

Abstract
Purpose of review
By establishing mechanisms that deliver oxygen to sustain cells and tissues, fight life-threatening pathogens and harness the immune system to eradicate cancer cells, hematopoietic stem and progenitor cells (HSPCs) are vital in health and disease. The cell biological framework for HSPC generation has been rigorously established, yet recent single-cell transcriptomic analyses have unveiled permutations of the hematopoietic hierarchy that differ considerably from the traditional roadmap. Deploying mutants that disrupt specific steps in hematopoiesis constitutes a powerful strategy for deconvoluting the complex cell biology. It is striking that a single transcription factor, GATA2, is so crucial for HSPC generation and function, and therefore it is instructive to consider mechanisms governing GATA2 expression and activity. This review focuses on an essential GATA2 enhancer (+9.5) and how +9.5 mutants inform basic and clinical/translational science.
Recent findings
+9.5 is essential for HSPC generation and function during development and hematopoietic regeneration. Human +9.5 mutations cause immunodeficiency, myelodysplastic syndrome and acute myeloid leukemia. Qualitatively and quantitatively distinct contributions of +9.5 cis-regulatory elements confer context-dependent enhancer activity. The discovery of +9.5 and its mutant alleles spawned fundamental insights into hematopoiesis, and given its role to suppress blood disease emergence, clinical centers test for mutations in this sequence to diagnose the etiology of enigmatic cytopenias.
Summary
Multi-disciplinary approaches to discover and understand cis-regulatory elements governing expression of key regulators of hematopoiesis unveil biological and mechanistic insights that provide the logic for innovating clinical applications.
Keywords: GATA2, enhancer, hematopoiesis, progenitor, stem cell, leukemia, bone marrow failure, immunodeficiency
INTRODUCTION
The diversification of hematopoietic stem cells (HSCs) into multipotent and lineage-committed progenitors, which generate the essential blood cells, involves complex cellular transitions that have been extensively studied. Applying single-cell transcriptomics to hematopoietic stem and progenitor cells (HSPCs) extracted from their microenvironments has introduced new permutations [1,2] of the classical hematopoietic cell developmental hierarchy [3]. This work has led to a model in which HSCs generate a continuous stream of differentiated progeny, an entirely different paradigm relative to models invoking well-defined cellular intermediates. Though the cell biological complexity of hematopoiesis is formidable, it is striking that a single transcription factor, GATA2, is so crucial for HSPC generation and function. Given this vital role, it is instructive to consider how GATA2 expression and activity are controlled in the distinct cellular contexts of the hematopoietic system.
The transcription factor GATA2 is essential for developmental and regenerative hematopoiesis, including hematopoietic stem cell (HSC) emergence, maintenance of HSC activity, myeloid and myelo-erythroid progenitor cell differentiation and erythroid precursor cell maintenance. Gata2-null mice exhibit impaired multi-lineage hematopoiesis and die at ~E10.5 [4]. GATA2 triggers the endothelial to hematopoietic transition in the mouse embryo and a comparable transition has been modelled with human induced-pluripotent cells (iPSCs) [5–7]. GATA2 depletion reduces hematopoietic stem and progenitor cell levels and function in mice, human cord blood [8] and iPSC [5,9] systems. Hypomorphic Gata2fGN/fGN mice with Gata2 expression 5-fold lower in bone marrow mononuclear cells versus wild type cells [10,11] have thrombocytopenia, hyperchromic and macrocytic erythrocytes, and upon aging, develop leukocytosis [12].
Considering the vital GATA2 functions in multiple sectors of the hematopoietic hierarchy, there are many opportunities for even small changes in GATA2 levels/activity to derail hematopoietic processes. We proposed that GATA2 levels/activity need to be maintained within a restricted physiological window to establish and maintain context-dependent and fragile GATA2-dependent genetic networks. Thus, high or low GATA2 levels/activities corrupt circuits and networks that sustain steady-state hematopoiesis and enable the hematopoietic system to regenerate in physiological and pathological contexts. Supporting this concept, ectopic GATA2 overexpression in bone marrow suppresses hematopoiesis [13], and high GATA2 mRNA is associated with poor prognosis of AML in adult [14] and pediatric [15] patient cohorts. Human GATA2 mutations, which are loss-of-function or gain-of-function, dependent upon context, cause a disease termed GATA2 deficiency syndrome that is discussed later in this review.
CIS-ELEMENT REQUIREMENTS FOR +9.5 ENHANCER FUNCTION
Using erythroid cells to discover principles of genetics and epigenetics and to understand how GATA factors control erythropoiesis, we identified five conserved GATA2- and GATA1- occupied chromatin sites upstream (−77, −3.9, −2.8, and −1.8 kb relative to the transcription start site) and within an intron (+9.5 kb) of the murine Gata2 locus [16–19]. GATA2 occupancy of its own locus implies positive autoregulation, and GATA1 displacement of GATA2 instigates a GATA switch that represses Gata2 transcription [19–21]. While these GATA1/2-occupancy sites exhibit enhancer attributes, −1.8 [22] or −2.8 [23] deletions revealed they are not essential for Gata2 expression and hematopoiesis, albeit they contribute to Gata2 expression in progenitors, and −1.8 maintains Gata2 repression in maturing erythroblasts [22]. The −3.9 deletion was inconsequential for Gata2 expression and hematopoiesis [24]. However, the −77 and +9.5 deletions revealed their essential functions to support embryogenesis and hematopoiesis. The −77 deletion is embryonic lethal after E15.5, and myelo-erythroid progenitor cell fate is corrupted, despite normal HSC levels [25]. By contrast, +9.5 deletion is lethal at ~E14.5 and abrogates HSC generation in the AGM region [26,27]. The +9.5 also increases Gata2 transcription at other developmental stages and in the adult during hematopoietic regeneration following stress [28]. Based on the extraordinarily important +9.5 activity to control GATA2 levels in HSPCs and erythroid precursors, and its direct role in, and utility for diagnosis of blood diseases, this review focuses principally on +9.5 structure, function and dysfunction.
+9.5 sequence conservation suggests the presence of multiple transcription factor-binding motifs that might function additively, synergistically or redundantly (Figure 1). In addition, certain motifs might operate only in restricted physiological and/or pathological contexts. Considerable progress has been made in testing these models utilizing +9.5 mutant alleles in mice (Figure 2). Studies with strains harboring a mutant E-box-spacer-AGATAA composite element (“composite element”) [26,27] and Ets [28] (Figure 1 and 2) indicate that individual motifs can exert qualitatively and quantitatively distinct activities. While the E-box-8bp spacer-AGATAA composite element structure is highly conserved, the spacer sequence differs among species, with the mouse and human spacers differing by 2 bp. ChIP-seq data revealed GATA2 occupancy at composite elements with 6- to 14-bp spacers, and the 8-bp spacer is overrepresented [29]. The 8-bp spacer allows maximal enhancer activity in a transient transfection assay [30] and complex assembly in vitro is abrogated by E-box or GATA motif mutations [31].
