

Abstract
Breast cancer tumorigenesis and response to therapy is regulated by cancer cell interactions with the tumor microenvironment (TME). Breast cancer signaling to the surrounding TME results in a heterogeneous and diverse tumor microenvironment, which includes the production of cancer-associated fibroblasts, macrophages, adipocytes, and stem cells. The secretory profile of these cancer-associated cell types results in elevated chemokines and growth factors that promote cell survival and proliferation within the tumor. Current co-culture approaches mostly rely on transwell chambers to study intercellular signaling between adipose-derived stem cells (ASCs) and cancer cells; however, these methods are limited to endpoint measurements and lack dynamic control. In this study, a 4-channel, “flow-free” microfluidic device was developed to co-culture triple-negative MDA-MB-231 breast cancer cells and ASCs to study intercellular communication between two distinct cell types found in the TME. The device consists of two layers: a top PDMS layer with four imprinted channels coupled with a bottom agarose slab enclosed in a Plexiglas chamber. For dynamic co-culture, the device geometry contained two centered, flow-free channels, which were supplied with media from two outer flow channels via orthogonal diffusion through the agarose. Continuous fresh media was provided to the cell culture channel via passive diffusion without creating any shearing effect on the cells. The device geometry also allowed for the passive diffusion of cytokines and growth factors between the two cell types cultured in parallel channels to initiate cell-to-cell crosstalk. The device was used to show that MDA-MB-231 cells co-cultured with ASCs exhibited enhanced growth, a more aggressive morphology, and polarization toward the ASCs. The MDA-MB-231 cells were found to exhibit a greater degree of resistance to the drug Paclitaxel when co-cultured with ASCs when compared to single culture studies. This microfluidic device is an ideal platform to study intercellular communication for many types of cells during co-culture experiments and allows for new investigations into stromal cell-mediated drug resistance in the tumor microenvironment.
Graphical Abstract
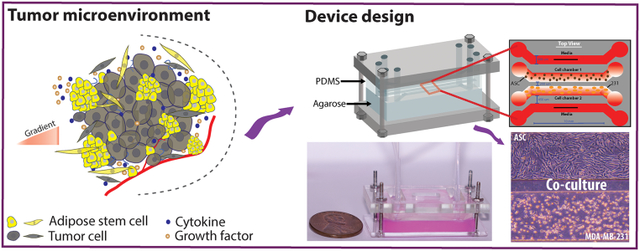
Introduction
Cancer cells coexist in the tumor microenvironment (TME) with stromal cells (e.g., fibroblasts or endothelial cells), adipose-derived stem cells (ASCs), and immune cells (e.g., macrophages) that communicate with each other by secreting cytokines, hormones, and growth factors that traverse the TME via passive diffusion, creating a complex, diverse, and heterogeneous environment.1,2 ASCs are the critical component of the breast tumor microenvironment, and they are well-documented mediators of breast cancer progression that stimulate tumor growth, metastasis, and drug resistance by secreting excess growth factors, cytokines (adipocytokines, IL-6, TNFα).3–5 Inflamed adipose tissue recruits increased macrophages which are associated with pro-inflammatory cytokines while expressing genes associated with cancer development, such as secreted phosphoprotein1(SPP1) and matrix metalloproteinase (MMP9).6 The crosstalk between cancer cells and ASCs utilizes paracrine signaling that induces both phenotypic and genotypic alterations in both cell types.7–9 ASCs can also create oxidative stress in the TME by synthesizing reactive oxygen species (ROS), resulting in DNA double-stranded breaks and reduced repair of damaged DNA, leading to increased mutations while secreted adipokines function as an encounter with the oxidative stress, eventually increasing chemoresistance.4,5 Gaining a comprehensive understanding of the intercellular communication between ASCs and breast cancer cells is of vital significance for the diagnosis and treatment of breast cancer patients. This needs to be further investigated by the continuous dynamic co-culture of cancer cells and ASCs.
Many in vitro studies have been conducted to understand cellular communication with disease progression, including intercellular communication of stem cells and cancer cells using the Transwell assay. A Transwell assay creates two distinct culture chambers separated by a semi-permeable insert composed of a porous membrane to physically separate two different cell types while allowing for indirect communication of soluble chemicals (e.g., cytokines and growth factors) via passive diffusion.10,11 This approach has facilitated numerous co-culture studies; however, it has several limitations including (i) its inability to monitor the cells during the experiment by light microscopy, (ii) Infiltration of the cells from the top chamber into the bottom one, and (iii) no infusion of fresh growth media into the system, resulting in a batch system with decreasing amounts of nutrients and an accumulation of waste as the experiment progresses. Recently, microfluidic devices have become an attractive alternative to perform dynamic co-culture studies.12,13 Microfluidic devices have several advantages including their (i) ability to precisely control the physical and chemical microenvironment, (ii) compatibility with light and fluorescent microscopy techniques, (iii) reproducibility, (iv) negligible impact on cellular viability, (v) capability to incorporate different biomaterials resembling physiological 3D matrices, and (vi) dramatic reduction in reagent volume.14–16 Several types of microfluidic devices had been reported to study cell-to-cell communication, including cell trapping and pairing with or without hydrodynamic trapping in a microfluidic channel, droplet-based cell encapsulation, microwell array culture systems, acoustic wave-based co-culture and digitally equipped microfluidics (electrophoretic assays) to interrogate interactions of microorganisms and mammalian cells.17–22 These different types of microfluidic co-culture devices have been summarized in several recent review articles.10,14,16,23–25 Hydrodynamic cell trapping and pairing with other cells on-chip is a popular method to study cell-to-cell communication at the single-cell level; however, it is complicated, produces shearing force, and lacks efficiency. Though these devices showed higher trapping efficiency and long-term culture capability, there was a limitation in media supply when both cells require distinct media for their growth since the channel design permitted only a single media. Cells co-cultured using the hydrodynamic technique is a promising approach to overcome the batch culture limitations; however, it requires complex fluidic control schemes to ensure cell isolation and media infusion.26 Another co-culture method that is ideally suited for single-cell studies is based on droplet encapsulation, but this method is limited by low overall cell numbers in a single droplet associated with low nutrient-to-cell mass transfer. Microfluidic droplet trapping arrays possess the potential for analysing cells at the single-cell level; however, due to the small size of the droplets they are limited in their ability to perform long-term co-culture experiments. Microwells allow for gravity-based settling of cells where the well size can control the number of cells and result in a greater degree of intercellular interactions by direct contact; however, they lacks dynamic control of the system and can result in non-steady culture conditions (for the devices without flow) or shearing effects (for the devices with the flow).23 Electrophoretic immunoassays integrate electrochemical features with on-chip microfluidic channels to monitor cell secretions due to changes in the biophysical properties of the medium. However, device setup and operation was complex, expensive, and did not allow for long-term co-culture experiments. Another key pitfall in these microfluidic devices was the formation of shearing forces by the media flow.
An alternative approach to microfluidic co-culture incorporates hydrogels to allow for passive diffusion of biomolecules and indirect cell-to-cell communication. Thomsen and co-workers developed an agarose hydrogel-based microwell co-culture platform where breast cancer cells (MDA-MB-231) were co-cultured with human mesenchymal stromal cells for a long term static experiment.27 They demonstrated the enhanced proliferation of cancer cells in the presence of mesenchymal stromal cells, verifying the exchange of secreted biomolecules between both types of cells through agarose hydrogel while cells remained in the agarose microwell as aggregates. The device was capable of making indirect cell communication while cells were restricted to the confined space of a static culture condition. Similarly, Liu and co-workers co-cultured stromal cells, fibroblasts, endothelial cells, and macrophages separately by incorporating a hydrogel barrier in a microfluidic device to simulate the tumor microenvironment.28 The incorporation of hydrogels into fluidic channels has offered the greatest promise with microfluidic co-culture applications.
In this work, a microfluidic co-culture device was developed to allow for the flow-free, indirect intercellular communication between ASCs and triple-negative breast cancer cells (MDA-MB-231) under steady-state conditions. The device consisted of four parallel fluidic channels imprinted into a polydimethylsiloxane (PDMS) replica, which was placed on top of an agarose slab, with the entire system encased in a Plexiglas chamber. The main design principle of the device is the high permeability of the bottom layer of agarose, which allows for the rapid and facile diffusion of small molecules and proteins. The two outer ‘flow’ channels were continuously supplied with culture29,30 media while the two inner “flow-free” channels were used to culture both cancer cells and ASCs. As a result, biomolecules would passively diffuse perpendicular to the direction of flow across the entire length of the culture channels to (i) continually supply cells with fresh media, (ii) remove cellular waste, and (iii) allow for the passive diffusion of paracrine signals between two different cell types. A similar approach has been used to study the chemotactic movement of neutrophils and bacteria in a three-channel microfluidic device in which the middle channel was for cell seeding, and the outer channels were for media infusion. Cheng et al. developed the agarose-based microfluidic device for the diffusion of biomolecules to study the chemotactic response of E. coli and HL-60 cells in separate experiments.31 This device was for the study of cell migration and did not show the capability for dynamic co-culture applications. In this presented work, dynamic co-culturing of adherent cell lines on agarose in the four-channel device required cell attachment on the agarose surface, which was facilitated by treating the agarose with cationic poly-d-lysine to induce the adhesion of the negatively charged mammalian cell membrane.32 The device was compatible with light microscopy to allow for the regular collection of images to track cell growth, orientation, and morphology. Additionally, the device allowed for on-chip viability staining and immunostaining. Each of these features offered by the presented device possesses the suitability for long term co-culturing of different cell lines comparing to the available devices in literature. MDA-MB-231 cells cultured in the presence of ASCs demonstrated enhanced growth and proliferation and an altered, more aggressive morphology, and they were found to polarize towards the ASCs. Moreover, cancer cells, when cultured with ASCs, were found to exhibit enhanced resistance and greater viability when exposed to Paclitaxel. These findings suggest a potential role of ASCs in both cancer progression and drug resistance.
