

Abstract
The electrophoretic deposition process (EPD) was utilized to produce bioactive hydroxyapatite/chitosan (HAP/CS) and hydroxyapatite/chitosan/gentamicin (HAP/CS/Gent) coatings on titanium. The bioactivity of newly synthesized composite coatings was investigated in the simulated body fluid (SBF) and examined by X-ray diffraction, Fourier transform infrared spectroscopy, and field emission scanning electron microscopy. The obtained results revealed carbonate-substituted hydroxyapatite after immersion in SBF, emphasizing the similarity of the biomimetically grown HAP with the naturally occurring apatite in the bone. The formation of biomimetic HAP was confirmed by electrochemical impedance spectroscopy and polarization measurements, through the decrease in corrosion current density and coating capacitance values after 28-day immersion in SBF. The osseointegration ability was further validated by measuring the alkaline phosphatase activity (ALP) indicating the favorable osseopromotive properties of deposited coatings (significant increase in ALP levels for both HAP/CS (3.206 U mL–1) and HAP/CS/Gent (4.039 U mL–1) coatings, compared to the control (0.900 U mL–1)). Drug-release kinetics was investigated in deionized water at 37 °C by high-performance liquid chromatography coupled with mass spectrometry. Release profiles revealed the beneficial “burst-release effect” (∼21% of gentamicin released in the first 48 h) as a potentially promising solution against the biofilm formation in the initial period. When tested against human and mice fibroblast cells (MRC-5 and L929), both composite coatings showed a noncytotoxic effect (viability >85%), providing a promising basis for further medical application trials.
1. Introduction
Orthopedics is nowadays turning to tissue engineering and its tools to develop solutions for serious tissue damage and extensive trauma of the bone tissue. The main requirement in the bone implants field is to develop a functional, extended-life biomaterial that should possess excellent biocompatibility, good mechanical properties, corrosion resistance, antibacterial activity, and bioactivity.1 Orthopedic implants are expected to induce bonding with surrounding bone tissue, without causing harmful effects to the host body.2 Considering the mechanical and load-bearing requirements that orthopedic implants have to endure, metals and metal alloys are a primary choice due to the excellent combination of stiffness, toughness, and strength. However, metallic implants are usually prone to corrosion, which in some cases can cause adverse effects on the host organism.3 Moreover, the lack of the osseointegration process when metallic implants are used limits their application. A promising approach for developing improved medical devices, orthopedic implants in particular, could be applying thin bioactive films on the metal surface.4 Orthopedic implants designed in such a manner can overcome the corrosion issues by creating biocompatible, bioactive layers that would enhance implant bonding with damaged bone.5,6 Due to the excellent mechanical properties and high corrosion resistance, titanium is often the metal of choice even though it lacks osseopromotive properties.7 Metal surface modification by applying composite coatings could overcome the problem, enhancing the biocompatibility at the same time. The electrophoretic deposition process (EPD) is a convenient technique that allows obtaining high-purity composite coatings with controlled topography and morphology at room temperature.8 The production of biocompatible materials including drugs and/or biologically active molecules, maintaining the overall composite safety and nontoxicity for medical use, is the main advantage of the EPD technique. The capability of processing drugs and therapeutic agents at room temperature singled out EPD as a prospective technique for obtaining biomaterials, especially due to drug sensitivity to high temperatures.9,10
Due to its similarity to the natural bone and ability to promote osteoblast adhesion, i.e., formation of a new bone, synthetic hydroxyapatite (HAP) could contribute to the bone implant’s bioactivity improvement.11−13 Certain in vivo experiments confirmed improved adhesion and differentiation of osteoprogenitor cells and the growth of a new bone, when porous synthetic HAP was used.14 In combination with natural polymers, it enables the production of improved composite materials aimed for bone implants. Chitosan (CS) is a natural, biodegradable polysaccharide possessing intrinsic antibacterial activity, good adhesion properties, and high potential as a drug carrier. Its role as a safe, reliable biomaterial has been recognized by the Food and Drug Administration (FDA), which approved its use in tissue engineering and drug delivery.15,16 Moreover, CS exhibits unique polycationic nature that enables its processing using the electrophoretic deposition process. Since composite HAP/CS and HAP/CS/gentamicin (Gent) coatings are intended for use in orthopedic implants, it is crucial to mimic the structure of natural bone (HAP and collagen). Among different polymers used in the biomaterials field, chitosan is singled out as the best solution due to its ability to induce cell adhesion, differentiation, and proliferation supporting tissue repair.17 It has been demonstrated that in in vivo conditions, some proteases (mostly lysozyme) are responsible for CS degradation and formation of nonharmful oligosaccharides, which can later be excreted from the organism.18 Chitosan was included as a component of the composite coatings to improve several crucial properties of HAP, including enhanced adhesion of the coating to the titanium surface, as well as decreased brittleness and improved gentamicin binding and drug-delivery properties. Further, chitosan also possesses well-documented intrinsic antibacterial activity18 that could help in maintaining the sterility of the implant even beyond the period of active antibiotic release. Chitosan has also been shown to increase the success rate of electrophoretic deposition of hydroxyapatite coatings19 and has been demonstrated to significantly influence the release of vancomycin and gentamicin from composite chitosan/bioactive glass coatings.20,21 Patel et al.22 also pointed to the efficacy of chitosan as a drug carrier that enables slow and sustained antibiotic ampicillin release from the composite chitosan–bioglass coating, which is highly opportune for intended biomedical use. Moreover, the polar groups (−OH and −NH2) carried by chitosan enable secondary interactions with other components of the composite and, at the same time, improve the hydrophilic properties.23 When used in the composite combined with HAP, CS contributes to the improvement of adhesive and antibacterial properties, serving as an organic matrix for the hydroxyapatite and antibiotic incorporation.24 The high risk of bacterial infection after orthopedic surgeries often demands antibiotic administration (locally and systemically).25,26 Particularly, the attachment of bacteria to the implant surface and biofilm formation are the most sensitive issues that have to be addressed. Once formed, the biofilm is difficult to eradicate and it can affect bacterial resistance to antibiotic therapy.27 Various studies verified gentamicin as a powerful antibacterial agent for the treatment of bone infections, due to its well-known antibacterial activity.28,29 Gentamicin is an aminoglycoside, water-soluble antibiotic that is commonly used in both prophylaxis and existing orthopedic bacterial infections.30 It is particularly relevant for future applications to use drugs such as Gent that are thermally stable since coated implants should be kept sterile till the implantation surgery. When designed as part of the orthopedic device, gentamicin could improve host protection in the first days of recovery, preventing bacterial adhesion and ensuring prolonged release. Gentamicin was already commercially used for local treatment of orthopedic bacterial infection, as a part of gentamicin-loaded poly(methyl methacrylate) (PMMA) beads. However, there is a necessity for a second surgery after 2 weeks since PMMA beads are not biodegradable and can become a fertile ground for bacteria adherence, developing biofilm.31
The host response to the biomaterial presence is usually unpredictable, and it depends on various factors, mainly surface properties and the ability to induce the osseointegration process.32 In vitro bioactivity of orthopedic implant materials is usually investigated in simulated body fluid (SBF), due to its similarity with human blood plasma. This method facilitates stronger bonding to the affected bone, i.e., bioactivity through the formation of a new apatite layer in the SBF solution.33
The HAP/CS/Gent coating deposition was carried out in a single step, i.e., by co-electrodeposition from a three-component aqueous suspension.26 This achievement represents a significant improvement over the methods available thus far in the field of biomedical implants with antibiotic-loaded bioactive coatings, which are usually produced in several steps21,34,35 since the drug is incorporated within the coating rather than on the coating surface.
This paper deals with the bioactivity and cytotoxicity of electrophoretically deposited HAP/CS and HAP/CS/Gent composite coatings on Ti substrate and drug-release profiles modeling.
2. Results and Discussion
2.1. Formation of Biomimetic HAP on the HAP/CS and HAP/CS/Gent Coatings during Immersion in SBF
In vitro bioactivity of composite HAP/CS and HAP/CS/Gent coatings was investigated by immersion in 20 mL of an SBF solution at 37 °C. The concentrations of ions in the SBF solution obtained after dissolution of reagent-grade salts were as follows: Na+ (142.0 mM), K+ (5.0 mM), Mg2+ (1.5 mM), Ca2+ (2.5 mM), HCO3– (4.2 mM), Cl– (147.8 mM), HPO42– (1.0 mM), and SO42– (0.5 mM). The final solution was buffered in Tris and pH-adjusted to 7.40 with 1.0 M HCl. After exposure to the SBF solution, the coatings were taken out, dried at room temperature, and characterized by X-ray diffraction (XRD), Fourier transform infrared (FT-IR) spectroscopy, and field emission scanning electron microscopy (FE-SEM).
2.1.1. XRD
Figure 1 shows the XRD patterns for biomimetic HAP obtained on HAP/CS (Figure 1a) and HAP/CS/Gent (Figure 1b) coatings after soaking in SBF at 37 °C for 7 days.
Figure 1.

XRD patterns for biomimetic HAP obtained on (a) HAP/CS and (b) HAP/CS/Gent coatings after 7-day immersion in SBF at 37 °C.
