

Abstract
Importance
Cytochrome P450 oxidoreductase deficiency (PORD) is a rare disease exhibiting a variety of clinical manifestations. This condition specifically leads to disordered steroidogenesis, which can affect the development of the reproductive system, skeleton, and other parts of the body. The severe form of PORD is difficult to differentiate with Antley‐Bixler syndrome (ABS). The genetic characters and clinical evaluation of PORD are still unclear in China.
Objective
To perform an exome analysis and identify the pathogenic cause in order to assist clinicians to obtain a proper evaluation on the genetic condition.
Methods
The proband underwent detailed physical evaluations. DNA of the proband and his parents was isolated and whole‐exome sequencing (WES) was performed. Variants were analyzed and evaluation according to the ACMG guideline.
Results
A 1‐year‐old Chinese boy with midface hypoplasia, choanal stenosis, multiple joint contractures, micropenis and right cryptorchidism was misdiagnosed with Crouzon syndrome. By trio‐whole‐exome sequencing, we identified an unreported compound heterozygous mutation (c.667C>T, p.R223* and c.1370G>A, p.R457H) in POR in the proband. This mutation was inherited from healthy heterozygous parents, supporting the diagnosis of PORD, which was further confirmed by biochemical characteristics.
Interpretation
We have identified a pathogenic variant with an unreported compound heterozygous POR mutation, which expands the clinical and genetic spectra of PORD and emphasizes the usefulness of WES for genetic diagnosis.
Keywords: Compound heterozygous mutation, Cytochrome P450 oxidoreductase, Disordered steroidogenesis, POR deficiency, Whole‐exome sequencing
INTRODUCTION
POR deficiency manifests with a wide range of clinical signs and symptoms. Mild cases can include primary amenorrhea, infertility in both men and women, and polycystic ovarian syndrome (PCOS). People with moderate cases of PORD may have ambiguous genitalia, and they usually do not have skeletal abnormalities. The severe form of PORD is sometimes called Antley‐Bixler syndrome (ABS) with genital anomalies and disordered steroidogenesis. Hormonal changes in affected males and females lead to the development of ambiguous genitalia or other genital abnormalities, as well as infertility. Severe cases are also characterized by midface hypoplasia, choanal stenosis or atresia, and multiple joint contractures.1
Exclusively skeletal form of ABS without genital anomalies or disordered steroidogenesis can be caused by heterozygous mutation in the gene encoding for fibroblast growth factor receptor 2 (FGFR2).2, 3
However, mutations in cytochrome P450 oxidoreductase (POR) were identified as the cause of ABS with genital anomalies and disordered steroidogenesis.4, 5 These mutations affect steroidogenesis by disrupting the ability of POR to transfer electrons from nicotinamide adenine dinucleotide phosphate (NADPH) to 17α‐hydroxylase/17,20‐lyase (P450c17), steroid aromatase (P450aro) and 21‐hydroxylase (P450c21), resulting in deficiency of P450c17, P450aro and P450c21.5, 6
Here, we report a 1‐year‐old Chinese male patient who first manifested head, skeletal and genitalia anomalies. He was misdiagnosed with Crouzon syndrome due to craniosynostosis and frontal bossing. We performed whole‐exome sequencing (WES) on the patient and his unaffected parents, and found unreported compound heterozygous mutations in POR in the proband.
METHODS
Clinical examination and testing
Informed consents were obtained and clinical examinations were performed. The proband underwent detailed physical evaluations by an experienced pediatrician, including radiographs of limbs, chest and spine, endoscope for ENT (ear, nose and throat), computerized tomography (CT) of accessory nasal sinuses, magnetic resonance imaging (MRI) of the skull, and other necessary tests. Karyotyping were the preliminary examinations conducted. At 2 months and 10 months of age, serum samples were sent for steroid analysis.