Experimental
Materials and Methods
The microfluidic device contained two separate layers: a bottom layer of 3% (w/v) agarose (Invitrogen, USA) to facilitate chemical diffusion and a top polydimethylsiloxane (PDMS, Sylgard 184, DOW) layer into which four parallel fluidic channels were imprinted (Figure 1A). The PDMS slab consisted of four 600-μm-wide parallel channels with a total operating length of 10 mm and a channel height of 150 μm. The outermost channels were designed to accommodate flow to perfuse the system with fresh media continually so that the device could reach an approximate steady state. The innermost channels were designed to be “flow-free” so that the cells did not experience any direct shear but could still be exposed to fresh media and cell-to-cell communication. The outermost channels were spaced 450 μm from the innermost “flow-free” culture channels. The two culture channels were spaced 200 μm apart to allow for facile diffusion of excreted factors for the two cell lines to communicate with each other. The PDMS and agarose components were fixed in place using two rectangular pieces of Plexiglas (McMaster-Carr) held together by four screws at the four corners of the device (Figure 1B). Windows were cut out of the top layer of Plexiglas to allow for the insertion of tubing into the device. The thickness of the agarose layer was ~5 mm, and the thickness of the PDMS layer was ~5 mm, as measured by a caliper both to allow for greater reproducibility and to ensure that the two layers remained fluid-tight. The top PDMS layer was fabricated using a combination of soft lithography and PDMS replication. The geometry was designed in AutoCAD (Autodesk, USA) to generate a transparency mask (CAD/Art) of the four fluidic channels. A silicon master wafer was fabricated by depositing SU-8 2050 (MicroChem), a negative photoresist polymer, onto a 3” silicon wafer (University Wafer) using a spin coater (WS-650MZ-23NPP, Laurell, USA) to generate a final channel height of 150 μm by utilizing a two-step process. First, a 75-μm-thick layer of SU-8 2050 was deposited onto the wafer, followed by baking at 65°C and 95°C for 10 min and 20 min, respectively. Next, the second layer of SU-8 2050 (also 75-μm-thick) was deposited on the wafer, followed by baking at the same conditions. After the wafer was cooled to room temperature, the transparency mask was placed on top of the wafer, followed by exposure to UV light (1.4 mW/cm2) for 128 seconds in a custom-built UV exposure set-up using a B100-AP lamp (VWR). The wafer was baked at 65°C for 15 min and at 95°C for 2 h after UV exposure. After cooling at room temp for 30 min, the SU-8 developer (MicroChem) was used to remove all uncrosslinked SU-8. The wafer was hard-baked at 150°C for 1 h to increase durability and then was treated with a silane (tridecafluoro-1, 1, 2, 2-tetrahydrooctyl trichlorosilane, Sigma-Aldrich, USA) in a vacuum to deposit a thin layer on the surface of the wafer, which facilitates detachment of the PDMS replicas. PDMS replicas were generated by mixing 25 g of the base and 2.5 g of the curing agent in a 10:1 ratio followed by degassing in a vacuum chamber to create a bubble-free mixture. This PDMS was poured on the silicon master wafer and was allowed to cure for at least 12 h at 65°C. Cured replicas were removed from the wafer and individual devices were cut out with an X-Acto knife, followed by punching the inlet and the outlet ports using a blunted 18-gauge needle.
Figure 1:

Design of the microfluidic co-culture device. (A) The flow-free microfluidic device consisted of four fluidic channels imprinted into a PDMS slab placed on top of 3 wt% agarose. The device contains four parallel channels: the two outermost ‘flow’ channels are constantly supplied with media while the two innermost “flow-free” channels are used to culture the MDA-MB-231 cells and adipose stem cells. All channels are 600 μm wide with a contact length of 10 mm and a height of 150 μm. The spacing between the media channels and the culture channels is 450 μm, while the spacing between the two cell culture channels is 200 μm to facilitate cellular crosstalk. (B) Image of a completely assembled device with agarose stained red for enhanced visualization. (C) Representative image of MDA-MB-231 cells (bottom) and ASCs (top) cultured in the device after 72 h.
Computational simulations of mass transfer in the microfluidic device
COMSOL simulations were performed to model the chemical and oxygen mass transfer within the device to (i) ensure the cells were supplied with sufficient media, (ii) confirm that the soluble factors released from cells were able to diffuse across the “flow-free” channels, and (iii) validate that the oxygen was able to diffuse into the culture channels, thus avoiding hypoxic conditions. The mass transfer of the media into the culture channels was simulated in the two outermost flow channels to mimic the constant infusion of growth media while there was no flow in the two innermost, “flow-free” culture channels. The Reynold’s number was calculated to be <10 in the outermost flow channels and was considered to be that of an isothermal and incompressible Newtonian liquid with the properties of water (density 995.6 kg-m−3, viscosity 7.97 × 10−4 Pa-s).20 The general steady-state diffusion–convection equation was solved by using the velocity results from the laminar flow to determine the concentration gradient of media and mass transfer. No-slip boundary conditions and a two-dimensional device profile were assumed. The porosity of 3% (w/v) agarose was approximated as 0.97.33 Diffusion of growth factors and other biomolecules through the agarose slab were approximated using the diffusion coefficient for SDF-1α (a model growth factor) in 3% (w/v) agarose set which was 110 μm2/s.34 All simulations approximated an initial concentration of the biomolecule as 140 nM in the outer channel. The fluid material was considered as water and a flowrate of 15 μL/min in the outermost channels was specified for optimum mass transfer. A similar approach, with the same assumptions, was taken when modeling the mass transfer across the two cell culture channels with the exception that these channels were simulated with no flow. For the oxygen mass transfer simulations, the height for oxygen transfer to the cells on the agarose layer was approximated at 5 mm. The top Plexiglas was cut to have a 10 mm by 3.5 mm window above the culture channels to allow for better mass transfer of oxygen into the device.
Cell culture and reagents
Triple-negative breast cancer cell line MDA-MB-231 was acquired from ATTC. Cells were cultured with Dulbecco’s modified eagle’s medium (DMEM, Corning) and supplemented with 10% v/v HyClone Cosmic Calf Serum (VWR Life Sciences Seradigm), 1% MEM Essential Amino Acids (Quality Biological Inc.), 1% MEM Non-Essential Amino Acids (Quality Biological Inc.), and 50 ng/mL insulin (Sigma- Aldrich, ST. Louis, MO). Cells were grown at 37°C with 5% humidified CO2 and sub-cultured every 3 days. Abdominal human adipose-derived stem cells (ASCs) were purchased directly from LaCell (New Orleans, LA), and these cells were grown in α-minimum essential medium (α MEM; Gibco, NY), 10% fetal bovine serum (FBS, Atlanta Biologicals), and 1% Antibiotic-Antimycotic (Anti-Anti, Gibco). ASCs were split at 60–70% confluence. The stem cell donor was Caucasian, female, and 24 years old with a body mass index (BMI) of 28. The stem cell medium was changed every third day.
