

Abstract
The increasing focus on lipid metabolism has revealed a need for analytical techniques capable of structurally characterizing lipids with a high degree of specificity. Lipids can exist as any one of a large number of double bond positional isomers, which are indistinguishable by single-stage mass spectrometry alone. Ozonolysis reactions coupled to mass spectrometry have previously been demonstrated as a means for localizing double bonds in unsaturated lipids. Here we describe an online, solution-phase reactor using ozone produced via a low-pressure mercury lamp, which generates aldehyde products diagnostic of cleavage at a particular double bond position. This flow-cell device is utilized in conjunction with structurally selective ion mobility-mass spectrometry. The lamp-mediated reaction was found to be effective for multiple lipid species in both positive and negative ionization modes, and the conversion efficiency from precursor to product ions was tunable across a wide range (20–95%) by varying the flow rate through the ozonolysis device. Ion mobility separation of the ozonolysis products generated additional structural information and revealed the presence of saturated species in a complex mixture. The method presented here is simple, robust, and readily coupled to existing instrument platforms with minimal modifications necessary. For these reasons, application to standard lipidomic workflows is possible and aids in more comprehensive structural characterization of a myriad of lipid species.
Graphical Abstract
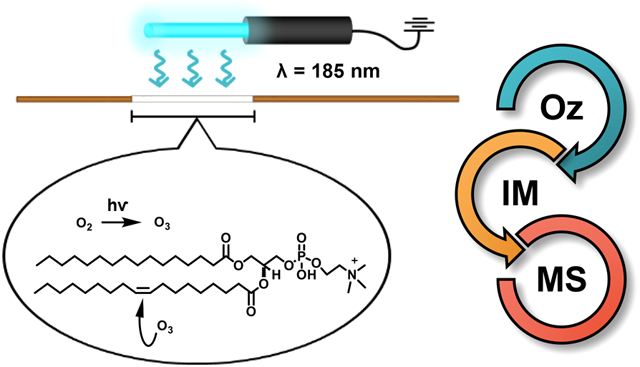
Mass spectrometry-based lipidomic approaches are emerging as an increasingly important analytical strategy for characterizing the varied and critical roles lipids play in biological systems.1-3 Like other omics techniques, lipidomic studies aim to generate a comprehensive snapshot of the lipid content of a given system, identifying and quantifying individual components and attempting to interpret the intermolecular interactions in coordination with relevant biological functions. Common lipidomic workflows often combine liquid chromatography and tandem mass spectrometry (LC-MS/MS) in broad-scale, untargeted analyses.2,4,5 However, the immense structural diversity of the lipidome often precludes putative identification of species, especially given the potential presence of a myriad of isomeric and isobaric species at a given mass-tocharge ratio. For example, phosphatidylcholine (PC) 14:0/20:1 and PC 18:0/16:1 are alkyl chain isomers that have identical chemical formulas (C42H82NO8P) and thus cannot be differentiated via mass measurement alone. Nevertheless, these two isomeric species can be distinguished by tandem MS/MS experiments via product ion scanning for diagnostic fatty acid fragments in negative mode electrospray ionization.6 Other classes of lipid isomers, such as sn-regioisomers and double bond positional isomers, are much more challenging to assign a specific chemical structure.7 As a result, there is no single analytical method currently available that can resolve all the possible configurations of lipid species that may exist in a given sample. For this reason, the combination and integration of several analytical techniques into a single workflow is often necessary to identify isomeric lipid species and expand lipidome coverage.8
Ozonolysis is a chemical reaction that has long been utilized by organic chemists to cleave carbon–carbon double bonds and has more recently been adopted by analytical chemists for the localization of lipid double bonds by mass spectrometry.9-11 The reaction may be performed in one of several ways, including online in the solution phase or directly in the ionization source of the instrument.12,13 Most prominently, the Blanksby group at the Queensland University of Technology has developed a family of techniques known as “OzID”, in which ozonolysis is performed on mass-selected ions in the gas phase in either a collision cell or a trapping region of the instrument.14,15 In recent publications, Blanksby and coworkers have demonstrated the capabilities of ozonolysis combined with MS on a variety of instrument platforms.15-17 Advantages of the technique include high reaction yield (product ion intensity ca. 20% of the precursor ion in the most recent iteration) and the ability to combine successive collision-induced dissociation (CID) and OzID experiments for increased structural characterization and lipid coverage.14-17 One drawback preventing widespread adoption of OzID is the instrument modifications that are necessary to perform the experiments, which typically include the addition of a gas manifold and ozone generator to the mass spectrometer.
Other chemical reactions that cleave carbon–carbon double bonds have also been utilized for double bond localization in mass spectrometry. Of recent note is the Paternò–Büchi reaction, which the Xia group at Purdue coupled to MS via UV irradiation of the tip of the nano-ESI emitter containing a lipid sample in the presence of an acetone cycloaddition reagent.18-21 A low-pressure mercury lamp is used to generate UV radiation to activate the acetone (primarily via a 254 nm band), resulting in the addition of an acetone radical to the double bond and forming an oxetane.18 Collision-induced dissociation of the oxetane is then used to enact retro-PB, resulting in two product ions spaced 26 Da apart that are diagnostic of the double bond position in the original lipid molecule.18 Given the relatively low quantum yield of acetone, the PB reaction yields are typically less than 50%. The nonquantitative chemical conversion can increase spectral complexity for mixture analysis and limit the identification of low-abundance unsaturated lipids, in particular for lipid species containing multiple sites of unsaturation, although careful choice of solvent composition can minimize side reaction products.19,20,22 In recent work by Murphy and coworkers, double bond cleavage products were observed for 9,12,15-octadecatrienoic acid and 6,9,12-octadecatrienoic acid prior to enacting retro-PB via CID.22 They hypothesized that these cleavage products were a result of ozonolysis due to the fact that the peaks were no longer observed upon degassing of sample solution prior to analysis but did not explore these observations further.