Figure 1:
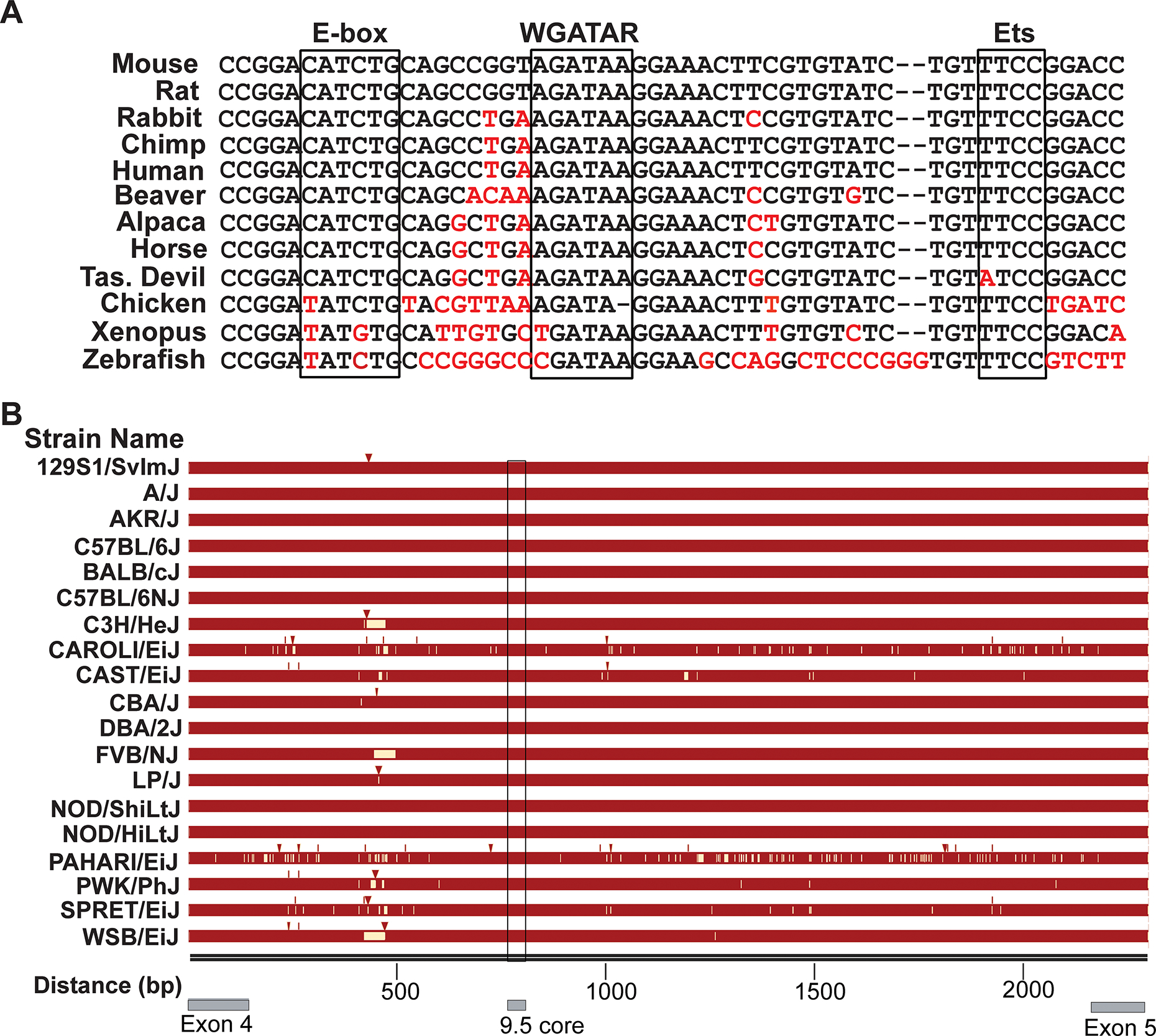
+9.5 enhancer sequence conservation. (A) Alignment of the +9.5 core. Deviation from the consensus sequence is indicated by red text. (B) Alignment of Gata2 intron 4 among 19 mouse strains. Conserved sequences are indicated in maroon. Insertions indicated by arrowheads and deletions or mismatches with white.
Figure 2:
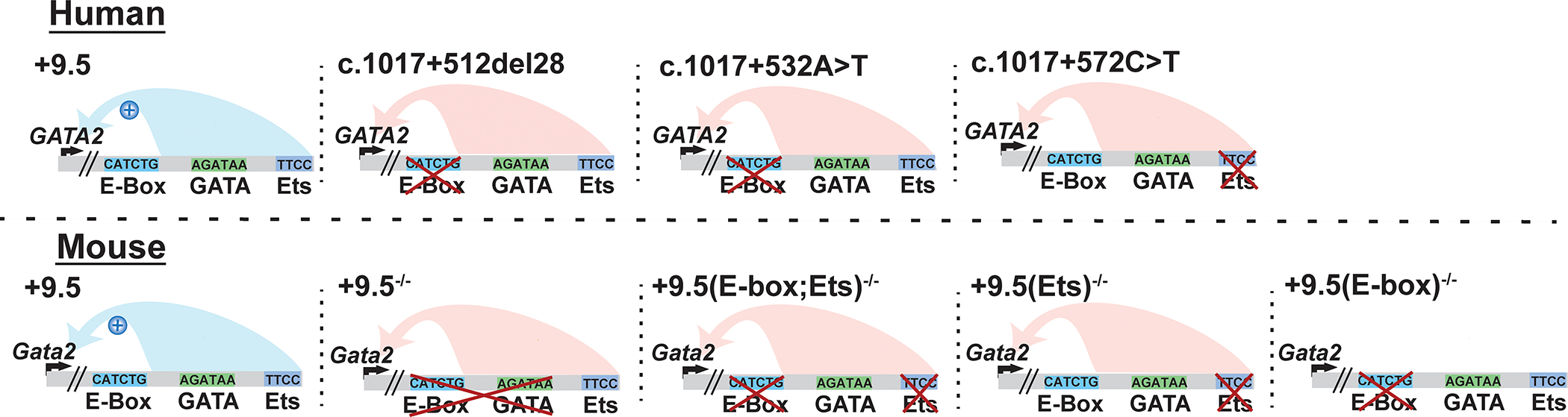
Mouse Gata2 +9.5 mutant alleles.
The +9.5 cis-elements described represent canonical binding motifs for the hematopoietic transcription factors SCL/TAL1 (E-box: CANNTG) [32,33] and GATA2 [4] or GATA1 [34,35] (WGATAR), as well as Ets transcription factor (GGAW) [36] family members, e.g. ERG [37], FLI1 [38] and ETV2 [39] that also have cell-type-specific expression patterns. GATA2 and SCL/TAL1 commonly co-occupy chromatin sites [40–42], and the coregulator LMO2 can also be present [43]. Deletion of the composite element in fetal liver cells prevents GATA2 and SCL/TAL1 chromatin occupancy [24]. +9.5 enhancer activity requires E-box, GATA, and Ets motifs in cell- and transgenic mouse-based reporter assays [30,40,44], and E-box deletion from a +9.5-containing transgene abolishes expression in mouse embryo endothelium [45]. Forced expression of GATA2, TAL1, LMO2 and ETV2 in iPSCs promotes the endothelial to hematopoietic transition [46], although whether these proteins function through the +9.5 in this context was not described.
