

ABSTRACT
Introduction
Congenital ichthyosiform erythroderma (CIE ) is characterized by fine, whitish scales on a background of erythematous skin over the whole body; it is reportedly caused by mutations in ABCA12, ALOX12B, ALOXE3, CERS3, CYP4F22, NIPAL4, PNPLA1, and TGM1 genes.
Case presentation
A 15‐month‐old girl presented with CIE associated with compound heterozygous ABCA12 mutations, a known missense mutation c.4139A>G (p.Asn1380Ser) from her father, and a novel missense mutation c.4300A>G (p.Thr1434Ala) from her mother.
Conclusion
This is the first report to indicate that compound heterozygous missense mutations in the first ATP‐binding cassette of ABCA12 could contribute to the onset of CIE.
Keywords: Congenital ichthyosiform erythroderma, ABCA12, Gene mutation
INTRODUCTION
Mutations in ABCA12 have been described in patients with autosomal recessive congenital ichthyoses including congenital ichthyosiform erythroderma (CIE), harlequin ichthyosis (HI), and lamellar ichthyosis (LI). The most severe phenotype, with the largest numbers of truncation or deletion mutations, has been observed in patients with HI. The presence of at least one non‐truncating mutation (predictive of residual protein function) typically causes less severe congenital ichthyosis (CIE or LI). The patients with these mutations are typically born with a collodion membrane, which is sheds in the first few weeks of life. Subsequently, CIE manifests as fine, whitish scales on a background of erythematous skin; while conversely, LI is characterized by the onset of large, thick, dark scales over the surface of the entire body, without serious background erythroderma. 1 , 2 , 3
ABCA12 mutations are reported to underlie autosomal recessive congenital ichthyoses in patients of European, African, Pakistani, Saudi Arabian, Israeli, and Spanish origin; in patients of Japanese origin, ABCA12 mutations are frequently associated specifically with CIE. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 However, such associations have rarely been reported in Chinese patients. 4 Here, we describe a Chinese girl with CIE who carried two ABCA12 mutations, one of which was novel and not previously described.
CASE REPORT
This study was approved by the Ethical Committee of Beijing Children’s Hospital, Capital Medical University and was conducted in accordance with the principles of the Declaration of Helsinki. Informed consent was obtained from the patient’s parents.
A 15‐month‐old girl with whitish scales on an erythematous background with slight ectropion and the absence of palmoplantar keratoderma was admitted to our hospital. Her hair, eyebrows, and eyelashes were noticeably sparse (Figure 1). She exhibited hypohidrosis and mild heat intolerance. Her hearing and vision were normal, and she showed no abnormalities in the heart or abdomen, based on ultrasound examinations. She was born with a collodion membrane; notably, no other members of her family had similar skin manifestations. She was finally diagnosed with CIE. Accordingly, emollients were used to control scaling and to improve the skin barrier function.
FIGURE 1.
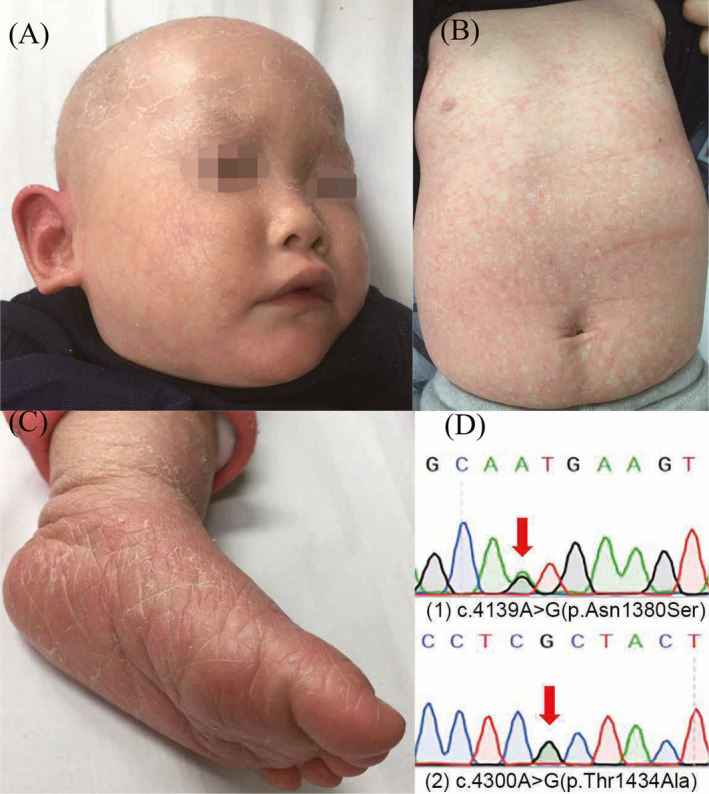
Clinical features and genetic analysis of a 15‐month‐old girl with congenital ichthyosiform erythroderma. (A) Her hair, eyebrows, and eyelashes were clearly sparse with slight ectropion. (B) Whitish scales on an erythematous background. (C) Absence of palmoplantar keratoderma. (D) Compound heterozygous ABCA12 mutations were detected in the patient.