Assembly and operation of the two-layer microfluidic device
The top Plexiglas layer was placed over the PDMS replica, followed by pre-plumbing the inlet and outlet ports with Tygon tubing (0.5588 mm, Cole Palmer) to prevent leakage during experimentation. The bottom agarose layer was prepared by pipetting 12 mL of 3% (w/v) agarose into a 60 mm petri dish and was allowed to solidify for a minimum of 1 h. The agarose was pre-treated with 0.1 mg/mL poly-d-lysine (Sigma Aldrich) to facilitate the attachment of the cells to the surface. Poly-d-lysine was resuspended in 1X phosphate-buffered saline (PBS; 137 mM NaCl, 10 mM Na2HPO4, 27 mM KCL, and 1.75 mM KH2PO4 at pH 7.4) for 10 min at room temperature. The poly-d-lysine solution was aspirated, and the agarose was incubated at room temperature for 2 h in a sterile environment followed by 2X washing with MDA-MB-231 culture media. A 20 × 30 mm agarose slab was cut from the dish using a razor blade and placed on top of the bottom Plexiglas layer. The pre-plumbed PDMS replica, coupled with the top Plexiglas piece, was placed on top of the agarose slab to generate a complete device. The device was rendered fluid-tight using four screws and hex nuts (18–8SS ¾, McMaster-Carr) at the four corners of the device. The screws were tightened gently (to ensure the same vertical spacing at all four corners), followed by leakage testing by injecting media through all four-fluidic channels at a rate of 15 μL/min using a syringe pump (KD Scientific). A digital slide caliper (Fisher Scientific) was used to measure the vertical spacing between the top and bottom Plexiglas layers. The assembled device was immersed in complete growth media supplemented with 1% (v/v) P/S (Penicillin and Streptomycin, Gibco) and primed for 4 h by injecting MDA-MB-231 culture media into the two outermost flow channels at a rate of 15 μL/min in a 37°C, 5% CO2 incubator (Symphony, VWR). After the priming period, cells were injected into the two innermost channels using a syringe pump at a rate of 5 μL/min. MDA-MB-231 cells were injected at a density of 5×105 cells/mL, and ASCs were injected at a density of 1×106 cells/mL. The cellular suspension was added sequentially (MDA-MB-231 cells first, ASC cells second) to avoid compromising the fluid-tight channels. The Tygon tubing connected to the inlet and outlet ports of the innermost cell culture channels were sealed with blunted 23G syringes to prevent contamination and media loss. The outermost flow channels were hooked up to 10 mL syringes (BD, VWR) containing complete growth media, which was infused continuously into the device at a rate of 15 μL/min. During single culture experiments, the media infused into the device was the same as described above for MDA-MB-231 cells and ASCs. During co-culture, MDA-MB-231 culture media was infused into the flow channel adjacent to the cancer cell culture channel, while ASC culture media was infused into the flow channel adjacent to the ASC culture channel. The device and syringe pumps were placed in a 37°C incubator for the extent of the experiment. Light microscopy images were collected every 12 h during the 72 h experiment using a Zeiss Primo Vert phase-contrast microscope equipped with an Axiocam105 color digital camera. All images were further processed in ImageJ (NIH, USA) software.
On-chip fluorescent labeling of cells
Immunofluorescent (IF) staining was performed at the end of an experiment. The cells were fixed by flowing 4% paraformaldehyde (v/v, in 1X PBS) into the adjacent media channel of the microfluidic device for 15 min at a flow rate of 15 μL/min, followed by washing with 1X PBS at the same flow rate for 15 min at room temperature. Cell permeabilization was initiated by infusing 0.5% Triton X-100 (v/v) in 1X PBS at room temperature for 15 min, followed by washing with PBS for 15 min. To eliminate non-specific binding, cells were incubated with 0.1% BSA (v/v in PBS) for 45 min. To stain for proliferation, the fluorescently-labeled primary antibody (Alexa Fluor 488 Mouse anti-Human Ki-67, BD Pharmingen) was diluted at 1:20 in the 0.1% BSA solution and incubated in the device for 6 h by continuous pumping using a syringe pump at room temperature. For actin cytoskeleton staining, phalloidin (BD pharmingen) was diluted 1:200 and incubated for the same time period. Immediately prior to imaging, the nuclei were stained by using 4′, 6-diamidino-2-phenylindole (DAPI, Invitrogen), diluted 1:500 in PBS. Cellular fluorescence was visualized using a Leica DMi8 inverted microscope outfitted with a 10× objective (Leica HC PL FL L, 0.4× correction), phase contrast, and brightfield applications. Digital images were acquired using the Flash 4.0 high-speed camera (Hamamatsu) with a fixed exposure time of 100 ms for FITC, DAPI, and Rhodamine filters, and 25 ms for brightfield. Image acquisition was controlled using the Leica Application Suite software. All images were recorded using the same parameters. At the end of a 72 h experiment, cells were co-incubated on-chip with 2.5 μM Calcein AM (Life Technologies, Carlsbad, CA) and 4 μM Ethidium homodimer-1 (Life Technologies, Carlsbad, CA) in 1X PBS for 2 h at a flow rate of 15 μL/min through the adjacent media channel at room temperature. Fluorescence images were taken using the same Leica DMi8 inverted microscope.
On-chip drug evaluation and cell viability studies
Paclitaxel (Adipogen Life Sciences, USA), an anticancer drug, was used to study the drug resistance of co-cultured and single-cultured MDA-MB-231 cells. MDA-MB-231 cells were exposed to paclitaxel at a final concentration of 20 nM. This solution was placed in Luer-Lok syringes to be infused into the device at a rate of 15 μL/min using the syringe pump. The Paclitaxel-DMEM media was introduced to the device after a 24 h pre-incubation period, which allowed the cells to settle and adhere to the surface prior to drug interrogation. The microfluidic device was infused with the drug solution for 72 h. After the termination of the experiment, cell viability was assessed by Calcein AM (live) and Ethidium homodimer-1 (dead) as described in the previous section. The fluorescence images were analyzed to distinguish and count live cells and dead cells using a custom Python algorithm called FluoroCellTrack developed by Vaithiyanathan et. al.35
Image analysis and processing
All microscopy images were processed by ImageJ (NIH, USA) to perform cell counts, measure cell aspect ratio, and determine cellular angular orientation. The cells were counted at each time point (0, 24, 48, and 72 h) from the images collected across the entire culture channel. Cell growth was normalized to the initial cell seeding density to remove any potential bias from slightly different cell counts at the 0 h time point. Normalized cell growth was obtained by dividing the number of cells of each time point by the 0 h cell number following equation (1):
| Eqn. 1 |
The cellular aspect ratio was determined by measuring the ratio of the major axis length to minor axis length of individual cells. A minimum of 300 cells was analyzed for each time point to determine the cellular aspect ratio. The angular orientation (directionality of the cells) was calculated by measuring the angle created by the cell’s major axis line with respect to the horizontal line. The angles were assigned to be 0° for a horizontally aligned cell and 90° for a vertically aligned cell. Any angles calculated between 91–180° were converted to values between 0–89° to simplify the analysis. A minimum of 500 cells was analyzed for each time point to measure cell polarization.
Statistical analysis
The statistical differences between two groups of single culture and co-culture cells were determined by the student’s t-test with a statistically significant value set at p<0.05, using Origin software (USA). All data presented in this study is formatted as (mean ± standard deviation).
Results and discussion
COMSOL simulations of biomolecule and oxygen mass transfer across the device
Time-dependent mass transfer diffusion studies of both biomolecules (representing nutrients, cytokines, and growth factors) and oxygen were simulated across the two center culture channels perpendicular to the direction of flow for 24 h. Small molecules’ concentration profiles across the channels were obtained by a line scan across the width of the center culture channel (Figure 2). First, COMSOL simulations were performed to confirm the mass transfer from the outer flow channel into the inner flow-free culture channels and that the culture channels reached a steady-state after ~24 h (Figure 2A). The simulations used the physical parameters of the growth factor SDF-1α to mimic biomolecule (e.g., cytokines, growth factors) diffusion. The pore size of 3% (w/v) agarose is ~100 nm, which should facilitate the diffusion of growth factors or cytokines, as the diameter of most growth factors is ~50 nm.36 A similar simulation was performed to approximate the diffusive mass transfer of soluble biomolecules across the flow-free cell culture channels to confirm that the microfluidic device facilitates intercellular communication during experimentation (Figure 2B). Again, an approximate steady-state was observed after ~24 h. Finally, the oxygen diffusion within the microfluidic device was simulated to ensure the cells were not exposed to hypoxic conditions (Figure 2C). PDMS is highly permeable to gases and allows diffusion of CO2 and oxygen, while agarose is capable of transporting biomolecules through infused medium inside microfluidic channels.37 For the oxygen diffusion simulation, the static ambient air was modeled to diffuse through an open window in the top layer of Plexiglas. These simulations indicate that the cell culture channels reached an approximate steady-state within 5 h. These results confirm that the two-layer microfluidic device allowed for indirect communication between two different cell types cultured in the two “flow-free” cell culture channels to facilitate co-culture studies of breast cancer cells and ASCs.