Ion mobility (IM) is an analytical technique that is gaining widespread use toward the structural analysis of lipids.23,24 When interfaced with a mass spectrometer, IM allows for the separation of isomeric lipid species based on differences in their collision cross section (CCS) which provides a structurally selective measurement that is orthogonal to mass analysis. For example, using IM, Baker and coworkers were able to distinguish sn-positional isomers and cis/trans isomers for several ceramide standards, and Groessl et al. demonstrated IM separation of phosphatidylcholine acyl-chain regioisomers via silver-cationized forms of the natural lipid species present in a porcine brain extract.25,26 Double bond positional isomers have also been resolved via IM-MS; however, the analytical specificity was only demonstrated for fatty acids and more biologically uncommon species such as PC 18:1(9Z)/18:1(9Z) and PC 18:1(6Z)/18:1(6Z).25,26 Moreover, lipid isomers in a complex mixture may have CCS differences that are too small to exhibit baseline separation using current instrument platforms.27 For these reasons, it was hypothesized that combining ozonolysis and IM-MS would provide for multiple dimensions of isomer structural discrimination. Ozonolysis provides the basis for determination of double bond position, while IM-MS facilitates separation of the ozonolysis products based upon differences in CCS and mass. Together, these analytical tools have the potential for enabling high confidence in identification and discrimination of lipid isomers in complex samples. Recently, the combination of IM-MS with OzID has been demonstrated by the Blanksby group in collaboration with PNNL, highlighting the potential of the combined techniques for the analysis of isomeric lipid species.28 Our work aims to further examine the benefits of utilizing IM in conjunction with ozonolysis, but rather than modifying the mass spectrometer for gas-phase OzD, we have implemented ozonolysis on a low-cost, portable solution-phase device.
In this work, we have developed an ozonolysis device that combines the immediacy of double bond cleavage with the simplicity of online ozone generation without instrument modification. The device is a continuation of our previous work29 and contains a flow cell, in which analyte solution is irradiated by the mercury lamp, producing solution-phase ozonolysis products that then are ionized in the source of the mass spectrometer. As in the ozonolysis and Paternò–Büchi experiments described previously, these products are diagnostic in that they can be used to determine the double bond position in the lipid precursor species when analyzed via mass spectrometry. Following ozonolysis in the device, IM-MS allows for separation of the ozonolysis products to aid in the deconvolution of isomeric signals. Here, we explore the utility of ozonolysis combined with ion mobility-mass spectrometry for the analysis of three distinct lipid systems: (1) a PC standard containing a single double bond, (2) a fatty acid standard containing multiple double bonds, and (3) a complex mixture in the form of chicken egg PC lipid extract. Additionally, given the importance of separation via liquid chromatography in many lipidomic workflows, the utilization of LC in combination with the ozonolysis device was demonstrated using a simple mixture of three isomeric PC standards.
■ EXPERIMENTAL METHODS
Sample Preparation.
Phosphatidylcholine standards 1-hexadecanoyl-2-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine (PC 16:0/18:1(9Z)), 1-palmitoyl-2-stearoyl-sn-glycero-3-phosphocholine (PC 16:0/18:0), 1,2-di(6Z-octadecenoyl)-sn-glycero-3-phosphocholine (PC 18:1(6Z)/18:1(6Z)), 1,2-di(9Z-octadecenoyl)-sn-glycero-3-phosphocholine (PC 18:1(9Z)/18:1(9Z)), 1,2-di(9E-octadecenoyl)-sn-glycero-3-phosphocho-line (PC 18:1(9E)/18:1(9E)), and a total TLC fraction of a phosphatidylcholine lipid extract (PC, chicken egg) were purchased from Avanti Polar Lipids. Docosahexaenoic acid (FA 22:6) was purchased from Cayman Chemical. Prior to analysis, individually infused samples were diluted to working concentrations ranging from 0.1 to 10 μM in 7:3 acetonitrile/water (Optima LC/MS grade, Fisher Scientific), while the three isomeric PC lipids were combined in equimolar ratios and diluted to approximately 6 μM in the solvent composition utilized at the beginning of the RPLC separation gradient. Formic acid (0.1%, Optima grade, Fisher Scientific) was added to PC lipids analyzed in positive ion mode to facilitate the formation of protonated species. A saturated phosphatidylcholine standard, PC 16:0/18:0, was spiked into the lipid extract at 1 μM due to low abundance of the fully saturated species inherent in the sample fraction. For mass calibration, a mixture of hexakis-(fluoroalkoxy) phosphazines (ESI-Low Concentration Tuning Mixture, Agilent Technologies) was purchased and diluted by a factor of 10 in a 59:1 (v/v) solution of acetonitrile/water.
Instrumentation and Parameters.