Diffraction maxima of biomimetic coating, obtained on the top of both HAP/CS and HAP/CS/Gent coatings after soaking in SBF, were assigned to HAP (JCPDS 09-0432) and the underlying titanium substrate (JCPDS 89-2762). The HAP diffraction maxima broadening suggested fine crystallite size of the biomimetic coatings. The differences in the diffraction patterns of biomimetic HAP grown on the HAP/CS (Figure 1a) and HAP/CS/Gent (Figure 1b) coatings at the 2θ values of 30–40° can be explained by the differences between the as-deposited HAP/CS and HAP/CS/Gent coatings, i.e., before soaking in SBF, that were caused by different mechanisms of their formation, as discussed in detail in our previous paper.26 Briefly, at a suspension pH of 4.4 for both coatings deposition, primary amino groups of chitosan become protonated (CS–NH3+). At the same time, HAP acquires a positive charge due to the protonation of hydroxyl groups. During EPD, protonated CS and HAP molecules migrate toward the cathode (Ti) and react with OH– ions evolved in the water electrolysis reaction, thus forming insoluble HAP/CS deposit on the cathode. Additionally, hydrogen bonding occurs between the amino and hydroxyl groups from CS with hydroxyl groups from HAP. In the case of HAP/CS/Gent coating, gentamicin molecules are also positively charged due to protonation of hydroxyl and amino groups, enabling the reaction of positively charged CS, HAP, and Gent molecules with OH– ions at the cathode, as explained above, thus forming an insoluble HAP/CS/Gent deposit on the cathode surface. The amino and hydroxyl groups in chitosan and gentamicin along with hydroxyl groups in HAP can form more hydrogen bonds compared to HAP/CS coating.
The calculated diffraction data including crystallite domain size, cell volume, V, and unit cell parameters, a and c, as well as d-spacing for HAP (002), (211), (112), and (300) planes, are presented in Table 1. A slightly smaller crystallite domain size of biomimetic HAP on the HAP/CS/Gent coating (374 Å), compared to biomimetic HAP on the HAP/CS coating (387 Å), could be the consequence of a smaller HAP/CS/Gent coating crystallite domain size before soaking (397 Å), with respect to HAP/CS coating before soaking (511 Å), as reported in our previous paper.26
Table 1. Calculated Values of d-Spacing, Unit Cell Parameters (a and c), Cell Volume (V), and Crystallite Domain Size for Biomimetic HAP Formed on the HAP/CS and HAP/CS/Gent Coatings after Immersion in SBF for 7 days.
| coating | HAP/CS | HAP/CS/Gent | ||
|---|---|---|---|---|
| crystal planes | (002) | d-spacing (Å) | 3.4247 | 3.4122 |
| (211) | 2.8046 | 2.8125 | ||
| (112) | 2.7642 | 2.7878 | ||
| (300) | 2.7379 | 2.7504 | ||
| parameters | a (Å) | 9.440 | 9.491 | |
| c (Å) | 6.881 | 6.820 | ||
| V (Å3) | 531.1 | 532.1 | ||
| crystallite domain size (Å) | 387 | 374 | ||
According to the presented results of unit cell parameters, the biomimetic HAP (grown on both HAP/CS and HAP/CS/Gent coatings) exhibited a slight a-axis increase (9.440 and 9.491 Å, respectively) compared to the theoretically reported value of 9.418 Å for HAP hexagonal crystal structure with a P63/m space group.36 On the other hand, a slight c-axis decrease (6.881 and 6.820 Å, respectively, compared to the literature value of 6.884 Å36) suggested carbonate group incorporation in the hydroxyapatite structure. Hydroxyl- and phosphate-ion sites in the biological apatites can be substituted by various anions. When the OH– or PO43– sites become occupied by carbonate ions, A- or B-type carbonated hydroxyapatites, respectively, can be achieved.37 Replacing the linear OH– or tetrahedral PO43– ions with planar CO32– ions causes changes in unit cell parameters (expansion or contraction of a or c-axis).38 Biological apatites, e.g., bone mineral, usually belong to the so-called AB-type mineral, meaning that simultaneous carbonate substitution of OH– and PO43– sites occurs, while greater changes in lattice parameters can point to a higher carbonate substitution degree in the hydroxyapatite structure.39 The changes in unit cell parameters (Table 1) suggested that biomimetically grown HAP on the top of HAP/CS and HAP/CS/Gent coatings is in fact carbonate-substituted hydroxyapatite. Greater changes in a and c parameters with respect to the theoretical ones36 were observed in the case of biomimetic HAP on HAP/CS/Gent coating, suggesting that more OH– and PO43– ions were substituted by CO32–, compared to biomimetic HAP on HAP/CS coating.
Considering that apatites with a Ca/P ratio lower than 1.67 are usually considered as carbonate-substituted,40,41 energy-dispersive spectrometry (EDS) analysis (data not shown) additionally proved that biomimetic HAP grown on the top of both HAP/CS and HAP/CS/Gent coatings is carbonate-substituted HAP since the Ca/P ratios were calculated to be 1.64 and 1.46, respectively. The obtained results are encouraging since substituted HAP is known to improve the bioactivity and osteoconductivity and increase adhesion, growth, and differentiation of osteoblast cells compared to the HAP that retains the stoichiometric ratio.42,43
2.1.2. FT-IR Spectroscopy
Figure 2 shows the FT-IR spectrum for biomimetic HAP obtained on HAP/CS and HAP/CS/Gent (Figure 2a) coatings after soaking in SBF at 37 °C for 7 days, as well as the details of carbonate band positions (Figure 2b).
Figure 2.
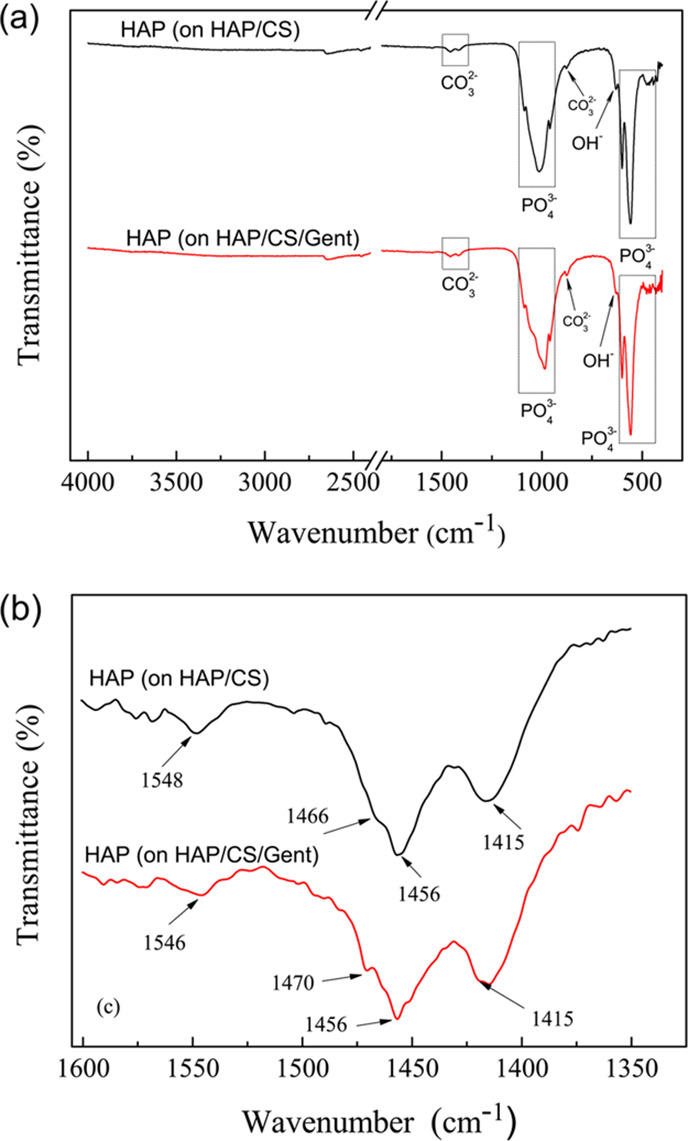
FT-IR spectra of biomimetic HAP obtained on (a) HAP/CS and HAP/CS/Gent coatings on titanium after 7-day immersion in SBF at 37 °C, and (b) details of carbonate band positions.
Intense bands in the 960–1200 cm–1 region (i.e., 960, 1014, and 1087 cm–1 for HAP/CS coating, and 959, 985, and 1087 cm–1 for HAP/CS/Gent coating) were assigned to (P–O) stretching vibrations of the PO43– group (Figure 2a). The phosphate group band position shifts can be explained by the deviation of phosphate ions from their ideal tetrahedral structure.44 The O–P–O bending modes were detected as bands at 558 and 600 cm–1 for both coatings, confirming the formation of calcium phosphate phases.44,45 The structural OH– groups in HAP gave rise to a band at around 630 cm–1 in the spectra of both coatings.46,47
Usually, in the HAP structure, a sharp band at about 3570 cm–1 can be detected and assigned to the OH– stretching from the HAP structure.26,46 However, this band was absent from the FT-IR spectra for both HAP coatings (Figure 2a), suggesting the existence of substituted, i.e., biomimetic HAP, occurring through the carbonate groups occupying the OH– sites. The biomimetic nature of HAP grown on both coatings was thus confirmed, as this particular band is also usually absent from the natural bone apatite spectrum.39
There are also several bands whose positions indicated the AB-type nature of carbonate substitution in biomimetic HAP.26,48−50 Various vibrational modes of the CO32– group were detected in the FT-IR spectra of both coatings (Figure 2a,b): 878 cm–1 (O–C–O), 1415 and 1456 cm–1 (O–C), “shoulder” at 1466 cm–1 (HAP/CS) and ∼1470 cm–1 (HAP/CS/Gent), and finally ∼1546 cm–1 (HAP/CS) and ∼1548 cm–1 (HAP/CS/Gent). These carbonate band locations suggested that the structure of biomimetically obtained HAP was similar to the structure of all biological apatites.48
For both biomimetic coatings, XRD and EDS results are in accordance with the FT-IR analysis, confirming that biomimetic HAP corresponds to carbonate-substituted hydroxyapatite.