Figure 2:
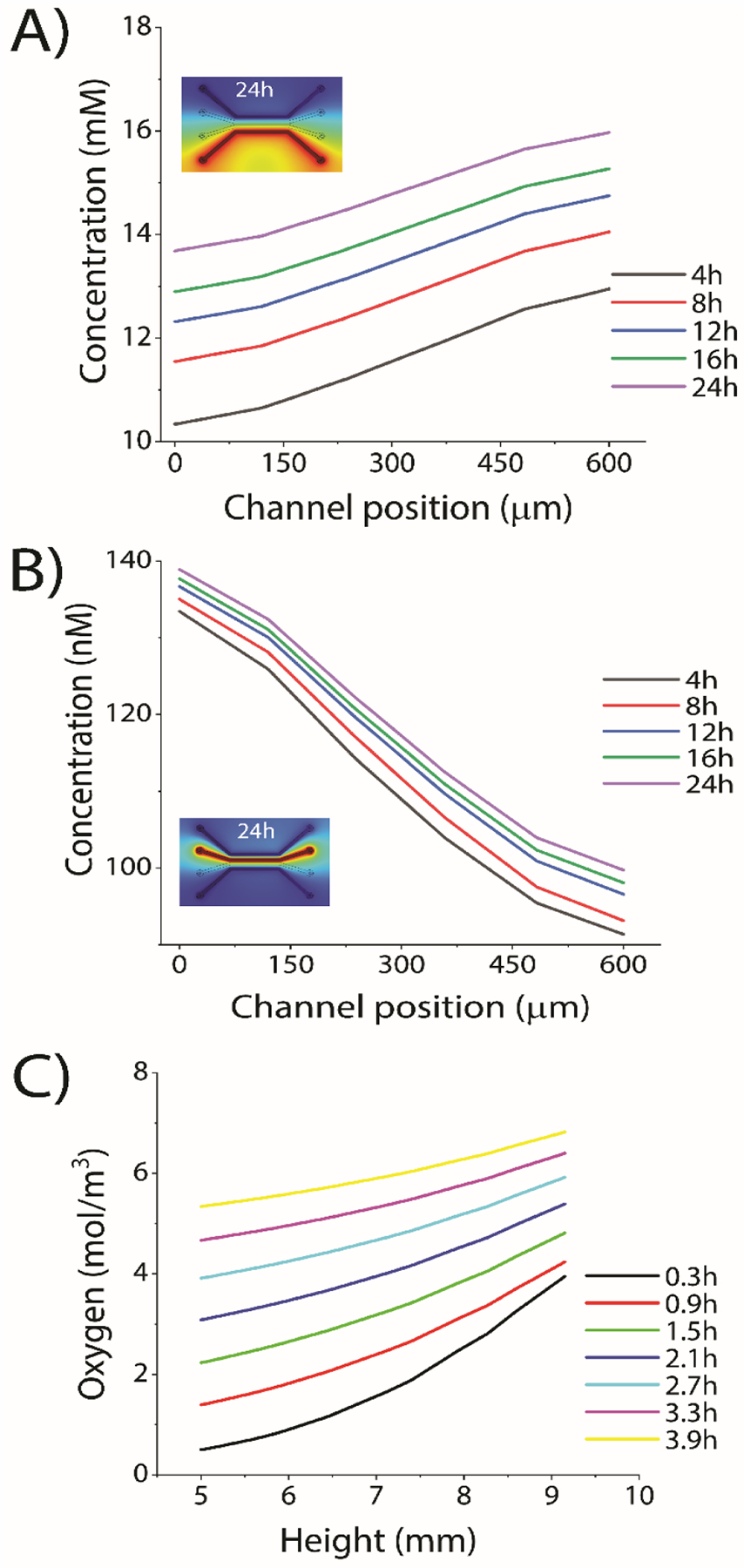
COMSOL simulation of mass transfer in “flow-free” cell culture channels. (A) Diffusion of a model biomolecule from the outer flow channel into the center culture channels. (Inset image) Visual representation of the COMSOL simulations showing diffusion across the device. (B) Simulation of biomolecules between the two flow-free culture channels. (Inset image) Visual representation of the COMSOL simulation. In both (A) and (B) biomolecules were approximated at 140 nM SDF-1α in the source channels(C) Simulation of the oxygen diffusion from outside of the device, through PDMS to cell culture channel. External oxygen was approximated to be ≈21%.
Simultaneous co-culture of cancer cells and stem cells increases the cellular growth rate
Prior to on-chip testing, it was important to confirm that adherent cell lines could be cultured on agarose and that there was no difference in cellular growth or morphology. To confirm this, MDA-MB-231 cells were cultured on polystyrene-coated Petridishes (herein called tissue culture plastic, TCP) or Petridishes filled with ~5 mm agarose coated with 0.1 mg/mL poly-d-lysine. There was no observable difference in the growth for the MDA-MB-231 cell cultures on TCP and on agarose for up to 48 h; however, there was a slight decrease in growth for the cells cultured on agarose after 72 h of incubation, as compared to cells cultured on TCP (Figure 3A). This was attributed to the culture media diffusing into the agarose layer from the cells being cultured on top of it, which minimized the available pool of nutrients for the cells to access. This loss of available nutrients was minimal and only affected cells after 72 h of continuous culture. It was assumed that this would not be an issue with the microfluidic device since it was continuously infused with fresh media, whereas the off-chip batch study was not. Once it was confirmed that adherent cells could be cultured on poly-d-lysine-coated agarose, single-culture on-chip experiments (e.g., the same cell type in both center culture channels) were performed with both MDA-MB-231 cells and ASCs (Figure S1). Both cell types were observed to reach ~80–90% confluency after 72 h of experimentation. Figure S1 confirmed the growth of both MDA-MB-231 cells and ASCs during the 72 h incubation. Both cell types exhibited similar morphology to cells cultured off-chip both on TCP and agarose (data not shown). This confirmed that both cancer cells and ASCs could be cultured in the two-layer microfluidic device. Terminal immunostaining of the actin cytoskeleton using phalloidin was performed to show the structure of the actin cytoskeleton in ASCs and MDA-MB-231 cells (Figure S2). The results from the cytoskeleton staining shows a similar behavior with respect to cellular spreading for both cell types, which also resembles the cytoskeletal behavior of these cell lines when cultured on TCP.38 Terminal proliferation staining of single-cultured MDA-MB-231 using Ki-67 (protein marker of cellular proliferation) confirmed that the cells were still actively proliferating after 72 h of incubation in the device (Figure S3A).
Figure 3:
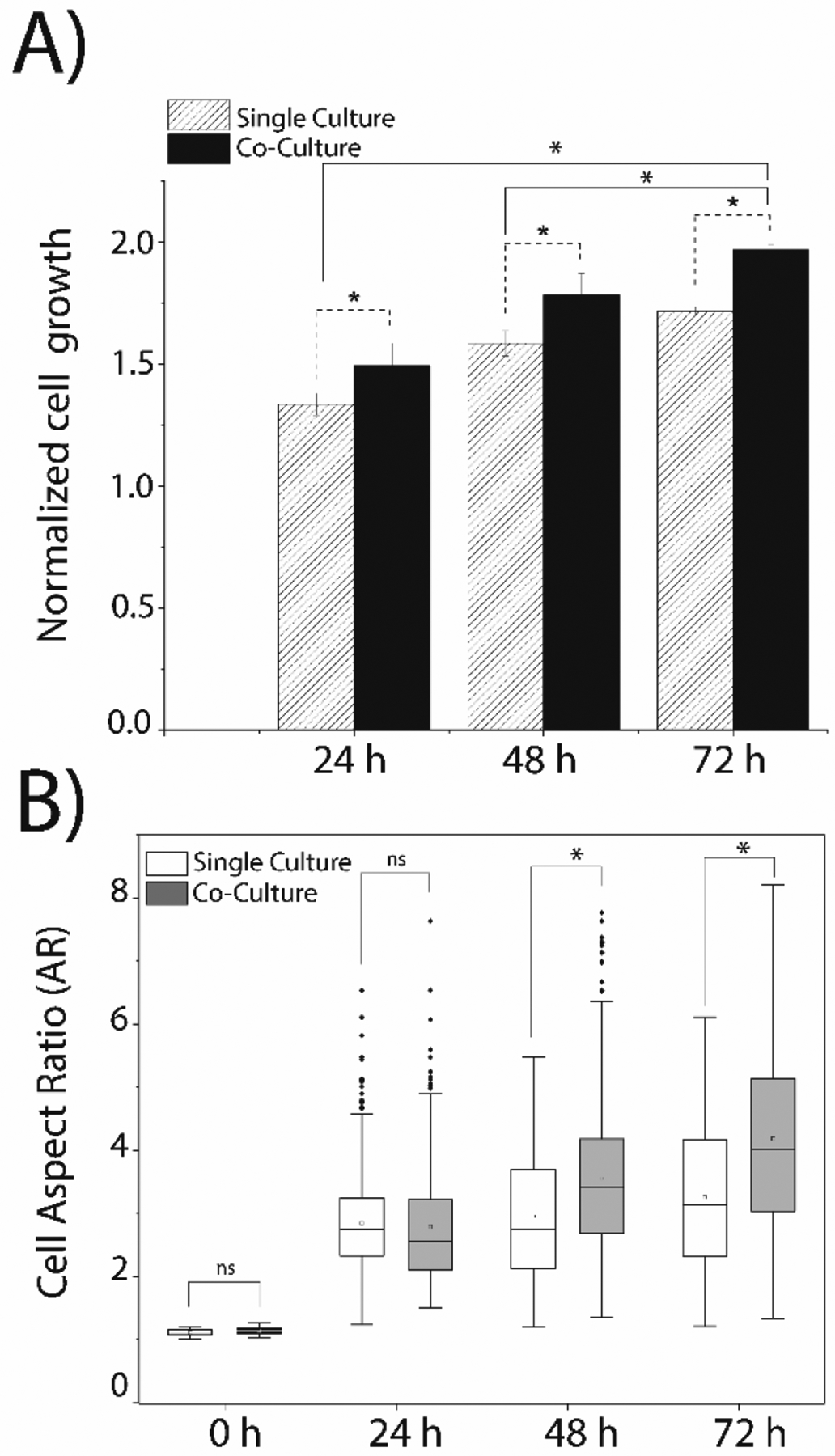
Simultaneous co-culture of MDA-MB-231 cells and ASCs alters breast cancer cell growth and morphology. (A) The growth of the MDA-MB-231 cells was observed in single and co-culture using an initial cell density of 0.5×105 cells/mL. (B) Morphological changes in MDA-MB-231 cells in single culture and co-culture was determined by calculating the aspect ratio: measuring the ratio of the major cell axis length to minor cell axis length of individual cells. A minimum of 500 MDA-MB-231 cells was analyzed for each time point. Single culture and co-culture data were compared by student’s t-test with a statistically significant value set at p<0.05.