Experiments were performed on a uniform field drift tube IM-MS (6560, Agilent) with nitrogen as the drift gas in both positive and negative ionization modes.30 The IM-MS instrument was tuned for high sensitivity in standard mass mode (m/z 50–1700) and mass calibrated prior to analysis using the Agilent tuning mixture. The drift tube was operated at ca. 300 K with an electric field strength of 14.7 V/cm, which represents conditions yielding optimal IM resolving power.31 For analysis of the fatty acid standard which displayed low ionization efficiency in negative mode, the ion mobility trap release time was increased from 150 to 450 μs, increasing signal with a slight loss in mobility resolving power (ca. 2% for m/z 261.1). Samples were directly infused into the electrospray source (ESI, Agilent Jet Stream) at various flow rates ranging from 5 to 500 μL/min. The source was operated with a capillary voltage of 3500 V and nozzle voltage of 2000 V. Nitrogen sheath and drying gases were heated to 275 and 325 °C at flow rates of 12 and 8 L/min, respectively. For LC experiments, 5–10 μL of sample was injected onto a C-18 2.1 mm × 50 mm (1.8 μm) analytical column with the flow rate set to 250 μL/min. Mobile phase A consisted of water with 0.1% formic acid, and mobile phase B consisted of 3/2 isopropanol:acetonitrile with 0.1% formic acid. The gradient elution profile was based upon conditions for analyzing hydrophobic compounds as described by Cruickshank-Quinn et al.32
Ozonolysis reactions were carried out in the solution-phase prior to ionization in the ESI source. A custom built, mechanically stable device was constructed in-house to support and align the low-pressure mercury lamp (81–1057–51, BHK Inc.), which is used to catalyze the relevant reaction (Figure 1). This particular lamp produces several emission bands spanning from 185 to 579 nm; however, previous studies have shown that the 185 nm band is responsible for the production of ozone necessary for these experiments.33,34 The custom support housing allows up to two lamps to be used, although only one is utilized in this current study. The device is constructed such that the entire length of the lamp(s) irradiates a polyimide-coated fused silica line (ID = 220 μm, OD = 363 μm, SGE Analytical Science) positioned in the center of the device parallel to the lamp(s). This silica line has been “windowed” by thermally removing the polyimide coating along the length of the lamp to allow UV transmission into the inner capillary. The fused silica is fixed securely in the device using PEEK microtight fittings (1/32″ = OD, Upchurch). Although not shown in Figure 1, the device is covered during operation such that UV radiation is blocked from sight for safety. The distance between the device and the IM-MS instrument (~20 cm) contributes to an observed “dead time” before reaction products are detected. Experiments were performed such that data acquisition was initiated concurrent to the lamp being turned on.
Figure 1.

(A) Diagram of the ozonolysis apparatus utilizing a low-pressure mercury lamp and fused silica line with a portion of the polyimide coating removed to permit transmission of UV radiation. (B) A rendered schematic of the mechanically stable housing for the ozonolysis device. (C) Photograph of the completed device with inclusion of thumb screws and capillary fittings for precise alignment of the silica line relative to the lamp.
Data Analysis.
Data was analyzed using MassHunter IMS Browser and Qualitative Analysis software (B.07.02 and B.07.00, Agilent Technologies). For the variable flow rate studies, precursor and ozonolysis product peaks at a given flow rate were extracted as ion chromatograms and normalized as a percent conversion over time. Replicates at each flow rate, collected in triplicate, were time-aligned, and percent conversions were averaged. The equilibrium conversion was determined using the maximal conversion reached after precursor/product ion ratios ceased to vary with time. For LC experiments, relevant extracted ion chromatograms (EICs) were extracted, smoothed, and integrated. Background subtracted mass spectra were subsequently extracted from each of the integrated LC peaks.
■ RESULTS AND DISCUSSION
Conversion of a Glycerophospholipid Standard.
For initial characterization purposes, a commonly studied phosphatidylcholine standard, PC 16:0/18:1(9Z), was directly infused through the device and into the instrument source at a flow rate of 10 μL/min. It was observed that the precursor protonated lipid peak at m/z 760.6 underwent a reaction to form a product aldehyde species at m/z 650.4, which is representative of double bond cleavage at the ninth carbon atom of the 18 carbon length alkyl chain (Figure 2A). Extracted mass spectra averaged over 0.3 min before and after the reaction confirm a transition of approximately ~110.1 Da, which is indicative of an ozonolysis reaction corresponding to a loss of 18 hydrogen and 9 carbon atoms from the alkyl chain as well as an oxygen atom gained during the cycloaddition of ozone as per the Criegee mechanism.35 Although a detailed analysis of the mechanism that gives rise to the diagnostic aldehyde product ion is not within the scope of this paper, the results are consistent with solution-phase ozonolysis, in which an aldehyde and carbonyl oxide are produced following reaction with ozone.36 The unstable carbonyl oxide subsequently reacts with water and rearranges to form additional aldehyde product and hydrogen peroxide, such that the aldehyde product is the only product detectable via MS. Similar rearrangements have also been demonstrated using methanol as a solvent in other ozonolysiscoupled MS studies.12 In the experiments described here where no methanol is used, the presence of a single primary diagnostic ozonolysis product ion simplifies spectral interpretation.29 However, as seen in Figure 2B, other minor product ions are also observed. These minor side products primarily include sodiated and potassiated forms of the aldehyde at m/z 672.4 and 688.4, respectively, as well as other observed peaks in the mass spectrum possibly indicative of solvent adduction processes that have been observed in earlier experiments where ozonolysis occurs in the solution phase (see Figure S1).11,12,37 Nonetheless, the major process observed involves conversion of the glycerophosphocholine precursor to an aldehyde product representative of cleavage at the 9Z position, a process that approaches 95% completion at 10 μL/min (Figure 2C). Even at this relatively low flow rate, the ozonolysis process is rapid and reaches a steady-state equilibrium in less than a minute. The high degree of reaction conversion efficiency, fast reaction kinetics, and low degree of reaction side products highlight several analytical advantages of this online, solution-phase method.
