expression of LAMP-1 gene was significantly reduced in FLV-SNED-treated cells (Figure 5A). These findings indicate that FLV-SNED disturb lysosomal activation and fusion of autophagy vesicles and lysosomes.
Finally, an important finding of the current work is FLV-mediated proinflammatory cytokine secretion in treated cells. Our results show that both FLV and FLV-SNED arrest the production of both IL-6 and TNF-α in a time-dependent manner. It is thought that IL-6 and TNF-α are critical proinflammatory cytokines regulated by CD22 or CD40, and they induce inflammatory events during development of cancer. In breast cancer, TNF-α enhances cell growth via upregulation of oncoprotein hepatitis B X-interacting protein (HBXIP).47 Breast cancer cells show high levels of IL-6 secretion, which plays a crucial role in tumor growth, metastasis, and therapeutic resistance. Therefore, IL-6 and its receptor may be attractive targets for breast cancer therapy.48
Conclusions
FLV-SNED treatment stimulates autophagosome formation and inhibits mTORC1 signaling. Furthermore, FLV-SNED-treated cells show increasing levels of both autophagosome formation and AMPK cascade components that competitively inhibit lysosomal activity and proinflammatory events. FLV-SNED is thus more effective than FLV for regulation of cancer cell viability via activation of autophagy vesicle formation along with depletion of lysosomal activity. The mechanism may involve programmed cell death.
Materials and Methods
Materials
Fluvastatin sodium, [7-(3-(4-fluorophenyl)-1-(1-methylethyl)-1H-indol-2-yl)-3,5-dihydroxy-6-heptenoate], was purchased from SPIMACO (Al-Qassim, SA). Polyoxyethylene (20) sorbitan monooleate (Tween 80) and poly(ethylene glycol) 200 (PEG 200) were purchased from Sigma-Aldrich (St. Louis, MO). Olive oil was purchased from Acros Organics (Morris Plains, NJ). Tissue culture media was from Invitrogen Life Technologies (Burlington, ON). Plasmid tandem red fluorescent protein (mRFP)-LC3B was purchased from Addgene (catalog no. 21075).49 Electrophoresis reagents were from Bio-Rad Laboratories (Mississauga, ON). Rabbit anti-microtubule-associated protein light chain 3B (LC3; catalog no. 2775), monoclonal rabbit anti-phospho-AMP-activated protein kinase (AMPK) (Thr-172; catalog no. 2535), rabbit anti-AMPK (catalog no. 2532), rabbit anti-lysosome-associated membrane protein 1 (LAMP-1) (catalog no. 9091), rabbit anti-phospho-S6 kinase (Thr389; 9205), and rabbit anti-phospho-p44/42 MAPK (Thr-202/Tyr-204) antibodies were from Cell Signaling Technology (Danvers, MA). Rabbit anti-S6 kinase (C-18, sc-230) antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal anti-α-tubulin was from Sigma. FITC-conjugated rabbit anti-sheep IgG (catalog no.31509) was from Zymed Laboratories Inc. (South San Francisco, CA). Enzyme-linked immunosorbent assay (ELISA) kits for tumor necrosis factor (TNF-α) and interleukin-6 (IL-6) were from Abcam (catalog nos. 181421 and 46042, respectively).
Preparation of Fluvastatin-SNED
FLV-SNED was prepared using the same protocol as reported previously.26 Olive oil was used as an oil phase, Tween 80 as a surfactant, and PEG 200 as a cosurfactant with percentages of 10, 66, and 24%, respectively. Briefly, 1 g of the SNED was prepared by combining the three components in an Eppendorf tube, vortexing the mixture for 1 min with a fixed dose of FLV (50 mg) to achieve complete solubilization of FLV in the formulation components.
