

Abstract
The G6PC1, G6PC2 and G6PC3 genes encode distinct glucose-6-phosphatase catalytic subunit (G6PC) isoforms. In mice, germline deletion of G6pc2 lowers fasting blood glucose (FBG) without affecting fasting plasma insulin (FPI) while, in isolated islets, glucose-6-phosphatase activity and glucose cycling are abolished and glucose-stimulated insulin secretion (GSIS) is enhanced at submaximal but not high glucose. These observations are all consistent with a model in which G6PC2 regulates the sensitivity of GSIS to glucose by opposing the action of glucokinase. G6PC2 is highly expressed in human and mouse islet beta cells, however, various studies have shown trace G6PC2 expression in multiple tissues raising the possibility that G6PC2 also affects FBG through non-islet cell actions. Using real time PCR we show here that expression of G6pc1 and/or G6pc3 are much greater than G6pc2 in peripheral tissues whereas G6pc2 expression is much higher than G6pc3 in both pancreas and islets with G6pc1 expression not detected. In adult mice, beta cell-specific deletion of G6pc2 was sufficient to reduce FBG without changing FPI. In addition, electronic health record-derived phenotype analyses showed no association between G6PC2 expression and phenotypes clearly unrelated to islet function in humans. Finally, we show that germline G6pc2 deletion enhances glycolysis in mouse islets and that glucose cycling can also be detected in human islets. These observations are all consistent with a mechanism by which G6PC2 action in islets is sufficient to regulate the sensitivity of GSIS to glucose and hence influence FBG without affecting FPI.
Introduction
Glucose-6-phosphatase catalyzes the hydrolysis of glucose-6-phosphate (G6P) to glucose and inorganic phosphate and is located in the endoplasmic reticulum (ER) membrane (Hutton and O’Brien 2009). It is thought to exist as a multi-component enzyme system in which a G6P transporter, encoded by the SLC37A4 gene, delivers substrate from the cytosol to the active site of a glucose-6-phosphatase catalytic subunit (G6PC) in the lumen of the ER with transporters for inorganic phosphate and glucose returning the reaction products back to the cytosol (Hutton and O’Brien 2009). The G6PC gene family is comprised of three members, G6PC1, G6PC2 and G6PC3 (Hutton and O’Brien 2009). G6PC1, also known as G6PC or G6Pase, is predominantly expressed in liver and kidney where it catalyzes the terminal step in the gluconeogenic and glycogenolytic pathways (Hutton and O’Brien 2009). G6PC3 is widely expressed, with especially high expression in kidney, testis, skeletal muscle and brain (Hutton and O’Brien 2009). While G6PC3 catalyzes G6P hydrolysis in vitro (Shieh, et al. 2003), its key substrate in vivo is thought to be the G6P analog 1,5-anhydroglucitol-6-phosphate (Veiga-da-Cunha, et al. 2019). Based on Northern blotting and immunohistochemistry, G6PC2, also known as IGRP, was thought to be only expressed in pancreatic islets in mice (Arden, et al. 1999) and humans (Martin, et al. 2001). However, human RNA-Seq data, available through the GTEx database, suggest very low expression of G6PC2 in other tissues, including liver, skeletal muscle, and hypothalamus, raising the possibility that G6PC2 is functionally important in non-islet tissues.
The beta cell glucose sensor, glucokinase, catalyzes the formation of G6P from glucose and was thought to be the key factor that determines the rate of beta cell glycolytic flux (Iynedjian 2009; Matschinsky and Wilson 2019). This glycolytic rate determines the sensitivity of glucose-stimulated insulin secretion (GSIS) to glucose and hence the influence of beta cells on fasting blood glucose (FBG) (Iynedjian 2009; Matschinsky and Wilson 2019). However, in isolated germline G6pc2 knockout (KO) islets glucose-6-phosphatase activity (Pound, et al. 2013) and glucose cycling (Wall, et al. 2015), calculated as the percentage of the G6P generated from glucose that is converted back to glucose, are abolished. This results in a leftward shift in the dose response curve for GSIS (Pound et al. 2013) such that under fasting conditions, insulin levels are the same in wild type (WT) and germline G6pc2 KO mice but FBG is reduced in KO mice (Pound et al. 2013; Wang, et al. 2007). These data challenge the existing dogma and suggest a new paradigm in which a glucokinase/G6PC2 futile cycle, rather than glucokinase alone, determines the rate of beta cell glycolytic flux and hence the sensitivity of GSIS to glucose. Molecular studies have shown that the ‘A’ allele of the rs560887 single nucleotide polymorphism (SNP) in the G6PC2 gene allele reduces G6PC2 RNA splicing (Baerenwald, et al. 2013) and hence expression of full length G6PC2. Consistent with the new paradigm, genome wide association studies (GWAS) have linked the rs560887 ‘A’ allele to reduced FBG (Bouatia-Naji, et al. 2008; Chen, et al. 2008).