Figure 2.

(A) Conversion of a glycerophospholipid to a diagnostic aldehyde product ion indicative of double bond position was demonstrated using PC 16:0/18:1 (9Z). (B) Following irradiation, the IM-MS spectrum contained a representative peak at m/z 760.6 that decreased in intensity as the sample was infused through the device. Another peak, m/z 650.4, increased in intensity in response to turning the lamp on, the latter corresponding to cleavage of a double bond at the ninth carbon atom of one alkyl chain. (C) A plot of percent conversion over time that indicates that conversion to the aldehyde product at 10 μL/min reached ca. 95% completion after about 1 min following observed onset of reaction.
Influence of Flow Rate on Conversion Efficiency.
Using the same phosphatidylcholine standard, the transition from m/z 760.6 to 650.4 was monitored as a function of time for flow rates ranging from 5 to 500 μL/min. The conversion–time profile displayed sigmoidal behavior, with an observed dead time (Figure 2C) corresponding to the length of tubing between the exit of the ozonolysis device and the source of the mass spectrometer. At sufficiently high flow rates, e.g. ~ 75 μL/min with 20 cm of tubing, this dead time becomes negligible (ca. 5 s or less), with observed product being formed and detected almost immediately after turning the lamp on. As the reaction reaches equilibrium (Figure 2C), the observed conversion from precursor to product ion plateaus to a fixed conversion rate. The extent to which the reaction proceeds is thus dependent on flow rate, with lower flow rates engendering greater maximum conversion (Figure 3A). As the flow rate through the device is increased, the observed conversion percentage from precursor to product decreases due to the fact that the reaction can no longer proceed to completion during the limited time spent in the cell. This inverse relationship between sample flow rate and product conversion is demonstrated in Figure 3B. The measured conversion—flow rate relationship of the device has been divided into three characteristic regions based upon suggested useful operating conditions. In the nanoflow regime (defined here as flow rates less than 10 μL/min), the reaction approaches 100% completion. From 10 to 300 μL/min (i.e., direct infusion conditions), the conversion efficiency decreases as a function of increasing flow rate. Finally, at the upper end of flow rates surveyed (the flow injection regime, ≥ 400 μL/min), the observed maximum conversion varies very little with flow rate. In this region of behavior, the high flow rate limits the observed conversion to approximately ~25%; however, the generation of the maximal conversion at a given flow rate in this regime is nearly instantaneous, allowing for fast “on–off’ switching of the device. This characteristic fast response of the device at high flow rates has the greatest implication for integration with liquid chromatography approaches. The reaction kinetics are such that the device could be toggled on and off during the elution of a chromatographic peak, facilitating identification via IM-MS of both the precursor and the product ion for each LC time bin, with the observed product allowing for determination of double bond position in the precursor.
Figure 3.

Performance of the ozonolysis device was evaluated as a function of flow rate. (A) The conversion of phosphatidylcholine precursor to an aldehyde product characteristic of double bond position was monitored, with the lamp switched on at a time corresponding to zero minutes. Five representative flow rates are shown here with error bars representing triplicate measurements. (B) Across the range of flow rates measured (5 to 500 μL/min) conversion efficiency was observed to decrease as flow rate increased.
Conversion in a System Containing Multiple Double Bonds.
The fatty acid docosahexaenoic acid (FA 22:6 (Δ4, 7, 10, 13, 16, 19)) was chosen for evaluating the device for localizing multiple double bonds, as this lipid contains six sites of unsaturation at positions Δ4, 7, 10, 13, 16, and 19 in the carbon chain (Figure 4B). To address the increased complexity of this system, ion mobility was utilized after the ozonolysis reaction to gain an additional dimension of separation of the ozonolysis products prior to mass analysis. Figure 4A contains a negative mode IM-MS spectrum for ozonolysis of deprotonated FA 22:6 at 10 μL/min. In contrast to the PC 16:0/18:1 described previously, the ozonolysis reaction is distributed across multiple possible reaction channels leading to a series of diagnostic product ions but a decreased conversion efficiency for any single product ion channel. Additionally, fatty acids appear less reactive than glycerophosphocholines, as it was observed that while 10 μL/min was sufficient to convert nearly 100% of the PC 16:0/18:1 precursor to its single product ion, the same flow rate for FA 22:6 only converted about 45% of the precursor to the six possible product ions. However, upon increasing the device flow rate, the conversion percentage decreased in a manner analogous to the phosphatidylcholine described in Figure 3 (Figure 4C) The differential reactivity of various lipid species has been noted by Blanksby and coworkers and has been suggested as a possible means for quantitation via ozonolysis but has not been further explored in this work due to the sensitivity of product generation via this technique to flow rate.15 Additionally, as discussed in the group’s OzESI work, because sample ionization occurs after ozonolysis, ion abundances are biased due to differences in ionization efficiency, making quantitation challenging, particularly in complex mixtures where ion suppression is prevalent.10,12
Figure 4.