Whole‐exome sequencing
DNA was isolated from 200 μL blood samples from the proband and his parents. Exon capture was performed using the Agilent SureSelect Human All Exome V6 kit (Agilent Technologies Inc. Mississauga, ON, Canada). Whole‐exome sequencing was performed on an Illumina Hiseq X Analyzers (Illumina, San Diego, CA, USA) using standard protocols for 150‐bp paired‐end runs.
Analysis and evaluation of variants
The sequences obtained were aligned to the GRCh37/hg19 human reference genome. Variant frequency was analyzed using three SNP databases (db‐SNP147, gnomAD and 1000 Genomes Project Database) and three disease databases (ClinVar, OMIM, and HGMD). We filtered all common variants with MAF ≥0.005. In order to identify the potential impact of the identified mutations on protein function, we used five applications: SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html), Polyphen‐2 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster (http://www.mutationtaster.org), Mutation Assessor (http://mutationassessor.org/) and CADD (http://cadd.gs.washington.edu/). Loss‐of‐function (LoF) mutations, such as nonsense/splicing mutations and frameshift indels, were considered to be deleterious.
Sanger sequencing
The variants detected by whole‐exome sequencing were verified through Sanger sequencing. Primers for PCR and Sanger sequencing were designed to cover mutated bases and flanking sequence. DNA samples from the proband and his parents were amplified. Direct sequencing was performed using the BigDye™ Terminator Cycle Sequencing kit and analyzed on a 3730xl automated sequencer (Applied Biosystems, Foster City, CA, USA).
RESULTS
Clinical findings
A newborn boy was first evaluated in the neonatal intensive care unit because of congenital dysplasia. He was born at 38 weeks gestation via Cesarean because of oligohydramnios for a week. Apgars were 10 at 1 min. Birth weight was 2.9 kg. On examination, the baby was dysmorphic with frontal bossing, bilateral proptosis, hypertelorism, depressed nasal bridge, micropenis and right cryptorchidism (Table 1). Mouth breathing and triretraction sign were observed in this baby indicating choanal or airway stenosis. Extremities had bilateral decreased extension and supination of the elbows and clinodactyly of the fourth toe. Bilateral lower extremities were hypotonic. The phallic stretched length was 1.5 centimeter (<−2.5 SD in Chinese). Testicular volume was 1 mL. There was no family history of these disorders. The mother was a 35‐year‐old primigravida with Hashimoto's thyroiditis for eight years. The mother developed some signs of virlization during her pregnancy such as hirsutism and acne.
Table 1.
Phenotypes of the proband fitting the diagnostic criteria of PORD
| Head | Skeletal | Genitalia |
|---|---|---|
| frontal bossing | craniosynostosis | micropenis |
| bilateral proptosis | radiohumeral synostosis | right cryptorchidism |
| hypertelorism | radioulnar synostosis | |
| depressed nasal bridge | chest wall deformity | |
| low‐set ears choanal stenosis |
The radiographs of upper limbs displayed fusion of certain adjacent bones of the arms, including bilateral radiohumeral synostosis (Figure 2A). The radiographs of chest and spine displayed scoliosis (Figure 2B). Endoscope for ENT (Figure 1) and CT of accessory nasal sinuses at 3 months of age confirmed a bony or thin layer of tissue blocking the passageway between the nose and throat, leading to difficulties in breathing. MRI of the skull at 10 months of age revealed brachycephaly with a flat occiput, frontal bossing, and broad forehead, suggesting craniosynostosis (Figure 3B).
Figure 2.