The experiments described here provide further support for this new model by showing that glucose cycling also exists in human islets and that germline deletion of G6pc2 enhances glycolysis in mouse islets. In addition, gene expression analyses, electronic health record-derived phenotype analyses, and studies in a novel mouse model in which G6pc2 is specifically deleted in beta cells all suggest that G6PC2 is not active in non-islet tissues, and instead imply that an islet-specific action of G6PC2 is sufficient to mediate the effect of G6PC2 on FBG.
Materials and Methods
Animal Care
The Vanderbilt University Medical Center Animal Care and Use Committee approved all protocols used. Mice were maintained on a standard rodent chow diet (calorie contributions: 28% protein, 12% fat, 60% carbohydrate (14% disaccharides); LabDiet 5001; PMI Nutrition International). Food and water were provided ad libitum.
Generation of Germline G6pc2 KO Mice
The generation of germline G6pc2 KO mice on a pure C57BL/6J genetic background has been previously described (Pound et al. 2013; Wang et al. 2007). The generation of beta cell-specific (BCS) G6pc2 KO mice is described in the Supplemental Material.
Mouse and Human Islet Isolation
Mouse islets were isolated by the Vanderbilt Islet Procurement and Analysis Core as previously described (Syring, et al. 2016). Human islets were obtained by A.C.P. through the NIDDK-funded Integrated Islet Distribution Program (https://iidp.coh.org/) and handled and assessed as previously described (Kayton, et al. 2015). Human islets designated as ‘Group 1’ (Kayton et al. 2015) based on islet perifusion analyses (Supplemental Fig. 4) were used for these studies.
Analysis of Gene Expression
Tissue RNA was isolated using the ToTALLY RNA kit whereas islet RNA was isolated using the RNAqueous kit (Ambion, Carlsbad, CA). The Turbo DNA-free DNAse Treatment Kit (Ambion, Carlsbad, CA) was then used to remove trace genomic DNA followed by cDNA generation using the iScript DNA Synthesis Kit (Bio-Rad, Hercules, CA). Gene expression was then quantitated by PCR using the dUTP-containing FastStart SYBR Green Master Mix in conjunction with Uracil-DNA-Glycosylase (Roche, Nutley, NJ). Fold induction of gene expression was calculated using the 2(-ΔΔC(T)) method (Livak and Schmittgen 2001). PCR primer sequences are provided in Supplemental Material.
Phenotypic Analysis of Fasted Germline and BCS G6pc2 KO Mice
Mice were fasted for 5 hours and then weighed. After an additional hour of fasting, mice were anesthetized using isoflurane and blood samples were isolated from the retro-orbital venous plexus. Glucose concentrations were measured in whole blood using a glucose monitor (Accu-Check Advantage; Roche, Indianapolis, USA). EDTA (5 μl; 0.5 M) was then added to blood samples prior to isolation of plasma by centrifugation. Insulin samples were assayed using RIA (Morgan and Lazarow 1963) by the Vanderbilt Hormone Assay and Analytical Services Core.
Hematoxylin and eosin analysis of tissue morphology
Mice were euthanized at 8 months of age and the pancreata were formalin-fixed. Pancreata were then paraffin-embedded and sections cut at a depth of 5 μm, dewaxed and stained in hematoxylin and eosin by the Vanderbilt Translational Pathology Shared Resource. Details on the analysis of pancreatic cryo-sections by immunofluorescence are provided in Supplemental Material.
