

Abstract

Photoinduced thiol-epoxy click polymerization possesses both the characteristics and advantages of photopolymerization and click reactions. However, the photopolymerization of pigmented or highly filled thiol-epoxy thick composites still remains a great challenge due to the light screening effect derived from the competitive absorption, reflection, and scattering of the pigments or functional fillers. In this article, we present a simple and versatile strategy to prepare thick composites via delayed thiol-epoxy photopolymerization. The irradiation of a small area with a light-emitting diode (LED) point light source at room temperature leads to the decomposition of a photobase generator and the released active basic species can uniformly disperse throughout the whole system, including unirradiated areas, via mechanical stirring. No polymerization was observed at room temperature and therefore the liquid formulations can be further processed with molds of arbitrary size and desired shapes. It is only by increasing the temperature that base-catalyzed thiol-epoxy polymerization occurs and controllable preparation of thick thiol-epoxy materials can be achieved. The formed networks display excellent uniformity in different radii and depths with comparable functionality conversions, similar Tg values, and thermal decomposition temperatures. The presented strategy can be applied to prepare thick composites with glass fibers possessing improved mechanical properties and dark composites containing 2 wt % carbon nanotubes.
Introduction
Base-catalyzed thiol-epoxy click polymerization possesses numerous advantages including fast rate, excellent stereoselectivity, and high yields.1−7 Moreover, this type of reaction leads to the formation of a β-hydroxythio-ether, and the formed hydroxyl groups improve adhesion to metallic surfaces or can be widely used in the synthesis and modification of polymers.8 In general, the base-catalyzed thiol-epoxy reaction follows an anionic nucleophilic polymerization mechanism:9 in the presence of a base catalyst, the thiolate anions, formed via the deprotonation of thiols, couple with the epoxide groups through a nucleophilic ring-opening reaction. The amine is a real thermal catalyst that is not consumed during the addition reaction. Therefore, the amine can catalyze a great number of addition reactions. This unique mechanism makes thiol-epoxy polymerization less sensitive to oxygen and impurities except acidic compounds. Moreover, the cured products are characterized by lower shrinkage, better adhesion, and superior chemical resistance.10,11
Multifunctional thiol-epoxy composites with superior properties have recently attracted wide attention.12−15 It was reported that recoverable self-healing thiol-epoxy elastomer composites with high thermal conductivity were obtained by introducing micron boron nitride fillers into the thiol-epoxy resin matrix.12 On the other hand, distinctly improved thermomechanical properties of the cross-linked network can be realized via designing thiol-epoxy organic–inorganic hybrid nanocomposites containing a monofunctionalized polyhedral oligomeric silsesquioxane (POSS).15 Until now, the preparation of thiol-epoxy composites mostly employed a thermally active catalyst with fast initial curing rates; despite this, it may lead to premature gelation and undesirably short storage stability.16−18 Employing latent thermal catalysts can increase the shelf life, but higher temperatures are required for the activation. Compared to thermal-induced thiol-epoxy polymerization, photoactivated thiol-epoxy polymerization has enormous advantages such as high energy utilization efficiency and enhanced temporal and spatial control.19−23 Since the employed photobase generator is only activated under irradiation, it can produce a conventional base catalyst reacting at a low curing temperature while providing a long shelf life. However, in photosensitive composites, the added pigments and functional fillers would cause competitive absorption, reflection, and scattering of light, therefore resulting in reduced polymerization rate and depth.24,25 In a dark composite with carbon materials such as carbon nanotubes or graphene, the penetrating ability of light into the resin and the final polymerization depth are further decreased. Therefore, achieving deep photopolymerization to prepare thick composites still remains a great challenge.26,27
Several strategies have been developed to improve the photocuring efficiency of thick composites. Using acyl phosphine oxide as a photobleaching initiator, thick urethane acrylate samples (30 μm) comprising carbon black were prepared by Decker.26 Taking advantage of the long induction period of cationic photopolymerization at a low temperature, Nie et al.28 developed a temperature-controlled cationic photocuring strategy, involving the migration of cationic active species into the deep unilluminated areas. Photocured epoxy resins with a thickness greater than 3.5 cm could be achieved by this technology even in the presence of 2 wt % carbon black as functional fillers dispersed in the resins. Frontal photopolymerization, essentially a phototriggered thermal or redox polymerization, is another efficient approach to prepare thick composites such as dental filling materials and opaque composites with functional fillers.29,30 Using near-infrared light with a strong penetration ability, our group has realized photopolymerization of a 22.5 mm acrylate sample with 0.5 wt % red pigment via upconversion nanoparticle-assisted photochemistry.31 The aforementioned approaches have been successfully applied only to free radical and cationic photopolymerization. To the best of our knowledge, no efficient strategy has been reported for preparing thick composites through thiol-epoxy anionic photopolymerization.
In this article, we present a simple and generic method to prepare thick composites based on delayed thiol-epoxy photopolymerization (DTEP). When a small area of the photosensitive resin is irradiated with a light-emitting diode (LED) point light source at room temperature, the exposed photobase generator decomposes and the released active basic species can uniformly disperse throughout the whole system, including unirradiated areas, either by simple diffusion or accelerated via mechanical stirring. For this proceeding, it is an advantage that efficient curing does not start immediately upon irradiation at ambient temperature; thus, uniform dispersion of the active basic species can be achieved in a low-viscous medium. The liquid formulations can be further processed with molds of arbitrary size and desired shapes. It is only by increasing the temperature that the base-catalyzed thiol-epoxy polymerization occurs, and controllable preparation of thick thiol-epoxy materials with uniform properties can be achieved (Scheme 1). We also demonstrate that the proposed method can be applied to the preparation of thick composites containing glass fibers or dark composites with carbon nanotubes.
Scheme 1. Schematic Diagram of Delayed Thiol-Epoxy Photopolymerization.

Result and Discussion
The photocuring process of thiol-epoxy films initiated by photobase generators has been extensively investigated. Compared to thermal-induced thiol-epoxy polymerization, which requires high temperature for acceptable polymerization speed and suffers from short pot life, photoactivate thiol-epoxy polymerization can occur at low curing temperatures while providing a long shelf life because the active species are only released under irradiation. However, due to the limited penetration of the UV light and the competitive absorption, reflection, and scattering of light by pigments and functional fillers, it is still difficult to prepare thick samples via thiol-epoxy anionic photopolymerization. A possible advantage of a photolatent amine catalyst is the fact that the liberated amine is a stable compound, a real thermal catalyst that is not consumed during the addition reaction. Thus, even if the catalyst can only be activated in the topmost layers of a composite material, it can subsequently diffuse into deeper layers, thereby inducing cross-linking by a thermal process in regions that were not or insufficiently irradiated.
In this methodology, the preparation of thiol-epoxy thick composites via DTEP can be divided into two stages. Initially, when using a LED point light source to irradiate a small area of the photosensitive resin, we activated the latent amine photochemically to form the amine 1,5,7-triazabicyclo[4.4.0] dec-5-ene (TBD), which is only moderately active at room temperature in the thiol-epoxy formulation used. This is different from most other known photopolymerization systems, such as free radical photopolymerization, where the cross-linking reaction is designed to start immediately after the active species is generated. The pot life of the activated formulation is long enough to allow the activation step to be performed in the first step that is decoupled from the proper curing reaction. Thus, activation by light can be performed in a separate vessel with stirring.