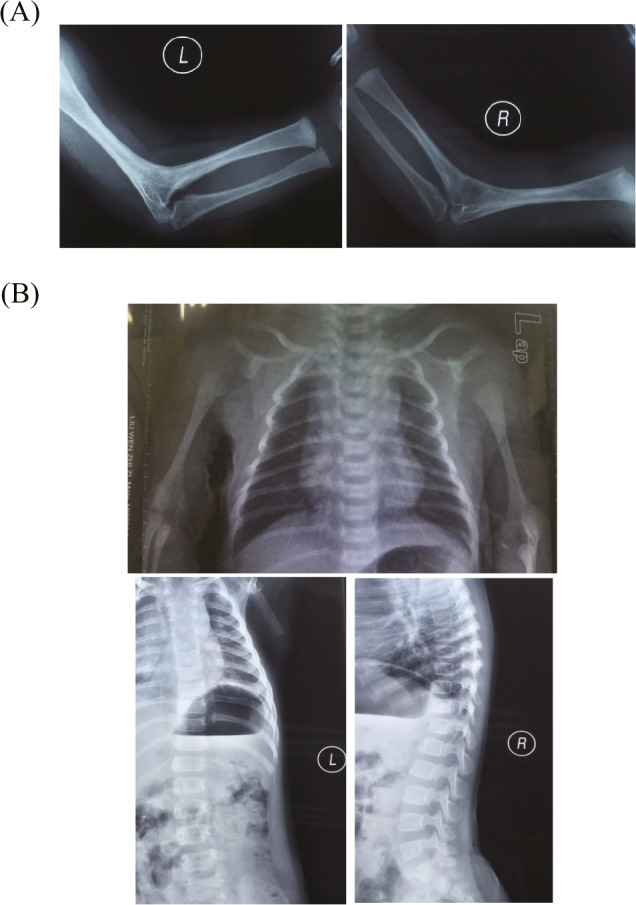
Skeletal abnormalities of arms and chest. A, The radiographs of arms showed bilateral radiohumeral synostosis. B, The radiographs of chest and spine showed scoliosis.
Figure 1.
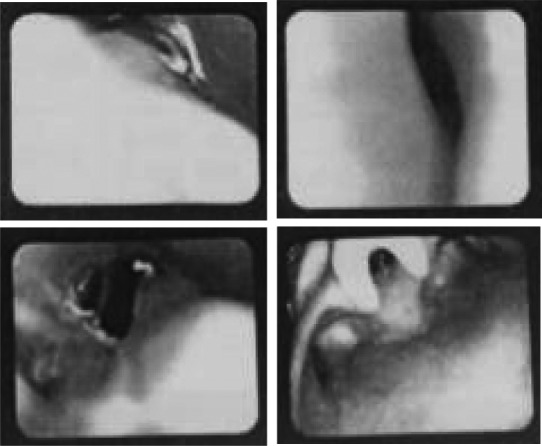
Endoscope for ENT showed choanal stenosis.
Figure 3.
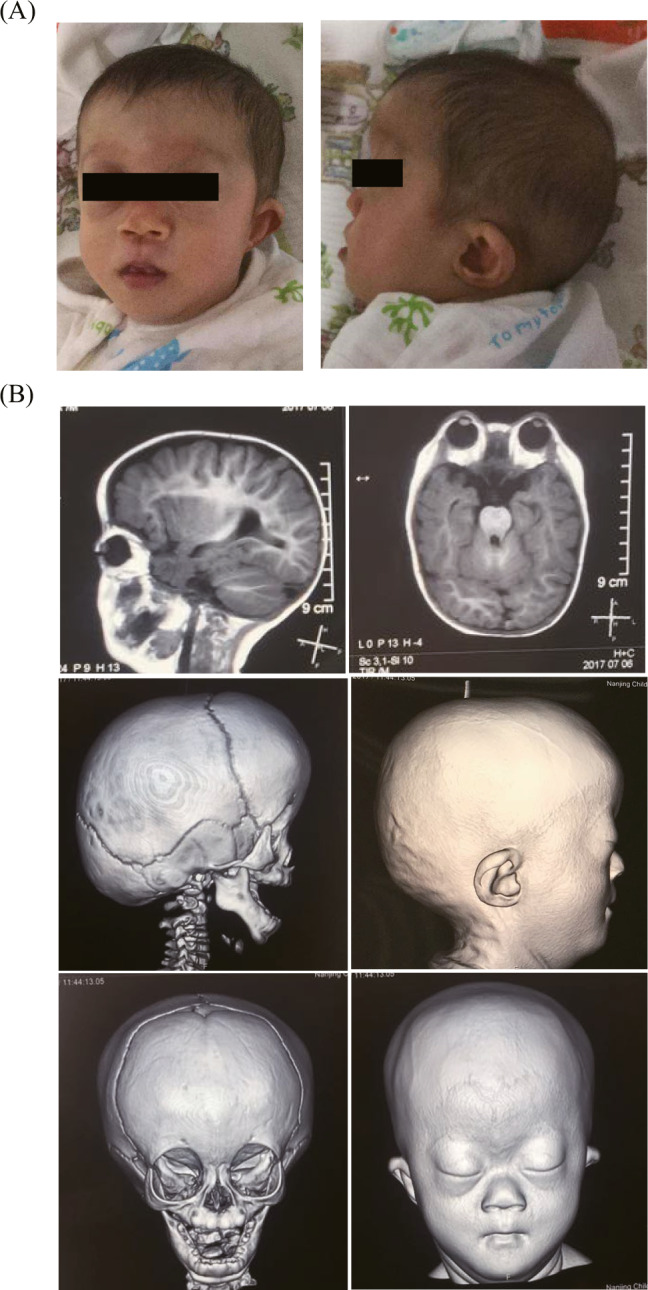
Skeletal abnormalities of head a nd face. A, Facial characteristics at age of 8 months. B, MRI of the skull revealed premature fusion of the skull bones (craniosynostosis), a flattened mid‐face, frontal bossing, broad forehead and low‐set ears.
Laboratory abnormalities
The results of laboratory examinations were shown in Table 2. At 2 months of age, baseline cortisol (COR) was normal, at 15.40 μg/dL (normal range 5–25 μg/dL), and adrenocorticotropic hormone (ACTH) was elevated, at 253.0 pg/mL (normal range 0–46 pg/mL). At 10 months of age, the patient had high levels of ACTH (116.6 pg/mL) and progesterone (PGN) (13.30 ng/mL; normal ≤2 ng/mL), with normal values for baseline COR, luteinizing hormone (LH), follicle‐stimulating hormone (FSH), testosterone (T) and prolactin (PRL).
Table 2.