2.1.3. FE-SEM
The FE-SEM microphotographs of bare Ti plate, HAP/CS, and HAP/CS/Gent coatings on Ti before and after 7-day soaking in SBF at 37 °C are presented in Figure 3. It is important to point out that the gentamicin encapsulation did not change the coating’s morphology—spherical agglomerates of different sizes were observed at the surfaces of both coatings (Figure 3a,3b). After 7-day immersion in SBF, spherical agglomerates of biomimetic HAP were observed on the HAP/CS coating surface (Figure 3c), whereas nonspherical particles appeared in the case of biomimetic HAP on the HAP/CS/Gent coating (Figure 3d). Besides changes in the shape of the newly formed particles, the size of the agglomerates of biomimetic HAP shrank in the case of drug-loaded coatings (HAP/CS/Gent) with respect to the coating without gentamicin (HAP/CS). This morphology change can be attributed to the presence of gentamicin, encapsulated inside the chitosan/hydroxyapatite coating, confirming the successful incorporation of the antibiotic during deposition. Namely, during the immersion in SBF, water penetrates into the coating and dissolves the drug. In this initial drug dissolution period, a pronounced burst-release effect was observed in the first 48 h, which will be discussed in detail in Section 2.2. As a consequence of drug dissolution, change of coating topography occurs, leading to precipitation of nonspherical hydroxyapatite agglomerates.
Figure 3.

SEM microphotographs of (a, c) HAP/CS and (b, d) HAP/CS/Gent coatings on titanium (a, b) before and (c, d) after soaking in SBF at 37 °C. Inset in (a): bare Ti.
2.1.4. Electrochemical Properties
Electrochemical measurements were also performed to investigate the bioactivity of HAP/CS and HAP/CS/Gent coatings and the growth of a new apatite phase during exposure to the SBF at 37 °C. Titanium is generally not very prone to corrosion, due to the thermodynamically favorable formation of a stable oxide layer on the metal surface in a very wide range of potentials and pH values.51 This oxide layer is passive and serves to protect the metal underneath from corrosion. However, electrochemical investigation of corrosion parameters for coated titanium samples can provide insight into the bioactivity of the coating itself.47,52 Different electrochemical tests were conducted to elucidate the apatite growth over a prolonged period of exposure to SBF as a biomineralization-inducing environment.
2.1.4.1. Open-Circuit Potential
Figure 4 shows the time dependence of the open-circuit potential (Eocp) for both coated samples during the investigated 28-day period of immersion in SBF at 37 °C. The initial values were negative for both samples (−80 mV for HAP/CS and −23 mV for HAP/CS/Gent), but the Eocp values quickly became more positive during exposure to the SBF solution. The Eocp increased during the first 5 days and reached almost constant values in the latter time period. This could indicate rapid apatite growth upon initial exposure to SBF, after which the newly formed apatite layer remained virtually unchanged until the end of the 28-day investigation. This apatite layer formation was further investigated by electrochemical impedance spectroscopy (EIS) and potentiodynamic sweep (PDS) measurements.
Figure 4.
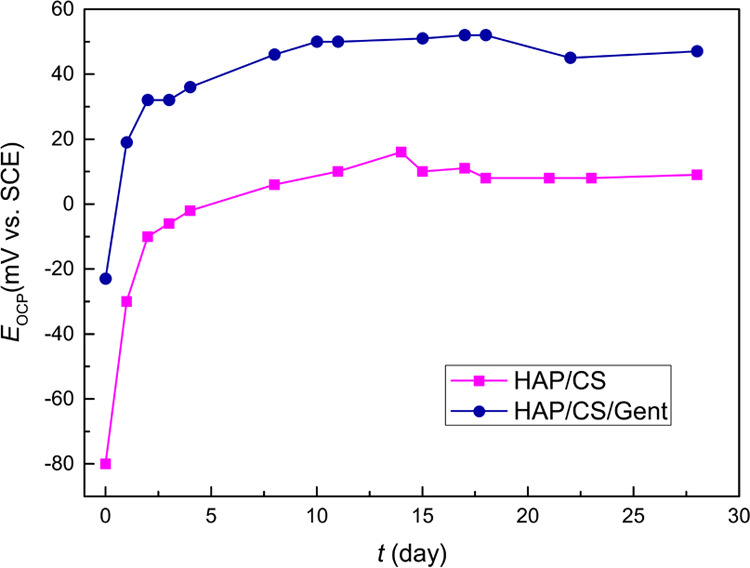
Time dependence of open-circuit potentials (Eocp) for HAP/CS and HAP/CS/Gent coatings during 28-day immersion in SBF at 37 °C.
2.1.4.2. EIS
EIS spectra for both coatings after different immersion periods (up to 28 days) in SBF at 37 °C are shown in Figure 5. Bode modulus plots (Figure 5a,b) indicated increased overall impedance for both HAP/CS and HAP/CS/Gent coatings that confirmed the formation and growth of a new apatite layer during immersion in SBF. The largest impedance increase was observed between initial immersion and 4 days, suggesting that the fastest apatite formation occurred upon initial contact with SBF, as also noted from Eocp measurements (Figure 4). Theoretically, the biomineralization process is a reversible one, where the dissolution of the coating and apatite precipitation occur simultaneously and the dominant reaction depends on Ca2+- and PO43–-ions concentrations.53 However, the mechanisms of dissolution and precipitation are different—ion exchange governs the dissolution process, while the concentration gradient and particle solubility influence the precipitation.53,54 The quick impedance increase during the first 4 days indicated that the predominant reaction was precipitation of calcium phosphate and the formation of a new HAP layer. Further apatite growth could slow down upon reaching equilibrium since the impedance increase was much slower between days 11 and 28 (Figure 5a,b).
Figure 5.

EIS Bode diagrams for (a) HAP/CS and (b) HAP/CS/Gent coatings during 28-day immersion in SBF at 37 °C and (c) the equivalent electrical circuit (EEC) used for EIS spectra fitting.
With the aim to investigate the electrochemical behavior of the samples, impedance spectra were fitted with an equivalent electrical circuit (EEC) presented in Figure 5c. The EEC contained ohmic resistance (RΩ), coupled in series with a parallel RC circuit, depicting the coating behavior (with CPEc—coating capacitance and Rp—coating pore resistance). The constant phase element (CPE) was used instead of a pure capacitor to account for deviations from ideal dielectric behavior. Another parallel RC circuit was coupled in series with Rp and contained double-layer capacitance (CPEdl) and charge-transfer resistance (Rct). This second time constant described charge-transfer processes and electrochemical reactions on the metal/electrolyte interface and was present due to the high porosity of the coating, which enabled the electrolyte to enter the pores and reach the metal underneath.
The impedance of a CPE element (ZCPE) can be expressed using eq 1,55 where Y0–1 is the admittance, ω is the angular frequency, α is a parameter that quantifies the deviation from an ideal capacitor, and i2 = −1. The capacitance value can be calculated using eq 2, where ωmax is the angular frequency at which the imaginary component of the impedance reaches its maximum.55
| 1 |
| 2 |
It is obvious that for α values close to unity, CCPE = Y0. As in our case α was always higher than 0.8, the Y0 values obtained from the fit were used as the coating and double-layer capacitances without further calculations. The goodness-of-fit values (χ2) were always in the range of 10–6–10–4.
The Cc and Cdl values over the investigated immersion period are presented in Figure 6. Both Cc and Cdl exhibited decreasing trends over the investigated 28-day period, which is in line with improving the anticorrosion performance of the metal/coating system. The lower Cc values indicated the thicker coating layer obtained due to the biomimetic HAP precipitation that obtruded the coating pores and prevented electrolyte penetration. The initial behavior of log Cc curves (Figure 6a) is in line with Eocp measurements (Figure 4) as well as Bode plots (Figure 5), showing the most significant changes between days 0 and 4, followed by a period of relative stagnation. This would indicate that the crucial period in the formation of a new apatite layer is the one upon short-term initial exposure to the biomineralization-inducing environment. The Cdl values (Figure 6b) also decreased following a similar trend. It is known that the double-layer capacitance value is proportional to the surface area of the metal directly exposed to the electrolyte. Therefore, the decrease in the Cdl values correlates with the decreased area of the metal/electrolyte interface, either through the new apatite layer deposition or due to the formation of a thin passive titanium oxide layer on the metal surface. Regarding the coating pore and charge-transfer resistances, similar behavior was observed. The Rp values increased for both coatings after 4 days (27.8 kΩ cm2 for HAP/CS and 33.1 kΩ cm2 for HAP/CS/Gent), compared to initial immersion (17.6 kΩ cm2 for HAP/CS and 25.9 kΩ cm2 for HAP/CS/Gent), indicating the coating thickness increase due to the precipitation of the newly formed HAP layer. After this initial 4-day phase, further increase was slower, reaching 38.7 kΩ cm2 for HAP/CS and 32.4 kΩ cm2 for HAP/CS/Gent after 28 days. As discussed above, HAP dissolution and precipitation are parallel processes and their simultaneous occurrence was expected during exposure to SBF. The coatings with smaller crystallites are more prone to dissolution as the specific contact area with the solution is higher in this case.53 Thus, the smaller Rp values of HAP/CS/Gent coating could be attributed to the smaller crystallite domain size before soaking, as determined from XRD (397 Å for HAP/CS/Gent compared to 511 Å for HAP/CS).26 Charge-transfer resistances, Rct, did not change significantly upon initial contact between 0 and 4 days (0.087–0.12 GΩ cm2 for HAP/CS and 2.97–2.68 GΩ cm2 for HAP/CS/Gent) as the electrolyte made contact with the metal through coating pores, suggesting that a thin oxide layer was already present on the metal surface that prevented direct contact of bare metal and electrolyte. Further, Rct increased toward the end of the monitored period (1.24 GΩ cm2 for HAP/CS and 13.1 GΩ cm2 for HAP/CS/Gent), indicating passivation of the metal surface hindering the corrosion process. Moreover, the Rct values were always in the order of magnitude of 107–109 Ω cm2, suggesting low overall proneness of Ti to corrosion in SBF.