This proceeding brings considerable technical advantages: first, due to the stirring effect, we can activate the catalyst homogenously in bulk material. It is possible to activate rather large amounts of resin materials, as required in the final application. Even if the photochemical activation takes part only in the topmost layer of the reaction vessel, the stirring effect assures that the activated catalyst will finally be homogeneously distributed throughout the entire formulation, and the latent form is transported close to the surface, which allows for full activation of the whole amount of photolatent catalyst. Second, the very slow curing after activation at room temperature allows the “activated” formulation to be applied by any technique. As an example, it is possible to pour the activated formulation into a mold of arbitrary size and desired shapes.
In the second stage, heat is applied to trigger the consequent, amine-catalyzed thermal polymerization. Although slow at room temperature, the active catalyst is active at elevated temperatures, leading to fast polymerization of the formulations, including functional fillers.
Generation and Migration of the Active Base Species under Irradiation at Room Temperature
TBD·HBPh4 was selected because it is an efficient photolatent base generator, which is capable of releasing the superbase TBD in situ upon UV irradiation at room temperature.32 However, the limited absorption, mainly below 300 nm, has restricted its applications (Figure 1).33 It has been reported that the addition of 2-isopropylthioxanthone (ITX) can increase the photosensitivity of TBD·HBPh4 in the long-wavelength regions.34
Figure 1.
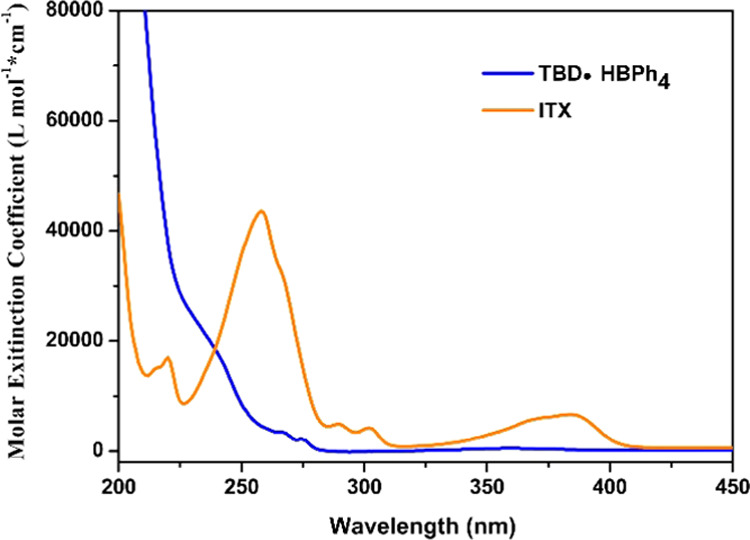
Ultraviolet–visible (UV–vis) absorption spectra of TBD·HBPh4 and ITX.
Figure 2 shows the photolysis mechanism of ITX-assisted TBD·HBPh4.3232 The photosensitive formulation, which contains ITX as a photosensitizer and TBD·HBPh4 as a photobase generator, is irradiated with LED light of 385 nm wavelength. ITX absorbs light of these wavelengths and undergoes a transition from the ground state to an excited triplet state. The energy of the excited state is then transferred from ITX to TBD·HBPh4. The generated BPh4– ion undergoes rearrangement extracting a proton from the neighboring TBD·H+ cation to release the superbase TBD.
Figure 2.
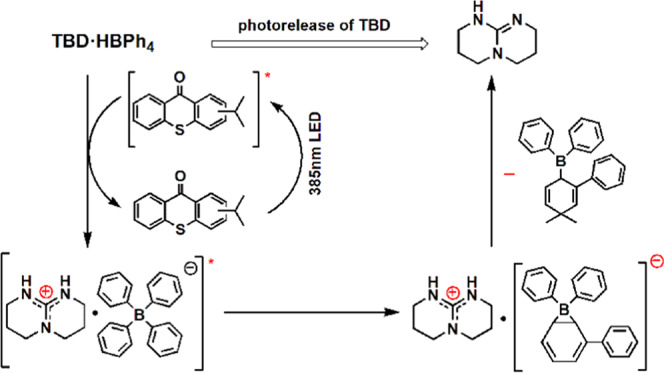
Photolysis mechanisms of the photobase generator.
To prove the base production of TBD·HBPh4 in the presence of ITX upon irradiation with a 385 nm LED lamp, the UV–vis absorption spectra of a ITX/TBD·HBPh4 mixture containing phenol red as a pH indicator were measured after irradiation for different irradiation times, as shown in Figure 3. A new absorption band appears at 560 nm. This band can be attributed to the deprotonated phenol red, indicating the base generation of the ITX/TBD·HBPh4 system under irradiation. Moreover, the intensity of the absorption peak increases upon increasing the irradiation time, and this is consistent with previous literature reports.33
Figure 3.
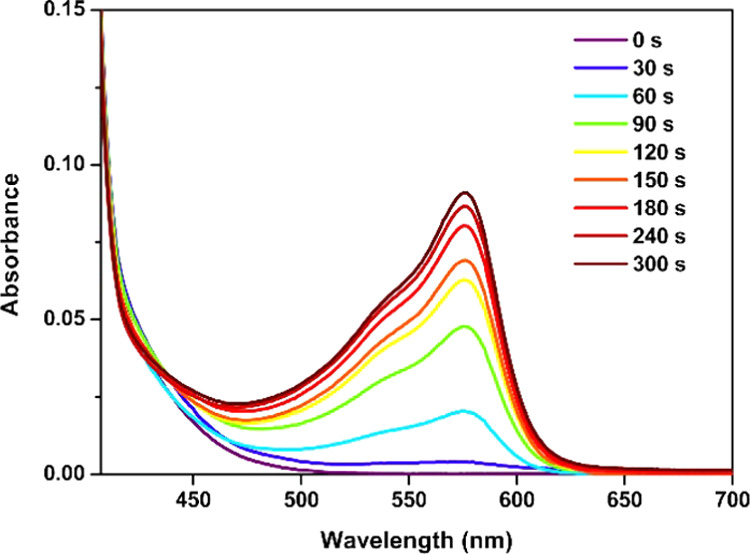
Changes in the UV–vis absorption spectra of a phenol red solution containing TBD·HBPh4/ITX irradiated over time.
After confirming the generation of active base species under irradiation, we continued to study the migration ability of the formed bases. Since the mobility of the active species is significantly affected by the viscosity of the resin, a rheometer was employed to measure the viscosity of the sample after 5 min illumination intervals. The commercial bisphenol A epoxy resin bisphenol A diglycidyl ether (BADGE) was selected since it is a low-cost epoxide with widespread use providing excellent mechanical properties of the cured materials. The secondary thiol PE-1 was used because of its lower odor and superior storage stability as compared to primary thiols.35 As shown in Figure 4a, with the extension of the irradiation time up to 20 min, only a marginal change in viscosity (7.6–7.8 Pa·s) of the sample at different shear rates was observed. The result indicates that the system did not undergo significant polymerization under irradiation at room temperature. This assumption is supported by the absence of polymers with increased molecular weight in gel permeation chromatography (GPC) tests performed after different irradiation times (Figure 4b). The marginally changed viscosity of the samples is beneficial for the uniform distribution of the generated active base species through the whole system and for further processing and assembly of the samples.