Visual Characterization of FLV-SNED
The FLV-SNED formulation was centrifuged at 4000 rpm for 20 min and exposed to three freeze–thaw cycles at −20 and +25 °C. Samples (100 mg) of the developed FLV-SNED were diluted with 200 mL of distilled water. Nanoemulsions (NEs) were visually inspected for transparency and emulsification tendency.50
Robustness to Dilution
FLV-SNED was evaluated for its ability to maintain its properties upon 50, 100, and 1000 times dilution with double-distilled water. Diluted NEs were kept for 12 h and inspected for any drug precipitation or phase separation features.51
Determination of FLV-SNED Size and ζ-Potential Distribution
FLV-SNED was diluted with 20 mL of distilled water. The resultant dispersion was examined for both hydrodynamic diameter and ζ-potential using a Malvern Zetasizer Nano ZSP, Malvern Panalytical Ltd. (Grovewood Road, United Kingdom) at 22 °C.
Surface Morphology of the FLV-SNED
The morphological features of FLV-SNED were characterized by light microscopy (Leica DM300 optical microscope, Wetzlar, Germany), as well as transmission electron microscopy (TEM;100CX; JEOL, Tokyo, Japan). FLV-SNED was photographed at 40 000× magnification with a digital camera (HTC M8; light microscopy). For TEM, a drop of diluted FLV-SNED formulation was added onto a microscope carbon-coated grid, left to dry for 3 min, and then stained with 1% phosphotungstic acid before the examination.
Cell Lines
MCF-7 cells, kindly provided by Dr. Suhad Ali (Department of Medicine, Cancer Research Program, McGill University Health Center, Canada), were cultured on plastic substratum in Dulbecco’s modified Eagle’s medium supplemented with 100 U/mL penicillin/streptomycin and 10% fetal bovine serum. The cells were maintained at 37 °C in a humidified 5% CO2 incubator.52 Other MCF-7 cells were purchased from (VACSERA, Giza, Egypt) and maintained in RPMI medium with 4 mM sodium pyruvate, 4 mM l-glutamine, 100 U/mL penicillin/streptomycin, and 2.5% bovine serum albumin (BSA). The cells were incubated at 37 °C in a humidified 5% CO2 incubator and were regularly checked for mycoplasma contamination.
Transfection Protocol
MCF-7 cells were placed in six-well plates at 2 × 105 cells/well and cultured in the medium for 18–24 h before transfection. Transient transfection of MCF-7 cells with RFP-LC3B was performed using the Lipofectamine 2000 reagent (Invitrogen Life Technologies, Burlington, Canada), according to the manufacturer’s instructions. Briefly, for each well, 1 μg of plasmid DNA and 3 μL of lipofectamine 2000 reagent were diluted in Opti-MEM. Diluted DNA was added to diluted Lipofectamine (1:1 ratio) and incubated for 20 min at 22 °C. The transfection mixture was then added to cultured MCF-7 cells. Plates were incubated in a CO2 incubator at 37 °C for 18–24 h. Lysates were then prepared for immunoblotting, as described below. For small interference RNA (siRNA) transfection, MCF-7 cells cultured in six-well plates were transfected with the respective siRNA against Atg7 (5′-GUACUCAACUGGGUCUUCUUCGA-3′) or luciferase (5′-AACUUACGCUGAGUACUUCGA-3′), which served as control. The transfection was performed for each well by mixing 200 ng of siRNA with 20 μL of HyperFect (Qiagen) in 500 μL of Opti-MEM media. The transfected cells were then incubated for 48 h, and the knockdown efficiency of Atg7 and the relative expression of P53 and Caspase 3 (Casp3) were monitored using qRT-PCR (see below).53
Fluvastatin Treatment
Transfected and untransfected MCF-7 cells were treated with either 10 μM FLV or FLV-SNED per mL of medium. Other cells were treated with 0.1% (v/v) DMSO, which served as control. The cells were incubated for 4, 8, 16, and 24 h.
Cell Viability and Cytotoxicity Assay
Cell death was assessed by measuring the release of lactate dehydrogenase (LDH), as described previously.29 LDH was monitored in a 96-well plate using an LDH production kit, according to the manufacturer’s instructions. Equal volumes of collected culture media and LDH buffer were incubated for 2 h, followed by a 1 h incubation with LDH substrate. LDH production was measured and calculated. Increased absorbance at 440 nm from the production of NADH reflects LDH activity.29 Cells treated with 10 μL of Triton X-100 were used as positive control.