Highly unsaturated fatty acid docosahexaenoic acid (FA 22:6) was examined using the flow-cell ozonolysis device in negative ion mode. (A) IM-MS spectrum of FA 22:6 following ozonolysis at a flow rate of 10 μL/min. The six products detected displayed a linear trend in both mass and drift time dimensions and correspond to fragmentation at double bond locations as depicted in (B). Isolation of this particular mobility-mass correlation increases the signal of the ozonolysis fragments versus the background noise. (C) Conversion versus flow rate plots for each of the products trend similarly to the single double bond containing species depicted in Figure 3.
Because the double bonds are symmetrically distributed along the fatty acid acyl tail (spaced three carbons apart), the ozonolysis products generated are linearly spaced in 40 Da intervals in the mass spectrum. These product ions each contain a negatively charged carboxylic acid moiety on one end and are truncated with an aldehyde corresponding to ozonolysis cleavage at a given double bond position. From the IM-MS spectra in Figure 4A, it was observed that there exists a mobility-mass correlation for the ozonolysis fragments and the fatty acid precursor, which allows for the isolation of product-specific mass and mobility spectra and thus improves the signal-to-noise ratio for the ions of interest, especially for that of the low-intensity products resulting from cleavage at the Δ4, 7, and 10 positions. This highlights the utility of IM separation for localization of double bond position in complex samples, as previous studies have discussed the difficulty of double bond assignment in polyunsaturated fatty acids due to low signal-to-noise of product fragments.19,20,22 Murphy et al. in particular have highlighted the challenges of PUFA analysis, and it is herein demonstrated that the additional dimension of separation afforded by IM alleviates the difficulty of double bond assignment in these species. Moreover, the intensity of each product fragment correlates to the propensity for the reaction at each double bond position. Specifically, the product ion intensities decrease relative to a given double bond’s proximity to the fatty acid headgroup, such that in this example, the ozonolysis reaction occurs most readily at the 19 carbon position (farthest from the headgroup) and least readily at the 4 carbon position (closest to the headgroup). We hypothesize that this observed decreased reaction efficiency near the lipid headgroup is likely due to the slight electron-withdrawing character of the carboxylate group of the fatty acid decreasing the nucleophilicity of the double bond near the headgroup of the molecule, which decreases the probability of ozonolysis. The mechanism of ozonolysis is generally thought to occur by an electrophilic 1,3-addition of ozone, with reaction rates a function of electron density around the double bond, which supports our hypothesis.38-40
Analysis of a Lipid Extract.
The utility of the ozonolysis device was further explored by analyzing a phosphatidylcholine lipid extract that contains a distribution of PC lipids of various degrees of saturation. Due to the low natural abundance of saturated lipids, a fully saturated lipid standard, PC 16:0/18:0, which should not undergo ozonolysis, was spiked into the extract prior to analysis. In this role, PC 16:0/18:0 acts as a negative control for ozonolysis in a biological sample. The resulting IM-MS spectra for the lamp on and off are contained in Figure 5. The two IM-MS spectra highlight a region corresponding to the series of phosphatidylcholines containing 34 alkyl chain carbon atoms and 0–4 alkyl chain double bonds (PC 34:x, x = 0–4). When the lamp is off (Figure 5A), all five of the species are readily detectable, and their peak centroids display a characteristic mobility-mass correlation consistent with previous IM lipid studies.41-43 This observed decrease in drift time with an increase the number of double bonds is due to changes in the gas-phase packing efficiency of the lipid as the degree of unsaturation increases. The addition of a cis double bond to the alkyl chain of a lipid introduces a kink in the structure that allows the lipid tail to wrap around the molecule, resulting in a more compact gas-phase structure.26 The further addition of a second or third double bond causes additional compaction and further reduces the drift time; however, this phenomena has diminishing returns as additional double bonds (≥3) result in more modest compaction of the gas-phase structure.
Figure 5.

Phosphatidylcholine lipid extract from chicken egg spiked with a saturated lipid standard (PC 34:0) was irradiated using the device. (A) The lipid signals observed in the IM-MS spectrum correspond to a series of phosphatidylcholines containing 34 alkyl chain carbons with 0–4 alkyl chain double bonds. (B) The four species containing double bonds were found to deplete upon irradiation, with low-intensity species completely converted. However, the fully saturated lipid standard at m/z 762.6 did not decrease in intensity, clearly identifying saturated versus unsaturated lipids within a complex lipid sample.
When the lamp is switched on (Figure 5B), all four of the unsaturated lipid species in this IM-MS region deplete as expected due to ozonolysis. Two of the lipid precursors (PC 34:4 and PC 34:3) are no longer detectable, and the other two lipids containing sites of unsaturation (PC 34:2 and PC 34:1) are depleted to the extent that the fully saturated species PC 34:0 at m/z 762.6 becomes the dominant species in this region of the spectrum. Hence, as earlier hypothesized, the ozonolysis device readily identifies fully saturated lipid species in a mixture by converting only the unsaturated species to product ions. While techniques such as high-resolution mass spectrometry (HRMS) can readily distinguish the degree of saturation for lipids, the above example demonstrates the utility of differentiating saturated from unsaturated lipids for lower-resolution mass spectrometry measurements.