Laboratory tests of the proband
| 2 months | 10 months | Reference ranges | |
|---|---|---|---|
| ACTH 8AM (pg/mL) | 253.00 | 116.60 | 0–46 |
| COR 8AM (μg/dL) | 15.40 | 7.33 | 5–25 |
| T (ng/dL) | ‐ | 58.90 | 0–106.90 |
| PGN (ng/mL) | ‐ | 13.30 | ≤2 |
| LH (IU/L) | ‐ | 1.47 | ≤4.10 |
| FSH (IU/L) | ‐ | 1.27 | ≤5.50 |
| PRL (ng/mL) | ‐ | 24.30 | 0.60–29.00 |
ACTH: adrenocorticotropic; COR: cortisol; T: testosterone; PGN: progesterone; LH: luteinizing hormone; FSH: follicle‐stimulating hormone; PRL: prolactin.
Pathogenicity of two POR variants
Karyotyping showed a normal male 46, XY. The mean coverage of WES was 98.4% and the average sequencing depth of the exomes was above 100×. A heterozygous nonsense variant c.667C>T (p.R223*) and a heterozygous missense variant c.1370G>A (p.R457H) in POR were identified in the patient (Table 3), which was inherited from his father and mother, respectively. The two variants of POR were confirmed by Sanger sequencing in the proband and his parents (Figure 4). They were also evaluated using five in silico predictive algorithms (Table 4) and most of them showed pathogenic prediction.
Table 3.