Figure 4.
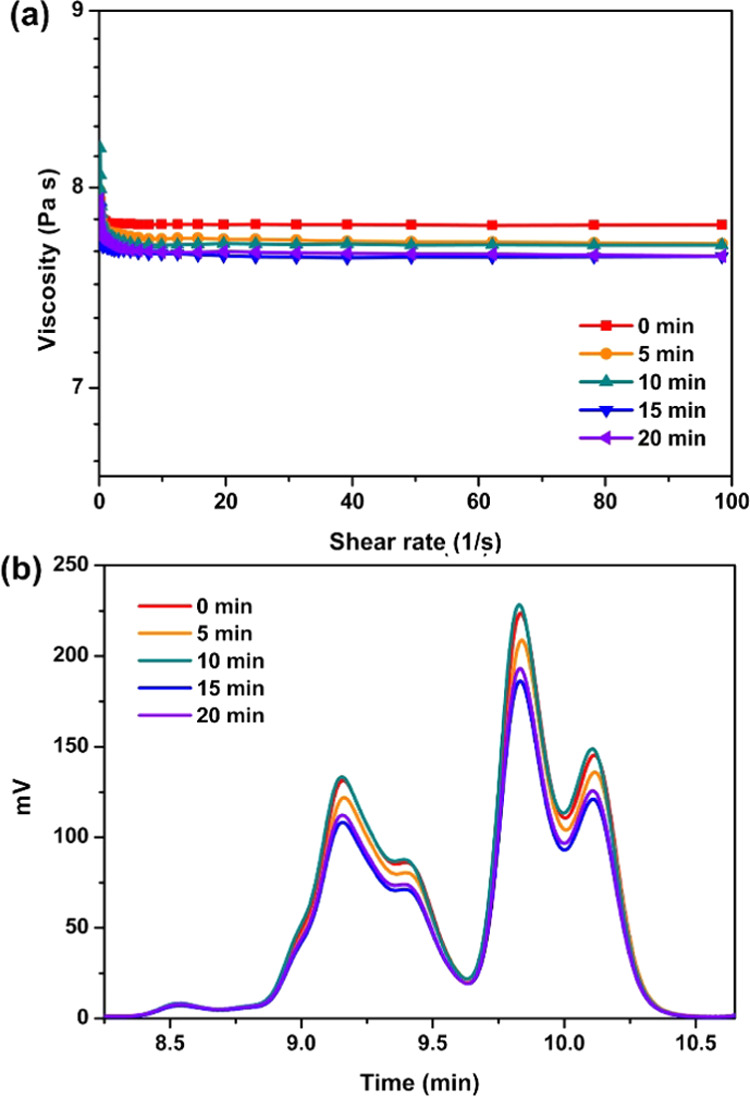
(a) Viscosity and (b) molecular weight changes in the thiol-epoxy samples with different irradiation times.
Thiol-Epoxy Polymerization at Increasing Temperature
To investigate influence of the photobase generator content, irradiation time, as well as postbaking temperature on the curing efficiency of a thiol-epoxy formulation, the thiol-epoxy resin samples containing the photobase generator were irradiated with a 385 nm LED lamp with a light intensity of 196 mW/cm2 while stirring for a defined time, and a small amount of the sample was withdrawn for isothermal and nonisothermal differential scanning calorimeter (DSC) measurements.
With the aim to elucidate the influence of the TBD·HBPh4 content on the curing rate of the thiol-epoxy system, four samples containing 1, 2, 3, and 4 wt % TBD·HBPh4 (the ITX content increased in parallel with TBD·HBPh4) in the thiol-epoxy formulations were prepared. The change in the heat flow peaks was monitored via nonisothermal DSC, as shown in Figure 5. After 20 min irradiation time and upon increase of the TBD·HBPh4 content in the thiol-epoxy formulation, the initial curing temperature of the samples gradually decreases. Moreover, a different shape of the DSC heat flow curve is observed for different contents of TBD·HBPh4. This phenomenon is most prominent for higher concentrations of TBD·HBPh4. The larger the amount of the produced base is at a given irradiation time, the stronger is the ability to initiate the polymerization. When the 1 or 2 wt % TBD·HBPh4 formulation is used, a shoulder peak can be observed in the DSC curves. This feature may indicate the presence of a second curing process of the thiol-epoxy under noncatalytic conditions.19 Moreover, this would indicate insufficient curing in the presence of a very low amount of TBD·HBPh4. When the TBD·HBPh4 content is increased to 3 wt %, the shoulder peak disappears. Additionally, as reported by other authors, higher TBD·HBPh4 content leads to a premature release of the heat from the formulation. This observation is consistent behavior of containing 4 wt % TBD·HBPh4, which exhibits a smaller heat flow peak area. From the results of the study, a TBD·HBPh4 content of 3 wt % was found to give optimum results and was selected to carry out the subsequent thiol-epoxy curing reactions.
Figure 5.
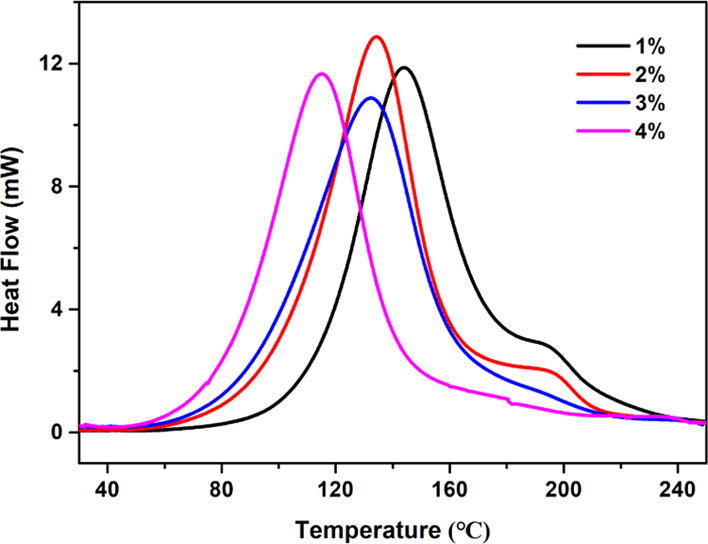
DSC heat flow curves of the thiol-epoxy samples with different TBD·HBPh4 contents after irradiation for 20 min.
The irradiation time is critical for the concentration of the base-active species produced via the TBD·HBPh4 photolysis, since this concentration affects the speed and the degree of the thiol-epoxy curing reaction. To evaluate the effect of the irradiation time on the photoactivation of TBD·HBPh4 (3 wt %), thiol-epoxy formulations were irradiated while stirring for different times and subsequently, the curing process was monitored via nonisothermal DSC, as shown in Figure 6. The initial curing temperature of the nonirradiated sample is higher than 120 °C because thermal activation of TBD·HBPh4 takes place, as it has been demonstrated in the literature. Due to the active base released during the photolysis process, a clear decrease of the initial curing temperature upon extending the irradiation time is observed. Moreover, after a rather short irradiation time (5 min), the DSC curve exhibits a broad shape of low intensity accompanied by a large shoulder peak at high temperature, indicating that the irradiation time was not long enough to completely photoactivate TBD·HBPh4 and hence thermal decomposition of the nonreacted TBD·HBPh4 is observed. By increasing the irradiation time up to 20 min, the shoulder peak disappears, indicating that almost complete photodecomposition of TBD·HBPh4 was achieved. When the irradiation time becomes longer than 20 min, the curing reaction starts at near-ambient temperature, but this would be undesirable for applications requiring a longer shelf life. Based on the results, an irradiation time of 20 min was selected to balance stability and reactivity for studying the subsequent thiol-epoxy curing reactions.