Electronic Health Record (EHR)-Based Analyses of Human Research Subjects
EHR-based analyses were conducted using data on human subjects in the Vanderbilt University Medical Center (VUMC) BioVU DNA databank. Genotyping data in BioVU is linked to the Synthetic Derivative (SD), a de-identified version of the VUMC EHR repository. Detailed descriptions of program operations, ethical considerations, and continuing oversight and patient engagement have been published (Pulley, et al. 2010; Roden, et al. 2008). The methods used to perform phenome-wide association studies (PheWAS) have been previously published (Denny, et al. 2010; Denny, et al. 2013). The methods used to perform laboratory value-wide association studies (LabWAS) are described in the GitHub repository [https://bitbucket.org/juliasealock/labwas/src/master/].
The BioVU sample was restricted to a homogenous European American (EA) population (N = 50,115) and an African American (AA) population (N = 9,640), based on global genetic ancestry estimated from principal components. We used the PheWAS package in R to perform 1,212 logistic regressions to determine whether any Phecodes (hierarchical clustering of International Classification of Disease (ICD) codes) were significantly associated with our SNPs of interest after adjusting for sex, age (defined for each individual as median age across their medical record) and the top four principal components of ancestry (Denny et al. 2010; Denny et al. 2013). For each phenotype, we required a minimum number of 100 cases for inclusion in the PheWAS. We used the LabWAS package to perform 453 linear regressions to determine whether any laboratory values were associated with SNP genotype. The SNPs tested were common in both EA and AA populations.
Glucose Cycling
Glucose cycling was analyzed as previously described (Wall et al. 2015). Briefly, aliquots of ~100 islets were incubated for 24 or 72 hr at 28°C in RPMI-1640 medium containing 5 mM or 11 mM glucose in a volume of 175 μl. Islets were incubated in either naturally labeled glucose or [1,2,3,4,5,6,6-2H7]glucose (D7-glucose) (98% isotopic purity per site; Cambridge Isotope Laboratories, Inc., Andover, MA). Following the 24 or 72 hr incubation, islets were resuspended by pipetting and pelleted by centrifugation. The supernatant was retained for GC-MS analysis of glucose concentration and mass isotopomer distribution, with glucose concentration determined through comparison to a standard curve. Glucose cycling and glucose uptake rates were calculated as described previously (Wall et al. 2015).
Glycolysis
Glycolysis was analyzed as described (Ashcroft, et al. 1972; Tamarit-Rodriguez, et al. 1998; Wang and Iynedjian 1997). Briefly, aliquots of ~100 WT and germline G6pc2 KO islets were incubated for 2 hrs at 37°C in Krebs bicarbonate buffer containing 5.6 mM glucose spiked with 1 μl D-[5-3H]glucose (Perkin Elmer, Waltham, MA; 10Ci/mmol; 1 mCi/ml) in a volume of 100 μl. Following the incubation, islets were pelleted by centrifugation. The supernatant was retained for analysis of 3H2O generation whereas DNA content of the cell pellet was measured using the Hoechst reagent (Sigma, St. Louis, MO) as described (Ashcroft et al. 1972).
Statistical Analyses
Mouse data were analyzed either using a Student’s t-test, two sample assuming equal variance or a one-way ANOVA, assuming normal distribution and equal variance, as indicated. Post hoc analyses were performed using the Bonferroni correction for multiple comparisons. P values indicating the level of significance are shown in the Figure Legends. EHR associations were analyzed with logistic and linear regressions using the PheWAS package in R and the LabWAS package in R. PheWAS and LabWAS results were deemed significant if the p-value of the association passed a Bonferroni multiple testing correction to account for 1,212 logistic and 453 linear regressions that were performed, respectively.