Two genetic variants of POR in the proband
| Gene | Transcript | cDNA | Protein Heterozygosity | Co‐segregation | gnomAD East Asian | 1000 Genomes | dbSNP number |
|---|---|---|---|---|---|---|---|
| POR | NM_000941.2 | c.667C>T | p.R223* | Heterozygous Proband and his father | 0 | ‐ | rs782677940 |
| POR | NM_000941.2 | c.1370G>A | p.R457H | Heterozygous Proband and his mother | 0.0004688 | 0.0002 | rs28931608 |
Figure 4.
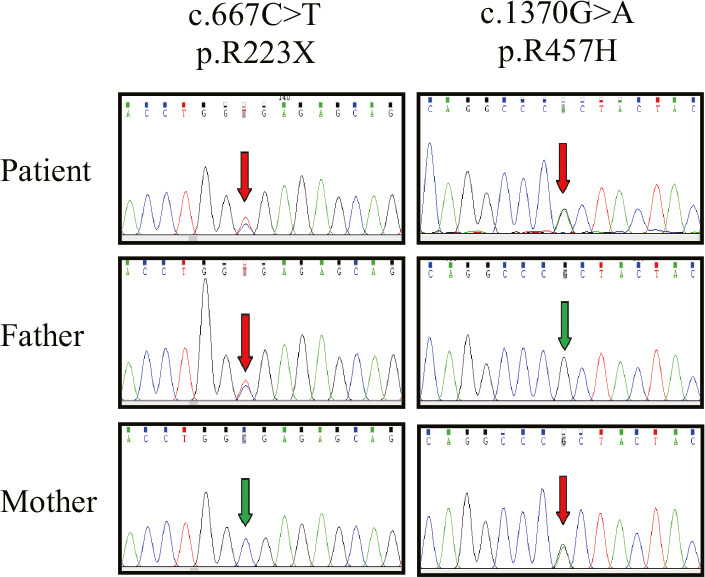
Sanger sequencing analysis of POR in the patient and his parents. The arrows indicated the mutated nucleotides. The patient carries a compound heterozygous nonsense mutation (p.R223*) and a missense mutation (p.R457H) in POR. p.R223* was inherited from patient's heterozygous father and p.R457H was inherited from patient's heterozygous mother.
Table 4.
Predicted pathogenicity of the two genetic variants of POR
| Gene | cDNA, Protein | SIFT | Polyphen‐2 | Mutation Taster | Mutation Assessor | CADD |
|---|---|---|---|---|---|---|
| POR | c.667C>T, p.R223* | ‐ | ‐ | Disease‐causing | ‐ | ‐ |
| POR | c.1370G>A, p.R457H | Damaging | Probably damaging | Disease‐causing | High impact | 32 |
The nonsense mutation (c.667C>T, p.R223*) in exon 6 were inherited from the proband's father, which creates a premature stop codon at codon 223. The in silico predictive algorithms on the MutationTaster server predicted it to be pathogenic. The missense mutation (c.1370G>A, p.R457H) in exon 11 is a single‐nucleotide polymorphism (SNP) in dbSNP150 (rs28931608), which was inherited from the proband's mother. The allele frequency (AF) among East Asian people is as low as 0.0004688 (8/17 066) according to gnomAD (Table 3). Multiple lines of computational evidence (SIFT, PolyPhen‐2, MutationTaster, Mutation Assessor and CADD) supported a deleterious effect of this variant on the gene or gene product (Table 4).
DISCUSSION
The patient was suspected of having Crouzon syndrome due to craniosynostosis and frontal bossing and came to our hospital for a genetic test. Crouzon syndrome is an autosomal dominant disorder is an autosomal dominant disorder caused by FGFR2 mutations.7, 8 However, using whole‐exome sequencing, we found a compound heterozygous nonsense mutation (p.R223*) and a missense mutation (p.R457H) in POR in the proband. This mutation inherited from healthy heterozygous parents and is an unreported compound heterozygous mutation in POR.