Figure 6.

Time dependence of (a) coating capacitance (Cc) and (b) double-layer capacitance (Cdl) for HAP/CS and HAP/CS/Gent coatings, obtained from EIS measurements during 28-day exposure to SBF at 37 °C.
2.1.4.3. PDS
Polarization measurements were conducted to evaluate the bioactivity of HAP/CS and HAP/CS/Gent coatings, after predetermined periods (0, 7, and 28 days) of exposure to SBF. Generally, PDS measurements provide information about corrosion parameters, but these could also be used to evaluate the coating behavior regarding its biomineralization. The polarization curves shown in Figure 7 indicated increasing corrosion stability with prolonged exposure to SBF for both samples, which suggested improved coating properties due to biomimetic HAP formation, as already discussed in Section 2.1.4.2. Corrosion potentials (Ecorr) were obtained from PDS curves, and corrosion current densities (jcorr) were calculated by extrapolating the cathodic curves, which were linear over at least a decade of current, to Ecorr. The obtained data are presented in Table 2. A steady decrease in jcorr values was observed for both samples confirming the improved corrosion resistance as a consequence of the newly formed apatite layer. Cathodic Tafel slopes suggested a multistep mechanism of the cathodic reaction, whereas the anodic ones could not be calculated at all due to the changing nature of anodic reactions.
Figure 7.

Potentiodynamic polarization curves of (a) HAP/CS and (b) HAP/CS/Gent coatings during 28-day immersion in SBF at 37 °C.
Table 2. PDS Parametersa.
| Ecorr (mV vs SCE) | jcorr (μA cm–2) | bc (mV dec–1) | ||
|---|---|---|---|---|
| day 0 | HAP/CS | –145 | 0.061 | –220 |
| HAP/CS/Gent | –238 | 0.52 | –220 | |
| day 7 | HAP/CS | –351 | 0.012 | –150 |
| HAP/CS/Gent | –365 | 0.035 | –140 | |
| day 28 | HAP/CS | –258 | 0.011 | –180 |
| HAP/CS/Gent | –128 | 0.017 | –200 | |
Corrosion potentials, corrosion current densities, and cathodic Tafel slopes for HAP/CS and HAP/CS/Gent coatings during exposure to SBF at 37 °C.
The PDS results agreed well with EIS data, suggesting bioactivity of both HAP/CS and HAP/CS/Gent coatings and growth of biomimetic hydroxyapatite layer that should greatly improve the osseointegration process of hard tissue implants.
2.2. Gentamicin-Release Study
Local drug administration offers a lot of advantages in comparison to conventional drug therapies. Such a drug-delivery route offers the ability to lower the administered dose while achieving a satisfactory therapeutic effect. When designing drug-eluting orthopedic devices, a lot of aspects have to be considered, such as coating’s composition, thickness, and porosity and the medium in which the drug should be released. Moreover, the released antibiotic concentration should be equal to or higher than the minimum inhibitory concentration (MIC) so as not to cause bacterial resistance and, in turn, should not be too high to cause toxic effects on the human cells.
Both the amount of loaded gentamicin in the composite coating and the corresponding release profile were obtained by high-performance liquid chromatography coupled with mass spectrometry (HPLC-MS). The total gentamicin loading was determined by scraping off the coating and dissolving it in slightly acidified dH2O, before measuring the amount of Gent in the HPLC-MS instrument. Thus, the measured amount of gentamicin loaded on 1 cm2 of the HAP/CS/Gent composite coating was 9.4 μg. Gentamicin release from composite HAP/CS/Gent coating was studied during 21-day immersion in deionized water. The cumulative release profile for HAP/CS/Gent coating in predetermined periods of time (24 h, 48 h, 7 days, 14 days, and 21 days) is represented in Figure 8 as an average of three samples. Gentamicin-release profile verified the initial burst-release effect of gentamicin from the composite coating, i.e., more than 50% of the loaded antibiotic was released within 7 days. A pronounced burst-release effect (∼21%) was also observed in the first 48 h, which could be very useful in preventing biofilm formation. This initial 7-day period was followed by a slower release pattern, where only 13% of the drug was released until day 21. Considering the gentamicin-release profiles, the obtained composite coatings could have potential use as prolonged drug-delivery systems for treating orthopedic infections.
Figure 8.
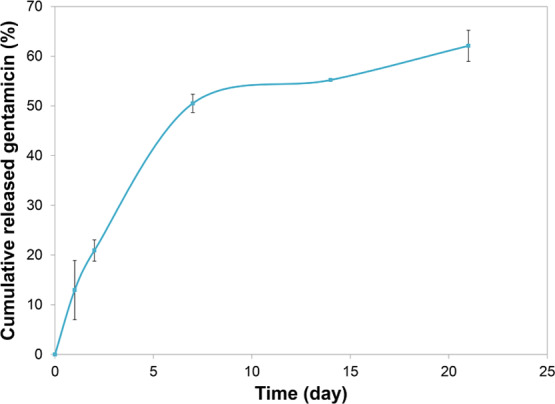
Cumulative gentamicin release from HAP/CS/Gent coating during 21 days in deionized water at 37 °C.
To investigate the governing mechanisms and diffusion coefficient of release, experimental data fitting was performed.
The first model used was the Korsmeyer–Peppas model,56 described by eq 3, where mt represents the mass of gentamicin released at time t, mtot represents the initial (total) gentamicin mass in the composite coating before experiment, kKP is the Korsmeyer–Peppas constant, which encompasses the particular carrier system properties, and n is a coefficient that depends on the diffusion type (n ∼ 0.5 for Fickian and n > 0.5 for non-Fickian diffusion) and is supposed to imply the mechanism of mass transport during the release process. The Korsmeyer–Peppas model was applied in its linear form, where the data were transformed to the logarithmic scale (eq 4).56
| 3 |
| 4 |
Thus, we were able to calculate exponent n from the slope of the Korsmeyer–Peppas graph (Figure 9a). A value of 0.528 was obtained for HAP/CS/Gent. Since n was close to 0.5, it was safe to conclude that the mechanism of gentamicin release from HAP/CS/Gent was governed by Fick’s diffusion law.
Figure 9.

Results of gentamicin-release profile fitting for HAP/CS/Gent coating: (a) Korsmeyer–Peppas model and (b) early time approximation (ETA).
As the time exponent from the Korsmeyer–Peppas model was approximately 0.5 (0.528), another model was applied, which assumes the linear correlation of the amount of drug released and the square root of time. This is the early time approximation (ETA) that enables us to calculate the diffusion coefficient of gentamicin during its release from composite coatings (eq 5). According to Ritger and Peppas,57 this model is appropriate for the one-dimensional release from thin films.
| 5 |
The parameters of eq 5 are as follows: mt—the mass of gentamicin released at the time, mtot—the initial (total) gentamicin mass in the composite coating before the experiment, D—the diffusion coefficient of gentamicin, t—time, and δ—the thickness of the coating. ETA is the approximation that refers to the Fickian diffusion model for release from thin polymer/composite samples, and it is valid for the first 60% of release57 (which was exactly achieved in this experiment). The ETA model was applied to the release profile of composite HAP/CS/Gent coating and is shown in Figure 9b. The diffusion coefficient, D, for composite HAP/CS/Gent coating was calculated to be 2.4 × 10–14 cm2 s–1, indicating a slow release from the coating over the course of 21 days. The slow 21-day release could point to good binding of gentamicin to the coating matrix and successful EPD transfer of the antibiotic. These results are also in agreement with gentamicin-release studies, by Pishbin et al.,21 where it was found that ∼70% of gentamicin was released after 56 days from EPD-deposited bioglass coating. This slow and sustained release could be beneficial for the intended application as orthopedic implants, where postoperative infection rate is high and systemic antibiotics administration bears many risks, such as the emergence of resistant bacterial strains. Therefore, the antibiotic-loaded coatings could be a good solution, as they provide quick and targeted drug delivery. The burst release, as seen in the Gent release profile (Figure 8), is required to curb the initial bacterial infection and prevent the formation of biofilm. However, it would not be acceptable if all of the loaded drugs were to be released immediately, as this could cause dose-induced toxicity to the surrounding tissue and there would be no antibiotic left for treatment in the subsequent period. With sustained release up to 21 days, and potentially beyond this period, protection of the implant site against infection is ensured in the critical several-week period after implantation.
These results are particularly interesting compared to the antibacterial activity of HAP/CS/Gent coating, which was evaluated in our previous paper.26 There, we have shown that HAP/CS/Gent coating caused complete eradication (∼5 logarithmic units) against Staphylococcus aureus and a significant reduction (∼2 logarithmic units) against Escherichia coli, indicating potent antibacterial activity in a short period of time, i.e., in the first 24 h of incubation. This effect was particularly strong against S. aureus—a common postoperative implant infection culprit, causing a 100% decrease in viable bacteria number within 3 h,26 which is in line with the observed initial burst-release effect.
2.3. Biological Testing
2.3.1. Dye Exclusion Test (DET)
Trypan blue DET is a commonly used method for estimating the proportion of viable cells.58 Tested cells are simply mixed with a solution of a dye; stained cells are assumed to be damaged, and those unstained are assumed to be undamaged.
The percent of stained/unstained cells or inhibition of growth (% K) was expressed as a percent of the control. The results of the DET test for MRC-5 (human fibroblasts) and L929 (mice fibroblasts) when tested in the presence of HAP/CS and HAP/CS/Gent coatings on Ti are given in Figure 10.
Figure 10.