Apoptotic cells were evaluated by the pattern of nuclear staining with Hoechst H33342 (see below). In this assay, nuclei of apoptotic cells show chromatin condensation and/or fragmentation and stain brightly with H33342. All other Hoechst H33342-stained cells were designated as “normal”.54
Immunoblotting
Cells were lysed in ice-cold lysis buffer containing 1% Triton X-100, 10 mM Tris, pH 7.4, 1 mM EGTA, 125 mM NaCl, 2 mM Na3VO4, 25 mM NaF, and 10 mM sodium pyrophosphate, supplemented with a protease inhibitor mixture (Bioshop). Cell lysates were centrifuged at 13 000 rpm for 10 min, and protein concentrations were measured with the Bradford assays. Cytosolic proteins were solubilized in Laemmli buffer and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were electrophoretically transferred to polyvinylidene difluoride membranes and blocked at 22 °C for 1 h with 5% BSA in 1× Tris-buffered saline with 0.1% Tween 20 (TBST). Membranes were then incubated with primary antibodies in 5% BSA in 1× TBST overnight, followed by goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody in 1× TBST for 1 h at 22 °C. Membranes were then developed with ECL.3,29 Quantitative densitometry of protein bands was performed using ImageJ software (National Institutes of Health, Bethesda, MD).
Immunofluorescence Microscopy
MCF-7 cells were seeded on coverslips at a density of 1 × 105 cells/slip in a 12-well plate and transfected with 0.5 μg of plasmids encoding cDNAs fused with fluorescent reporters. After 24 h of transfection, the cells were either left untreated or treated with 10 μM FLV or FLV-SNED, and the cells were fixed with 4% paraformaldehyde/phosphate-buffered saline (PBS) for 15 min. The cells were then washed three times with 1× PBS, and nuclei were stained with Hoechst H33342 and then rinsed with 1× PBS before being mounted on glass slides. Fixed cells were imaged using Zeiss Axio Observer fluorescence microscope with visual output connected to an AxioCam MRm monochrome camera.
In another set of experiments, 1 × 105 cells were cultured on coverslips in a 12-well plate for 24 h and treated with 10 μM FLV or FLV-SNED for 24 h. The cells were then fixed with ice-cold 100% methanol for 15 min at −20 °C. The cells were washed three times with cold 1× PBS and subsequently incubated with anti-LAMP-1 monoclonal antibody (1:200 dilution in 1× PBS supplemented with 1% BSA and 0.3% Triton X-100) for 18 h at 4 °C. Secondary antibody incubation consisted of a 1:500 dilution of Alexa-Fluor 488 goat anti-rabbit in 1× PBS supplemented with 1% BSA. Nuclei were stained with Hoechst H33342, and the cells were rinsed with cold PBS before mounting on glass slides. Fixed cells were examined as described above.
Total RNA Isolation and cDNA Synthesis
Cells were harvested from culture plates in RNase-free tubes. Total RNA was isolated using TriZol (Invitrogen) and chloroform methods. After dissolving RNA in RNase-free water, the samples were adjusted to final concentrations of 100 ng/mL. Each sample of purified total RNA (10 μL) was used to produce cDNA using a cDNA synthesis kit (Qiagen). Based on the manufacturer’s protocol, total RNA was incubated with reverse transcriptase and oligo-(dT) primers at 45 °C for 1 h followed by a 5 min incubation at 95 °C. cDNA was stored at −20 °C until usage.29
Quantitative Real-Time PCR (Q-RT-PCR) Assay
Q-RT-PCR was used to assess the expression levels of LC3B and LAMP-1 genes in treated and untreated cells. cDNA was employed as a template for PCR amplification with DNA polymerase and specific primers for LC3B and LAMP-1. The relative gene expression of LC3B and LAMP-1 was determined using a Quanti-Tect SYBR Green PCR Kit (Qiagen). Oligonucleotides specific for each gene were designed with Primer Express software (version 2.0) (Table 1). Levels of GAPDH were used for normalization. The real-time PCR was performed in an Applied Biosystems device at a temperature of 94 °C for 3 min followed by 35 cycles of 20 s at 95 °C, annealing at 58 °C (15 s), and extension at 72 °C (45 s). Data analyses of threshold cycle (CT) values were performed using SDS 2.2.7
Table 1. Oligonucleotides Sequence Used for Detection of Steady-State mRNA of Indicated Genes.