Results
G6pc Isoforms Exhibit Tissue Specific Expression in Male Mice
While published data strongly suggest that G6PC2 is active in islets, a major unanswered question is whether it is also active in other tissues. The observation from GTEx data that G6PC2 is expressed at low levels in multiple tissues raise this possibility, though it is important to note that there is currently no evidence for detectable G6PC2 protein expression outside of islets. Whether G6PC2 is functionally active in these tissues is likely to depend in part on its expression relative to other G6PC isoforms. We therefore used real time PCR to compare the tissue specific expression of mouse G6pc1 (Fig. 1A), G6pc2 (Fig. 1B), and G6pc3 (Fig. 1C), as well as Slc37a4 (Fig. 1D). Of the tissues tested, G6pc2 expression was only detected in pancreas (Fig. 1B). In contrast, G6pc1 was expressed predominantly in liver and kidney (Fig. 1A), whereas G6pc3 expression was detected in all tissues examined (Fig. 1C). Similarly, expression of Slc37a4, which is known to couple with G6PC1 and G6PC3 (Pan, et al. 2011), was detected in all tissues examined (Fig. 1D). These real time PCR data are similar to the results of expression analyses for G6pc1 (Shelly, et al. 1993), G6pc2 (Arden et al. 1999), G6pc3 (Boustead, et al. 2004) and Slc37a4 (Gerin, et al. 1997) performed using Northern blotting. These results may indicate that the pattern of G6pc2 expression differs between mouse and human or, more likely, that this apparent difference simply reflects the increased sensitivity of RNA-Seq versus real time PCR with the former method being able to detect extremely low expression. For example, RNA-Seq data demonstrate that G6PC2 is expressed in liver at ~0.018% of the level in islets (Ku, et al. 2012), a difference too small to easily detect using real time PCR. Even if G6pc2 is expressed at trace levels in these non-pancreatic tissues, because both G6PC1 (Lei, et al. 1993) and G6PC3 (Shieh et al. 2003) have much higher catalytic activity than G6PC2 (Petrolonis, et al. 2004), these results suggest that G6PC2 is highly unlikely to directly affect metabolism in these tissues.
Figure 1. Comparison of Tissue-Specific G6pc Isoform Expression in Male Mice.
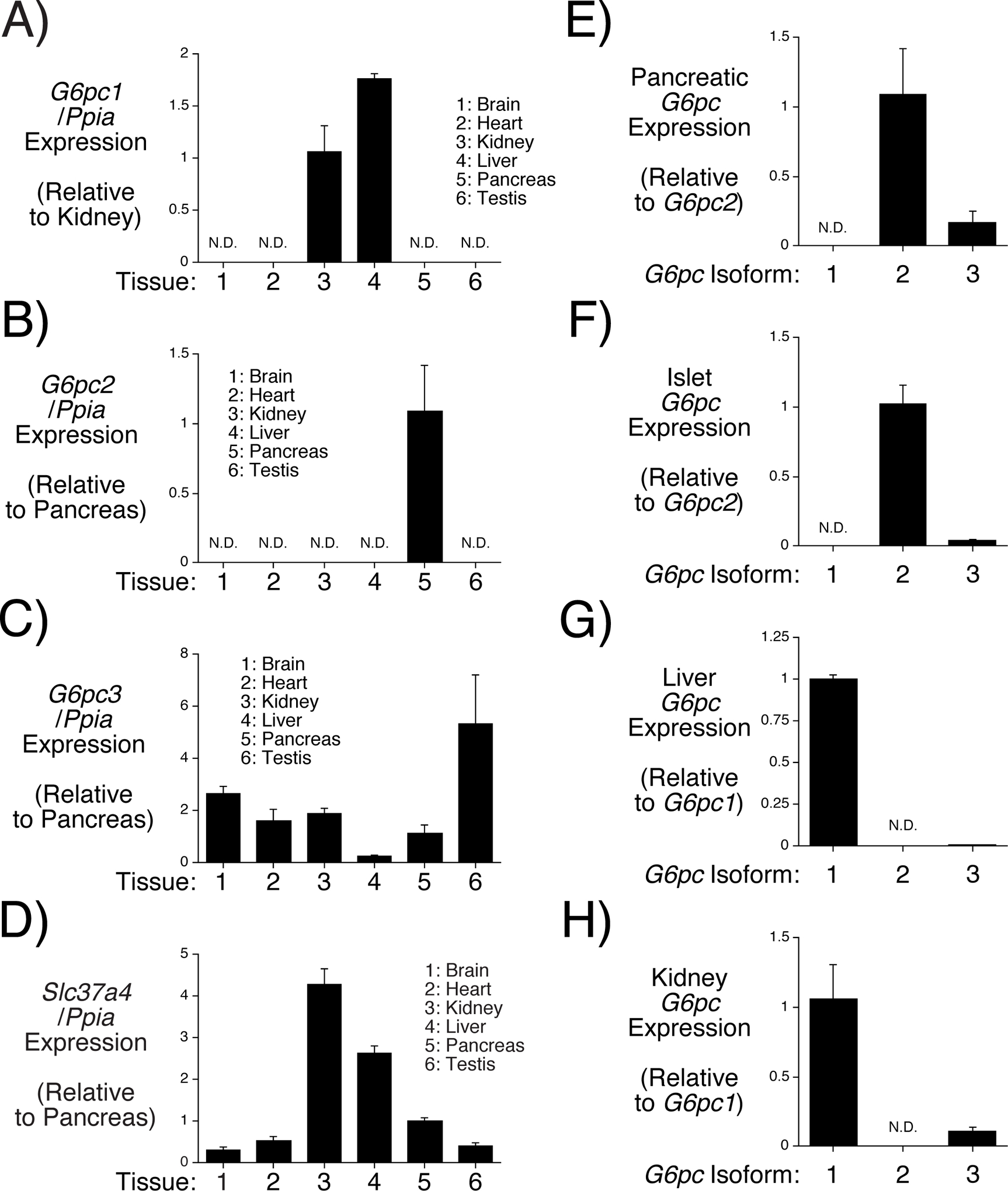
Comparison of G6pc1 (Panel A), G6pc2 (Panel B), G6pc3 (Panel C) and Slc37a4 (Panel D) expression in brain, heart, kidney, liver, pancreas and testis from 2 month old non-fasted male mice. G6pc and Slc37a4 expression were quantitated relative to Ppia (cyclophilin A) expression in the indicated tissue and then expressed relative to that in either kidney (Panel A) or pancreas (Panels B-D). Comparison of G6pc1, G6pc2 and G6pc3 expression in pancreas (Panel E), islets (Panel F), liver (Panel G) and kidney (Panel H) from 2 month old non-fasted male mice. G6pc expression was quantitated relative to G6pc2 expression in pancreas (Panel E) or islet (Panel F) or relative to G6pc1 expression in liver (Panel G) or kidney (Panel H). Results represent the mean data ± S.E.M. derived from tissues isolated from 3 male mice.