Blood was extracted from the patient and her parents to perform genetic diagnosis. Genomic DNA was isolated using standard procedures. The sequencing library constructed from the DNA sample of the patient was subjected to next‐generation sequencing (NGS) for a panel of genes involved in ichthyosis (designed by Beijing Pediatric Research Institute). Coding regions of the target genes and 10‐bp portions of adjacent introns were sequenced using custom‐designed capture probes (Agilent Technologies, Santa Clara, CA, USA). Sequence variants identified in the patient by NGS were verified by Sanger sequencing in the patient and her parents. The results revealed compound heterozygous ABCA12 mutations including a known paternal missense mutation c.4139A>G (p.Asn1380Ser), and a novel maternal missense mutation c.4300A>G (p.Thr1434Ala); the maternal mutation was predicted to be pathogenic by SIFT, Polyphen and Mutation taster. To the best of our knowledge, it has not been previously reported (based on searches of HGMD, ClinVar and PubMed) and is absent from the population (based on searches of 1000 Genomes, ExAC and dbSNPs).
DISCUSSION
HI, LI, and CIE are autosomal recessive congenital ichthyoses, which form a heterogeneous group of non‐syndromic disorders of keratinization. The genotype‐phenotype correlation is not very clear in patients with these disorders, because mutations in the same genes may lead to divergent phenotypes, while a single phenotype may be associated with any of several underlying mutations. CIE can be caused by mutations in the ABCA12, ALOX12B, ALOXE3, CERS3, CYP4F22, NIPAL4, PNPLA1, and TGM1 genes, as well as mutations in other unidentified genes. 1 , 2 , 3 , 4
ABCA12 belongs to a subfamily of the ATP‐binding cassette transporter protein superfamily, which carries out energy‐dependent transport of substrate molecules. Notably, ABCA12 can bind and hydrolyze ATP to facilitate the transport of lipids into the trans‐Golgi network and lamellar granules, thereby enabling the accumulation of lipids that are necessary in formation of the skin barrier. 5 , 6 Other findings suggest an additional essential role for ABCA12 in transferring proteolytic enzymes that are required for normal desquamation. 7 ABCA12 contains two transmembrane domains, each with approximately six membrane‐spanning segments, and two ATP‐binding cassettes in the cytoplasm (Figure 2). Importantly, ABCA12 is expressed in keratinocytes. Therefore, ABCA12 deficiency causes disruptions in lamellar granule lipid transport, which lead to defective intercellular lipid layers in the stratum corneum and altered integrity of the skin barrier function; these changes result in extreme compensatory hyperkeratosis. 1 , 2
FIGURE 2.
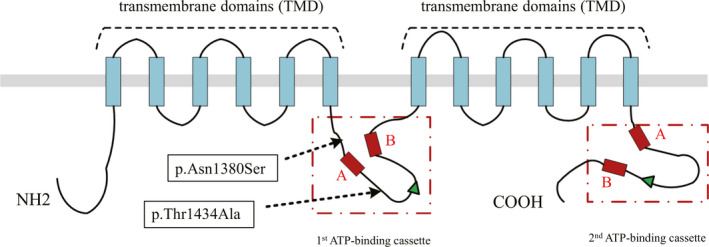
Structure of the ABCA12 protein and the two mutations detected in this patient. The ATP‐binding cassettes contain three characteristic highly conserved motifs: Walker A motif (the red box A), Walker B motif (the red box B) and active transport signature (the green array).
Distinct mutations in ABCA12 may produce markedly disparate phenotypes. More than 93% of patients with HI reportedly exhibit homozygous or heterozygous truncation and deletion mutations in ABCA12, which lead to considerable loss of function. The most severe phenotype is present in patients with HI, which is characterized by thick, fissured, armor‐plate hyperkeratosis; severe eclabium and ectropion; auricle malformation; and early complications. Patients with less severe congenital ichthyoses, such as CIE or LI, carry at least one non‐truncating mutation, although a few have two truncation mutaions. 1 , 2 , 3 , 4 Lefevre et al 8 reported that the mutations (homozygous p.Asn1380Ser, p.Gly1381Glu, p.Arg1514His, p.Glu1539Lys, and p.Gly1651Ser; compound heterozygous p.Asn1380Ser and p.Gly1651Ser) were clustered in the region of the first ATP‐binding cassette (an active site essential for function); they presumed that these mutations underlie the LI phenotype in Africa. In CIE, which is prevalent in Japan, more than half of the mutations in ABCA12 (e.g., missense, nonsense, deletion, and splice site mutations) are present in the transmembrane and protein‐binding domains. 9 , 10 , 11 , 12 Akiyama 9 suggested that at least one missense mutation in transmembrane domains might underlie the CIE phenotype in Japan. In Spain, two unrelated patients with CIE demonstrated homozygous p.Asn1380Ser mutations, and a founder effect was reported. 13
In conclusion, to the best of our knowledge, this is the first report to suggest that compound heterozygous missense mutations (p.Asn1380Ser and p.Thr1434Ala) in ABCA12 may produce a partial loss of function leading to a mild ichthyosis phenotype (CIE), despite their location in a functional domain (i.e., the first ATP‐binding cassette) (Figure 2). Our finding expands the phenotypic and genotypic spectrum of ABCA12 mutations. In addition, it will aid in the prenatal genetic diagnosis when the patient’s mother gives birth to another baby.