The POR gene encodes a flavoprotein that transfers electrons from NADPH to all microsomal cytochrome P450 type II (including 21‐hydroxylase, 17α‐hydroxylase/17, 20‐lyase and aromatase), which is fundamental for their enzymatic activity.5 POR mutations cause variable impairments in steroidogenic enzyme activities that result in wide phenotypic variability ranging from mild to severe. Signs and symptoms of mild‐to‐moderate PORD include primary amenorrhea, polycystic ovarian syndrome (PCOS), infertility in both men and women, and ambiguous genitalia. The severe form of POR mutations is called Antley‐Bixler syndrome with genital anomalies and disordered steroidogenesis. The main anomalies of PORD are characterized by craniofacial, skeletal, extremity and urogenital anomalies. Recently, a female Chinese patient with a homozygous missense mutation (p.R457H) was described, who presented with amenorrhea and recurrence of large ovary cyst but had not a typical skeletal deformity.6
Our patient presented severe craniofacial, skeletal, extremity malformation with frontal bossing, depressed nasal bridge, craniosynostosis, radiohumeral synostosis, scoliosis and camptodactyly in feet. Urogenital anomalies were also observed as micropenis and right cryptorchidism. In addition, POR mutations can lead to an apparent combined 17‐hydroxylase/21‐hydroxylase deficiency.
The p.R457H mutation is the most frequent mutation in Japanese PORD patients, and it is observed in approximately 65.4% of unrelated alleles.9 Other ethnic groups also have the R457H mutation.5 This mutation destroyed Bsa XI and TstI sites, but these enzymes recognize degenerate 33–37 base sequences and cut both strands twice.5 Two PORD cases have been reported as compound heterozygous carriers including p.R223* in POR 10, 11 and was predicted to induce a large splicing change.12
Antley‐Bixler syndrome (ABS) is a variant of the autosomal dominant group of craniosynostosis syndromes caused by mutant FGF receptors, usually FGFR2 gene. PORD, sometimes called ABS with disordered steroidogenesis, is an autosomal recessive disorder caused by mutations in POR.3, 5 It is clinically important to distinguish patients with PORD from those with ABS, because patients with POR deficiency are vulnerable to different risk factors and require different management and different teams of physicians.5 In this case, ABS could also be suspected, based on the head and skeletal anomaly. However, the anomaly of genitalia could be a target for the differentiation between ABS and PORD, which demonstrated the clinical value of physical examination of genitalia. POR plays an important role in biological activities such as biosynthesis, cholesterol metabolism, sex hormone metabolism and the metabolism of drugs and toxins.6 Patients with POR mutations may require steroid hormone supplementation and may be at risk for adrenal insufficiency and Addisonian crisis, especially at severe febrile illness or major surgery.3 The mutation of POR gene or associated SNPs may affect drug metabolism in human body.13, 14 For example, the R457H mutation in this patient can inactivate the P450 enzymes including CYP1A2, CYP2C19, CYP2D6 and CYP3A4, which are important for drug metabolism.15, 16 Thus, there may be possible risks if the patients with R457H mutations are treated with medications metabolized by these enzymes.
CONCLUSIONS
We have identified a pathogenic variant with an unreported compound heterozygous POR mutation. This case report expands the clinical and genetic spectra of PORD and emphasizes the usefulness of WES for genetic diagnosis.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Hao C, Guo J, Guo R, Qi Z, Li W, Ni X. Compound heterozygous variants in POR gene identified by whole‐exome sequencing in a Chinese pedigree with cytochrome P450 oxidoreductase deficiency. Pediatr Invest. 2018;2:90‐95. 10.1002/ped4.12035
Funding source
National Natural Science Foundation of China (81670789; 31401171); Ministry of Science and Technology of China (2016YFC1000306); The Beijing Municipal Science and Technology Commission Foundation (Z141100002114009); Beijing Municipal Commission of Health and Family Planning Foundation (PXM2017_026274_000001); Beijing Municipal Administration of Hospitals Foundation (QML20161201).