The utility of the ozonolysis device to analyze a complex lipid mixture is also demonstrated in the resulting narrower IM peak profiles observed following the reaction. Normally, the overlapping of isotopic envelopes between adjacent lipid species introduces drift profile peak broadening and can lead to shifts in the drift peak’s measured centroid (Figure S2). As such, this isotopic overlap can result in inaccurate calculations of collision cross section for closely related lipid species. As increasing efforts are made to establish collision cross section as a discriminating identifier in metabolomic and lipidomic workflows, the generation of CCS databases that are both comprehensive and accurate is becoming increasingly important.42-45 Ozonolysis in this setting provides an additional analytical tool for minimizing isobaric interferences and thus resulting in more accurate CCS measurements.
Although not depicted in Figure 5, several other series of phosphatidylcholines were also detected in the lipid extract, such as PC 38:x (x = 3–8) and PC 18:x (x = 0–4). In contrast to traditional postmobility collision-induced dissociation (IM/CID-MS) where product and precursor ions are drift time correlated, the ozonolysis reactions in this work occur prior to source ionization. Thus, the product ions created via the mercury lamp cannot be time-aligned to a lipid precursor ion, resulting in challenging fragment association and identification. In fact, multiple precursors could generate a given product ion, and additionally, if a precursor contains multiple double bonds, it can generate multiple product ions, as demonstrated in Figure 4. For these reasons, the resulting postozonolysis product spectrum for a mixture is challenging to analyze, and double bond positions in the precursor are difficult to assign (Figure S3). These findings indicate that chromatographic separation prior to ozonolysis (LC-Oz-IM-MS) would facilitate improved characterization of double bond position in complex mixtures. A recent review by Hancock et al. concludes that initial separation by chromatography, followed by online ozonolysis and IM-MS, should be sufficient not only for determination of double bond position, but for near complete structural characterization of the lipidome.7 This assertion lends further credence to the strength and potential impact of our online ozonolysis approach via the low-pressure mercury lamp, especially when coupled to chromatography for broad-scale lipidomic analyses.
Demonstration of a Simple Chromatography Experiment in Conjunction with Ozonolysis.
As described in the preceding section, chromatographic separation is crucial to the application of the ozonolysis technique to the identification of double bond position in complex mixtures because it allows for the precise alignment of ozonolysis precursor and product species. To illustrate the analytical utility of performing ozonolysis following LC separation, a mock mixture of three isomeric glycerophosphocholines (PC 18:1 (9E)/18:1 (9E), PC 18:1 (9Z)/18:1 (9Z), and PC 18:1 (6Z)/18:1 (6Z)) was prepared and analyzed by LC-Oz-IM-MS. The results of this experiment are depicted in Figure 6. The chromatographic separation consisted of a simple linear gradient, and in lieu of interleaving on–off experiments, the analysis was performed twice: in the first, the ozonolysis device remained off, and in the second, it was activated for the duration of the experiment. Performing two parallel experiments facilitated simpler correlation of precursors and products and thus allowed easier interpretation of the data. Note that in both experiments, the degasser apparatus was bypassed so as to not remove oxygen from the solvent and decrease ozonolysis reaction efficiency. Low inter-run variability allowed for the alignment of the two resulting extracted ion chromatograms (EICs) for the isomer species at m/z 786.6, although only the EIC corresponding to the second experiment with the lamp on is displayed in Figure 6B for simplicity. With the device off (Figure 6A), it is unclear from the EIC of m/z 786.6 that three isomeric species are present, since only two chromatographic peaks are observed. However, once the ozonolysis device is switched on (Figure 6B), the background subtracted extracted mass spectra corresponding to each chromatographic peak reveal the existence of an additional third isomer present in the second chromatographic peak. Evidence supporting the assignment of two isomers in the second peak arises from the diagnostic ozonolysis fragments at m/z 634.4 and 676.6 corresponding to aldehyde products resulting from cleavage at the 6Z and 9Z positions on the fatty acid chain. However, other ozonolysis products in addition to the aldehyde product are also detected at masses approximately 22 and 56 Da above that of the aldehyde and are thought to correspond to sodiated and solvent adducted species (Figure S4).
Figure 6.

A mock mixture of three isomeric glycerophosphatidylcholines was analyzed via LC-Oz-IM-MS. (A) With the device turned off, the EIC trace of m/z 786.6 shows separation into cis and trans isomers, and no diagnostic ozonolysis fragments are detected. (B) Activation of the ozonolysis device reveals the presence of two distinct cis double bond positional isomers at the 6Z and 9Z positions, with diagnostic aldehyde fragments at m/z 634.4 and 676.6, respectively, underneath the second LC peak in the EIC trace.
Interestingly, this experiment highlights how neither chromatographic nor ozonolysis approaches alone are sufficient to resolve this system of isomers: liquid chromatography separates the cis and trans isomers via differences in their retention time, and ozonolysis produces fragments diagnostic of double bond location to identify the 6Z and 9Z position isomers. As a proof of concept case, the experiment demonstrates how the combination of ozonolysis and liquid chromatography is able to improve the confidence of identification for lipid isomers and should also be broadly applicable to real biological samples in lipidomic workflows. Moreover, though not specifically illustrated in this particular example, ion mobility separation provides an additional dimension of separation to further increase the signal-to-noise of the ozonolysis fragments. In this way, multiple techniques serve complementary roles within a combined analytical platform for more comprehensive lipid coverage.