CONSENT FOR PUBLICATION
Consent was obtained from the patient’s parents.
Funding Source
National Natural Science Foundation of China (No. 81673042)
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Acknowledgments
We would like to thank the patient and her family.
Yang Z, Qi Z, Xu Z, Li W, Ma L. Congenital ichthyosiform erythroderma with a novel variant in ABCA12 in a Chinese patient. Pediatr Invest. 2020;4:51–54. 10.1002/ped4.12182
REFERENCES
- 1. Fischer J. Autosomal recessive congenital ichthyosis. J Invest Dermatol. 2009;129:1319–1321. [DOI] [PubMed] [Google Scholar]
- 2. Akiyama M. Novel ABCA12 missense mutation p.Phe2144Ser underlies congenital ichthyosiform erythroderma. J Dermatol. 2013;40:581–582. [DOI] [PubMed] [Google Scholar]
- 3. Pigg MH, Bygum A, Gånemo A, Virtanen M, Brandrup F, Zimmer AD, et al. Spectrum of autosomal recessive congenital ichthyosis in Scandinavia: Clinical characteristics and novel and recurrent mutations in 132 Patients. Acta Derm Venereol. 2016;96:932–937. [DOI] [PubMed] [Google Scholar]
- 4. Yang Z, Xu Z, Xing H, Ma L. Novel ABCA12 compound heterozygous mutations identified in a patient with congenital ichthyosiform erythroderma and aortopulmonary window. Eur J Dermatol. 2019;29:83–85. [DOI] [PubMed] [Google Scholar]
- 5. Akiyama M, Sugiyama‐Nakagiri Y, Sakai K, et al. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115:1777–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sakai K, Akiyama M, Sugiyama‐Nakagiri Y, McMillan JR, Sawamura D, Shimizu H. Localization of ABCA12 from Golgi apparatus to lamellar granules in human upper epidermal keratinocytes. Exp Dermatol. 2007;16:920–926. [DOI] [PubMed] [Google Scholar]
- 7. Zhang L, Ferreyros M, Feng W, Hupe M, Crumrine DA, Chen J, et al. Defects in stratum corneum desquamation are the predominant effect of impaired ABCA12 function in a novel mouse model of harlequin ichthyosis. PLoS One. 2016;11:e0161465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lefévre C, Audebert S, Jobard F, Bouadjar B, Lakhdar H, Boughdene‐Stambouli O, et al. Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Hum Mol Genet. 2003;12:2369–2378. [DOI] [PubMed] [Google Scholar]
- 9. Akiyama M. ABCA12 mutations and autosomal recessive congenital ichthyosis: A review of genotype/phenotype correlations and of pathogenetic concepts. Hum Mutat. 2010;31:1090–1096. [DOI] [PubMed] [Google Scholar]
- 10. Shimizu Y, Sugiura K, Aoyama Y, Ogawa Y, Hitomi K, Iwatsuki K, et al. Novel ABCA12 missense mutation p.Phe2144Ser underlies congenital ichthyosiform erythroderma. J Dermatol. 2013;40:581–582. [DOI] [PubMed] [Google Scholar]
- 11. Wada Y, Kusakabe M, Nagai M, Yamamoto M, Imai Y, Ide YH, et al. Mild case of congenital ichthyosiform erythroderma with periodic exacerbation: Novel mutations in ABCA12 and upregulation of calprotectin in the epidermis. J Dermatol. 2017;44:e282–e283. [DOI] [PubMed] [Google Scholar]
- 12. Murase C, Takeichi T, Sugiura K, Kobayashi M, Shiomi K, Ikebuchi K, et al. Hearing impairment: A secondary symptom in a congenital ichthyosiform erythroderma patient with ABCA12 mutations. J Dermatol. 2018;45:e303–e304. [DOI] [PubMed] [Google Scholar]
- 13. Esperón‐Moldes U, Ginarte M, Rodríguez‐Pazos L, Fachal L, Pozo T, Aguilar JL, et al. ABCA12 mutations in patients with autosomal recessive congenital ichthyosis: evidence of a founder effect in the Spanish population and phenotype‐genotype implications. J Dermatol Sci. 2018;91:328–331. [DOI] [PubMed] [Google Scholar]