Homozygous deletion of the composite element (+9.5−/−) [26] or the E-box and Ets motifs, while retaining the GATA motif [+9.5(E-box;Ets)−/−] [28], permits embryonic development beyond the stage at which Gata2−/− embryos die (E14.5 vs. E10.5) [4]. However, HSC emergence is abrogated in +9.5−/− aorta-gonad-mesenephros (AGM) [26–28]. Since AGM-derived HSCs populate the fetal liver [47], hematopoietic stem and progenitor cells (HSPCs) are severely depleted in the mutant fetal livers. Deletion of a single +9.5 allele decreases Gata2 expression in fetal liver by ~50% and HSPC levels by ~30% [26]. Although Gata2 expression in +9.5+/- AGM is not significantly altered, colony forming activity is 2-fold lower [27]; given the small percentage of GATA2-expressing hemogenic endothelial cells in the AGM, one would not expect to see mRNA changes with bulk RNA measurements. Competitive transplantation of heterozygous +9.5+/- fetal liver or AGM revealed ~3-fold decreases in long-term repopulating activity [26,27]. The unique phenotypes of −77 and +9.5 mutant alleles informed hematopoiesis mechanisms.
Combining different enhancer mutant alleles in compound heterozygous mice provides an innovative strategy to generate unique mouse models with phenotypes distinct from conventional perturbations and elucidate mechanisms underlying enhancer function. The combination of a single +9.5 mutant allele with a single −77 mutant allele, either of which elicit only minor phenotypes by themselves, to yield compound heterozygous mice allows development to a later stage (~E15.5) than +9.5−/− mutants [48]. This extended developmental window revealed +9.5 activity to regulate GATA2 expression and function in progenitor cells, which was not detected in the +9.5−/− context in which HSC emergence is ablated. While all myeloid progenitor populations are lower in the compound heterozygous mutants, megakaryocyte erythrocyte progenitors (MEPs) are essentially eliminated, disproportionately relative to other myeloid progenitors [48].
By contrast to the multi-motif mutations of the +9.5−/− allele [26] and the compound heterozygous mutant described above [48], a homozygous single-nucleotide Ets motif mutant, which models a human GATA2 deficiency syndrome mutation, is not embryonic lethal [28]. In this mutant, HSC emergence in the AGM decreases 2-fold, and HSCs in the fetal liver decrease 4-fold. In the steady-state, these defects do not persist in adults, although multipotent progenitors modestly decrease. However, following 5-flurouracil treatment, +9.5(Ets)−/− mice are defective in expanding HSPCs, and HSCs are reduced in a competitive transplantation assay [28]. Thus, the Ets motif promotes regeneration after hematopoietic injury. These studies revealed differences between +9.5 function in regenerative versus developmental hematopoiesis, and given the +9.5 cis-element complexity and context-dependent activities, future studies will almost certainly further transform concepts.
MECHANISTIC INSIGHTS DERIVED FROM “+9.5-LIKE” ENHANCERS
Among the reported Gata2 enhancers, only +9.5 contains a conserved composite element. Within the human genome, nearly 9000 composite elements with CATCTG-(N8-N14)-AGATAA permutations exist [29,49]. Limiting the spacer length to 8 reduces the number to 797 in mice, 62 of which are GATA2-occupied; 34 of these were GATA2/SCL/TAL1-co-occupied. Composite elements at Bcl2l1, Dapp1, and Samd14 were functionally validated by gene editing in G1E-ER-GATA1 proerythroblast cells [29]. “+9.5-like” elements at the Kit promoter, Runx1 intron, Smad1 intron, Klf1 promoter, Ebp4.2 promoter, Gata1 promoter and Smad5 promoter exhibit enhancer activities in transfection and transgenic mouse assays but have not been functionally analyzed at their endogenous loci [49]. SCL/TAL1, GATA2, the Ets factor PU.1 and coregulators LMO2 and LDB1 occupy the Runx1 intronic enhancer [50]. As SCL/TAL1 occupancy can occur even without an E-box adjacent to the GATA motif [51], how different configurations of GATA motif-containing cis-elements translate into unique functions remains elusive. Composite elements also reside at select GATA1-occupied chromatin sites [29,41] and are likely to be broadly important in GATA2 and GATA1 contexts.
Based on the Ets motif (GGAW) sequence simplicity [36] and existing mechanistic knowledge, it is impossible to predict whether a particular Ets motif is essential, contributory or inconsequential for enhancer function. Furthermore, the rules governing the specificity of how Ets factor family members occupy chromatin in vivo are unknown. Although comparative genomics involving the integration of diverse datasets, including conservation, ChIP-seq data, natural genetic variation and patient mutations, can be used to stratify sites to unveil important insights, genetic editing is essential to establish important functions at endogenous loci [29].
DEVELOPMENTAL VERSUS REGENERATIVE +9.5 ENHANCER FUNCTIONS
A mechanism that controls hematopoiesis during embryogenesis and in the adult may have mechanistic components that are essential in only one of these contexts. This concept is exemplified by the critical GATA2 activity to control hematopoiesis in multiple contexts, while the essential +9.5 and −77 activities to regulate GATA2 expression and hematopoiesis are context-dependent. It is instructive therefore to compare and contrast +9.5 functions during development and regeneration.
The transcription factors SCL/TAL1 and GATA2 and the coregulator LMO2 are expressed in HSPCs and in certain lineage-committed progenitor cells. In the distinct regulatory milieus, presumably, these factors assemble on composite elements at target gene ensembles in a cell type-specific manner, although many questions remain unanswered regarding mechanisms governing multimeric complex assembly and function [33,49]. GATA2-mediated transcriptional activation is enhanced in a context-dependent manner by multi-site phosphorylation [52,53], and other components of the complex are also phosphorylated [54–58]. In principle, post-translational modifications might impact complex assembly and/or function but the importance of these mechanisms in vivo and whether they operate similarly or distinctly during development and regeneration has not been described.
Differential +9.5 functions in development and regeneration may reflect differences in the levels/activities of its transcription factor components in these contexts. The Ets factor ETV2 is transiently expressed during embryogenesis and silenced once definitive HSPCs are generated [59–61]. Conditional Etv2 deletion using Tie:Cre or Vav:Cre does not impact steady-state hematopoiesis [59]. By contrast, treatment of the conditional mutant mice with polyinosinic:polycytidylic acid (pIpC), which activates interferon signaling, or 5-fluorouracil (5-FU), which kills proliferating cells and activates quiescent HSCs, causes rapid HSC depletion. ETV2 occupies +9.5 in embryonic stem cells and is required for Gata2 induction post-5-FU treatment [28]. It is likely therefore that ETV2 induction by hematopoietic stress contributes to +9.5 regenerative functions. As developmental hematopoiesis is relatively normal in +9.5(Ets)−/− mice [28], the Ets motif activity exemplifies a context-dependent permutation of the +9.5 mechanism.