The Relative Expression of G6pc Isoforms Varies within Specific Tissues in Male Mice
We next examined the relative expression of G6pc isoforms within individual tissues. Within pancreas (Fig. 1E) and islets (Fig. 1F) G6pc2 expression is higher than that of G6pc3, and G6pc1 expression is not detected. These results suggest that G6PC3 is unlikely to compensate for the absence of G6PC2 in pancreatic islets. Indeed, glucose-6-phosphatase activity (Pound et al. 2013) and glucose cycling (Wall et al. 2015) are abolished rather than reduced in germline G6pc2 KO islets. Similarly, G6pc1 expression is higher than that of G6pc3 in both liver (Fig. 1G) and kidney (Fig. 1H) suggesting that G6PC3 is unlikely to be able to compensate for the absence of G6PC1 in either tissue, explaining why inactivating mutations in G6PC1 result in glycogen storage disease type 1a despite the presence of G6PC3 (Chou and Mansfield 2008).
G6pc Isoform Expression, FBG and FPI Change with Aging in Male Mice
To potentially further establish a critical role for pancreatic G6PC2 in the regulation of FBG, we examined the correlation between the induction of G6pc2 expression during development with the ability of G6PC2 to regulate FBG. G6pc2 expression is markedly induced during the weaning period between days 12 and 21 after birth relative to Slc2a2, which encodes the GLUT2 glucose transporter (Fig. 2A). Ins2 expression shows a modest induction over this same time period (Fig. 2B). G6pc2 expression changes little between day 21 and day 56 (2 months) (Fig. 2A) and remains unchanged even in old mice (17 months) (Fig. 2C). In contrast, expression of G6pc1 (Fig. 2D) and G6pc3 (Fig. 2E) decline markedly in liver and kidney in old mice. G6pc3 expression remains unchanged in other tissues in old mice (Supplemental Figure 1) while Slc37a4 expression is selectively reduced in kidney (Fig. 2). The induction of G6pc2 expression at P21 did not correlate with a reduction in FBG in germline G6pc2 KO relative to WT mice; a reduction was only apparent at P56 (Fig. 2G). Since FBG (Fig. 2G) and especially FPI (Fig. 2H) increase markedly between P21 and P56, we hypothesize that the absence of a difference in FBG between WT and germline G6pc2 KO mice at P21 is simply because the dose response curve for GSIS is at a low point where it is not affected by G6PC2 (Supplemental Figure 2). A trend towards reduced blood glucose levels is also observed in non-fasted G6pc2 KO mice (Fig. 2I), with no change in plasma insulin (Fig. 2J) or glucagon (Fig. 2K). Since the weaning period is associated with the switch to a relatively high carbohydrate diet, we hypothesize that this induction of G6pc2 represents part of the beta cell maturation process (Stolovich-Rain, et al. 2015) and is important for enabling the independent offspring to tightly regulate blood glucose levels.