Simultaneous co-culture experiments were then performed: the ASCs were cultured in the “top” channel and the MDA-MB-231 cells were cultured in the “bottom” channel for 72 h (Figure 3B). The terms “top” and “bottom” do not refer to the z-direction in the two-layer microfluidic device, but instead, refer to the manner in which the microscope images were collected and presented. Terminal proliferation staining using Ki-67 confirmed that the MDA-MB-231 cells were still actively proliferating after 72 h of co-culture (Figure S3B). Interestingly, when compared to the proliferative behavior of the MDA-MB-231 cells in single culture versus co-culture, it was determined that cancer cells in co-culture with ASCs exhibited a statistically significant increase in proliferation (Figure 3C). This suggested that the ASCs provide a supportive environment for the cancer cells and facilitate enhanced growth and proliferation. Prior studies have identified that the genetic expression of leptin was altered in ASCs when they were co-cultured with another model breast cancer cell line (MCF7), resulting in an increase in the proliferative behavior of the cancer cells.4,39 The results presented here are in alignment with prior studies, which suggest that ASCs enable enhanced cancer progression.40,41 Terminal viability staining of the MDA-MB-231 cells confirmed >95% of cells were alive after the 72 h co-culture experiment (Figure S3C).
A normalized growth rate of the MDA-MB-231 cells was approximated by direct counts of cancer cells across the entire length of the channel under both single culture and co-culture conditions to determine how the presence of the ASCs enhanced cellular growth. A statistically significant increase in the growth rate of MDA-MB-231 cells was observed in the presence of ASCs when compared to single-culture studies (Figure 4A). Similar findings were observed in a previous co-culture studies where Muehleberg et al. investigated the effects of ASCs on breast cancer cells and reported that the cancer cells induced the secretion of SDF-1α from the ASCs, which activated paracrine signaling between the two cell types, resulting in enhanced proliferation, invasion, and metastasis of the cancer cells.42 Interestingly, Sabol et al. found that ASCs promoted metastasis in MCF7 cells (another model breast cancer cell line) but did not result in a significant increase in tumor growth.4
Figure 4:
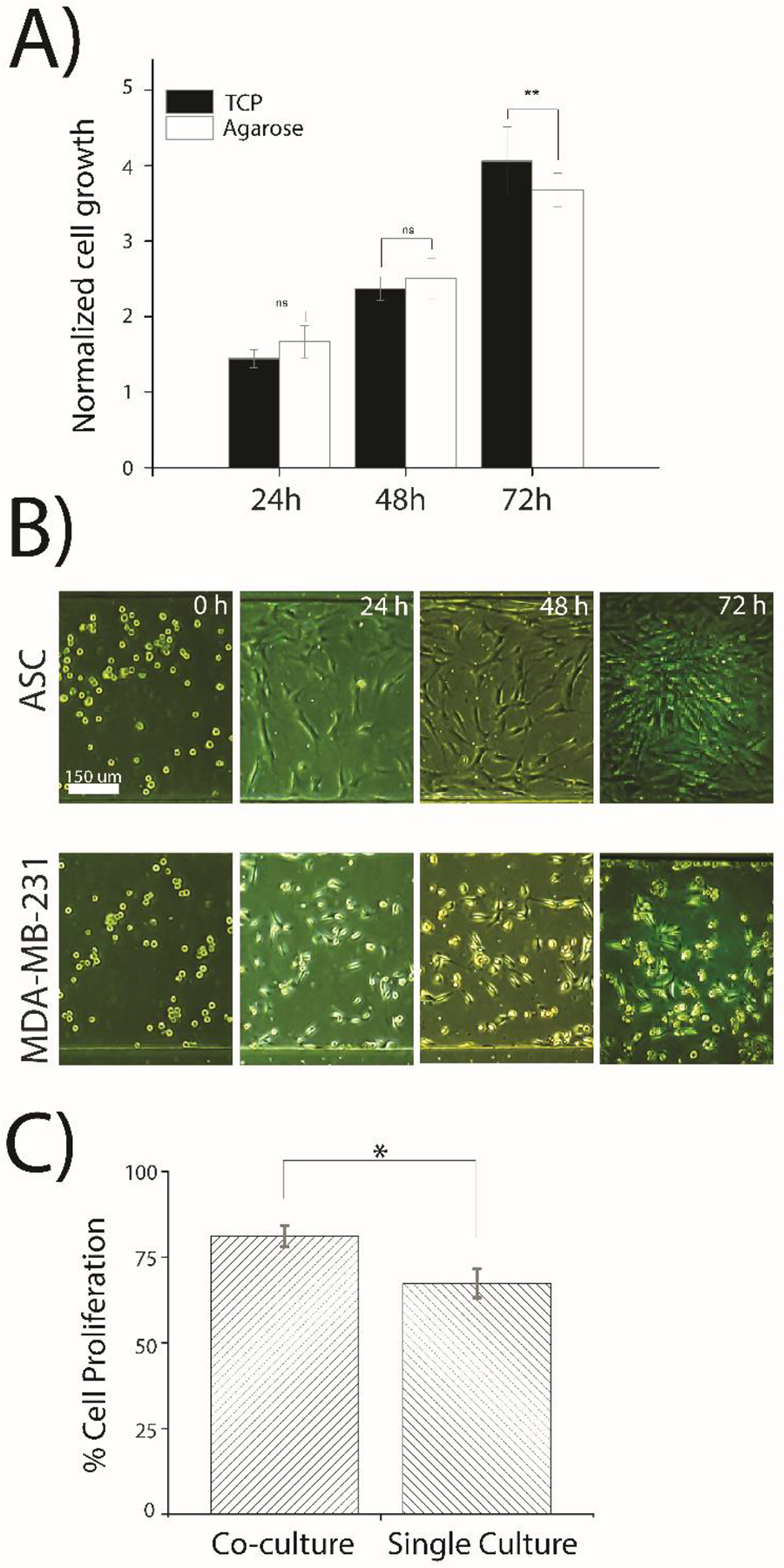
Cell growth and proliferation cultured off-chip and on microfluidic device. (A) Normalized growth of MDA-MB-231 cells cultured off-chip on either TCP or agarose. Cells were cultured in a 100 mm petri dish for 72 hours with an initial total number of 700,000 cells. Cell growth was normalized by dividing the number of cells of each time point by the 0-hour cell numbers. The statistical significance test between cell growth on TCP and agarose with ** representing p<0.05 and ns indicating no significance. (B) Transmitted light (brightfield) images of ASCs (top culture channel) and MDA-MB-231 cells (bottom culture channel) in the device during a 72 h experiment. Images are representative of cell cultures across the entire channel and of triplicate experimentation. (C) Cell proliferation was quantified in the microfluidic device for MDA-MB-231 cells using anti Ki-67 antibodies was compared among cells in co-culture and single culture experiments. Experiments were conducted in duplicate and the statistical differences between two groups were determined by the student’s t-test with a statistical confidence interval value set at p<0.05.
Biophysical changes in MDA-MB-231 cells due to their co-culture with ASCs
The analysis of the physical properties of cells provides insight into their phenotypic characteristics and is associated with heterogeneity in their migratory behavior, the transformation of their stiffness, and their morphology. Cell morphology plays an essential role in cancer progression, which depends on various factors, including metastatic cell density, TME, and cell-to-cell contacts.43,44 EMT induces epithelial cells to develop a more invasive phenotype with increased migratory potential, which is detectable by observing the transformation of cell morphology from a circular shape to an elongated mesenchymal shape. Previous studies have found vimentin expression and E-cadherin dysfunction (considered hallmarks of EMT), where transforming growth factor (TGF β) pathways induce mesenchymal transition phenotype.45–47 The relationship of cancer metastasis and morphological properties, such as cellular area, shape, and aspect ratio, have been studied in pancreatic cancer cells and breast cancer cells, suggesting that the metastatic potential of cancer cells increases while losing phenotyping heterogeneity.48 The two-layer microfluidic platform was utilized to observe changes in the biophysical properties of the MDA-MB-231 cells in the presence of ASCs with respect to both cellular morphology and orientation of the cancer cells. The cellular morphology was quantified by calculating the aspect ratio of the MDA-MB-231 cells cultured in the presence and in the absence of ASCs to identify how ASCs potentially affect EMT in cancer cells. A higher aspect ratio is indicative of a mesenchymal phenotype, while a lower aspect ratio denotes a cancer cell that has an epithelial phenotype. Initially, there was no difference in the average aspect ratio of MDA-MB-231 cells after 24 h of single and co-culture; however, after 48 h the aspect ratio of MDA-MB-231 cells increased significantly in cells co-cultured with ASCs, as compared to single-cultured cells (Figure 4B). Interestingly, the extremes in the heterogeneity of the aspect ratios (denoted by the number of outliers beyond the interquartile range in Figure 4B) across the population of cancer cells co-cultured with ASCs decreased over time, suggesting that distinct subpopulations exist across the entire population of the MDA-MB-231 cells. This would infer that some cancer cells are more sensitive to the presence of the ASCs and react much more quickly to intercellular communication than others; however, the majority of the population eventually recognizes and responds to the presence of the ASCs. This is in line with several single-cell analysis studies highlighting the heterogeneity of cellular behavior with respect to growth, behavior, and enzyme activity.49 These findings suggest that breast cancer cells co-cultured with ASCs adopt a more mesenchymal phenotype, which gradually increases over three days co-culture.