Figure 6.
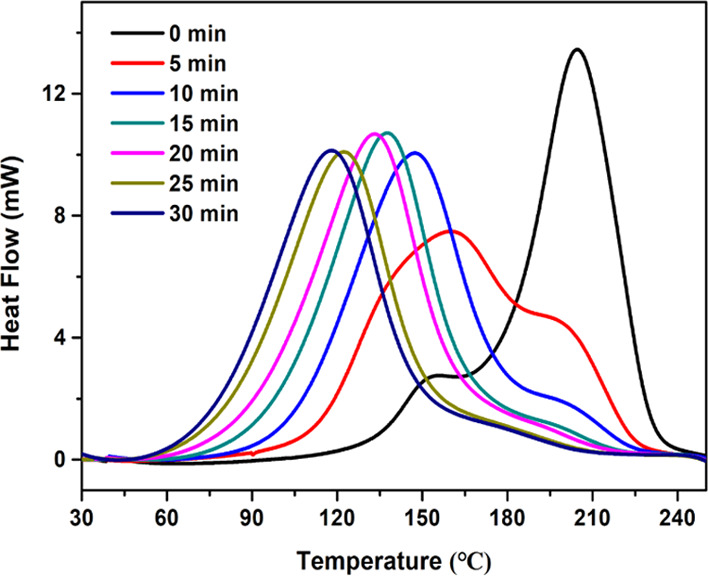
DSC heat flow curve of thiol-epoxy samples containing 3 wt % TBD·HBPh4 with different irradiation times.
Since the temperature promotes, to a certain extent, the release and diffusion of the active base, increasing the temperature can accelerate the curing reaction. Therefore, the effect of the postbaking temperature on the time to achieve completely cured thiol-epoxy formulations was investigated via isothermal DSC. Figure 7 shows the DSC heat flow as a function of the reaction time for the thiol-epoxy samples at different isothermal temperatures. By increasing the isothermal temperature, the time for full cure of the samples is shortened. This result proves that the postbaking temperature of the sample significantly affects the rate of the curing reaction. When the isothermal temperature is 70 °C, the sample remains insufficiently cured after 60 min at isothermal temperature (the heat flow peak is still present). By increasing the isothermal temperature to 90 °C, the DSC heat flow curve gradually narrows and the time for complete cure is shortened to 30 min. When the isothermal temperature is increased to higher than 100 °C, the heat flow curve becomes sharp and the integral area of the peak is significantly reduced, indicating that a large amount of heat has already been released during the heating process. Consequently, a postbaking temperature of 90 °C was selected to balance the energy consumption and reactivity for subsequent investigations of the thiol-epoxy curing reaction.
Figure 7.
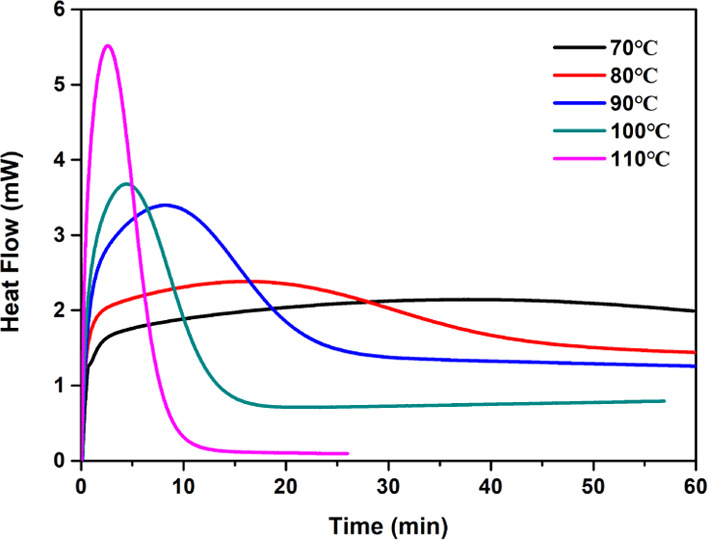
DSC plot of the heat flow as a function of the reaction time for the thiol-epoxy samples at different isothermal temperatures.
Analysis of the Properties of Thiol-Epoxy Samples of High Thickness
Thick cylindric thiol-epoxy samples (r = 1.5 cm, h = 4 cm) were prepared using the optimized parameters (3 wt % photobase generator TBD·HBPh4, 20 min of irradiation, and 90 °C postbaking temperature). In principle, thiol-epoxy samples with arbitrary size can be prepared via DTEP with molds of desired shape and adjusted parameters. The videotaped curing process and a picture of a thick cured sample are shown in the Supporting Information Movie 1 and Figure S2, respectively.
To evaluate the uniformity of the cross-linked network of the thick cured sample, the conversion of the functional groups (epoxy and thiol) in the samples was investigated via attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy. To this end, the cured sample columns were cut into slices of 0.5 cm thickness, and the ATR-FTIR spectra were measured at the surfaces. Figure 8 shows the thiol and epoxy group conversion in the thick sample at different radii and depths. The medium thiol conversions at different radii and depths are 88.2 ± 1.9 and 87.4 ± 2.0%, respectively. The medium epoxy conversions are 87.6 ± 2.0 and 87 ± 2.1%, respectively. These results are almost identical. This shows that the degree of the cross-linking in the cured samples at different positions is equivalent. Moreover, the samples show a relatively excellent network uniformity, which is due to the uniform dispersion of the base-active species throughout the formulation system (including the unirradiated area). This high degree of uniformity was achieved by irradiating and stirring the sample and it caused the curing reaction to initiate simultaneously all over the sample thickness. By comparing the conversions of the thiol and the epoxide for a fixed radius and depth, the results are roughly equivalent. This suggests that the thiol-epoxy curing reaction catalyzed by the photobase generator was carried out in a stoichiometric 1:1 ratio.
Figure 8.