Contributor Information
Wei Li, Email: liwei@bch.com.cn.
Xin Ni, Email: nixin@bch.com.cn.
REFERENCES
- 1. McGlaughlin KL, Witherow H, Dunaway DJ, David DJ, Anderson PJ. Spectrum of Antley‐Bixler syndrome. J Craniofac Surg. 2010;21:1560‐1564. [DOI] [PubMed] [Google Scholar]
- 2. Chun K, Siegel‐Bartelt J, Chitayat D, Phillips J, Ray PN. FGFR2 mutation associated with clinical manifestations consistent with Antley‐Bixler syndrome. Am J Med Genet. 1998;77:219‐224. [DOI] [PubMed] [Google Scholar]
- 3. Reardon W, Smith A, Honour JW, et al. Evidence for digenic inheritance in some cases of Antley‐Bixler syndrome? J Med Genet. 2000;37:26‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fluck CE, Tajima T, Pandey AV, et al. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley‐Bixler syndrome. Nat Genet. 2004;36:228‐230. [DOI] [PubMed] [Google Scholar]
- 5. Huang N, Pandey AV, Agrawal V, et al. Diversity and function of mutations in p450 oxidoreductase in patients with Antley‐Bixler syndrome and disordered steroidogenesis. Am J Hum Genet. 2005;76:729‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bai Y, Li J, Wang X. Cytochrome P450 oxidoreductase deficiency caused by R457H mutation in POR gene in Chinese: case report and literature review. J Ovarian Res. 2017;10:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gorry MC, Preston RA, White GJ, et al. Crouzon syndrome: mutations in two spliceoforms of FGFR2 and a common point mutation shared with Jackson‐Weiss syndrome. Hum Mol Genet. 1995;4:1387‐1390. [DOI] [PubMed] [Google Scholar]
- 8. Jabs EW, Li X, Scott AF, et al. Jackson‐Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2. Nat Genet. 1994;8:275‐279. [DOI] [PubMed] [Google Scholar]
- 9. Fukami M, Horikawa R, Nagai T, et al. Cytochrome P450 oxidoreductase gene mutations and Antley‐Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: molecular and clinical studies in 10 patients. J Clin Endocrinol Metab. 2005;90:414‐426. [DOI] [PubMed] [Google Scholar]
- 10. Idkowiak J, O'Riordan S, Reisch N, et al. Pubertal presentation in seven patients with congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab. 2011;96:E453‐E462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krone N, Reisch N, Idkowiak J, et al. Genotype‐phenotype analysis in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab. 2012;97:E257‐E267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347:1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pandey AV, Fluck CE. NADPH P450 oxidoreductase: structure, function, and pathology of diseases. Pharmacol Ther. 2013;138:229‐254. [DOI] [PubMed] [Google Scholar]
- 14. Subramanian M, Agrawal V, Sandee D, Tam HK, Miller WL, Tracy TS. Effect of P450 oxidoreductase variants on the metabolism of model substrates mediated by CYP2C9.1, CYP2C9.2, and CYP2C9.3. Pharmacogenet Genomics. 2012;22:590‐597. [DOI] [PubMed] [Google Scholar]
- 15. Agrawal V, Huang N, Miller WL. Pharmacogenetics of P450 oxidoreductase: effect of sequence variants on activities of CYP1A2 and CYP2C19. Pharmacogenet Genomics. 2008;18:569‐576. [DOI] [PubMed] [Google Scholar]
- 16. Sandee D, Morrissey K, Agrawal V, et al. Effects of genetic variants of human P450 oxidoreductase on catalysis by CYP2D6 in vitro . Pharmacogenet Genomics. 2010;20:677‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]