To further explore this invasive phenotype, the orientation of breast cancer cells cultured in the “bottom” channels was compared between cancer cells co-cultured with ASCs in the “top” channel versus cells in the “top” channel. Cellular orientation is an indicator of cellular movement, invasiveness, and intracellular signaling, as chemotactically-migrating cells will align vertically towards a chemical gradient.50,51 The angular orientation, or the polarization of cancer cells towards the ASCs, suggested that the MDA-MB-231 cells under co-culture conditions exhibited a bias towards the adjacent channel when ASCs were present (Figure 5). Perpendicular orientation of MDA-MB-231 cells developed over time with >50% of the population of cancer cells having angles of orientation ranging from 60°−90° after 72 h of co-culture with ASCs. Conversely, only 37% of the MDA-MB-231 cells exhibited this bias orientation when MDA-MB-231 cells were cultured in the adjacent channel. These findings suggest that the cancer cells were responding to paracrine signals emitted by the ASCs and were orienting themselves towards the source of this chemical gradient. A similar trend has been observed during the chemotactic response of breast cancer cells to the gradient of the EGF growth factor.52 This suggests that the breast cancer cells in the “bottom” channel co-cultured with ASCs were undergoing the initial steps of migration through chemosensing and polarization. No significant chemotactic movement was observed in this study; however, that can be attributed to several factors, including the lack of stiffness of the 2D agarose surface, the absence of extracellular matrix (ECM) components, and, most likely, a very shallow gradient of soluble chemokines. Prior studies on the chemotactic behavior of MDA-MB-231 cells show that the majority of migrating cells prefer a steep gradient across the cell with a minimal amount of cells able to respond to and migrate up a shallow gradient in some extent.53
Figure 5:
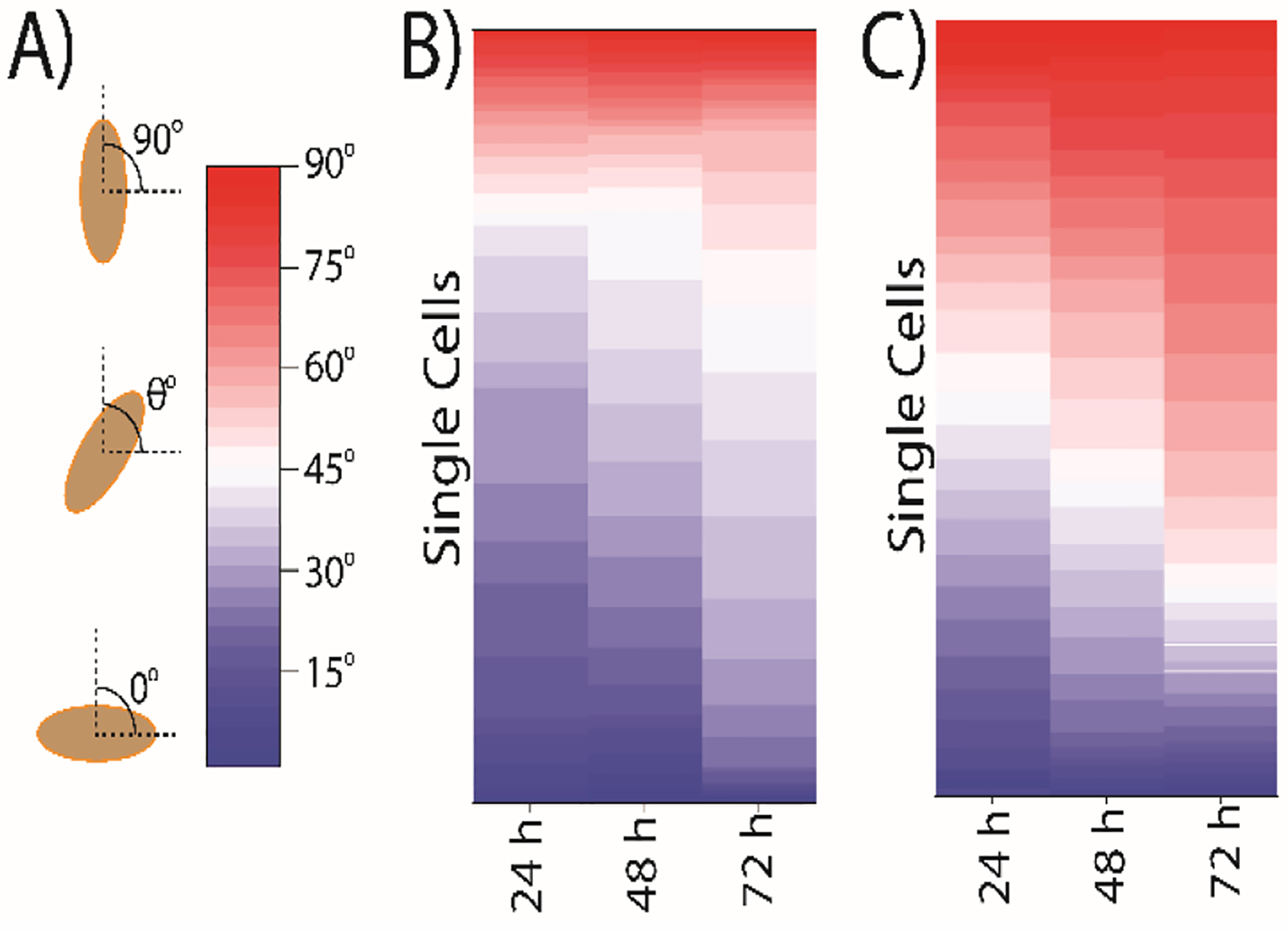
MDA-MB-231 cells orient themselves towards the ASC population during simultaneous co-culture. (A) The angle was calculated with the cell major axis line with respect to the horizontal line. The angles were taken within 0° to 90° where 0° refer no directionality of cells in the parallel position of the channel, and 90° indicates 100% directionality of cells in the perpendicular position of channels. All angles were measured (B) when MDA-MB-231 cells were cultured in single culture. (C) when MDA-MB-231 cells were cultured in Co-culture with ASCs for 72 h. The warm color represents angle in the range of 45° to 90°. A minimum 500 cells were analyzed for angle measurement.
The results presented here confirmed significant changes in cancer cell morphology due to their co-culture with ASCs and provided additional support for enhanced invasiveness of breast cancer cells. Significant increases in cell aspect ratio and a biased directional orientation towards nearby ASCs suggest that ASCs activated signaling pathways in breast cancer cells. Walter et al. verified that interleukin-6 (IL-6) secreted by ASCs stimulated breast cancer cell migration and invasion in the xenograft mouse model.54 This study also identified a cytoskeleton regulator, cofilin-1, that had an active role in stimulating ASCs to secrete excess IL-6. These results obtained from the two-layer microfluidic device demonstrated the stimulation of triple-negative MDA-MB-231 breast cancer cells by ASCs, which expands upon the prior studies and confirms the role of ASCs in cancer progression.
MDA-MB-231 cells exhibit enhanced drug resistance when co-cultured with ASCs
ASCs is not only associated with enhanced metastasis but is also correlated with enhanced drug resistance. Paclitaxel (Taxol) is a chemotherapeutic agent that is used in the treatment of breast cancer. Paclitaxel stabilizes the microtubules of cancer cells and works as an inhibitor of chromosomal replication.55 Cancer drug resistance is correlated with alterations of tubulin structure, microtubular drug-binding affinity, cell cycle deregulation, and over-expression of membrane P-glycoprotein during aerobic glycolysis (the Warburg effect).56 The excessive amount of glycolysis-generated ATP further consumed by drug-resistant cells renders the cells immune from lower doses of Paclitaxel.55,57 The two-layer microfluidic device was used to investigate a potential role in ASC-mediated Paclitaxel resistance of MDA-MB-231 cells under co-culture conditions. Since the drug was introduced to the cells via passive diffusion from the outer flow channel, it was first necessary to identify an approximate IC50 of Paclitaxel. Dose-response experiments were performed with three different concentrations of drug (10, 20, and 30 nM) infused into the device in the outer flow channel adjacent to the “bottom” culture channel, where the MDA-MB-231 cells were seeded (Figure S4). This experiment found that 20 nM Paclitaxel resulted in only ~50% viable cells remaining in the device after 72 h of treatment; thus, this dose was chosen for the drug resistance studies. A similar Paclitaxel concentration was used by Jihui et al. during a microfluidic combinatorial drug screen.58 Next, cellular viability was assessed in MDA-MB-231 cells cultured in the presence and absence of ASCs during a 72 h drug treatment using a terminal viability staining for live and dead cells (Figure S5). Figure 6 demonstrated a reduced effect of Paclitaxel on MDA-MB-231 cells co-cultured with ASCs with ~81% viable cancer cells found under co-culture conditions compared to 56% viable cancer cells remaining with single culture experiments. These findings support the concept that ASCs confer drug resistance in triple-negative breast cancer cells. While the mechanism of drug resistance is not fully understood, these studies suggest that paracrine signals do affect this phenomenon. Another study showed that multi-drug-resistant breast cancer cells expressed higher level of IL-6, where breast cancer cells that are sensitive to drug treatment didn’t produce IL-6.59 Strong et al. demonstrated high levels of IL-6 expressed in MCF7 cells co-cultured with ASCs, which indicates IL-6 could be responsible for drug resistance in cancer cells.4 Similar results have been identified in this study, suggesting that the cancer cells become drug-resistant in co-culture with ASCs, which suggests future studies may be directed at the investigation of IL-6 as a possible mechanism of crosstalking of ASCs and MDA-MB-231.