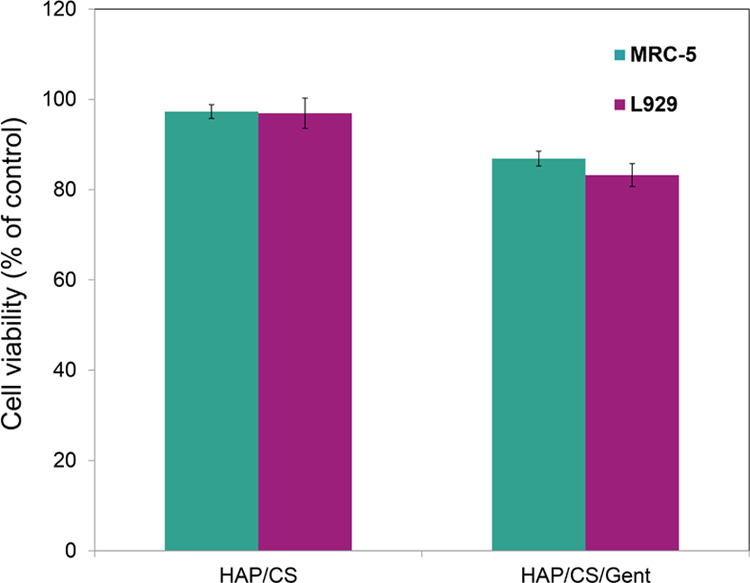
Cell viability (expressed as percent of control) of MRC-5 and L929 cell cultures in the presence of HAP/CS and HAP/CS/Gent coatings.
There was no inhibition of growth in the case of either cell line, MRC-5 and L929, for HAP/CS coating on Ti, since values were ≈100%. Gentamicin release resulted in decreased cell growth in the presence of HAP/CS/Gent-coated Ti samples (Figure 10), but percent viabilities were above 85% for both cell lines. According to the one-way analysis of variance statistical test, the differences in viability of both MRC-5 and L929 cell lines were statistically negligible (p > 0.01) between the two samples when DET testing was considered. Therefore, DET provided strong evidence to support the noncytotoxic effect for both HAP/CS and HAP/CS/Gent coatings, which could be thus considered a safe material for biomedical use.
2.3.2. Alkaline Phosphatase (ALP) Assay
Alkaline phosphatase (ALP) is the most widely recognized biochemical marker for osteoblast activity. Although its precise function is poorly understood, it is believed to play a role in skeletal mineralization. Alkaline phosphatase (ALP, EC 3.1.3.1) catalyzes the hydrolysis of phosphate esters in the alkaline buffer and produces an organic radical and inorganic phosphate. Changes in alkaline phosphatase level and activity are associated with various bone disease states. The differentiation of osteoblast-like cells is important in the healing process, and inducing biomineralization is required for a biomaterial to be considered bioactive. ALP is a membrane-bound biochemical marker of bone turnover that is secreted from osteoblasts. Usually, the ALP assay is performed on osteoblast cells or other cell lines that can differentiate into osteoblasts; however, within the scope of this study, we have decided to test the basic ALP expression potential of the coatings on the nonspecific cell lines first, with a twofold reason in mind: to confirm the basic biomineralization potential of HAP/CS and HAP/CS/Gent coatings before moving on to more tissue-specific cells such as osteoblasts, and to ensure consistency of the two biological assays using the same cell lines (MRC-5 and L929) that were used in the biocompatibility study (i.e., DET cytotoxicity test). Moreover, it was shown that MRC-5 fibroblasts respond to differentiating factors that promote osteoblastic phenotype in bone-derived cell cultures.59 The results of the ALP assay performed on the cell extract showed that HAP/CS/Gent coating had the highest levels of ALP in the cell extract (Figure 11).
Figure 11.
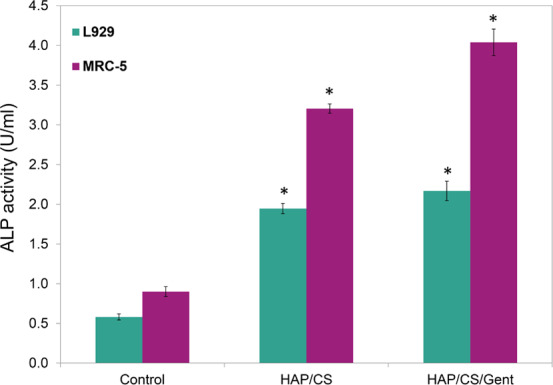
ALP assay results showing the levels of ALP in the MRC-5 and L929 cell extract in the presence of HAP/CS and HAP/CS/Gent coatings.
Both HAP/CS and HAP/CS/Gent coatings had significantly increased ALP activity compared to the control (3 and 4 times increase in ALP expression levels for MRC-5 cell line, respectively). However, HAP/CS/Gent (4.039 U mL–1) coating increased ALP expression levels of MRC-5 cells more than HAP/CS (3.206 U mL–1), suggesting improved biomineralization potential of the gentamicin-loaded coating. Due to cell compatibility, there was no surprise that the MRC-5 cell line overall performed better in this assay. The ALP assay results indicated that both of these materials could induce biomineralization and are therefore strong candidates for further studies with tissue-specific (i.e., osteoblasts) or stem cells, and could be considered as prospective bone implant coatings.
3. Conclusions
Bioactive composite coatings based on HAP and CS, without and with gentamicin (HAP/CS and HAP/CS/Gent, respectively), aimed for orthopedic implant materials were produced using electrophoretic deposition on titanium. Osseopromotive features were investigated in the SBF solution at 37 °C, using FE-SEM, XRD, and FT-IR spectroscopy that confirmed AB-type carbonate-substituted HAP on the top of both investigated coatings, giving insight into the similarity of biomimetically grown HAP with naturally occurring biological apatites and pointing to the highly promising medical applications.
The corrosion current density values obtained from polarization measurements exhibited a decreasing trend over the time of immersion in SBF (0.011 μA cm–2 for HAP/CS and 0.017 μA cm–2 for HAP/CS/Gent after 28 days, compared to 0.061 and 0.52 μA cm–2, respectively, before immersion). Similarly, the EIS measurements suggested increased impedance of both coatings, along with decreased coating capacitances and double-layer capacitances, indicating the formation of a new biomimetic HAP layer. These results, along with the XRD and FT-IR results, proved the strong potential for biomimetic mineralization of HAP/CS and HAP/CS/Gent coatings in SBF. The ability of both investigated composite coatings to induce new bone growth was undoubtedly confirmed by the ALP assay. Almost 50% increase in ALP activity compared to the control could classify these coatings as prospective materials for bone implant design.
The noncytotoxicity effect of both HAP/CS and HAP/CS/Gent coatings was confirmed, when tested toward MRC-5 and L929 cell lines. High survival rates were detected, i.e., percent viabilities of both cell lines were exceeding 85%. In vitro release studies confirmed the successful loading of gentamicin (9.4 μg cm–2) and the so-called “burst” effect in the first days of coating exposure to deionized water. The amount of released gentamicin in the first 7 days linearly increased (∼50%), followed by a slower release pattern in the next 14 days. Concerning the biofilm preventing issues, highly promising results were obtained for the first 48 h when ∼21% of the drug was released. Using the Korsmeyer–Peppas model, the diffusion coefficient of gentamicin, D, was calculated to be 2.4 × 10–14 cm2 s–1.
Considering all of the above-mentioned results and findings, it could be stated that bioactive HAP/CS/Gent composite coating electrophoretically deposited on titanium can be employed as a potential biomedical device with therapeutic impact, due to the ability to induce the growth of new hydroxyapatite.
4. Experimental Section
4.1. Materials
Electrophoretic deposition process was performed on pure titanium plates (15 mm × 10 mm × 0.25 mm, 99.7%). Hydroxyapatite powder (particles <200 nm), chitosan (medium molecular weight 190–310 kDa, 75–85% deacetylation degree), and gentamicin sulfate (Figure 12) solution (50 mg mL–1 in dH2O) were used for obtaining composite coatings on Ti substrate. All listed chemicals and Ti were purchased from Sigma-Aldrich. EPD was preceded by mechanical polishing and ultrasonication of Ti plates (15 min in acetone). Electrochemical measurements were performed on Ti specimens of 30 mm × 20 mm × 0.25 mm dimensions. For gentamicin-release studies, Ti plates were cut to 10 mm × 10 mm × 0.25 mm sizes and prepared in the same manner as for the other measurements. All solvents used for drug-release measurements were of HPLC grade from J.T. Baker or Sigma-Aldrich. Deionized water was obtained by passing distilled water through a GenPure ultrapure water system (TKA, Germany).
Figure 12.

Chemical structure of gentamicin sulfate.
4.2. Electrophoretic Deposition
Composite HAP/CS coating was deposited from an aqueous suspension with the following composition: 1 wt % HAP powder and 0.05 wt % chitosan in 1% acetic acid solution. HAP/CS/Gent was deposited from an aqueous suspension containing 1 wt % HAP powder, 0.05 wt % chitosan, and 0.1 wt % gentamicin sulfate. The measured pH values for both suspensions were 4.4. The suspensions were prepared according to the same protocol as reported in our previous research paper.26 Briefly, chitosan was dissolved in 1% acetic acid, after which HAP powder was added under vigorous stirring and sonicated for 30 min, and thus obtained suspension contained 1 wt % HAP and 0.05 wt % CS. HAP/CS/Gent was prepared in the same manner, then gentamicin sulfate solution was added, and thus the obtained suspension contained 1 wt % HAP powder, 0.05 wt % chitosan, and 0.1 wt % gentamicin sulfate. EPD was performed as a cataphoretic deposition process on a Ti plate, serving as a working electrode (cathode) at previously determined conditions of 12 min deposition time and 5 V constant voltage.26 As counter electrodes (anodes), two platinum panels were employed. Uniform coating on both sides of Ti was achieved by placing the working electrode (Ti) between the Pt anodes at an equal distance of 1.5 cm. The deposited coatings were air-dried at room temperature for 24 h, and the difference in cathode mass before and after deposition was taken as the composite coating mass. The cross-sectional thicknesses of the as-deposited HAP/CS and HAP/CS/Gent coatings, measured by a polarized light microscope (Reichert MeF3), were 3.3 and 3.1 μm, respectively.