| description | primer sequences (5′–3′) |
|---|---|
| LC3B-sense | CGTCCTGGACAAGACCAAGT |
| LC3B-anti-sense | CCATTCACCAGGAGGAAGAA |
| LAMP-1-sense | CCCACAAACCCCACTGTATC-3 |
| LAMP-1-anti-sense | GGTCACCGTCTTGTTGTCCT |
| GAPDH-sense | TGGCATTGTGGAAGGGCTCA |
| GAPDH-anti-sense | TGGATGCAGGGATGATGTTCT |
| Atg7-sense | CGTTGCCCACAGCATCATCTTC |
| Atg7-anti-sense | CACTGAGGTTCACCATCCTTGG |
| P53-sense | GCGAGCACTGCCCAACAACA |
| P53-anti-sense | GGTCACCGTCTTGTTGTCCT |
| Casp3-sense | GGACAGCAGTTACAAAATGGATTA |
| Casp3- anti-sense | CGGCAGGCCTGAATGATGAAG |
Enzyme-Linked Immunosorbent Assay
ELISA was used for quantitative measurement of TNF-α and IL-6 (human ELISA kits; Abcam 181421 and Abcam 46042, respectively). MCF-7 cells were seeded in a 96-well plate at a density of 10 000 cells per well. The cells were treated with 10 μM FLV or FLV-SNED per mL medium. In a time course experiment (4, 8, 16, and 24 h), the cells were lysed and lysates were collected to monitor the amounts of TNF-α and IL-6 produced at different time points. Standards and test samples (100 μL) were placed on an ELISA 96-well plate and incubated overnight at 22 °C. After washing, 100 μL of detection antibody was added to each well, followed by 1 h incubation at RT. Then, 200 μL of substrate solution was added to each well and incubated for 20 min in the dark at RT. Finally, 50 μL of stop solution was added. Color develops according to the amount of indicated cytokine bound. Color intensity was measured at 450 nm.55
Statistical Analysis
Graphs and histograms were prepared using Microsoft Excel and GraphPad Prism software (version 8.4, San Diego, CA). SDS2.2.2 software was used to analyze Q-RT-PCR data to derive Ct values according to delta–delta Ct equations (Delta Ct) = Ct-gene – Ct-GAPDH. (ΔΔCt) = ΔCt-sample – ΔCt-control. Relative gene expression (indicated by fold change) is equal to 2–ΔΔct.7,56 Data are presented as mean ± standard error of the mean (SEM). Comparisons between groups were performed using one- or two-way analysis of variance (ANOVA); the Bonferroni method was used for post hoc comparisons (GraphPad Prism, version 8.4.2). Probability values of <0.05 were considered statistically significant.
Acknowledgments
Dr. Cybulsky is the recipient of a grant from the Canadian Institutes of Health Research (MOP-133492) and holds the Catherine McLaughlin Hakim Chair. The authors acknowledge the assistance of Cloé L. Esposito with the graphical illustration.
Glossary
Abbreviations
- FLV
Fluvastatin
- SNED
self-nanoemulsifying delivery system
- LC3I/II
microtubule-associated protein 1A/1B-light chain 3
- LAMP-1
lysosomal-associated membrane protein 1
- MAPK
mitogen-activated protein kinase
- ERK
extracellular signal-regulated kinase
- AMPK
AMP-dependent protein kinase
- HMG-CoA
3-hydroxy-3-methylglutaryl coenzyme A
- MCF-7
human breast adenocarcinoma cells
- mTOR
mechanistic target of rapamycin
- TEM
transmission electron microscopy
- LDH
lactate dehydrogenase
- TSC1 and TSC2
tuberous sclerosis complex 1 and 2
- TNF-α
tumor necrosis factor
- IL-6
interleukin-6
- NE
nanoemulsion
The authors declare no competing financial interest.