Figure 6:
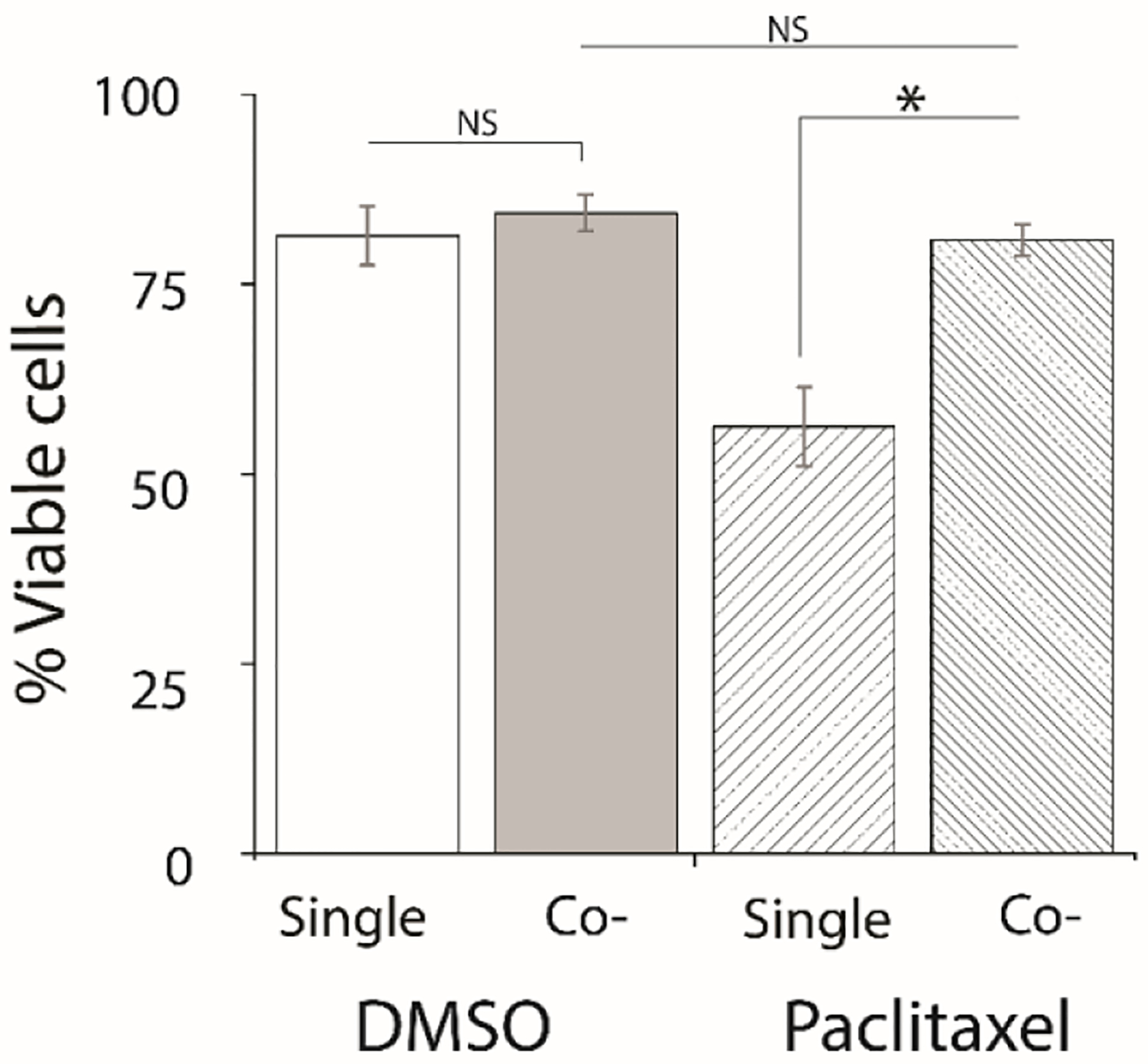
Co-cultured cancer cells show drug resistance to 20 nM Paclitaxel. The viability of MDA-MB-231 cells treated with either a DMSO control or Paclitaxel was measured on-chip after 3 days of Paclitaxel treatment in single cultured and co-cultured experiments. MDA-MB-231 cells were incubated with Calcein AM (2.5 μM) and Ethidium homodimer-1 (4 μM) after terminating the experiment. Viability experiments were conducted triplicate and the statistical differences between two groups were determined by the student’s t-test with a statistically confidence interval value set at p<0.05.
Conclusion
In this study, a two-layer, four-channel “flow-free” microfluidic device was developed to allow for the continuous co-culture of two distinct types of cells found in the tumor microenvironment. The microfluidic device allowed for passive diffusion of biomolecules from continuous infusion flow channels to flow-free culture channels to overcome limitations associated with other platforms, including batch culture conditions (e.g., media depletion and waste accumulation) or shear effects. The microfluidic device was used to study intercellular communication between triple-negative breast cancer cells (MDA-MB-231) and ASCs. Prior studies have shown a link between ASCs and cancer; however, there were no prior studies elucidating the impact of ASCs on cancer cell growth, proliferation, morphology, and drug resistance. The co-culture studies performed here confirmed that ASCs enhance cancer cell growth and proliferation, supporting the hypothesis of the importance of their role in cancer cell progression. Biophysical studies identified a role for ASCs in enhancing cancer cell invasiveness by encouraging MDA-MB-231 cells to adopt a more aggressive morphology, coupled with physical alignment towards ASCs as the first step in the invasion. Drug studies with Paclitaxel confirmed that ASCs confer resistance on cancer cells, also confirming their role in drug resistance during cancer treatment. Taken together, these findings highlight the role of dynamic paracrine signaling between cancer and stromal cells. Finally, the microfluidic device developed in this study offers the potential for promising future dynamic co-culture studies that could be an alternative to animal models, thus undertaking new experiments examining the role of intercellular communication of cancer progression and drug resistance.
Supplementary Material
Acknowledgements
This work was supported by start-up funds from Louisiana State University (SMR, ATM). JS and KR were both supported by undergraduate research grants from LSU Discover Day. Supported in part by U54 GM104940 from the National Institute of General Medical Sciences of the National Institutes of Health, which funds the Louisiana Clinical and Translational Science Center (ECM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to thank Matthew Burow (Tulane University School of Medicine) for generously providing the MDA-MB-231 cell line. The authors would also like to thank Mr. Jeffrey Anderson (LSU) for assistance with developing the poly-d-lysine treatment protocol and Ms. Manibarathi Vaithiyanathan (LSU) for assistance with the FluoroCellTrack algorithm.
Footnotes
Electronic Supplementary Information (ESI) available: [supplementary information includes five figures. See DOI: 10.1039/x0xx00000x
Conflicts of interest
“There are no conflicts to declare”.