PATHOGENIC HUMAN +9.5 ENHANCER MUTATIONS
Considering the essential GATA2 activities during embryogenesis and in the adult, GATA2 dysregulation would be expected to be at the forefront of at least certain blood diseases. Human heterozygous GATA2 germline mutations cause primary immunodeficiency, myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), pulmonary alveolar proteinosis, defects in the vasculature and lymphatic systems and additional complex phenotypes [62–65]. Although the penetrance is incomplete, human genetic analysis with multi-generational pedigrees implicates GATA2 dysregulation as the disease-instigating mechanism. Mutations include heterozygous point mutations, small insertions and deletions within the gene body or large deletions encompassing the gene. Predicted loss-of-function mutations in the C-terminal zinc finger can inhibit DNA binding [66,67] yet retain activity or exhibit hyperactivity, e.g. with the R307W N-finger mutant [67]. GATA2 expression levels are lower in the majority of normal karyotype-AML patients, based on RNA-seq analysis of CD34+ cells [68]. GATA2 expression can be lower in MDS/AML patients versus asymptomatic relatives harboring the mutation [69].
Strikingly, GATA2 deficiency syndrome germline mutations can reside in and disrupt the +9.5 [26,44]. These mutations include deletion (c.1017+513del28) [26] or substitution (c.1017+532T>A) [70] within the E-box, or, most frequently, a C>T transition in a 3’ Ets motif (c.1017+572C>T) [44,70–74]. GATA2 expression is reduced from the mutant allele [26,44]. Additional patient mutations have been detected, but based on emerging principles of +9.5 structure/function, their functional consequences are unclear (Figure 3).
Figure 3:
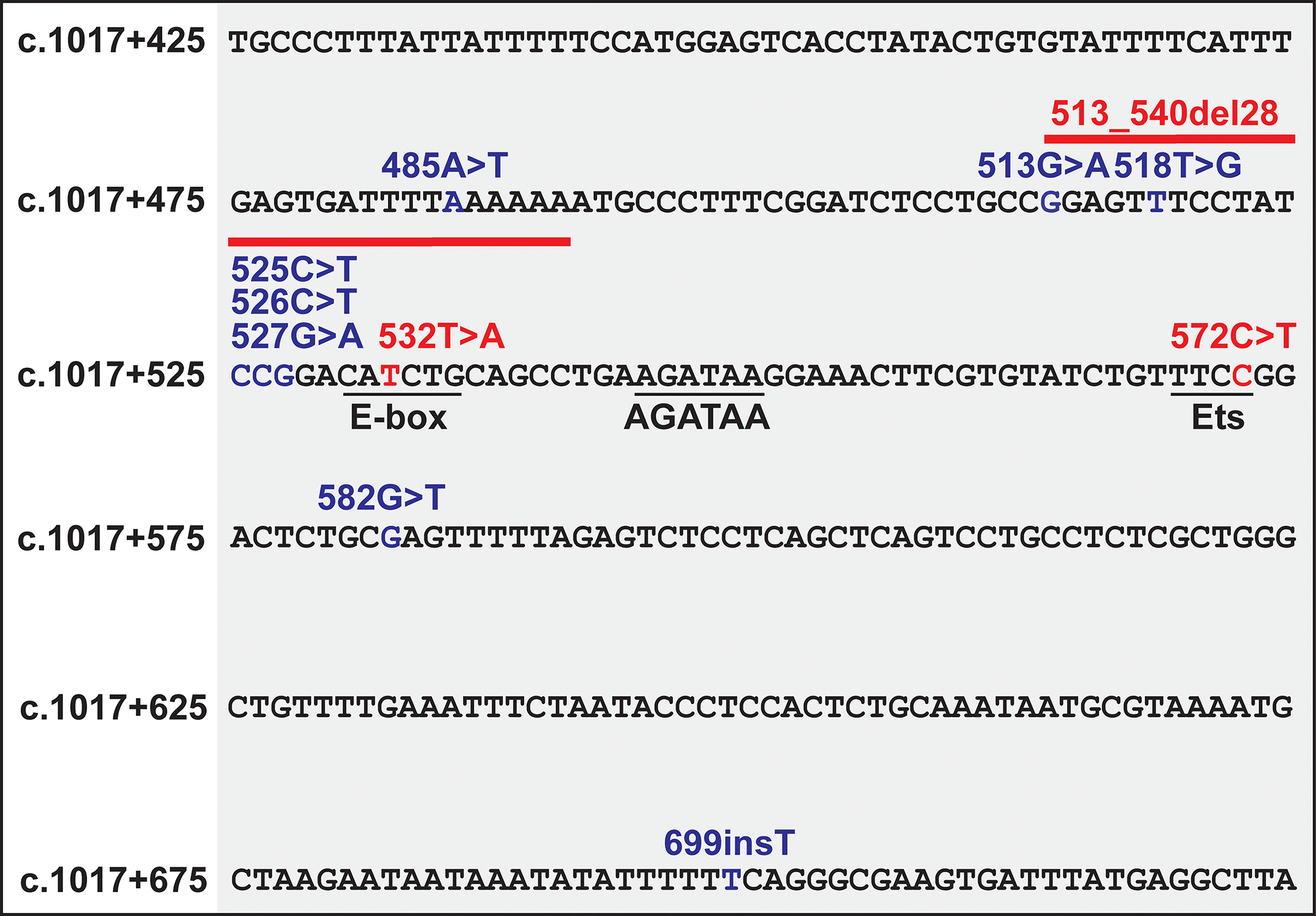
Human +9.5 variants. Red text indicates mutations predicted to be pathogenic. Blue text indicates variants of unknown significance. Mutations were described in ClinVar/ClinGen (https://www.clinicalgenome.org/data-sharing/clinvar/).
GATA2 +9.5 mutant patients exhibit decreased GATA2 mRNA expression, consistent with a GATA2 deficiency [44]. However, since GATA2 coding mutations can exert either loss-of-function or gain-of-function activity, depending upon the mutation and biological and molecular outputs analyzed [7,67,75], the evidence supports a model in which insufficient or excessive GATA2 activity is pathogenic. In certain patient cohorts, more severe aberrations, including frameshifts and deletions, correlate with vascular defects, including lymphedema [65,76,77]. In a European cohort, patients with missense mutations have a high risk of developing leukemia [73]. Missense mutations in the N-or C-terminal zinc fingers differ in disease presentation [78]. Much more evidence involving larger sample cohorts is required to rigorously assess potential genotype-phenotype correlations.
GATA2 deficiency syndrome patients frequently present with recurrent viral, mycobacterial, and fungal infections. Although infections may be secondary to immunodeficiency, the infectious agent may dysregulate cellular processes that control HSPCs. Inflammatory signals, including interferons, IL-1, and G-CSF, induce HSC proliferation while impairing self-renewal [79]. These alterations can skew differentiation, e.g. increase myelopoiesis and suppress erythropoiesis [80], increase granulocytes, macrophages, and dendritic cells and decrease B-cells [81], or favor monocytic differentiation at the expense of other lineages [82]. Metabolic byproducts may also alter HSPC function. Mutations of human genes encoding components of the Fanconi Anemia (FA) pathway, which resolves DNA crosslinks, lead to bone marrow failure. While FA mouse models do not spontaneously develop bone marrow failure, increasing acetaldehyde by mutation of Aldh2 elevates HSC DNA damage [83], inducing bone marrow failure and leukemogenesis. Inactivation of homologous recombination factors BRCA1, BRCA2, or RAD51 also renders cells hypersensitive to acetaldehyde [84]. It will be important to ascertain the contribution of extrinsic and intrinsic mechanisms to disease progression instigated by GATA2 mutation.