References
- Dhillon A. S.; Hagan S.; Rath O.; Kolch W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Bhalla U. S.; Ram P. T.; Iyengar R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 2002, 297, 1018–1023. 10.1126/science.1068873. [DOI] [PubMed] [Google Scholar]
- Elimam H.; Papillon J.; Takano T.; Cybulsky A. V. Complement-mediated activation of calcium-independent phospholipase A2gamma: role of protein kinases and phosphorylation. J. Biol. Chem. 2013, 288, 3871–3885. 10.1074/jbc.M112.396614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Guan T.; Zhang H.; Xia Y.; Liu F.; Zhang Y. Inhibitory crosstalk between ERK and AMPK in the growth and proliferation of cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2008, 368, 402–407. 10.1016/j.bbrc.2008.01.099. [DOI] [PubMed] [Google Scholar]
- Mihaylova M. M.; Shaw R. J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell. Biol. 2011, 13, 1016–1023. 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd El Maksoud A. I.; Taher R. F.; Gaara A. H.; Abdelrazik E.; Keshk O. S.; Elawdan K. A.; Morsy S. E.; Salah A.; Khalil H. Selective Regulation of B-Raf Dependent K-Ras/Mitogen-Activated Protein by Natural Occurring Multi-kinase Inhibitors in Cancer Cells. Front. Oncol. 2019, 9, 1220. 10.3389/fonc.2019.01220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil H.; Tazi M.; Caution K.; Ahmed A.; Kanneganti A.; Assani K.; Kopp B.; Marsh C.; Dakhlallah D.; Amer A. O. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics 2016, 11, 381–388. 10.1080/15592294.2016.1144007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A.; Kaarniranta K.; Kauppinen A. Beclin 1 interactome controls the crosstalk between apoptosis, autophagy and inflammasome activation: impact on the aging process. Ageing Res. Rev. 2013, 12, 520–534. 10.1016/j.arr.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Goldstein J. L.; Brown M. S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Jiang P.; Mukthavaram R.; Chao Y.; Nomura N.; Bharati I. S.; Fogal V.; Pastorino S.; Teng D.; Cong X.; Pingle S. C.; Kapoor S.; Shetty K.; Aggrawal A.; Vali S.; Abbasi T.; Chien S.; Kesari S. In vitro and in vivo anticancer effects of mevalonate pathway modulation on human cancer cells. Br. J. Cancer 2014, 111, 1562–1571. 10.1038/bjc.2014.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurnher M.; Nussbaumer O.; Gruenbacher G. Novel aspects of mevalonate pathway inhibitors as antitumor agents. Clin. Cancer Res. 2012, 18, 3524–3531. 10.1158/1078-0432.CCR-12-0489. [DOI] [PubMed] [Google Scholar]
- Chan K. K.; Oza A. M.; Siu L. L. The statins as anticancer agents. Clin. Cancer Res. 2003, 9, 10–19. [PubMed] [Google Scholar]
- Wong W. W.; Dimitroulakos J.; Minden M. D.; Penn L. Z. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 2002, 16, 508–519. 10.1038/sj.leu.2402476. [DOI] [PubMed] [Google Scholar]
- Bonovas S.; Filioussi K.; Tsavaris N.; Sitaras N. M. Use of statins and breast cancer: a meta-analysis of seven randomized clinical trials and nine observational studies. J. Clin. Oncol. 2005, 23, 8606–8612. 10.1200/JCO.2005.02.7045. [DOI] [PubMed] [Google Scholar]