4.3. Characterization
X-ray diffraction (XRD) analysis was performed by a powder diffractometer (Philips PW 1710, Philips, the Netherlands) with Ni-filtered Cu Kα radiation (λ = 1.5418 Å). The intensity of diffraction was recorded at room temperature, between 4 and 70°, at a step of 0.05°. PowderCell software was utilized for phase analysis. Fourier transform infrared (FT-IR) spectroscopy was performed by a Nicolet IS-50 (Thermo Fisher Scientific) in ATR mode (400–4000 cm–1 range). The achieved spectral resolution was 4 cm–1. LEO SUPRA 55 equipped with an In-Lens detector (Carl Zeiss AG, Germany) operating at a 10 kV voltage acceleration was employed for field emission scanning electron microscopy (FE-SEM) in a combined secondary electron–backscattered electron (SE–BSE) mode.
4.4. Gentamicin-Release Profiles
The total amount of gentamicin incorporated in the composite coating by the EPD process was estimated from the scrapped-off coated surfaces by resuspension and complete dissolution of the obtained powder in acidified deionized water. For the drug-release assay, coated samples were immersed in deionized water and kept at 37 °C. Gentamicin release was monitored for 21 days. All of the measurements were done in triplicate. The concentration of released gentamicin was determined using a high-performance liquid chromatograph (HPLC) (Thermo Fisher Scientific) coupled with ion trap (LCQ Advantage, Thermo Fisher Scientific) as a mass spectrometer (MS). The separation of analytes was performed on a reverse-phase Zorbax Eclipse XDB-C18 column (Agilent Technologies, 4.6 mm × 75 mm × 3.5 μm). A precolumn (4.6 mm × 12.5 mm × 5 μm) was placed in front of the column. The mobile phase consisted of methanol (A), deionized water (B), and 10% acetic acid (C). The optimal chromatographic separation of gentamicin compounds was achieved using the mobile-phase gradient shown in Table 3.
Table 3. HPLC and MS Operating Parameters for the Determination of Gentamicin Compoundsa.
| HPLC |
MS |
|||||||
|---|---|---|---|---|---|---|---|---|
| time (min) | flow (mL min–1) | A (%) | B (%) | C (%) | precursor ion (m/z) | quantification reaction | CE (%) | |
| gentamicin C1a | 0.00 | 0.3 | 50 | 49 | 1 | 450[M + H]+ | 450 → 322 | 22 |
| 3.00 | 0.3 | 50 | 49 | 1 | ||||
| gentamicin C2 | 3.01 | 0.5 | 100 | 0 | 0 | 464[M + H]+ | 464 → 322 | 23 |
| 8.00 | 0.5 | 100 | 0 | 0 | ||||
| gentamicin C1 | 8.01 | 0.3 | 50 | 49 | 1 | 478[M + H]+ | 478 → 322 | 23 |
| 15.0 | 0.3 | 50 | 49 | 1 | ||||
Mobile-phase gradient, analytes’ precursor ions, fragmentation reactions used for quantification, and optimal collision energies (CEs).
Gentamicin mass spectrum was recorded in the m/z 50–1000 range, using the electrospray ionization technique. Since gentamicin is a complex antibiotic consisting of a mixture of three major compounds, gentamicins C1a, C2, and C1, there were three most abundant ions in the MS spectrum that were chosen as the precursor ions for each compound. Their most sensitive transitions were selected for quantification purposes (Table 3). Presented concentration results are sums of the three determined gentamicins.
Experimental data were fitted using two kinetic models to investigate the gentamicin-release mechanism and to calculate the diffusion coefficient of gentamicin from composite coatings.
4.5. Biomimetic Mineralization
In vitro bioactivity of composite HAP/CS and HAP/CS/Gent coatings was investigated by immersion in 20 mL of an SBF solution, prepared according to a previously published protocol.60 The SBF solution, stored under thermostatic conditions at 37 °C, was refreshed every 24 h for 7 days. After exposure to the SBF solution, the coatings were taken out, dried at room temperature, and used for further characterization (XRD, FT-IR, and FE-SEM).
4.6. Electrochemical Measurements
Biomimetic apatite growth, monitored during the exposure of coating to SBF at 37 °C, up to 28 days, was evaluated by electrochemical impedance spectroscopy (EIS) and potentiodynamic sweep (PDS) measurements. Electrochemical measurements were conducted in a Plexiglas electrochemical cell with a three-electrode setup. The coated Ti plate served as the working electrode with a 1 cm2 testing surface area, whereas the counter electrode was a platinum mesh of a significantly larger surface area compared to the working electrode. Saturated calomel electrode (SCE) was utilized as the reference electrode. Reference 600 potentiostat/galvanostat/ZRA (Gamry Instruments, Inc.) was used for all electrochemical measurements. Before each experiment, the open-circuit potential (Eocp) was monitored until a stability of 0.01 mV s–1 was reached. Impedance data were collected over a wide frequency range, applying a 5 mV AC voltage amplitude at the open-circuit potential. Gamry Instruments Echem Analyst software was used for impedance data processing. PDS measurements were carried out over the ±600 mV DC potential range with respect to Eocp, with a 0.5 mV s–1 scan rate.
4.7. Cytotoxicity
Cytotoxicity evaluation was performed using the dye exclusion test (DET) assay toward mice fibroblast cell line L929 (ATCC CRL-636) following the Biological Evaluation Guidance of the U.S. Food and Drug Administration (FDA), as well as the international standard ISO 10993-1.61 These institutions recommend that the cytotoxicity testing of a new material for medical implant application should be carried out on the L929 cell line (ATCC CRL-6364). The human lung fibroblast cell line (MRC-5, ATCC CCL-171) was also examined, as it is the most similar to both L929 line and the connective tissue that is present at the target site and is often used in similar studies as a model system for in vitro cytotoxicity.62−64 Cells were cultured and propagated in the same manner as already published in our previous work.26 Viable cells were seeded in 12-well plates (Costar) on the examined coated Ti plates at a concentration of 1 × 105 mL–1. Control wells contained only cells, without coated samples. During the next 48 h, Petri dishes with disks and seeded cells were incubated at 37 °C under air flow with 5% CO2. The following step included separation of cells from coated samples by trypsinization treatment (addition of 0.1% solution of trypsin). Cell viability and number were measured using a trypan blue exclusion method. Inhibition of growth was expressed as a percent of control according to the formula: K = (Ns/Nk) × 100%, where Nk is the overall number of cells in the sample and Ns is the number of cells in the investigated sample.
4.8. Alkaline Phosphatase Activity
Colorimetric assessment of the release of alkaline phosphatase (ALP) by the cells was performed using an ALP detection kit (Abcam’s alkaline phosphatase assay kit) according to the manufacturer’s instructions. The assay was repeated three times to ensure the reproducibility of the test. The absorbance of the solution was measured at 405 nm using a bioanalyzer. Abcam’s alkaline phosphatase assay kit (colorimetric) (ab83369)65 is a highly sensitive, simple, direct, and high-throughput screening (HTS)-ready colorimetric assay designed to measure ALP activity in serum and biological samples. The kit uses p-nitrophenyl phosphate (pNPP) as a phosphatase substrate, which turns yellow (λmax = 405 nm) when dephosphorylated by ALP.
4.9. Statistical Analysis
Cytotoxicity and ALP results were subjected to statistical evaluation by one-way analysis of variance, followed by a multiple-comparison post hoc test. The results were considered statistically significant if the p-value was lower than 0.01.
Acknowledgments
This work was supported by the Ministry of Education, Science, and Technological Development of the Republic of Serbia (contract nos. 451-03-68/2020-14/200135 and 451-03-68/2020-14/200287) and the Basic Science Research Program of the Ministry of Education, Science and Technology of Korea (grant number: 2018R1A2B5A02023190).
The authors declare no competing financial interest.