The majority of GATA2 deficiency syndrome patients have additional mutations and/or cytogenetic abnormalities. Somatic mutations in ASXL1, KRAS/NRAS, SF3B1, SETBP1 and additional genes have been described [85–87]. Within the +9.5-mutant patient cohort, chromosomal abnormalities are common, including trisomy 1q, trisomy 21 [88], complex cytogenetics including der(Y)t(Y;1)(q11.23;q21) [73], +1, +8, der(1;7)(q10;p10) [71] and, most frequently, monosomy 7 [70,72,88]. Regardless of the mutation, GATA2 deficiency syndrome patients with infections, MDS/AML, and/or CMML can be treated with hematopoietic stem cell transplant (HSCT) [89]. Overall survival declines after disease presentation [90]. Critically, donor GATA2 genotype must be established, as matched related or haploidentical potential donors may carry a GATA2 mutation even if they are asymptomatic [88].
CONCLUSION
As a critical regulator of hematopoiesis, GATA2 levels must be established and maintained through enhancer-dependent mechanisms and almost certainly additional mechanisms remaining to be discovered. These mechanisms are highly context-dependent and cannot be extrapolated from one cellular context to another or a physiological to a pathological state. GATA2 dysregulation creates a predisposition to develop or instigate pathologies including immunodeficiency, bone marrow failure and leukemia. In addition to missense, frameshift and splice site mutations, GATA2 +9.5 germline mutations occur in patients with GATA2 deficiency syndrome. These mutations highlight the importance of the essential +9.5 function to control hematopoiesis and suppress the development of blood diseases. Accordingly, it will be crucial to discover and understand the factors and signals that confer cell type-specific +9.5 activities, identify the full ensemble of GATA2-regulated genes, proteins and small molecules and elucidate how these components constitute cell type-specific regulatory networks and circuits governing stem and progenitor cell genesis and/or function.
Key Points.
GATA2 is a critical regulator of hematopoiesis in diverse cellular contexts.
Context-dependent establishment of GATA2 levels/activity is achieved through multiple enhancers.
GATA2 enhancers consist of cis-regulatory elements that mediate overlapping and distinct functions during development and regeneration.
Dysregulation of GATA2 expression (decreased or increased) generates a predisposition to or instigates pathogenesis.
Acknowledgements
Financial Support and Sponsorship: The work was supported by NIH DK68634 and DK50107 (E.H.B.), Evans MDS Foundation (E.H.B.), and Leukemia and Lymphoma Society Career Development Program (A.A.S.).
Footnotes
Conflicts of interest: The authors have declared that no conflict of interest exists.
REFERENCES AND RECOMMENDED READING
▯ of special interest
▯▯ of outstanding interest
- 1.Laurenti E, Gottgens B: From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553:418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Review article describes a new paradigm for the cell biology of hematopoiesis.
- 2.Giladi A, Paul F, Herzog Y, Lubling Y, Weiner A, Yofe I, Jaitin D, Cabezas-Wallscheid N, Dress R, Ginhoux F, et al. : Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat Cell Biol 2018, 20:836–846. [DOI] [PubMed] [Google Scholar]; ▯Single progenitor cell transcriptomes provided evidence for a model of differentiation trajectories.
- 3.Orkin SH, Zon LI: Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008, 132:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai FY, Keller G, Kuo FC, Weiss M, Chen J, Rosenblatt M, Alt FW, Orkin SH: An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 1994, 371:221–226. [DOI] [PubMed] [Google Scholar]; ▯▯Foundational study describing GATA2 cloning.
- 5.Kang H, Mesquitta WT, Jung HS, Moskvin OV, Thomson JA, Slukvin II: GATA2 Is Dispensable for Specification of Hemogenic Endothelium but Promotes Endothelial-to-Hematopoietic Transition. Stem Cell Reports 2018, 11:197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Demonstrated a GATA2 requirement in ES cell-derived hemogenic endothelium.
- 6.de Pater E, Kaimakis P, Vink CS, Yokomizo T, Yamada-Inagawa T, van der Linden R, Kartalaei PS, Camper SA, Speck N, Dzierzak E: Gata2 is required for HSC generation and survival. J Exp Med 2013, 210:2843–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Conditional Gata2 knockout models demonstrated GATA2 role in HSC emergence and HSC survival.
- 7.Chong CE, Venugopal P, Stokes PH, Lee YK, Brautigan PJ, Yeung DTO, Babic M, Engler GA, Lane SW, Klingler-Hoffmann M, et al. : Differential effects on gene transcription and hematopoietic differentiation correlate with GATA2 mutant disease phenotypes. Leukemia 2018, 32:194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menendez-Gonzalez JB, Vukovic M, Abdelfattah A, Saleh L, Almotiri A, Thomas LA, Agirre-Lizaso A, Azevedo A, Menezes AC, Tornillo G, et al. : Gata2 as a Crucial Regulator of Stem Cells in Adult Hematopoiesis and Acute Myeloid Leukemia. Stem Cell Reports 2019, 13:291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung M, Cordes S, Zou J, Yu SJ, Guitart X, Hong SG, Dang V, Kang E, Donaires FS, Hassan SA, et al. : GATA2 deficiency and human hematopoietic development modeled using induced pluripotent stem cells. Blood Adv 2018, 2:3553–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoshino T, Shimizu R, Ohmori S, Nagano M, Pan X, Ohneda O, Khandekar M, Yamamoto M, Lim KC, Engel JD: Reduced BMP4 abundance in Gata2 hypomorphic mutant mice result in uropathies resembling human CAKUT. Genes Cells 2008, 13:159–170. [DOI] [PubMed] [Google Scholar]
- 11.Ainoya K, Moriguchi T, Ohmori S, Souma T, Takai J, Morita M, Chandler KJ, Mortlock DP, Shimizu R, Engel JD, et al. : UG4 enhancer-driven GATA-2 and bone morphogenetic protein 4 complementation remedies the CAKUT phenotype in Gata2 hypomorphic mutant mice. Mol Cell Biol 2012, 32:2312–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harada N, Hasegawa A, Hirano I, Yamamoto M, Shimizu R: GATA2 hypomorphism induces chronic myelomonocytic leukemia in mice. Cancer Sci 2019, 110:1183–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Persons DA, Allay JA, Allay ER, Ashmun RA, Orlic D, Jane SM, Cunningham JM, Nienhuis AW: Enforced expression of the GATA-2 transcription factor blocks normal hematopoiesis. Blood 1999, 93:488–499. [PubMed] [Google Scholar]
- 14.Vicente C, Vazquez I, Conchillo A, Garcia-Sanchez MA, Marcotegui N, Fuster O, Gonzalez M, Calasanz MJ, Lahortiga I, Odero MD: Overexpression of GATA2 predicts an adverse prognosis for patients with acute myeloid leukemia and it is associated with distinct molecular abnormalities. Leukemia 2012, 26:550–554. [DOI] [PubMed] [Google Scholar]
- 15.Luesink M, Hollink IH, van der Velden VH, Knops RH, Boezeman JB, de Haas V, Trka J, Baruchel A, Reinhardt D, van der Reijden BA, et al. : High GATA2 expression is a poor prognostic marker in pediatric acute myeloid leukemia. Blood 2012, 120:2064–2075. [DOI] [PubMed] [Google Scholar]
- 16.Grass JA, Boyer ME, Pal S, Wu J, Weiss MJ, Bresnick EH: GATA-1-dependent transcriptional repression of GATA-2 via disruption of positive autoregulation and domain-wide chromatin remodeling. Proc. Natl. Acad. Sci. U. S. A 2003, 100:8811–8816. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Discovered GATA1 and GATA2 occupancy sites at the Gata2 locus and provided evidence that GATA1 replaces GATA2 on chromatin during erythroid maturation, inducing a distinct transcriptional output.