- Van Wyhe R. D.; Rahal O. M.; Woodward W. A. Effect of statins on breast cancer recurrence and mortality: a review. Breast Cancer 2017, 9, 559–565. 10.2147/BCTT.S148080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuturk M.; Ersoz M.; Altiok N. Simvastatin induces apoptosis in human breast cancer cells: p53 and estrogen receptor independent pathway requiring signalling through JNK. Cancer Lett. 2007, 250, 220–228. 10.1016/j.canlet.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Lacerda L.; Reddy J. P.; Liu D.; Larson R.; Li L.; Masuda H.; Brewer T.; Debeb B. G.; Xu W.; Hortobagyi G. N.; Buchholz T. A.; Ueno N. T.; Woodward W. A. Simvastatin radiosensitizes differentiated and stem-like breast cancer cell lines and is associated with improved local control in inflammatory breast cancer patients treated with postmastectomy radiation. Stem Cells Transl. Med. 2014, 3, 849–856. 10.5966/sctm.2013-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clendening J. W.; Penn L. Z. Targeting tumor cell metabolism with statins. Oncogene 2012, 31, 4967–4978. 10.1038/onc.2012.6. [DOI] [PubMed] [Google Scholar]
- Tobert J. A. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat. Rev. Drug Discovery 2003, 2, 517–526. 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- Ahmed T. A.; El-Say K. M.; Ahmed O. A. A.; Zidan A. S.. Sterile Dosage Forms Loaded Nanosystems for Parenteral, Nasal, Pulmonary and Ocular Administration. In Nanoscale Fabrication, Optimization, Scale-Up and Biological Aspects of Pharmaceutical Nanotechnology; William Andrew Publishing, 2018; pp. 335–395. [Google Scholar]
- El-Say K. M.; El-Sawy H. S. Polymeric nanoparticles: Promising platform for drug delivery. Int. J. Pharm. 2017, 528, 675–691. 10.1016/j.ijpharm.2017.06.052. [DOI] [PubMed] [Google Scholar]
- Harhaji L.; Isakovic A.; Raicevic N.; Markovic Z.; Todorovic-Markovic B.; Nikolic N.; Vranjes-Djuric S.; Markovic I.; Trajkovic V. Multiple mechanisms underlying the anticancer action of nanocrystalline fullerene. Eur. J. Pharmacol. 2007, 568, 89–98. 10.1016/j.ejphar.2007.04.041. [DOI] [PubMed] [Google Scholar]
- Khan M. I.; Mohammad A.; Patil G.; Naqvi S. A.; Chauhan L. K.; Ahmad I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials 2012, 33, 1477–1488. 10.1016/j.biomaterials.2011.10.080. [DOI] [PubMed] [Google Scholar]
- Liu H. L.; Zhang Y. L.; Yang N.; Zhang Y. X.; Liu X. Q.; Li C. G.; Zhao Y.; Wang Y. G.; Zhang G. G.; Yang P.; Guo F.; Sun Y.; Jiang C. Y. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt-TSC2-mTOR signaling. Cell Death Dis. 2011, 2, e159 10.1038/cddis.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Yang W.; Man N.; Zheng F.; Shen Y.; Sun K.; Li Y.; Wen L. P. Autophagy-mediated chemosensitization in cancer cells by fullerene C60 nanocrystal. Autophagy 2009, 5, 1107–1117. 10.4161/auto.5.8.9842. [DOI] [PubMed] [Google Scholar]
- Ahmed T. A.; Elfaky M. A.; Fahmy U. A.; Aljaeid B. M.; Alshareef O. A.; El-Say K. M. Development of optimized self-nanoemulsifying lyophilized tablets (SNELTs) to improve finasteride clinical pharmacokinetic behavior. Drug Dev. Ind. Pharm. 2018, 44, 652–661. 10.1080/03639045.2017.1405977. [DOI] [PubMed] [Google Scholar]
- Limbach L. K.; Li Y.; Grass R. N.; Brunner T. J.; Hintermann M. A.; Muller M.; Gunther D.; Stark W. J. Oxide nanoparticle uptake in human lung fibroblasts: effects of particle size, agglomeration, and diffusion at low concentrations. Environ. Sci. Technol. 2005, 39, 9370–9376. 10.1021/es051043o. [DOI] [PubMed] [Google Scholar]
- Shi W.; Wang J.; Fan X.; Gao H. Size and shape effects on diffusion and absorption of colloidal particles near a partially absorbing sphere: implications for uptake of nanoparticles in animal cells. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2008, 78, 061914 10.1103/PhysRevE.78.061914. [DOI] [PubMed] [Google Scholar]