Figure 2. Analysis of Changes in G6pc Isoform Expression, FBG and FPI with Aging in Male Mice.
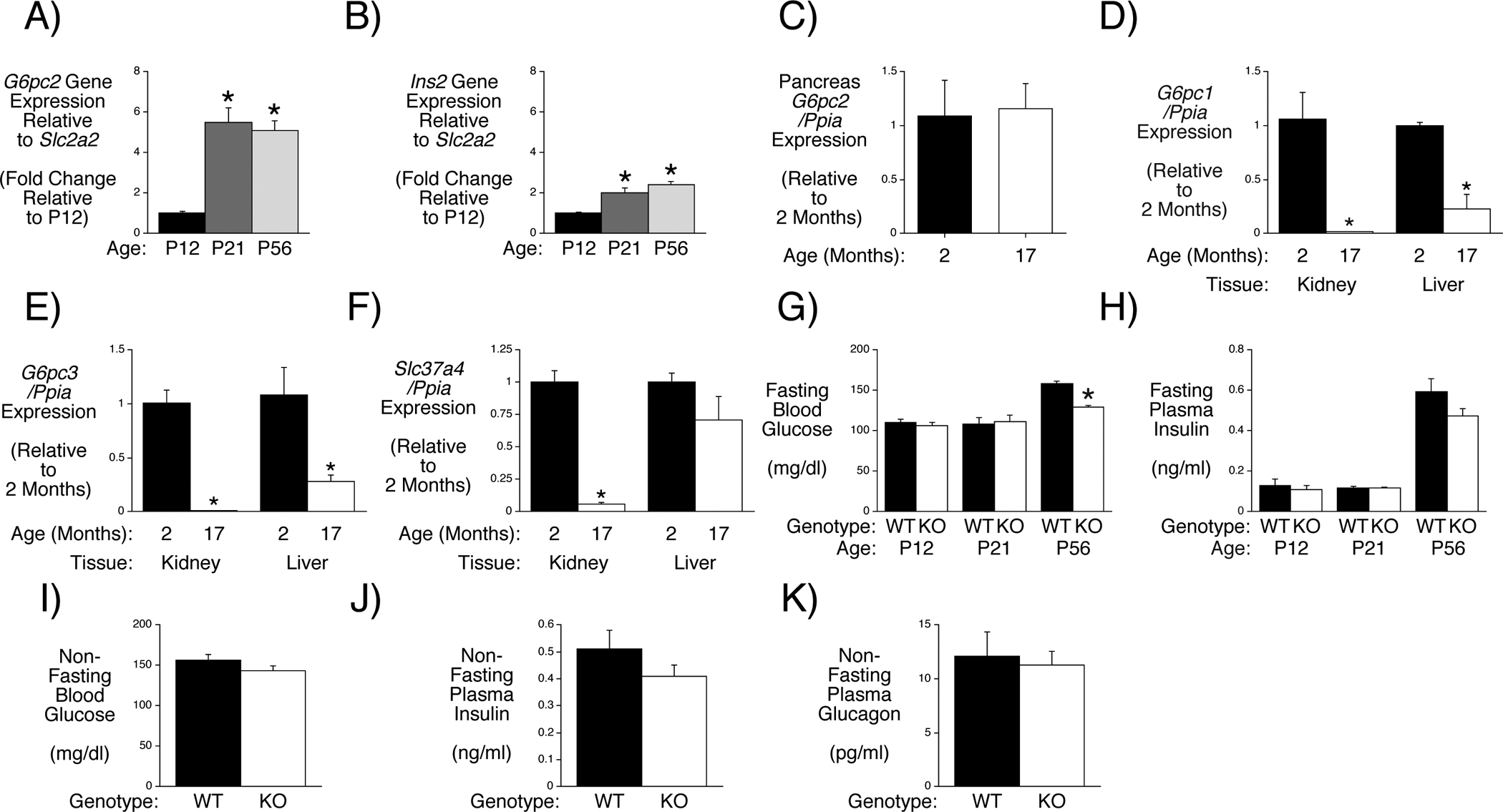
Comparison of pancreatic G6pc2 (Panel A) and Ins2 (Panel B) expression in P12 (non-fasted) and 6 hr fasted P21 and P56 male mice. G6pc2 and Ins2 expression were quantitated relative to Slc2a2 expression and then expressed relative to that at P12. Results represent the mean data ± S.E.M. derived from pancreata isolated from 3–5 mice. *p < 0.05 vs P12, one-way ANOVA. Comparison of G6pc2 (Panel C), G6pc1 (Panel D), G6pc3 (Panel E) and Slc37a4 (Panel F) expression in pancreas, kidney or liver in 2 versus 17 month old non-fasted male mice. G6pc and Slc37a4 expression were quantitated relative to Ppia (cyclophilin A) expression in the indicated tissue and then expressed relative to expression at 2 months. Results represent the mean data ± S.E.M. derived from tissues isolated from 3 male mice. *p < 0.05 vs 2 months. Comparison of FBG (Panel G) and FPI (Panel H) in P12 (non-fasted) and 6 hr fasted P21 and P56 male WT and germline G6pc2 KO mice. FBG data represent the mean ± S.E.M. derived from WT, n=10; KO, n=7 (P12), WT, n=9; KO, n=9 (P21) and WT, n=21; KO, n=24 (P56) mice. FPI data represent the mean ± S.E.M. derived from WT, n=10; KO, n=7 (P12), WT, n=9; KO, n=9 (P21) and WT, n=15; KO, n=23 (P56) mice. *p < 0.05 vs WT. Comparison of glucose (Panel I), insulin (Panel J) and glucagon (Panel K) in 14–16 week old non-fasted WT (n=7) and germline G6pc2 KO (n=7) mice. Data represent the mean ± S.E.M.
FBG is Reduced in Male Beta Cell-Specific (BCS) G6pc2 KO Mice