- 17.Grass JA, Jing H, Kim S-I, Martowicz ML, Pal S, Blobel GA, Bresnick EH: Distinct functions of dispersed GATA factor complexes at an endogenous gene locus. Mol. Cell. Biol 2006, 26:7056–7067. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯GATA2 chromatin occupancy analysis led to discovery of −77 and +9.5 Gata2 enhancers
- 18.Martowicz ML, Grass JA, Boyer ME, Guend H, Bresnick EH: Dynamic GATA factor interplay at a multi-component regulatory region of the GATA-2 locus. J. Biol. Chem 2005, 280:1724–1732. [DOI] [PubMed] [Google Scholar]
- 19.Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S: GATA switches as developmental drivers. J Biol Chem 2010, 285:31087–31093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bresnick EH, Martowicz ML, Pal S, Johnson KD: Developmental control via GATA factor interplay at chromatin domains. J Cell Physiol 2005, 205:1–9. [DOI] [PubMed] [Google Scholar]
- 21.Katsumura KR, Bresnick EH, Group GFM: The GATA factor revolution in hematology. Blood 2017, 129:2092–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snow JW, Trowbridge JJ, Fujiwara T, Emambokus NE, Grass JA, Orkin SH, Bresnick EH: A single cis element maintains repression of the key developmental regulator Gata2. PLoS Genet 2010, 6:e1001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snow JW, Trowbridge JJ, Johnson KD, Fujiwara T, Emambokus NE, Grass JA, Orkin SH, Bresnick EH: Context-dependent function of “GATA switch” sites in vivo. Blood 2011, 117:4769–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanalkumar R, Johnson KD, Gao X, Boyer ME, Chang YI, Hewitt KJ, Zhang J, Bresnick EH: Mechanism governing a stem cell-generating cis-regulatory element. Proc Natl Acad Sci U S A 2014, 111:E1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson KD, Kong G, Gao X, Chang Y-I, Hewitt KJ, Sanalkumar R, Prathibha R, Ranheim EA, Dewey CN, Zhang J, et al. : Cis-regulatory mechanisms governing stem and progenitor cell transitions. Science Advances 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Discovered an essential function of the Gata2 -77 enhancer to confer progenitor cell fate.
- 26.Johnson KD, Hsu AP, Ryu MJ, Wang J, Gao X, Boyer ME, Liu Y, Lee Y, Calvo KR, Keles S, et al. : Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity. J Clin Invest 2012, 122:3692–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Foundational study demonstrating essential +9.5 activity to generate hematopoietic stem and progenitor cells during embryogenesis and mutation of this enhancer in human patients with GATA2 deficiency syndrome.
- 27.Gao X, Johnson KD, Chang YI, Boyer ME, Dewey CN, Zhang J, Bresnick EH: Gata2 cis-element is required for hematopoietic stem cell generation in the mammalian embryo. J Exp Med 2013, 210:2833–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Demonstrated +9.5 requirement for HSC emergence in the AGM of mouse embryos.
- 28.Soukup AA, Zheng Y, Mehta C, Wu J, Liu P, Cao M, Hofmann I, Zhou Y, Zhang J, Johnson KD, et al. : Single-nucleotide human disease mutation inactivates a blood-regenerative GATA2 enhancer. J Clin Invest 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Developed +9.5 mutant alleles and demonstrated the +9.5 Ets motif promotes regenerative hematopoiesis.
- 29.Hewitt KJ, Kim DH, Devadas P, Prathibha R, Zuo C, Sanalkumar R, Johnson KD, Kang YA, Kim JS, Dewey CN, et al. : Hematopoietic Signaling Mechanism Revealed from a Stem/Progenitor Cell Cistrome. Mol Cell 2015, 59:62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Established an ensemble of “+9.5-like” enhancers, including one at Samd14, which was demonstrated to encode a stimulator of c-Kit signaling
- 30.Wozniak RJ, Boyer ME, Grass JA, Lee Y, Bresnick EH: Context-dependent GATA factor function: combinatorial requirements for transcriptional control in hematopoietic and endothelial cells. J Biol Chem 2007, 282:14665–14674. [DOI] [PubMed] [Google Scholar]; ▯Described +9.5 activity to confer transcriptional regulation of a transgene in endothelial and hematopoietic cells
- 31.Wadman IA, Osada H, Grutz GG, Agulnick AD, Westphal H, Forster A, Rabbitts TH: The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J. 1997, 16:3145–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯▯Developed a paradigm in which a GATA factor, SCL/TAL1 and associated factors form a multiprotein complex on composite elements.
- 32.Porcher C, Swat W, Rockwell K, Fujiwara Y, Alt FW, Orkin SH: The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell 1996, 86:47–57. Established a critical SCL/TAL1 function to promote multi-lineage hematopoiesis. [DOI] [PubMed] [Google Scholar]
- 33.Hoang T, Lambert JA, Martin R: SCL/TAL1 in Hematopoiesis and Cellular Reprogramming. Curr Top Dev Biol 2016, 118:163–204. [DOI] [PubMed] [Google Scholar]
- 34.Tsai SF, Martin DI, Zon LI, D’Andrea AD, Wong GG, Orkin SH: Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature 1989, 339:446–451. [DOI] [PubMed] [Google Scholar]; ▯▯Foundational study describing cloning of the first mammalian GATA factor, GATA1.
- 35.Evans T, Felsenfeld G: The erythroid-specific transcription factor Eryf1: a new finger protein. Cell 1989, 58:877–885. [DOI] [PubMed] [Google Scholar]; ▯▯Foundational study describing cloning of the first mammalian GATA factor, GATA1.
- 36.Hollenhorst PC, McIntosh LP, Graves BJ: Genomic and biochemical insights into the specificity of ETS transcription factors. Annu Rev Biochem 2011, 80:437–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loughran SJ, Kruse EA, Hacking DF, de Graaf CA, Hyland CD, Willson TA, Henley KJ, Ellis S, Voss AK, Metcalf D, et al. : The transcription factor Erg is essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. Nat Immunol 2008, 9:810–819. [DOI] [PubMed] [Google Scholar]
- 38.Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M, Watson DK: Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol 2000, 20:5643–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee D, Park C, Lee H, Lugus JJ, Kim SH, Arentson E, Chung YS, Gomez G, Kyba M, Lin S, et al. : ER71 acts downstream of BMP, Notch, and Wnt signaling in blood and vessel progenitor specification. Cell Stem Cell 2008, 2:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯▯Discovered that ER71 (ETV2) has a critical function downstream of developmental signaling pathways to regulate blood and vascular development.
- 40.Wozniak RJ, Keles S, Lugus JJ, Young K, Boyer ME, Tran TT, Choi K, Bresnick EH: Molecular hallmarks of endogenous chromatin complexes containing master regulators of hematopoiesis. Mol. Cell. Biol 2008, 28:6681–6694. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Demonstrated GATA2 and SCL/TAL1 co-occupy chromatin at new target genes.