References
- 1.Wirtz D, Konstantopoulos K and Searson PC, Nat. Rev. Cancer, 2011, 11 VN-r, 512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D and Weinberg RA, Cell, 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 3.Rhodes LV, Antoon JW, Muir SE, Elliott S, Beckman BS and Burow ME, Mol. Cancer, 2010, 9, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sabol RA, Beighley A, Giacomelli P, Wise RM, Harrison MAA, O’donnnell BA, Sullivan BN, Lampenfeld JD, Matossian MD, Bratton MR, Wang G, Collins-Burow BM, Burow ME and Bunnell BA, Int. J. Mol. Sci, 2019, 20, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strong AL, Ohlstein JF, Biagas BA, Rhodes LV, Pei DT, Tucker HA, Llamas C, Bowles AC, Dutreil MF, Zhang S, Gimble JM, Burow ME and Bunnell BA, Breast Cancer Res, 2015, DOI: 10.1186/s13058-015-0622-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donohoe CL, Lysaght J, O’Sullivan J and Reynolds JV, Trends Endocrinol. Metab, 2017, 10.1016/j.tem.2016.08.004 [DOI] [PubMed] [Google Scholar]
- 7.Lengyel E, Makowski L, DiGiovanni J and Kolonin MG, Trends in Cancer, 2018, 4, 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borsig L, Wolf MJ, Roblek M, Lorentzen A and Heikenwalder M, Oncogene, 2014, 33, 3217–24. [DOI] [PubMed] [Google Scholar]
- 9.Sakurai M, Miki Y, Takagi K, Suzuki T, Ishida T, Ohuchi N and Sasano H, Breast Cancer Res, 2017, 19, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freemont P, Polizzi KM and Goers L, J. R. Soc. Interface, 2014, DOI: 10.1098/rsif.2014.0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renaud J and Martinoli MG, J. Vis. Exp, 2016, 2016, 1–12. [DOI] [PubMed] [Google Scholar]
- 12.Rothbauer M, Zirath H and Ertl P, Lab Chip, 2017, 18, 249–270. [DOI] [PubMed] [Google Scholar]
- 13.Campbell JM, Balhoff JB, Landwehr GM, Rahman SM, Vaithiyanathan M and Melvin AT, Int. J. Mol. Sci, 2018, DOI: 10.3390/ijms19092731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vu TQ, de Castro RMB and Qin L, Lab Chip, 2017, 17, 1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landwehr GM, Kristof AJ, Rahman SM, Pettigrew JH, Coates R, Balhoff JB, Triantafillu UL, Kim Y and Melvin AT, Biomicrofluidics, 2018, DOI: 10.1063/1.5063824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tehranirokh M, Kouzani AZ, Francis PS and Kanwar JR, 2013, 051502, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ai X, Lu W, Zeng K, Li C, Jiang Y and Tu P, Analytical Chemistry, 2018, DOI: 10.1021/acs.analchem.7b04833. [DOI] [PubMed] [Google Scholar]
- 18.Zhu J, Wang Y, Chen P, Su H, Du W and Liu BF, Sensors Actuators, B Chem, 2019, 283, 685–692. [Google Scholar]
- 19.Zhang C, Barrios MP, Alani RM, Cabodi M and Wong JY, Exp. Cell Res, 2016, 342, 159–165. [DOI] [PubMed] [Google Scholar]
- 20.Burmeister A, Hilgers F, Langner A, Westerwalbesloh C, Kerkhoff Y, Tenhaef N, Drepper T, Kohlheyer D, Von Lieres E, Noack S and Grünberger A, Lab Chip, 2019, 19, 98–110. [DOI] [PubMed] [Google Scholar]
- 21.Yen H-W, Yang Y-C and Yu Y-H, J. Biosci. Bioeng, 2012, 114, 453–456. [DOI] [PubMed] [Google Scholar]
- 22.Estrada MF, Rebelo P, Davies EJ, Pinto MT, Pereira H, Santo VE, Smalley MJ, Barry ST, Gualda EJ, Alves PM, Anderson E and Brito C, Biomaterials, 2016, 78, 50–61. [DOI] [PubMed] [Google Scholar]
- 23.Peela N, Truong D, Saini H, Chu H, Mashaghi S, Ham SL, Singh S, Tavana H, Mosadegh B and Nikkhah M, Biomaterials, 2017, 133, 176–207. [DOI] [PubMed] [Google Scholar]
- 24.Rothbauer M, Zirath H and Ertl P, Lab Chip, 2018, 18, 249–270. [DOI] [PubMed] [Google Scholar]
- 25.Sakthivel K, O’Brien A, Kim K and Hoorfar M, TrAC - Trends Anal. Chem, 2019, DOI: 10.1016/j.trac.2019.03.026 [DOI] [Google Scholar]
- 26.Luo T, Fan L, Zhu R and Sun D, Micromachines, 2019, 10, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomsen AR, Aldrian C, Bronsert P, Thomann Y, Nanko N, Melin N, Rücker G, Follo M, Grosu AL, Niedermann G, Layer PG, Heselich A and Lund PG, Lab Chip, 2018, 18, 179–189. [DOI] [PubMed] [Google Scholar]
- 28.Liu PF, Cao YW, Zhang SD, Zhao Y, Liu XG, Shi HQ, Hu KY, Zhu GQ, Ma B and Niu HT, Oncotarget, 2015, DOI: 10.18632/oncotarget.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng S-Y, Heilman S, Wasserman M, Archer S, Shuler ML and Wu M, Lab Chip, 2007, 7, 763. [DOI] [PubMed] [Google Scholar]
- 30.Haessler U, Kalinin Y, Swartz MA and Wu M, Biomed. Microdevices, 2009, 11, 827–835. [DOI] [PubMed] [Google Scholar]
- 31.Cheng S-Y, Heilman S, Wasserman M, Archer S, Shuler ML and Wu M, Lab Chip, 2007, 7, 763–9. [DOI] [PubMed] [Google Scholar]
- 32.Wu MH, Bin Huang S and Bin Lee G, Lab Chip, 2010, 10, 939–956. [DOI] [PubMed] [Google Scholar]
- 33.Pluen A, Netti PA, Jain RK and Berk DA, Biophys. J, 1999, 77, 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim BJ, Hannanta-anan P, Chau M, Kim YS, Swartz MA and Wu M, PLoS One,2013, DOI: 10.1371/journal.pone.0068422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaithiyanathan M, Safa N and Melvin AT, PLoS One, 2019, DOI: 10.1371/journal.pone.0215337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peckys DB, Baudoin JP, Eder M, Werner U and De Jonge N, Sci. Rep, 2013, DOI: 10.1038/srep02626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halldorsson S, Lucumi E, Gómez-Sjöberg R and Fleming RMT, Biosens. Bioelectron, 2015, 63, 218–231. [DOI] [PubMed] [Google Scholar]
- 38.Huang YL, Tung CK, Zheng A, Kim BJ and Wu M, Integr. Biol. (United Kingdom), 2015, DOI: 10.1039/c5ib00115c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strong AL, Strong TA, Rhodes LV, Semon JA, Zhang X, Shi Z, Zhang S, Gimble JM, Burow ME and Bunnell BA, Breast Cancer Res, 2013, DOI: 10.1186/bcr3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strong AL, Pei DT, Hurst CG, Gimble JM, Burow ME and Bunnell BA, Stem Cells Int, 2017, DOI: 10.1155/2017/9216502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strong AL, Burow ME, Gimble JM and Bunnell BA, Stem Cells, 2015, 33, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muehlberg FL, Song YH, Krohn A, Pinilla SP, Droll LH, Leng X, Seidensticker M, Ricke J, Altman AM, Devarajan E, Liu W, Arlinghaus RB and Alt EU, Carcinogenesis, 2009, DOI: 10.1093/carcin/bgp036. [DOI] [PubMed] [Google Scholar]
- 43.Katira P, Bonnecaze RT and Zaman MH, Front. Oncol, 2013, 3, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugimoto M, Kitagawa Y, Yamada M, Yajima Y, Utoh R and Seki M, Lab Chip, 2018, 18, 1378–1387. [DOI] [PubMed] [Google Scholar]
- 45.Bidarra SJ, Oliveira P, Rocha S, Saraiva DP, Oliveira C and Barrias CC, Sci. Rep, 2016, 6, 27072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel D, Matharu Z, Gao Y, Revzin A, Diehl AM, Zhou Q, Haque A, Kwa T and Stybayeva G, Lab Chip, 2015, 15, 4467–4478. [DOI] [PubMed] [Google Scholar]
- 47.Fazilaty H, Gardaneh M and Bahrami T, 2019, 2019–2030. [DOI] [PubMed]
- 48.Wu PH, Phillip JM, Khatau SB, Chen WC, Stirman J, Rosseel S, Tschudi K, Van Patten J, Wong M, Gupta S, Baras AS, Leek JT, Maitra A and Wirtz D, Sci. Rep, 2015, 5, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Safa N, Pettigrew JH, Gauthier TJ and Melvin AT, Biochem. Eng. J, 2019, DOI: 10.1016/j.bej.2019.107320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneider IC and Haugh JM, Cell Cycle, 2006, 5, 1130–1134. [DOI] [PubMed] [Google Scholar]
- 51.Zheng Y, Wang S, Xue X, Xu A, Liao W, Deng A, Dai G, Liu AP and Fu J, Lab Chip, 2017, 17, 1948–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roussos ET, Condeelis JS and Patsialou A, Nat. Rev. Cancer, 2011, 11, 573–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raja WK, Gligorijevic B, Wyckoff J, Condeelis JS and Castracane J, Integr. Biol, DOI: 10.1039/c0ib00044b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walter M, Liang S, Ghosh S, Hornsby PJ and Li R, Oncogene, 2009, DOI: 10.1038/onc.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB, Ledoux SP and Tan M, Mol. Cancer, 2010, 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vander Heiden MG and DeBerardinis RJ, Cell, 2017, 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu A, Loutherback K, Lambert G, Estevez-Salmeron L, Tlsty TD, Austin RH and Sturm JC, Proc. Natl. Acad. Sci, 2013, 110, 16103–16108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun J, Liu W, Li Y, Gholamipour-Shirazi A, Abdulla A and Ding X, Microfluid. Nanofluidics, 2017, 21, 1–11. [Google Scholar]
- 59.Conze D, Weiss L, Regen PS, Rincón M, Weaver D, Bhushan A and Johnson P, Cancer Research, 2001, 61, 8851.–. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.