References
- Porter J. R.; Ruckh T. T.; Popat K. C. Bone Tissue Engineering: A Review in Bone Biomimetics and Drug Delivery Strategies. Biotechnol. Prog. 2009, 25, 1539–1560. 10.1002/btpr.246. [DOI] [PubMed] [Google Scholar]
- Albrektsson T.; Albrektsson B. Osseointegration of Bone Implants: A Review of an Alternative Mode of Fixation. Acta Orthop. 1987, 58, 567–577. 10.3109/17453678709146401. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Thouas G. A. Metallic Implant Biomaterials. Mater. Sci. Eng., R 2015, 87, 1–57. 10.1016/j.mser.2014.10.001. [DOI] [Google Scholar]
- Bosco R.; Van Den Beucken J.; Leeuwenburgh S.; Jansen J. Surface Engineering for Bone Implants: A Trend from Passive to Active Surfaces. Coatings 2012, 2, 95–119. 10.3390/coatings2030095. [DOI] [Google Scholar]
- Bosco R.; Edreira E. R. U.; Wolke J. G. C.; Leeuwenburgh S. C. G.; van den Beucken J. J. J. P.; Jansen J. A. Instructive Coatings for Biological Guidance of Bone Implants. Surf. Coat. Technol. 2013, 233, 91–98. 10.1016/j.surfcoat.2013.02.039. [DOI] [Google Scholar]
- Surmenev R. A.; Surmeneva M. A.; Ivanova A. A. Significance of Calcium Phosphate Coatings for the Enhancement of New Bone Osteogenesis - A Review. Acta Biomater. 2014, 10, 557–579. 10.1016/j.actbio.2013.10.036. [DOI] [PubMed] [Google Scholar]
- Kulkarni M.; Mazare A.; Schmuki P.; Iglič A.. Biomaterial Surface Modification of Titanium and Titanium Alloys for Medical Applications. In Nanomedicine;Seifalian A., de Mel A., Kalaskar D. M., Eds.; One Central Press: U.K., 2014; pp 111–136. [Google Scholar]
- Mehnath S.; Arjama M.; Rajan M.; Premkumar K.; Karthikeyan K.; Jeyaraj M. Mineralization of Bioactive Marine Sponge and Electrophoretic Deposition on Ti6Al4V Implant for Osteointegration. Surf. Coat. Technol. 2020, 392, 125727 10.1016/j.surfcoat.2020.125727. [DOI] [Google Scholar]
- Boccaccini A. R.; Keim S.; Ma R.; Li Y.; Zhitomirsky I. Electrophoretic Deposition of Biomaterials. J. R. Soc., Interface 2010, 7, S581–S613. 10.1098/rsif.2010.0156.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhshandeh S.; Amin Yavari S. Electrophoretic Deposition: A Versatile Tool against Biomaterial Associated Infections. J. Mater. Chem. B 2018, 6, 1128–1148. 10.1039/C7TB02445B. [DOI] [PubMed] [Google Scholar]
- Harun W. S. W.; Asri R. I. M.; Alias J.; Zulkifli F. H.; Kadirgama K.; Ghani S. A. C.; Shariffuddin J. H. M. A Comprehensive Review of Hydroxyapatite-Based Coatings Adhesion on Metallic Biomaterials. Ceram. Int. 2018, 44, 1250–1268. 10.1016/j.ceramint.2017.10.162. [DOI] [Google Scholar]
- Sumathra M.; Rajan M.; Amarnath Praphakar R.; Marraiki N.; Elgorban A. M. In Vivo Assessment of a Hydroxyapatite/κ-Carrageenan-Maleic Anhydride-Casein/Doxorubicin Composite-Coated Titanium Bone Implant. ACS Biomater. Sci. Eng. 2020, 6, 1650–1662. 10.1021/acsbiomaterials.9b01750. [DOI] [PubMed] [Google Scholar]
- Pan J.; Prabakaran S.; Rajan M. In-Vivo Assessment of Minerals Substituted Hydroxyapatite / Poly Sorbitol Sebacate Glutamate (PSSG) Composite Coating on Titanium Metal Implant for Orthopedic Implantation. Biomed. Pharmacother. 2019, 119, 109404 10.1016/j.biopha.2019.109404. [DOI] [PubMed] [Google Scholar]
- Wang W.; Yeung K. W. K. Bone Grafts and Biomaterials Substitutes for Bone Defect Repair: A Review. Bioact. Mater. 2017, 2, 224–247. 10.1016/j.bioactmat.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivethaa E. A. K.; Martin C. A.; Frank-Kamenetskaya O. V.; Kalkura S. N.. Chitosan and Chitosan Based Nanocomposites for Applications as a Drug Delivery Carrier: A Review. In Processes and Phenomena on the Boundary Between Biogenic and Abiogenic Nature; Frank-Kamenetskaya O., Vlasov D. Y., Panova E. G., Lessovaia S. N., Eds.; Lecture Notes in Earth System Sciences; Springer: Cham, 2020; pp 23–37. [Google Scholar]
- Lakshmi Prasanthi N.; Roy H.; Jyothi N.; Sri Vajrapriya V. A Brief Review on Chitosan and Application in Biomedical Field. Am. J. PharmTech Res. 2016, 6, 1–11. [Google Scholar]
- Wang W.; Meng Q.; Li Q.; Liu J.; Zhou M.; Jin Z.; Zhao K. Chitosan Derivatives and Their Application in Biomedicine. Int. J. Mol. Sci. 2020, 21, 487 10.3390/ijms21020487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croisier F.; Jérôme C. Chitosan-Based Biomaterials for Tissue Engineering. Eur. Polym. J. 2013, 49, 780–792. 10.1016/j.eurpolymj.2012.12.009. [DOI] [Google Scholar]
- Pang X.; Zhitomirsky I. Electrophoretic Deposition of Composite Hydroxyapatite-Chitosan Coatings. Mater. Charact. 2007, 58, 339–348. 10.1016/j.matchar.2006.05.011. [DOI] [Google Scholar]
- Ordikhani F.; Simchi A. Long-Term Antibiotic Delivery by Chitosan-Based Composite Coatings with Bone Regenerative Potential. Appl. Surf. Sci. 2014, 317, 56–66. 10.1016/j.apsusc.2014.07.197. [DOI] [Google Scholar]
- Pishbin F.; Mouriño V.; Flor S.; Kreppel S.; Salih V.; Ryan M. P.; Boccaccini A. R. Electrophoretic Deposition of Gentamicin-Loaded Bioactive Glass/Chitosan Composite Coatings for Orthopaedic Implants. ACS Appl. Mater. Interfaces 2014, 6, 8796–8806. 10.1021/am5014166. [DOI] [PubMed] [Google Scholar]
- Patel K. D.; El-Fiqi A.; Lee H. Y.; Singh R. K.; Kim D.-A.; Lee H. H.; Kim H.-W. Chitosan-Nanobioactive Glass Electrophoretic Coatings with Bone Regenerative and Drug Delivering Potential. J. Mater. Chem. 2012, 22, 24945–24956. 10.1039/c2jm33830k. [DOI] [Google Scholar]
- Rinaudo M. Chitin and Chitosan: Properties and Applications. Prog. Polym. Sci. 2006, 31, 603–632. 10.1016/j.progpolymsci.2006.06.001. [DOI] [Google Scholar]
- Molaei A.; Yousefpour M. Preparation of Chitosan-Based Nanocomposites and Biomedical Investigations in Bone Tissue Engineering. Int. J. Polym. Mater. Polym. Biomater. 2019, 68, 701–713. 10.1080/00914037.2018.1493683. [DOI] [Google Scholar]
- Zhao L.; Chu P. K.; Zhang Y.; Wu Z. Antibacterial Coatings on Titanium Implants. J. Biomed. Mater. Res., Part B 2009, 91B, 470–480. 10.1002/jbm.b.31463. [DOI] [PubMed] [Google Scholar]
- Stevanović M.; Đošić M.; Janković A.; Kojić V.; Vukašinović-Sekulić M.; Stojanović J.; Odović J.; Crevar Sakač M.; Rhee K. Y.; Misković-Stanković V. Gentamicin-Loaded Bioactive Hydroxyapatite/Chitosan Composite Coating Electrodeposited on Titanium. ACS Biomater. Sci. Eng. 2018, 4, 3994–4007. 10.1021/acsbiomaterials.8b00859. [DOI] [PubMed] [Google Scholar]
- Thompson K.; Petkov S.; Zeiter S.; Sprecher C. M.; Geoff Richards R.; Fintan Moriarty T.; Eijer H. Intraoperative Loading of Calcium Phosphate-Coated Implants with Gentamicin Prevents Experimental Staphylococcus aureus Infection in Vivo. PLoS One 2019, 14, e0210402 10.1371/journal.pone.0210402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima S.; Sato T.; Honda M.; Suetsugu Y.; Ozeki K.; Kikuchi M. Fabrication of Gentamicin-Loaded Hydroxyapatite/Collagen Bone-Like Nanocomposite for Anti-Infection Bone Void Fillers. Int. J. Mol. Sci. 2020, 21, 551 10.3390/ijms21020551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin E. J. Recent Coating Developments for Combination Devices in Orthopedic and Dental Applications: A Literature Review. Adv. Drug Delivery Rev. 2017, 112, 88–100. 10.1016/j.addr.2017.01.007. [DOI] [PubMed] [Google Scholar]
- Flores C.; Degoutin S.; Chai F.; Raoul G.; Hornez J. C.; Martel B.; Siepmann J.; Ferri J.; Blanchemain N. Gentamicin-Loaded Poly(Lactic-Co-Glycolic Acid) Microparticles for the Prevention of Maxillofacial and Orthopedic Implant Infections. Mater. Sci. Eng., C 2016, 64, 108–116. 10.1016/j.msec.2016.03.064. [DOI] [PubMed] [Google Scholar]
- Neut D.; van de Belt H.; Stokroos I.; van Horn J. R.; van der Mei H. C.; Busscher H. J. Orthopaedic Revision Surgery. J. Antimicrob. Chemother. 2001, 47, 885–891. 10.1093/jac/47.6.885. [DOI] [PubMed] [Google Scholar]
- Florea D. A.; Albuleţ D.; Grumezescu A. M.; Andronescu E. Surface Modification – A Step Forward to Overcome the Current Challenges in Orthopedic Industry and to Obtain an Improved Osseointegration and Antimicrobial Properties. Mater. Chem. Phys. 2020, 243, 122579 10.1016/j.matchemphys.2019.122579. [DOI] [Google Scholar]