- 41.Fujiwara T, O’Geen H, Keles S, Blahnik K, Linnemann AK, Kang YA, Choi K, Farnham PJ, Bresnick EH: Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol Cell 2009, 36:667–681. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Described the first GATA1 and GATA2 ChIP-seq datasets, which were used to generate mechanistic and biological insights.
- 42.Yu M, Riva L, Xie H, Schindler Y, Moran TB, Cheng Y, Yu D, Hardison R, Weiss MJ, Orkin SH, et al. : Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol Cell 2009, 36:682–695. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Described the first GATA1 ChIP-seq datasets and insights into GATA1-regulated activation and repression.
- 43.Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, et al. : Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 2010, 7:532–544. [DOI] [PubMed] [Google Scholar]; ▯Described GATA2 co-occupancy of chromatin with several transcription factors and coregulators.
- 44.Hsu AP, Johnson KD, Falcone EL, Sanalkumar R, Sanchez L, Hickstein DD, Cuellar-Rodriguez J, Lemieux JE, Zerbe CS, Bresnick EH, et al. : GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood 2013, 121:3830–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Provided evidence that heterozygous +9.5 mutation in patient samples lowers GATA2 transcription and proposed that GATA2 deficiency syndrome involves haploinsufficiency.
- 45.Khandekar M, Brandt W, Zhou Y, Dagenais S, Glover TW, Suzuki N, Shimizu R, Yamamoto M, Lim KC, Engel JD: A Gata2 intronic enhancer confers its pan-endothelial-specific regulation. Development 2007, 134:1703–1712. [DOI] [PubMed] [Google Scholar]; ▯Utilized a +9.5-containing transgene to demonstrate Gata2 expression in endothelial, endocardial and lymphatic cells.
- 46.Lange L, Hoffmann D, Schwarzer A, Ha TC, Philipp F, Lenz D, Morgan M, Schambach A: Inducible Forward Programming of Human Pluripotent Stem Cells to Hemato-endothelial Progenitor Cells with Hematopoietic Progenitor Potential. Stem Cell Reports 2020, 14:122–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dzierzak E, Medvinsky A: The discovery of a source of adult hematopoietic cells in the embryo. Development 2008, 135:2343–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mehta C, Johnson KD, Gao X, Ong IM, Katsumura KR, McIver SC, Ranheim EA, Bresnick EH: Integrating Enhancer Mechanisms to Establish a Hierarchical Blood Development Program. Cell Rep 2017, 20:2966–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Used compound heterozygous +9.5 cis-element mutants to demonstrate +9.5 function is not restricted to hemogenic endothelium and HSCs, but it is also active in progenitors and essential for generation of MEPs.
- 49.Hewitt KJ, Johnson KD, Gao X, Keles S, Bresnick EH: The Hematopoietic Stem and Progenitor Cell Cistrome: GATA Factor-Dependent cis-Regulatory Mechanisms. Curr Top Dev Biol 2016, 118:45–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liau WS, Ngoc PC, Sanda T: Roles of the RUNX1 Enhancer in Normal Hematopoiesis and Leukemogenesis. Adv Exp Med Biol 2017, 962:139–147. [DOI] [PubMed] [Google Scholar]
- 51.Tripic T, Deng W, Cheng Y, Vakoc CR, Gregory GD, Hardison RC, Blobel GA: SCL and associated protein distinguish active from repressive GATA transcription factor complexes. Blood 2008, 113:2191–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Provided evidence that SCL/TAL1 can co-localize with GATA1 at chromatin sites lacking an E-box.
- 52.Katsumura KR, Yang C, Boyer ME, Li L, Bresnick EH: Molecular basis of crosstalk between oncogenic Ras and the master regulator of hematopoiesis GATA-2. EMBO Rep 2014, 15:938–947. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Described an oncogenic Ras-dependent multi-site GATA2 phosphorylation mechanism that enhances GATA2-dependent transcriptional activation in a context-dependent manner.
- 53.Katsumura KR, Ong IM, DeVilbiss AW, Sanalkumar R, Bresnick EH: GATA Factor-Dependent Positive-Feedback Circuit in Acute Myeloid Leukemia Cells. Cell Rep 2016, 16:2428–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng JT, Cobb MH, Baer R: Phosphorylation of the TAL1 oncoprotein by the extracellular-signal- regulated protein kinase ERK1. Mol Cell Biol 1993, 13:801–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wadman IA, Hsu HL, Cobb MH, Baer R: The MAP kinase phosphorylation site of TAL1 occurs within a transcriptional activation domain. Oncogene 1994, 9:3713–3716. [PubMed] [Google Scholar]
- 56.Prasad KS, Jordan JE, Koury MJ, Bondurant MC, Brandt SJ: Erythropoietin stimulates transcription of the TAL1/SCL gene and phosphorylation of its protein products. J. Biol. Chem 1995, 270:11603–11611. [DOI] [PubMed] [Google Scholar]
- 57.Prasad KS, Brandt SJ: Target-dependent effect of phosphorylation on the DNA binding activity of the TAL1/SCL oncoprotein. J Biol Chem 1997, 272:11457–11462. [DOI] [PubMed] [Google Scholar]
- 58.Tang T, Arbiser JL, Brandt SJ: Phosphorylation by mitogen-activated protein kinase mediates the hypoxia-induced turnover of the TAL1/SCL transcription factor in endothelial cells. J Biol Chem 2002, 277:18365–18372. [DOI] [PubMed] [Google Scholar]
- 59.Xu CX, Lee TJ, Sakurai N, Krchma K, Liu F, Li D, Wang T, Choi K: ETV2/ER71 regulates hematopoietic regeneration by promoting hematopoietic stem cell proliferation. J Exp Med 2017, 214:1643–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯▯Established an essential ER71/ETV2 function to promote hematopoietic regeneration.
- 60.Liu F, Li D, Yu YY, Kang I, Cha MJ, Kim JY, Park C, Watson DK, Wang T, Choi K: Induction of hematopoietic and endothelial cell program orchestrated by ETS transcription factor ER71/ETV2. EMBO Rep 2015, 16:654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Developed molecular insights into ER71/ETV2-dependent hematopoietic regeneration.
- 61.Sumanas S, Choi K: ETS Transcription Factor ETV2/ER71/Etsrp in Hematopoietic and Vascular Development. Curr Top Dev Biol 2016, 118:77–111. [DOI] [PubMed] [Google Scholar]
- 62.Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, Lakey JH, Rahman T, Wang XN, McGovern N, et al. : Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood 2011, 118:2656–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯▯Foundational study demonstrating GATA2 mutations cause a human blood disease.
- 63.Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, Babic M, Lin M, Carmagnac A, Lee YK, et al. : Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet 2011, 43:1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯▯Foundational study demonstrating GATA2 mutations cause a human blood disease.
- 64.Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, Frucht DM, Vinh DC, Auth RD, Freeman AF, et al. : Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011, 118:2653–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯▯Foundational study demonstrating GATA2 mutations cause a human blood disease.
- 65.Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, Dafou D, Kilo T, Smithson S, Lunt P, et al. : Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet 2011, 43:929–931. [DOI] [PubMed] [Google Scholar]; ▯▯Foundational study demonstrating GATA2 mutations cause a human blood disease.