- Elimam H.; Papillon J.; Takano T.; Cybulsky A. V. Calcium-independent phospholipase A2gamma enhances activation of the ATF6 transcription factor during endoplasmic reticulum stress. J. Biol. Chem. 2015, 290, 3009–3020. 10.1074/jbc.M114.592261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elimam H.; Papillon J.; Kaufman D. R.; Guillemette J.; Aoudjit L.; Gross R. W.; Takano T.; Cybulsky A. V. Genetic Ablation of Calcium-independent Phospholipase A2gamma Induces Glomerular Injury in Mice. J. Biol. Chem. 2016, 291, 14468–14482. 10.1074/jbc.M115.696781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N.; Yoshimori T.; Levine B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh K. K.; Eskelinen E. L.; Scott C. C.; Malevanets A.; Saftig P.; Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007, 26, 313–324. 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P.; Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- Ma L.; Chen Z.; Erdjument-Bromage H.; Tempst P.; Pandolfi P. P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Paquette M.; El-Houjeiri L.; Pause A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18 10.3390/cancers10010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y.; Inoki K. The role of mechanistic target of rapamycin in maintenance of glomerular epithelial cells. Curr. Opin. Nephrol. Hypertens. 2016, 25, 28–34. 10.1097/MNH.0000000000000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goard C. A.; Chan-Seng-Yue M.; Mullen P. J.; Quiroga A. D.; Wasylishen A. R.; Clendening J. W.; Sendorek D. H.; Haider S.; Lehner R.; Boutros P. C.; Penn L. Z. Identifying molecular features that distinguish fluvastatin-sensitive breast tumor cells. Breast Cancer Res. Treat. 2014, 143, 301–312. 10.1007/s10549-013-2800-y. [DOI] [PubMed] [Google Scholar]
- Ruiz-Limon P.; Barbarroja N.; Perez-Sanchez C.; Aguirre M. A.; Bertolaccini M. L.; Khamashta M. A.; Rodriguez-Ariza A.; Almaden Y.; Segui P.; Khraiwesh H.; Gonzalez-Reyes J. A.; Villalba J. M.; Collantes-Estevez E.; Cuadrado M. J.; Lopez-Pedrera C. Atherosclerosis and cardiovascular disease in systemic lupus erythematosus: effects of in vivo statin treatment. Ann. Rheum. Dis. 2015, 74, 1450–1458. 10.1136/annrheumdis-2013-204351. [DOI] [PubMed] [Google Scholar]
- Garwood E. R.; Kumar A. S.; Baehner F. L.; Moore D. H.; Au A.; Hylton N.; Flowers C. I.; Garber J.; Lesnikoski B. A.; Hwang E. S.; Olopade O.; Port E. R.; Campbell M.; Esserman L. J. Fluvastatin reduces proliferation and increases apoptosis in women with high grade breast cancer. Breast Cancer Res. Treat. 2010, 119, 137–144. 10.1007/s10549-009-0507-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelaziz D. H.; Khalil H.; Cormet-Boyaka E.; Amer A. O. The cooperation between the autophagy machinery and the inflammasome to implement an appropriate innate immune response: do they regulate each other?. Immunol. Rev. 2015, 265, 194–204. 10.1111/imr.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C.; Klionsky D. J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elimam H.; Papillon J.; Guillemette J.; Navarro-Betancourt J. R.; Cybulsky A. V. Genetic Ablation of Calcium-independent Phospholipase A2gamma Exacerbates Glomerular Injury in Adriamycin Nephrosis in Mice. Sci. Rep. 2019, 9, 16229 10.1038/s41598-019-52834-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z.; Zang M.; Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 2010, 6, 457–470. 10.2217/fon.09.