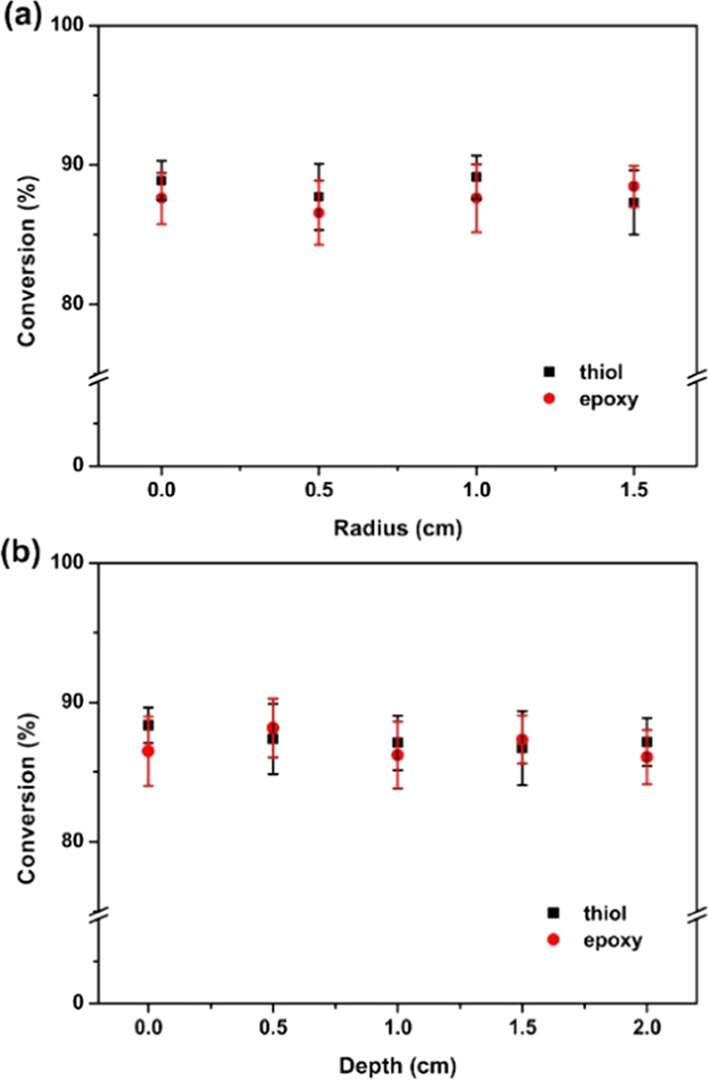
Conversions of the thiol-epoxy thick cured deep-solidified samples with (a) different depths and (b) radii.
Cured thiol-epoxy samples taken at different radii and depths obtained by cutting the cured column as described before were studied via thermogravimetric analysis (TGA) to evaluate their thermal stability. Figure 9 shows their weight loss curves and their quantitative thermal properties. Moreover, it shows the resultant 5% weight loss temperatures (Td5%) and the temperatures corresponding to the maximum weight loss (Tdmax) for different radii and depths. The degradation curves show almost identical shapes and the same amount of completely decomposed residual carbon. The cured samples taken at different radii and depths exhibit substantially the same Td5% and Tdmax (273.3 ± 2.8 and 317.6 ± 1.0 °C, respectively). These findings show that the thermal stability of the samples at different radii and depths is identical. This indicates that the curing occurred very homogeneously over the entire volume of the sample material, and this is consistent with the nearly equivalent thiol and epoxy conversions determined using ATR-FTIR (Figure 8). Undoubtedly, this result can be attributed to the unique curing method reported in this article, which consists of the irradiation of the formulation while stirring.
Figure 9.
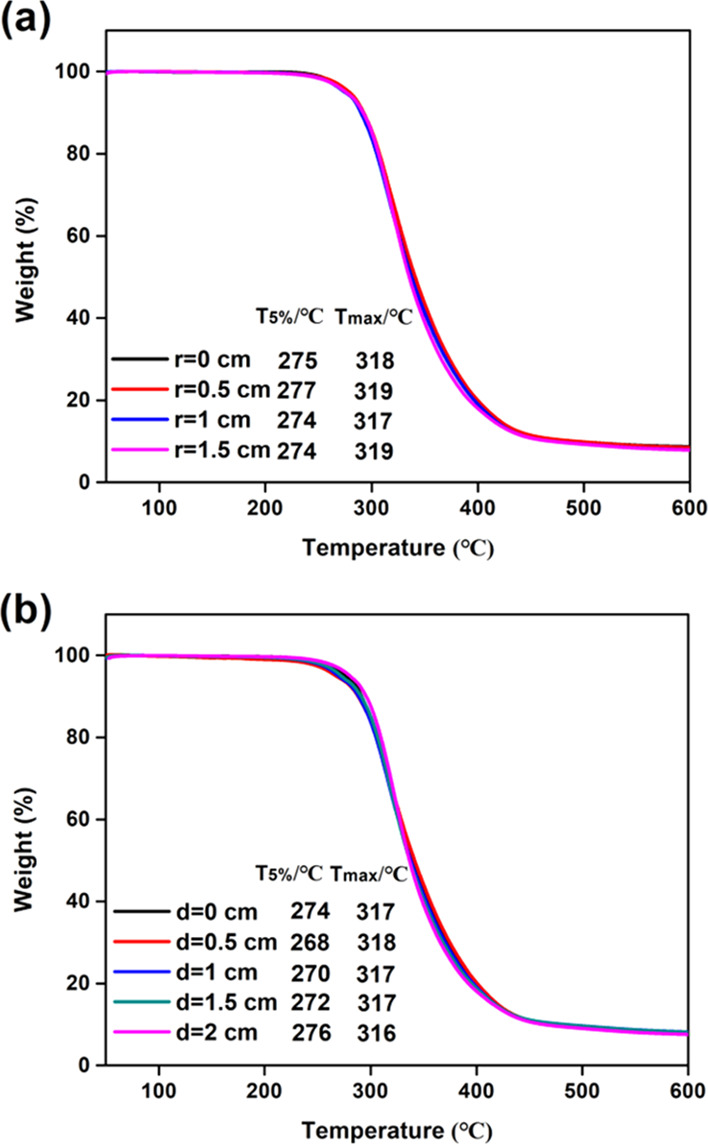
TGA curves of cured thiol-epoxy samples at (a) different radii and (b) different depths.
The glass transition temperature (Tg) of samples taken at different radii and depths of the cured thick sample was analyzed via differential scanning calorimetry to evaluate the thermal behavior of the material at different positions. Figure 10 shows the DSC curves of the thiol-epoxy samples at different radii and depths together with their corresponding Tg values. All of the samples exhibit a similar shape of the glass transition zone and almost identical Tg values, which fluctuate around 49 °C (Figure 10c). Again, these results show that the degree of cross-linking of the polymer network at different positions is almost equal, resulting in almost equal Tg values all over the entire volume of the sample.
Figure 10.

DSC curves of thick cured thiol-epoxy samples at (a) different depths, (b) radii, and (c) medium Tg at different depth and radii.
Feasibility and Performance Comparison of Curing Thick Samples of Thiol-Epoxy Composites
After demonstrating that a homogeneously cured thiol-epoxy sample can be prepared by delayed thiol-epoxy anionic photopolymerization, we continued to apply such a strategy to cure thick thiol-epoxy composites. The thiol-epoxy formulation activated by light and stirring was embedded with an amount of glass fiber powder and was cured by the temperature-controlled thiol-epoxy polymerization. Generally, glass fibers can effectively strengthen the mechanical properties of the materials due to their strength and stiffness values, which are higher than the ones of the matrices. Thus, tensile and flexural properties of the thiol-epoxy cured materials with 1 wt % glass fiber were investigated and compared to glass fiber-free samples of the same thickness. The results are summarized in Table 1. Since the cross-link density of the system is not affected, the addition of 1 wt % glass fiber powder increases the tensile strength and the flexural strength approximately by 15.4 and 31.9%, respectively. Moreover, Young’s modulus and the flexural modulus of the samples increase by approximately 8.4 and 51.9%, respectively. These findings show that thick thiol-epoxy composites can be prepared via DTEP and that this technique drastically improves the mechanical properties of the materials. It should be noted that the glass fiber powders used in the tensile and in the flexural tests were not modified. Further improvements could be achieved by employing fibers with optimized surface treatments, such as coupling agents specifically tailored to resin chemistry.36,37
Table 1. Mechanical Properties of the Thick Thiol-Epoxy Cured Materials with and without Glass Fiber Powder.
| sample | tensile strength (MPa) | Young’s modulus (MPa) | flexural strength (MPa) | flexural modulus (GPa) |
|---|---|---|---|---|
| 1a | 56.6 | 1666.2 | 128.8 | 7.9 |
| 2b | 65.3 | 1806.5 | 169.9 | 12 |
The thiol-epoxy sample without fiber.