■ CONCLUSIONS
The online flow-cell device developed here allows for determination of double bond position in multiple lipid species in both positive and negative ionization modes. Postozonolysis ion mobility separation imparts further structural information regarding the aldehyde product ions and may in the future aid in identification of other classes of lipid isomers. Characterization of the device’s performance revealed that the rate of production of diagnostic product ions as well as total observed conversion of precursor to product displayed a high degree of dependence on the sample flow rate. Moreover, preliminary experiments indicate that the flow rates achievable through the device are complementary with liquid chromatographic approaches. The LC-Oz-IM-MS separation of three lipid isomers was able to differentiate the presence of each isomer, thus demonstrating the utility of combining several analytical dimensions within a single experiment. Overall, the use of a low-pressure mercury lamp to induce ozonolysis in glycerophosphatidylcholines and fatty acids resulted in fast reaction kinetics and high product yield under the conditions studied. Moreover, the platform is low-cost and simple to implement on any instrument because it requires no instrument modifications and does not require an ozone generator. For these reasons, this device has the potential for wide adoption into existing MSbased lipidomic workflows.
Supplementary Material
■ ACKNOWLEDGMENTS
R.H. acknowledges the Harold Stirling Graduate Fellowship from the Vanderbilt University Graduate School. Financial support for aspects of this research was provided by The National Institutes of Health (NIH Grant R01GM092218) and under Assistance Agreement No. 83573601 awarded by the U.S. Environmental Protection Agency. This work has not been formally reviewed by EPA. The views expressed in this document are solely those of the authors and do not necessarily reflect those of the Agency. EPA does not endorse any products or commercial services mentioned in this publication. Furthermore, the content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies and organizations. This work was supported in part using the resources of the Center for Innovative Technology at Vanderbilt University.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b04007.
An expanded version of the PC 16:0/18:1 spectra following irradiation with the ozonolysis device at 10 μL/min; comparison of drift time profiles for two lipid species from a PC lipid extract with the ozonolysis device off and on; full IM-MS spectra of the PC lipid extract with the ozonolysis device off and on; comparison of extracted ion chromatograms (EICs) for three PC standards ran separately using the LC-Oz-IM-MS technique, with the ozonolysis device turned on (PDF)
The authors declare no competing financial interest.
■ REFERENCES
- (1).Wenk MR Nat. Rev. Drug Discovery 2005, 4 (7), 594–610. [DOI] [PubMed] [Google Scholar]
- (2).Navas-Iglesias N; Carrasco-Pancorbo A; Cuadros-Rodríguez L TrAC, Trends Anal. Chem 2009, 28 (4), 393–403. [Google Scholar]
- (3).Rolim AEH; Henrique-Araújo R; Ferraz EG; de Araújo Alves Dultra FK; Fernandez LG Gene 2015, 554 (2), 131–139. [DOI] [PubMed] [Google Scholar]
- (4).Pulfer M; Murphy RC Mass Spectrom. Rev 2003, 22 (5), 332–364. [DOI] [PubMed] [Google Scholar]
- (5).McDonald JG; Ivanova PT; Brown HA Approaches to Lipid Analysis In Biochemistry of Lipids, Lipoproteins and Membranes, 6th ed.; Ridgway N, McLeod R, Eds.; Elsevier: Amsterdam, 2016; pp 41–72. [Google Scholar]
- (6).Blanksby SJ; Mitchell TW Annu. Rev. Anal. Chem 2010, 3 (1), 433–465. [DOI] [PubMed] [Google Scholar]
- (7).Hancock SE; Poad BLJ; Batarseh A; Abbott SK; Mitchell TW Anal. Biochem 2017, 524, 45–55. [DOI] [PubMed] [Google Scholar]
- (8).May JC; McLean JA Annu. Rev. Anal. Chem 2016, 9 (1), 387–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Harrison KA; Murphy RC Anal. Chem 1996, 68 (18), 3224–3230. [DOI] [PubMed] [Google Scholar]
- (10).Brown SHJ; Mitchell TW; Blanksby SJ Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2011, 1811 (11), 807–817. [DOI] [PubMed] [Google Scholar]
- (11).Sun C; Zhao YY; Curtis JM Anal. Chim. Acta 2013, 762, 68–75. [DOI] [PubMed] [Google Scholar]
- (12).Thomas MC; Mitchell TW; Harman DG; Deeley JM; Murphy RC ; Blanksby SJ Anal. Chem 2007, 79 (13), 5013–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sun C; Black BA; Zhao YY; Ganzle MG; Curtis JM Anal. Chem 2013, 85 (15), 7345–7352. [DOI] [PubMed] [Google Scholar]
- (14).Thomas MC; Mitchell TW; Harman DG; Deeley JM; Nealon JR; Blanksby SJ Anal. Chem 2008, 80 (1), 303–311. [DOI] [PubMed] [Google Scholar]