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn D. M.; Shackelford D. B.; Egan D. F.; Mihaylova M. M.; Mery A.; Vasquez D. S.; Turk B. E.; Shaw R. J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008, 30, 214–226. 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X. B.; Liu G.; Liu F.; Yan Z. G.; Wang Z. Y.; Liu Z. P.; Wang L. Autophagy blockade and lysosomal membrane permeabilization contribute to lead-induced nephrotoxicity in primary rat proximal tubular cells. Cell Death Dis. 2017, 8, e2863 10.1038/cddis.2017.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K.; Li Y.; Wang S.; Wang X.; Liao C.; Hu X.; Fan L.; Kang Q.; Zeng Y.; Wu X.; Wu H.; Zhang J.; Wang Y.; He Z. Inhibition of autophagosome-lysosome fusion by ginsenoside Ro via the ESR2-NCF1-ROS pathway sensitizes esophageal cancer cells to 5-fluorouracil-induced cell death via the CHEK1-mediated DNA damage checkpoint. Autophagy 2016, 12, 1593–1613. 10.1080/15548627.2016.1192751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X.; Cao C.; Li J.; Chen F.; Zhang S.; Liu B.; Zhang W.; Zhang X.; Ye L. Inflammatory factor TNF-alpha promotes the growth of breast cancer via the positive feedback loop of TNFR1/NF-kappaB (and/or p38)/p-STAT3/HBXIP/TNFR1. Oncotarget 2017, 8, 58338–58352. 10.18632/oncotarget.16873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.; Huang J.; Zhong H.; Shen N.; Faggioni R.; Fung M.; Yao Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 2014, 141, 125–139. 10.1016/j.pharmthera.2013.09.004. [DOI] [PubMed] [Google Scholar]
- Kimura S.; Noda T.; Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Ahmed T. A.; El-Say K. M.; Hosny K. M.; Aljaeid B. M. Development of optimized self-nanoemulsifying lyophilized tablets (SNELTs) to improve finasteride clinical pharmacokinetic behavior. Drug Dev. Ind. Pharm. 2018, 44, 652–661. 10.1080/03639045.2017.1405977. [DOI] [PubMed] [Google Scholar]
- El-Say K. M.; Ahmed T. A.; Ahmed O. A. A.; Hosny K. M.; Abd-Allah F. I. Self-Nanoemulsifying Lyophilized Tablets for Flash Oral Transmucosal Delivery of Vitamin K: Development and Clinical Evaluation. J. Pharm. Sci. 2017, 106, 2447–2456. 10.1016/j.xphs.2017.01.001. [DOI] [PubMed] [Google Scholar]
- Sundararajan R.; Salameh T.; Camarillo I. G.; Prabu R. R.; Natarajan A.; Sankaranarayanan K.. Irreversible Electroporation: a Drug-Free Cancer Treatment. In Electroporation-Based Therapies for Cancer; Elsevier, 2014; pp. 219–243. [Google Scholar]
- Khalil H.; Abd El Maksoud A. I.; Alian A.; El-Hamady W. A.; Daif A. A.; Awad A. M.; Guirgis A. A. Interruption of Autophagosome Formation in Cardiovascular Disease, an Evidence for Protective Response of Autophagy. Immunol. Invest. 2020, 49, 249–263. 10.1080/08820139.2019.1635619. [DOI] [PubMed] [Google Scholar]
- Luhovy A. Y.; Jaberi A.; Papillon J.; Guillemette J.; Cybulsky A. V. Regulation of the Ste20-like kinase, SLK: involvement of activation segment phosphorylation. J. Biol. Chem. 2012, 287, 5446–5458. 10.1074/jbc.M111.302018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil H.; Arfa M.; El-Masrey S.; El-Sherbini S. M.; Abd-Elaziz A. A. Single nucleotide polymorphisms of interleukins associated with hepatitis C virus infection in Egypt. J. Infect. Dev. Countries 2017, 11, 261–268. 10.3855/jidc.8127. [DOI] [PubMed] [Google Scholar]
- Schmittgen T. D.; Jiang J.; Liu Q.; Yang L. A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Res. 2004, 32, e43 10.1093/nar/gnh040. [DOI] [PMC free article] [PubMed] [Google Scholar]