The thiol-epoxy sample with 1 wt % fiber.
Due to the favorable properties of carbon fiber reinforced materials in many applications, there is a high need in the industry for a simple, reliable, and economically and ecologically favorable process. Although “radiation curing” would be the process of choice in view of the balance between long pot life and fast cure speed, it is not applicable due to the light screening effect of the fillers.
Several strategies have been introduced to overcome the inherent shortcomings of radiation curing. One approach involves the stepwise curing of thin resin layers that may be impregnated with carbon fibers.38 The thin layers allow for sufficient penetration of light. A disadvantage of this multistep process is that it is rather time-consuming when fabricating thick layers. An additional disadvantage is that the final material is not homogeneously cured due to the stepwise buildup fashion resulting in lower adhesion between individual layers, which may cause undesirable physical properties of the cured materials.
Another approach proposed in the literature is frontal photopolymerization,39 where a thermal curing reaction is activated by light at the surface of the material and propagates, driven by its own polymerization exothermic, into deeper layers. While this concept is feasible and principally elegant, it is not widely applicable due to a difficult control of the curing condition. Moreover, the exothermic and thus the local temperature must be relatively high to ensure a full thorough cure. Such locally high temperatures are, however, not tolerated in many applications.
The presented DTEP strategy is a simple process with significant advantages for industrial applications. An outstanding advantage of the presented proceeding as compared to other processes aimed at the production of carbon fiber reinforced materials is its technical simplicity, which allows its realization in normal application labs without the need of special and costly equipment and severe reaction control. This is in contrast to the layerwise buildup of such materials, which requires a multistep proceeding partly performed by robots or the frontal polymerization process that requires exact control of the reaction parameters (e.g., temperature, speed of the proceeding of the polymerizing front).
Moreover, due to the fact that the photochemical activation of the catalyst is performed in a separate step, disconnected from the proper polymerization step, there are no limits regarding the thickness, the type of fillers or pigments to be included, and the dimensional shape of the final article (it is well known that three-dimensional objects are difficult to cure by simple photopolymerization due to geometrical restriction hindering uniform irradiation). As a proof of concept, a hard cylindrical (r = 1.5 cm, h = 4 cm) thick dark composite containing 2 wt % black carbon nanotubes was successfully obtained via DTEP. The digital picture and the videotaped curing process of this thiol-epoxy black composite are shown in Figure 11 and in the Supporting Information, Movie 2, respectively.
Figure 11.

Thiol-epoxy cured sample with 2 wt % black carbon nanotubes.
Conclusions
We present a simple and versatile strategy to prepare thick composites based on delayed thiol-epoxy photopolymerization (DTEP). The concentration of TBD·HBPh4, irradiation time, and postbaking temperature are variables that show a remarkable influence on the curing process and the properties of these properties in the cured material. The cured thick samples of the thiol-epoxy formulation exhibit comparable functionality conversions, similar Tg values, and thermal decomposition temperatures at different radii and depths, indicating an excellent uniformity of the cured materials. Significantly improved mechanical properties can be obtained when 1 wt % fiber of glass fiber is added to the thiol-epoxy formulation. The same strategy can also be applied to produce dark composites containing 2 wt % carbon nanotubes.
Materials and Methods
Materials
Pentaerythritol-tetrakis-3-mercaptopropionate (PE-1) was purchased from Japan Showa Denko, Chembridge International Corp (Taiwan), and used without any purification. Bisphenol A diglycidyl ether (BADGE) (E51) with an epoxy equivalent weight of 196 g/mol was obtained from Jiangsu Sanmu Group Co. Ltd. and dried in vacuum prior to use. 1,5,7-Triazabicyclo[4.4.0] dec-5-ene (TBD) and tetraphenylboron sodium (NaBPh4) were purchased from Adamas Reagent Co. Ltd., whereas hydrochloric acid (HCl) and methyl alcohol (MeOH) were purchased from Sinopharm Chemical Reagent Co., Ltd. 2-Isopropylthioxanthone (ITX) was supplied by BASF. Glass fibers with 9–13 μm particle size were purchased from Sigma-Aldrich Chemical Reagent Co. Ltd. A short arm carbon nanotube was purchased from Tokyo Chemical Industry Development Co., Ltd.$
Preparation and Photocuring of Thiol-Epoxy Photopolymerizable Samples
Since the photobase generator (TBD·HBPh4) and the photosensitizer (ITX) are sensitive to light, the thiol-epoxy formulations containing the initiating system were prepared in the dark. TBD·HBPh4 (1–3 wt %) and a certain amount of bisphenol A epoxy resin E51 were accurately weighed and inserted into a clean 100 mL blue cap bottle. They were mixed by stirring at 100 °C until the vials appeared to be clear and transparent. Typically, it takes about 2 h to obtain a clear formulation when using a 3 wt % photobase generator (PBG). At this point, the mixtures were cooled to room temperature. The PE-1 monomer was added to the mixture in a quantity where epoxy and thiol functional groups were present in equimolar amounts. To prevent any premature activation, ITX was added at the end and its amount was half the weight of TBD·HBPh4 in all formulations. The mixture was stirred continuously at 100 °C until a homogeneous, still low-viscous thiol-epoxy formulation was obtained. The ready-to-use formulation was stored in the dark at a low temperature. Subsequently, the thiol-epoxy formulations (Table S1) were poured into a glass culture dish (Φ = 3 cm, h = 4 cm). A 6 mm × 25 mm magnetic stir bar was added and a 385 nm LED spot lighting system (UVEC-4IIB, Shenzhen Lamplic Technology Co. Ltd.) was adjusted to ensure that the light spot (Φ = 1 cm) was in the center of the culture dish. The sample was then irradiated vertically. The irradiation intensity was adjusted to 196 mW/cm2 at the resin surface to activate TBD·HBPh4 via sensitization of ITX. Since the base-catalyzed thiol/epoxy addition proceeds very slowly at room temperature, the activated resin remains of low viscosity over a long time and can easily be stirred. After irradiation and stirring for a certain period of time, a small amount of the material was selected for the DSC tests. The remaining material was poured into tetrafluoroethylene molds (cylindrical, dumbbell, and rectangle) and heated up in an oven to temperatures of 70, 80, 90, 100, and 110 °C. The cured samples were subsequently removed from the molds and used to perform several tests. For the composite preparation, 1 wt % glass fiber powder or 2 wt % carbon nanotubes was added to the formulations prior to irradiation.
Characterization
UV–Vis Absorption Spectroscopy Measurements
A series of solutions containing different concentrations of TBD·HBPh4 and ITX in acetonitrile was prepared. The absorbance of the main absorption peak was measured using a UV–vis spectrophotometer to obtain the absorbance of the samples. Moreover, the acetonitrile solution of the TBD·HBPh4 and ITX mixture at a concentration of 2.0 × 10–3 mol/L was also irradiated. The content of the base released by TBD·HBPh4 was measured by adding a fixed amount of a phenol red solution after the irradiation experiment (see below).