- (15).Poad BLJ; Pham HT; Thomas MC; Nealon JR; Campbell JL; Mitchell TW; Blanksby SJ J. Am. Soc. Mass Spectrom 2010, 21 (12), 1989–1999. [DOI] [PubMed] [Google Scholar]
- (16).Marshall DL; Pham HT; Bhujel M; Chin JSR; Yew JY; Mori K; Mitchell TW; Blanksby SJ Anal. Chem 2016, 88 (5), 2685–2692. [DOI] [PubMed] [Google Scholar]
- (17).Poad BLJ; Green MR; Kirk JM; Tomczyk N; Mitchell TW; Blanksby SJ Anal. Chem 2017, 89 (7), 4223–4229. [DOI] [PubMed] [Google Scholar]
- (18).Ma X; Xia Y Angew. Chem., Int Ed 2014, 53 (10), 2592–2596. [DOI] [PubMed] [Google Scholar]
- (19).Ma X; Chong L; Tian R; Shi R; Hu TY; Ouyang Z; Xia Y Proc. Natl. Acad. Sci. U. S. A 2016, 113 (10), 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ma X; Zhao X; Li J; Zhang W; Cheng JX; Ouyang Z; Xia Y Anal. Chem 2016, 88 (18), 8931–8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Stinson CA; Xia Y Analyst 2016, 141 (12), 3696–3704. [DOI] [PubMed] [Google Scholar]
- (22).Murphy RC; Okuno T; Johnson CA; Barkley RM Anal. Chem 2017, 89 (16), 8545–8553. [DOI] [PubMed] [Google Scholar]
- (23).Kliman M; May JC; McLean JA Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2011, 1811 (11), 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Paglia G; Kliman M; Claude E; Geromanos S; Astarita G Anal. Bioanal. Chem 2015, 407 (17), 4995–5007. [DOI] [PubMed] [Google Scholar]
- (25).Groessl M; Graf S; Knochenmuss R Analyst 2015, 140 (20), 6904–6911. [DOI] [PubMed] [Google Scholar]
- (26).Kyle JE; Zhang X; Weitz KK; Monroe ME; Ibrahim YM; Moore RJ; Cha J; Sun X; Lovelace ES; Wagoner J; et al. Analyst 2016, 141 (5), 1649–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dodds JN; May JC; McLean JA Anal. Chem. 2017, 89 (1), 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Poad BLJ; Zheng X; Mitchell TW; Smith RD; Baker ES; Blanksby SJ Online Ozonolysis Combined with Ion Mobility-Mass Spectrometry Provides a New Platform for Lipid Isomer Analyses. Anal. Chem 2017, In press. 10.1021/acs.analchem.7b04091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Stinson CA; Zhang W; Xia Y UV Lamp as a Facile Ozone Source for Structural Analysis of Unsaturated Lipids via Electrospray Ionization-Mass Spectrometry. J. Am. Soc. Mass Spectrom 2017; In press. 10.1007/s13361-017-1861-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).May JC; Goodwin CR; Lareau NM; Leaptrot KL; Morris CB; Kurulugama RT ; Mordehai A; Klein C; Barry W; Darland E; et al. Anal. Chem 2014, 86 (4), 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).May JC; Dodds JN; Kurulugama RT ; Stafford GC; Fjeldsted JC; McLean JA Analyst 2015, 140 (20), 6824–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cruickshank-Quinn C; Quinn KD; Powell R; Yang Y; Armstrong M; Mahaffey S; Reisdorph R; Reisdorph N Multi-Step Preparation Technique to Recover Multiple Metabolite Compound Classes for In-Depth and Informative Metabolomic Analysis. J. Visualized Exp [Online] 2014. 10.3791/51670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zoschke K; Bornick H; Worch E Water Res. 2014, 52 (0), 131–145. [DOI] [PubMed] [Google Scholar]
- (34).Stinson CA UV-Induced Online Photochemical Reactions for Enhanced Biomolecule Structural Characterization on an ESI MS/MS Platform. Ph. D. Dissertation, Purdue University: West Layfayette, IN, 2015. [Google Scholar]
- (35).Criegee R Angew. Chem., Int. Ed. Engl 1975, 14 (11), 745–752. [Google Scholar]
- (36).Santrock J; Gorski RA ; O’Gara JF Chem. Res. Toxicol 1992, 5 (1), 134–141. [DOI] [PubMed] [Google Scholar]
- (37).Sun C; Zhao YY; Curtis JM Rapid Commun. Mass Spectrom 2012, 26 (8), 921–930. [DOI] [PubMed] [Google Scholar]
- (38).Klutsch G; Fliszár S Can. J. Chem 1972, 50, 2841–2844. [Google Scholar]
- (39).Pryor WA; Giamalva D; Church DF J. Am. Chem. Soc 1985, 107 (9), 2793–2797. [Google Scholar]
- (40).Fisher TJ; Dussault PH Tetrahedron 2017, 73 (30), 4233–4258. [Google Scholar]
- (41).Kim HI; Kim H; Pang ES; Ryu EK; Beegle LW; Loo JA; Goddard WA; Kanik I Anal. Chem 2009, 81 (20), 8289–8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Paglia G; Angel P; Williams JP; Richardson K; Olivos HJ; Thompson JW; Menikarachchi L; Lai S; Walsh C; Moseley A; et al. Anal. Chem 2015, 87 (2), 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Leaptrot KL; May JC; Dodds JN; Mclean JA Conformational Atlas of Sphingolipids and Glycerophospholipids Mapped by Uniform Field Ion-Mobility Mass Spectrometry. In preparation. [Google Scholar]
- (44).May JC; Morris CB; McLean JA Anal. Chem 2017, 89 (2), 1032–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Stow SM; Causon TJ; Zheng X; Kurulugama RT; Mairinger T; May JC; Rennie EE; Baker ES; Smith RD; McLean JA; et al. Anal. Chem 2017, 89 (17), 9048–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.