- 66.Ping N, Sun A, Song Y, Wang Q, Yin J, Cheng W, Xu Y, Wen L, Yao H, Ma L, et al. : Exome sequencing identifies highly recurrent somatic GATA2 and CEBPA mutations in acute erythroid leukemia. Leukemia 2017, 31:195–202. [DOI] [PubMed] [Google Scholar]
- 67.Katsumura KR, Mehta C, Hewitt KJ, Soukup AA, Fraga de Andrade I, Ranheim EA, Johnson KD, Bresnick EH: Human leukemia mutations corrupt but do not abrogate GATA-2 function. Proc Natl Acad Sci U S A 2018, 115:E10109–E10118. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Innovated an ex vivo genetic rescue system and demonstrated retention of activity, or hyperactivity, with certain GATA2 disease mutants that were assumed to be loss-of-function.
- 68.Celton M, Forest A, Gosse G, Lemieux S, Hebert J, Sauvageau G, Wilhelm BT: Epigenetic regulation of GATA2 and its impact on normal karyotype acute myeloid leukemia. Leukemia 2014, 28:1617–1626. [DOI] [PubMed] [Google Scholar]
- 69.Al Seraihi AF, Rio-Machin A, Tawana K, Bodor C, Wang J, Nagano A, Heward JA, Iqbal S, Best S, Lea N, et al. : GATA2 monoallelic expression underlies reduced penetrance in inherited GATA2-mutated MDS/AML. Leukemia 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Demonstrated reduced GATA2 expression in patients with clinical manifestations versus asymptomatic carriers.
- 70.Wlodarski MW, Hirabayashi S, Pastor V, Stary J, Hasle H, Masetti R, Dworzak M, Schmugge M, van den Heuvel-Eibrink M, Ussowicz M, et al. : Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 2016, 127:1387–1397; quiz 1518. [DOI] [PubMed] [Google Scholar]; ▯A large clinical study of GATA2-mutant pediatric patients.
- 71.Ganapathi KA, Townsley DM, Hsu AP, Arthur DC, Zerbe CS, Cuellar-Rodriguez J, Hickstein DD, Rosenzweig SD, Braylan RC, Young NS, et al. : GATA2 deficiency-associated bone marrow disorder differs from idiopathic aplastic anemia. Blood 2015, 125:56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Comparative analysis of GATA2-mutant instigated bone marrow failure and idiopathic aplastic anemia.
- 72.Koegel AK, Hofmann I, Moffitt K, Degar B, Duncan C, Tubman VN: Acute lymphoblastic leukemia in a patient with MonoMAC syndrome/GATA2 haploinsufficiency. Pediatr Blood Cancer 2016, 63:1844–1847. [DOI] [PubMed] [Google Scholar]
- 73.Donadieu J, Lamant M, Fieschi C, de Fontbrune FS, Caye A, Ouachee M, Beaupain B, Bustamante J, Poirel HA, Isidor B, et al. : Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica 2018, 103:1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯A large clinical study of 53 families with GATA2 mutations, including a +9.5 mutation.
- 74.McReynolds LJ, Yang Y, Yuen Wong H, Tang J, Zhang Y, Mule MP, Daub J, Palmer C, Foruraghi L, Liu Q, et al. : MDS-associated mutations in germline GATA2 mutated patients with hematologic manifestations. Leuk Res 2019, 76:70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang SJ, Ma LY, Huang QH, Li G, Gu BW, Gao XD, Shi JY, Wang YY, Gao L, Cai X, et al. : Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci U S A 2008, 105:2076–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar-Rodriguez J, Hsu AP, Dyack S, Fernandez CV, Chong CE, et al. : Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood 2012, 119:1283–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, Arthur DC, Gu W, Gould CM, Brewer CC, et al. : GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood 2014, 123:809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tien FM, Hou HA, Tsai CH, Tang JL, Chiu YC, Chen CY, Kuo YY, Tseng MH, Peng YL, Liu MC, et al. : GATA2 zinc finger 1 mutations are associated with distinct clinico-biological features and outcomes different from GATA2 zinc finger 2 mutations in adult acute myeloid leukemia. Blood Cancer J 2018, 8:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pietras EM: Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130:1693–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kato H, Igarashi K: To be red or white: lineage commitment and maintenance of the hematopoietic system by the “inner myeloid”. Haematologica 2019, 104:1919–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ueda Y, Kondo M, Kelsoe G: Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med 2005, 201:1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao JL, Ma C, O’Connell RM, Mehta A, DiLoreto R, Heath JR, Baltimore D: Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell 2014, 14:445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garaycoechea JI, Crossan GP, Langevin F, Mulderrig L, Louzada S, Yang F, Guilbaud G, Park N, Roerink S, Nik-Zainal S, et al. : Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553:171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Discovered that endogenous aldehyde overproduction induces DNA damage in HSCs.
- 84.Tacconi EM, Lai X, Folio C, Porru M, Zonderland G, Badie S, Michl J, Sechi I, Rogier M, Matia Garcia V, et al. : BRCA1 and BRCA2 tumor suppressors protect against endogenous acetaldehyde toxicity. EMBO Mol Med 2017, 9:1398–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu Y, Zhang T, Bao X, Wang Q, Zhang L, Hong Y, Zeng Z, Shen H, Wu D, Pan J, et al. : Combining gene variants with clinical characteristics improves outcome prediction in Chinese patients with myelodysplastic syndromes. Leuk Lymphoma 2019:1–8. [DOI] [PubMed] [Google Scholar]
- 86.West RR, Hsu AP, Holland SM, Cuellar-Rodriguez J, Hickstein DD: Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica 2014, 99:276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▯Identification of somatic mutations that may trigger pathogenesis in the context of germline GATA2 mutations.
- 87.Fisher KE, Hsu AP, Williams CL, Sayeed H, Merritt BY, Elghetany MT, Holland SM, Bertuch AA, Gramatges MM: Somatic mutations in children with GATA2-associated myelodysplastic syndrome who lack other features of GATA2 deficiency. Blood Adv 2017, 1:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parta M, Shah NN, Baird K, Rafei H, Calvo KR, Hughes T, Cole K, Kenyon M, Schuver BB, Cuellar-Rodriguez J, et al. : Allogeneic Hematopoietic Stem Cell Transplantation for GATA2 Deficiency Using a Busulfan-Based Regimen. Biol Blood Marrow Transplant 2018, 24:1250–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bogaert DJ, Laureys G, Naesens L, Mazure D, De Bruyne M, Hsu AP, Bordon V, Wouters E, Tavernier SJ, Lambrecht BN, et al. : GATA2 deficiency and haematopoietic stem cell transplantation: challenges for the clinical practitioner. Br J Haematol 2019. [DOI] [PubMed] [Google Scholar]
- 90.Simonis A, Fux M, Nair G, Mueller NJ, Haralambieva E, Pabst T, Pachlopnik Schmid J, Schmidt A, Schanz U, Manz MG, et al. : Allogeneic hematopoietic cell transplantation in patients with GATA2 deficiency-a case report and comprehensive review of the literature. Ann Hematol 2018, 97:1961–1973. [DOI] [PubMed] [Google Scholar]