Analysis of the Curing Process
Differential scanning calorimeter (DSC) Mettler DSC1/1100SF was used to determine the kinetics of the curing process. All of the thiol-epoxy samples (8.7 ± 0.1 mg) were analyzed under isothermal and nonisothermal conditions in a constant N2 atmosphere with a gas flow of 10 mL/min. The isothermal experiments were carried out in the temperature range of 30–250 °C, whereas the nonisothermal experiments were performed at temperatures of 70, 80, 90, 100, and 110 °C.
Testing of the Thiol-Epoxy Curing Degree during the Irradiation Process
The formulation containing 3 wt % TBD·HBPh4 and 1.5 wt % ITX was poured into a Petri dish prior to illumination. During irradiation over a period of 20 min, an appropriate amount of samples was withdrawn at intervals of 5 min. The molecular weight and the viscosity at 25 °C were measured via a GPC and a rheometer, respectively.
Thiol and Epoxy Conversion Measurements
After irradiation, the samples were heated to 90 °C for 20 min. The cured cylindric polymer samples (r = 1.5 cm, h = 4 cm) obtained in the deep curing experiment were cut into several parts of different radii (r = 0.5, 1, and 1.5 cm) and depths (h = 0.5, 1, 1.5, and 2 cm), as shown in Figure S1. The cross-section layer for different depths and r = 0 cm and the top layer at different radii and h = 1 cm were ground into fine powders and then mixed with KBr to give KBr pellets (5 wt %). A Fourier transform infrared spectrometer was used to measure the absorbance peaks of the uncured and cured samples (ATR-FTIR, Nicolet 6700, Thermo Fisher Scientific, wavelength range of 500–4000 cm–1, resolution of 16 cm–1, 4 scans per sample). The thiol and the epoxy group conversions were determined based on the ratio between the absorbance intensity of the thiol (peak at 2570 cm–1) and of the epoxy (peak at 760 cm–1), respectively, using the C=O peak (1717 cm–1) as a reference. The conversion can be described via the following formula
where At and A0 denote the absorbance intensities of the thiol or the epoxy before and after the curing process, respectively, whereas Arb and Ara represent the absorbance intensities of the C=O bond before and after the curing process, respectively.
Analysis of Thermal and Mechanical Properties
The glass transition temperatures of samples of the thiol-epoxy thick cured material taken at different radii and depths were determined using DSC-8000 (TA Instruments). All of the samples were heated up to 100 °C at a rate of 40 °C/min in a nitrogen atmosphere to erase their thermal history. This process was followed by cooling at a rate of 10 °C/min. The thermal stability of the samples taken at different radii and depths was investigated by thermogravimetric analysis (TGA-1100SF, Mettler Toledo International Trading Co. Ltd., Switzerland). The samples were heated from 30 to 600 °C under a continuous N2 flow of 50 mL/min and with a heating rate of 10 °C/min. The tensile and flexural properties of the thiol-epoxy samples with and without glass fibers were measured according to ASTM-D638 and ASTM-D790 using an Instron Universal testing machine (model 5967, Instron) with a crosshead speed of 10 mm/min. The sizes of the tensile and flexural samples were 75 mm × 12.5 mm × 3.6 mm and 75 mm × 3.7 mm × 2 mm, respectively. Both the tensile and the flexural test were carried out on at least five specimens to obtain a reliable average.
Acknowledgments
The authors acknowledge the financial support from the Natural Science Foundation of Jiangsu Province (BK20191336) and the National First-class Discipline Program of Food Science and Technology (JUFSTR20180301). One of the authors (K.D.) is indebted to China for providing a visiting scientist grant through the International Collaboration program (MOE & SAFEA, 111 Project (B13025)).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01170.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hwang J.; Lee D. G.; Yeo H.; Rao J.; Zhu Z.; Shin J.; Khan A.; et al. Proton transfer hydrogels: Versatility and applications. J. Am. Chem. Soc. 2018, 140, 6700–6709. 10.1021/jacs.8b03514. [DOI] [PubMed] [Google Scholar]
- Zivic N.; Kuroishi P. K.; Dumur F.; Gigmes D.; Dove A. P.; Sardon H. Recent advances and challenges in the design of organic photoacid and photobase generators for polymerizations. Angew. Chem., Int. Edit. 2019, 58, 10410–10422. 10.1002/anie.201810118. [DOI] [PubMed] [Google Scholar]
- Gadwal I.; Rao J.; Baettig J.; Khan A. Functionalized molecular bottlebrushes. Macromolecules 2014, 47, 35–40. 10.1021/ma402259q. [DOI] [Google Scholar]
- Huynh C. T.; Liu F.; Cheng Y.; Coughlin K. A.; Alsberg E. Thiol-epoxy “Click” chemistry to engineer cytocompatible PEG-based hydrogel for siRNA-mediated osteogenesis of hMSCs. ACS Appl. Mater. Interfaces 2018, 10, 25936–25942. 10.1021/acsami.8b07167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadwal I.; Stuparu M. C.; Khan A. Homopolymer bifunctionalization through sequential thiol–epoxy and esterification reactions: an optimization, quantification, and structural elucidation study. Polym. Chem. 2015, 6, 1393–1404. 10.1039/C4PY01453G. [DOI] [Google Scholar]
- De S.; Khan A. Efficient synthesis of multifunctional polymers via thiol–epoxy “click” chemistry. Chem. Commun. 2012, 48, 3130–3132. 10.1039/c2cc30434a. [DOI] [PubMed] [Google Scholar]
- Cengiz N.; Rao J.; Sanyal A.; Khan A. Designing functionalizable hydrogels through thiol–epoxy coupling chemistry. Chem. Commun. 2013, 49, 11191–11193. 10.1039/c3cc45859h. [DOI] [PubMed] [Google Scholar]
- Stuparu M. C.; Khan A. Thiol-epoxy “click” chemistry: Application in preparation and postpolymerization modification of polymers. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 3057–3070. 10.1002/pola.28195. [DOI] [Google Scholar]
- Carioscia J. A.; Stansbury J. W.; Bowman C. N. Evaluation and control of thiol-ene/thiol-epoxy hybrid networks. Polymer 2007, 48, 1526–1532. 10.1016/j.polymer.2007.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brändle A.; Khan A. Thiol–epoxy ‘click’polymerization: efficient construction of reactive and functional polymers. Polym. Chem. 2012, 3, 3224–3227. 10.1039/c2py20591b. [DOI] [Google Scholar]
- Nowak A. P.; Adjorlolo Rodringuez A. R.; Boundy T. I.; Pajel C. A.; Adjorlolo A. A.. Rapid Curing Thiol Epoxy Resin with Improved Compression Strength Performance. U.S. Patent US10035873B22018.