- Kokubo T.; Takadama H. How Useful Is SBF in Predicting in Vivo Bone Bioactivity?. Biomaterials 2006, 27, 2907–2915. 10.1016/j.biomaterials.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Ballarre J.; Aydemir T.; Liverani L.; Roether J. A.; Goldmann W. H.; Boccaccini A. R. Versatile Bioactive and Antibacterial Coating System Based on Silica, Gentamicin, and Chitosan: Improving Early Stage Performance of Titanium Implants. Surf. Coat. Technol. 2020, 381, 125138 10.1016/j.surfcoat.2019.125138. [DOI] [Google Scholar]
- Tian B.; Tang S.; Wang C. D.; Wang W. G.; Wu C. L.; Guo Y. J.; Guo Y. P.; Zhu Z. A. Bactericidal Properties and Biocompatibility of a Gentamicin-Loaded Fe3O4/Carbonated Hydroxyapatite Coating. Colloids Surf., B 2014, 123, 403–412. 10.1016/j.colsurfb.2014.09.050. [DOI] [PubMed] [Google Scholar]
- Ragu A.; Senthilarasan K.; Sakthivel P. Synthesis and Characterization of Nano Hydroxyapatite with Polyoxymethylene Nanocomposites for Bone Growth Studies. Int. J. Sci. Eng. Res. 2015, 3, 120–123. [Google Scholar]
- Rincón-López J. A.; Hermann-Muñoz J. A.; Giraldo-Betancur A. L.; De Vizcaya-Ruiz A.; Alvarado-Orozco J. M.; Muñoz-Saldaña J. Synthetic and Bovine-Derived Hydroxyapatite Ceramics: A Comparison. Materials 2018, 11, 333 10.3390/ma11030333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng F.; Veilleux E.; Schmidt M.; Wei M. Synthesis of Hydroxyapatite Nanoparticles with Tailorable Morphologies and Carbonate Substitutions Using a Wet Precipitation Method. J. Nanosci. Nanotechnol. 2012, 12, 2774–2782. 10.1166/jnn.2012.5714. [DOI] [PubMed] [Google Scholar]
- Yang W.; Xi X.; Li J.; Cai K. Comparison of Crystal Structure between Carbonated Hydroxyapatite and Natural Bone Apatite with Theoretical Calculation. Asian J. Chem. 2013, 25, 3673–3678. 10.14233/ajchem.2013.13709. [DOI] [Google Scholar]
- Victoria E. C.; Gnanam F. D. Synthesis and Characterisation of Biphasic Calcium Phosphate. Trends Biomater. Artif. Organs. 2002, 16, 12–14. [Google Scholar]
- Ishikawa K.; Ducheyne P.; Radin S. Determination of the Ca/P Ratio in Calcium-Deficient Hydroxyapatite Using X-Ray Diffraction Analysis. J. Mater. Sci.: Mater. Med. 1993, 4, 165–168. 10.1007/BF00120386. [DOI] [Google Scholar]
- Cacciotti I.Cationic and Anionic Substitutions in Hydroxyapatite. In Handbook of Bioceramics and Biocomposites; Antoniac I. V., Ed.; Springer International Publishing: Cham, 2016; pp 146–188. [Google Scholar]
- Bonfield W.; Gibson I. R. Novel Synthesis and Characterization of an AB-Type Carbonate-Substituted Hydroxyapatite. J. Biomed. Mater. Res. 2002, 59, 697–708. 10.1002/jbm.10044. [DOI] [PubMed] [Google Scholar]
- Müller L.; Müller F. A. Preparation of SBF with Different HCO3- Content and Its Influence on the Composition of Biomimetic Apatites. Acta Biomater. 2006, 2, 181–189. 10.1016/j.actbio.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Cromme P.; Zollfrank C.; Müller L.; Müller F. A.; Greil P. Biomimetic Mineralisation of Apatites on Ca2+ activated Cellulose Templates. Mater. Sci. Eng., C 2007, 27, 1–7. 10.1016/j.msec.2005.11.001. [DOI] [Google Scholar]
- Berzina-Cimdina L.; Borodajenko N.. Research of Calcium Phosphates Using Fourier Transform Infrared Spectroscopy. In Infrared Spectroscopy: Materials Science, Engineering and Technology;Theophanides T., Ed.; IntechOpen: London, 2012; pp 123–148. [Google Scholar]
- Janković A.; Eraković S.; Mitrić M.; Matić I. Z.; Juranić Z. D.; Tsui G. C. P.; Tang C. Y.; Mišković-Stanković V.; Rhee K. Y.; Park S. J. Bioactive Hydroxyapatite/Graphene Composite Coating and Its Corrosion Stability in Simulated Body Fluid. J. Alloys Compd. 2015, 624, 148–157. 10.1016/j.jallcom.2014.11.078. [DOI] [Google Scholar]
- Ren F.; Ding Y.; Leng Y. Infrared Spectroscopic Characterization of Carbonated Apatite: A Combined Experimental and Computational Study. J. Biomed. Mater. Res., Part A 2014, 102, 496–505. 10.1002/jbm.a.34720. [DOI] [PubMed] [Google Scholar]
- Bonadio T. G. M.; Sato F.; Medina A. N.; Weinand W. R.; Baesso M. L.; Lima W. M. Bioactivity and Structural Properties of Nanostructured Bulk Composites Containing Nb2O5 and Natural Hydroxyapatite. J. Appl. Phys. 2013, 113, 223505 10.1063/1.4809653. [DOI] [Google Scholar]
- Čolović B.; Milivojević D.; Babić-Stojić B.; Jokanović V. Pore Geometry of Ceramic Device: The Key Factor of Drug Release Kinetics. Sci. Sintering 2013, 45, 107–116. 10.2298/SOS1301107C. [DOI] [Google Scholar]
- Yu S. Y.; Scully J. R. Corrosion and Passivity of Ti-13% Nb-13% Zr in Comparison to Other Biomedical Implant Alloys. Corrosion 1997, 53, 965–976. 10.5006/1.3290281. [DOI] [Google Scholar]
- Đošić M.; Eraković S.; Janković A.; Vukašinović-Sekulić M.; Matić I. Z.; Stojanović J.; Rhee K. Y.; Mišković-Stanković V.; Park S.-J. In Vitro Investigation of Electrophoretically Deposited Bioactive Hydroxyapatite/Chitosan Coatings Reinforced by Graphene. J. Ind. Eng. Chem. 2017, 47, 336–347. 10.1016/j.jiec.2016.12.004. [DOI] [Google Scholar]
- Zhang Q.; Chen J.; Feng J.; Cao Y.; Deng C.; Zhang X. Dissolution and Mineralization Behaviors of HA Coatings. Biomaterials 2003, 24, 4741–4748. 10.1016/S0142-9612(03)00371-5. [DOI] [PubMed] [Google Scholar]
- Vahabzadeh S.; Roy M.; Bandyopadhyay A.; Bose S. Phase Stability and Biological Property Evaluation of Plasma Sprayed Hydroxyapatite Coatings for Orthopedic and Dental Applications. Acta Biomater. 2015, 17, 47–55. 10.1016/j.actbio.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C. H.; Mansfeld F. Concerning the Conversion of the Constant Phase Element Parameter Y0 into a Capacitance. Corrosion 2001, 57, 747–755. 10.5006/1.3280607. [DOI] [Google Scholar]
- Korsmeyer R. W.; Gurny R.; Doelker E.; Buri P.; Peppas N. A. Mechanisms of Solute Release from Porous Hydrophilic Polymers. Int. J. Pharm. 1983, 15, 25–35. 10.1016/0378-5173(83)90064-9. [DOI] [PubMed] [Google Scholar]
- Ritger P. L.; Peppas N. A. A Simple Equation for Description of Solute Release. J. Controlled Release 1987, 5, 23–36. 10.1016/0168-3659(87)90034-4. [DOI] [Google Scholar]
- Phillips H. J.Dye Exclusion Tests for Cell Viability. In Tissue Culture: Methods and Applications; Kruse P., Patterson M., Eds.; Academic Press, Inc.: Cambridge, MA, 1973; pp 406–408. [Google Scholar]
- Almeida M. J.; Pereira L.; Milet C.; Haigle J.; Barbosa M.; Lopez E. Comparative Effects of Nacre Water-Soluble Matrix and Dexamethasone on the Alkaline Phosphatase Activity of MRC-5 Fibroblasts. J. Biomed. Mater. Res. 2001, 57, 306–312. . [DOI] [PubMed] [Google Scholar]
- Kokubo T.; Takadama H.. Simulated Body Fluid (SBF) as a Standard Tool to Test the Bioactivity of Implants. In Handbook of Biomineralization: Biological Aspects and Structure Formation; Bäuerlein E., Ed.; John Wiley & Sons: Hoboken, NJ, 2008; pp 97–109. [Google Scholar]
- Food and Drug Administration (FDA) . Use of International Standard ISO 10993-1, “Biological Evaluation of Medical Devices—Part 1: Evaluation and Testing within a Risk Management Process”. Guidance for Industry and Food and Drug Administration Staff; U.S. Department of Health and Human Services, Food and Drug Administration, Center for Devices and Radiological Health, 2016. [Google Scholar]
- Lan M. Y.; Liu C. P.; Huang H. H.; Chang J. K.; Lee S. W. Diameter-Sensitive Biocompatibility of Anodic TiO2 Nanotubes Treated with Supercritical CO2 Fluid. Nanoscale Res. Lett. 2013, 8, 150 10.1186/1556-276X-8-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Ortega E.; Jos A.; Cameán A. M.; Pato-Mourelo J.; Segura-Egea J. J. In Vitro Evaluation of Cytotoxicity and Genotoxicity of a Commercial Titanium Alloy for Dental Implantology. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2010, 702, 17–23. 10.1016/j.mrgentox.2010.06.013. [DOI] [PubMed] [Google Scholar]
- Madathil B. K.; Lin Q.; Hew C. L.; Mohanty M. Hypoxia-like Effect of Cobalt Chromium Alloy Micro Particles on Fibroblasts in Vitro. J. Orthop. Res. 2010, 28, 1360–1367. 10.1002/jor.21133. [DOI] [PubMed] [Google Scholar]
- Abcam . Alkaline Phosphatase Assay Kit; Abcam, 2015; Vol. 1, pp 1–11. [Google Scholar]