- Yang X.; Guo Y.; Luo X.; Zheng N.; Ma T.; Tan J.; Li C.; Zhang Q.; Gu J. Self-healing, Recoverable Epoxy Elastomers and Their Composites with Desirable Thermal Conductivities by Incorporating Bn Fillers Via In-situ Polymerization. Compos. Sci. Technol. 2018, 164, 59–64. 10.1016/j.compscitech.2018.05.038. [DOI] [Google Scholar]
- Ellson G.; Di Prima M.; Ware T.; Tang X.; Voit W. Tunable thiol–epoxy shape memory polymer foams. Smart Mater. Struct. 2015, 24, 055001 10.1088/0964-1726/24/5/055001. [DOI] [Google Scholar]
- Isarn I.; Ramis X.; Ferrando F.; Serra A. Thermoconductive thermosetting composites based on boron nitride fillers and thiol-epoxy matrices. Polymers 2018, 10, 277 10.3390/polym10030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lungu A.; Ghitman J.; Cernencu A. I.; Serafim A.; Florea N. M.; Vasile E.; Iovu H. POSS-containing hybrid nanomaterials based on thiol-epoxy click reaction. Polymer 2018, 145, 324–333. 10.1016/j.polymer.2018.05.015. [DOI] [Google Scholar]
- Guzmán D.; Ramis X.; Fernández-Francos X.; Serra A. New catalysts for diglycidyl ether of bisphenol A curing based on thiol–epoxy click reaction. Eur. Polym. J. 2014, 59, 377–386. 10.1016/j.eurpolymj.2014.08.001. [DOI] [Google Scholar]
- Sanda F.; Kaizuka T.; Sudo A.; Endo T. A novel thermally latent anionic initiator. Polymerization of epoxide with hydroxylamide based on thermal dissociation. Macromolecules 2003, 36, 967–968. 10.1021/ma0257292. [DOI] [Google Scholar]
- Kirino M.; Tomita I. Aminimides derived from benzoylformic acid esters as thermally latent base catalysts. Macromolecules 2010, 43, 8821–8827. 10.1021/ma1013836. [DOI] [Google Scholar]
- Konuray A. O.; Fernández-Francos X.; Ramis X. Latent curing of epoxy-thiol thermosets. Polymer 2017, 116, 191–203. 10.1016/j.polymer.2017.03.064. [DOI] [Google Scholar]
- Sangermano M.; Vitale A.; Dietliker K. Photolatent amines producing a strong base as photocatalyst for the in-situ preparation of organic–inorganic hybrid coatings. Polymer 2014, 55, 1628–1635. 10.1016/j.polymer.2014.02.045. [DOI] [Google Scholar]
- Salmi H.; Allonas X.; Ley C.; Defoin A.; Ak A. Quaternary ammonium salts of phenylglyoxylic acid as photobase generators for thiol-promoted epoxide photopolymerization. Polym. Chem. 2014, 5, 6577–6583. 10.1039/C4PY00927D. [DOI] [Google Scholar]
- Seubert C. M.; Nichols E. M. Epoxy thiol photolatent base clearcoats: curing and formulation. J. Coat. Technol. Res. 2010, 7, 615–622. 10.1007/s11998-010-9248-3. [DOI] [Google Scholar]
- Yeo H.; Khan A. Photoinduced Proton-Transfer Polymerization: A Practical Synthetic Tool for Soft Lithography Applications. J. Am. Chem. Soc. 2020, 142, 3479–3488. 10.1021/jacs.9b11958. [DOI] [PubMed] [Google Scholar]
- Kardar P.; Ebrahimi M.; Bastani S. J. UV curing behavior and mechanical properties of unpigmented and pigmented epoxy acrylate/SiO2 nanocomposites. Therm. Anal. Calorim. 2016, 124, 1425–1430. 10.1007/s10973-016-5306-0. [DOI] [Google Scholar]
- Jahn R.; Jung T. Relationship between pigment properties and UV-curing efficiency. Prog. Org. Coat. 2001, 43, 50–55. 10.1016/S0300-9440(01)00221-1. [DOI] [Google Scholar]
- Decker C.; Zahouily K.; Decker D.; Nguyen T.; Viet T. Performance analysis of acylphosphine oxides in photoinitiated polymerization. Polymer 2001, 42, 7551–7560. 10.1016/S0032-3861(01)00221-X. [DOI] [Google Scholar]
- Jandt K. D.; Mills R. W. A brief history of LED photopolymerization. Dent. Mater. 2013, 29, 605–617. 10.1016/j.dental.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Yang L.; Yang J.; Nie J.; Zhu X. Temperature controlled cationic photo-curing of a thick, dark composite. RSC Adv. 2017, 7, 4046–4053. 10.1039/C6RA25346F. [DOI] [Google Scholar]
- Gugg A.; Gorsche C.; Moszner N.; Liska R. Frontal polymerization: polymerization induced destabilization of peracrylates. Macromol. Rapid Commun. 2011, 32, 1096–1100. 10.1002/marc.201100188. [DOI] [PubMed] [Google Scholar]
- He M.; Huang X.; Zeng Z.; Yang J. Phototriggered base proliferation: a highly efficient domino reaction for creating functionally photo-screened materials. Macromolecules 2013, 46, 6402–6407. 10.1021/ma400983t. [DOI] [Google Scholar]
- Li Z.; Chen H.; Wang C.; Chen L.; Liu J.; Liu R. Efficient photopolymerization of thick pigmented systems using upconversion nanoparticles-assisted photochemistry. J. Polym. Sci., Part A: Polym. Chem. 2018, 56, 994–1002. 10.1002/pola.28969. [DOI] [Google Scholar]
- Sun X.; Gao J. P.; Wang Z. H. Bicyclic guanidinium tetraphenylborate: a photobase generator and a photocatalyst for living anionic ring-opening polymerization and cross-linking of polymeric materials containing ester and hydroxy groups. J. Am. Chem. Soc. 2008, 130, 8130–8131. 10.1021/ja802816g. [DOI] [PubMed] [Google Scholar]
- Arimitsu K.; Endo R. Application to photoreactive materials of photochemical generation of superbases with high efficiency based on photodecarboxylation reactions. Chem. Mater. 2013, 25, 4461–4463. 10.1021/cm4022485. [DOI] [Google Scholar]
- Jian Y.; He Y.; Sun Y.; Yang H.; Yang W.; Nie J. Thiol–epoxy/thiol–acrylate hybrid materials synthesized by photopolymerization. J. Mater. Chem. C 2013, 1, 4481–4489. 10.1039/c3tc30360h. [DOI] [Google Scholar]
- Chen L.; Wu Q.; Wei G.; Liu R.; Li Z. Highly stable thiol–ene systems: from their structure–property relationship to DLP 3D printing. J. Mater. Chem. C 2018, 6, 11561–11568. 10.1039/C8TC03389G. [DOI] [Google Scholar]
- Ku H.; Wang H.; Pattarachaiyakoop N.; Trada M. A review on the tensile properties of natural fiber reinforced polymer composites. Composites, Part B 2011, 42, 856–873. 10.1016/j.compositesb.2011.01.010. [DOI] [Google Scholar]
- Fu S.-Y.; Lauke B.; Mäder E.; Yue C.-Y.; Hu X. Tensile properties of short-glass-fiber-and short-carbon-fiber-reinforced polypropylene composites. Composites, Part A 2000, 31, 1117–1125. 10.1016/S1359-835X(00)00068-3. [DOI] [Google Scholar]
- Marechal D.; Criqui A.; Allonas X.; Lecompere M.. Dual-Cure Cationically Polymerisablecomposition and Method for Producing a Coating or A Composite Material Implementing Saidcomposition. U.S. Patent US102145972019.
- Liska R.; Bomze D.; Kern W.; Knaack P.. Process for Frontal Polymerization of Cationically Polymerizable Monomers. U.S. Patent U.S15/7570372019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.