We reasoned that a second approach for addressing whether G6PC2 is also active in other tissues would be through the generation and analysis of beta cell-specific (BCS) G6pc2 KO mice. If the effect of G6pc2 deletion on FBG were retained it would argue that G6PC2 primarily regulates FBG through an islet-dependent mechanism. BCS G6pc2 KO mice were generated by breeding mice with a floxed G6pc2 allele in which LoxP sites surround G6pc2 exon 3, which encodes the third transmembrane domain (Arden et al. 1999), with MIP1-CreERT2 knock in mice in which expression of the CreERT2 recombinase gene is driven by the endogenous mouse insulin1 promoter (Thorens, et al. 2015). Treatment of these mice with tamoxifen allows for the removal of exon 3 from the floxed G6pc2 allele in beta cells in adult mice thereby inactivating the gene. Sequence analysis indicates that, were splicing of G6pc2 exon 2 to exon 4 to occur, an in-frame G6PC2 variant lacking only exon 3 encoded amino acids would not be generated.
Neither the presence of LoxP sites in the G6pc2 gene nor the presence of the MIP1-CreERT2 gene prior to tamoxifen treatment had major effects on pancreatic G6pc2, Slc37a4 or Ins2 RNA expression in homozygous floxed relative to WT mice (Figs. 3A–F). In contrast, tamoxifen treatment (by three days of IP injections) of homozygous floxed mice expressing the MIP1-CreERT2 gene led to a selective ~60% reduction in pancreatic G6pc2 RNA (Figs. 3G–I) and protein (Figs. 3J & K) expression in floxed mice relative to WT mice, similar to the efficiency of gene deletion reported by Thorens et al. (Thorens et al. 2015) using these MIP1-CreERT2 mice.
Figure 3. Analysis of G6pc2 RNA and G6pc2 protein expression in Floxed G6pc2 Male Mice.
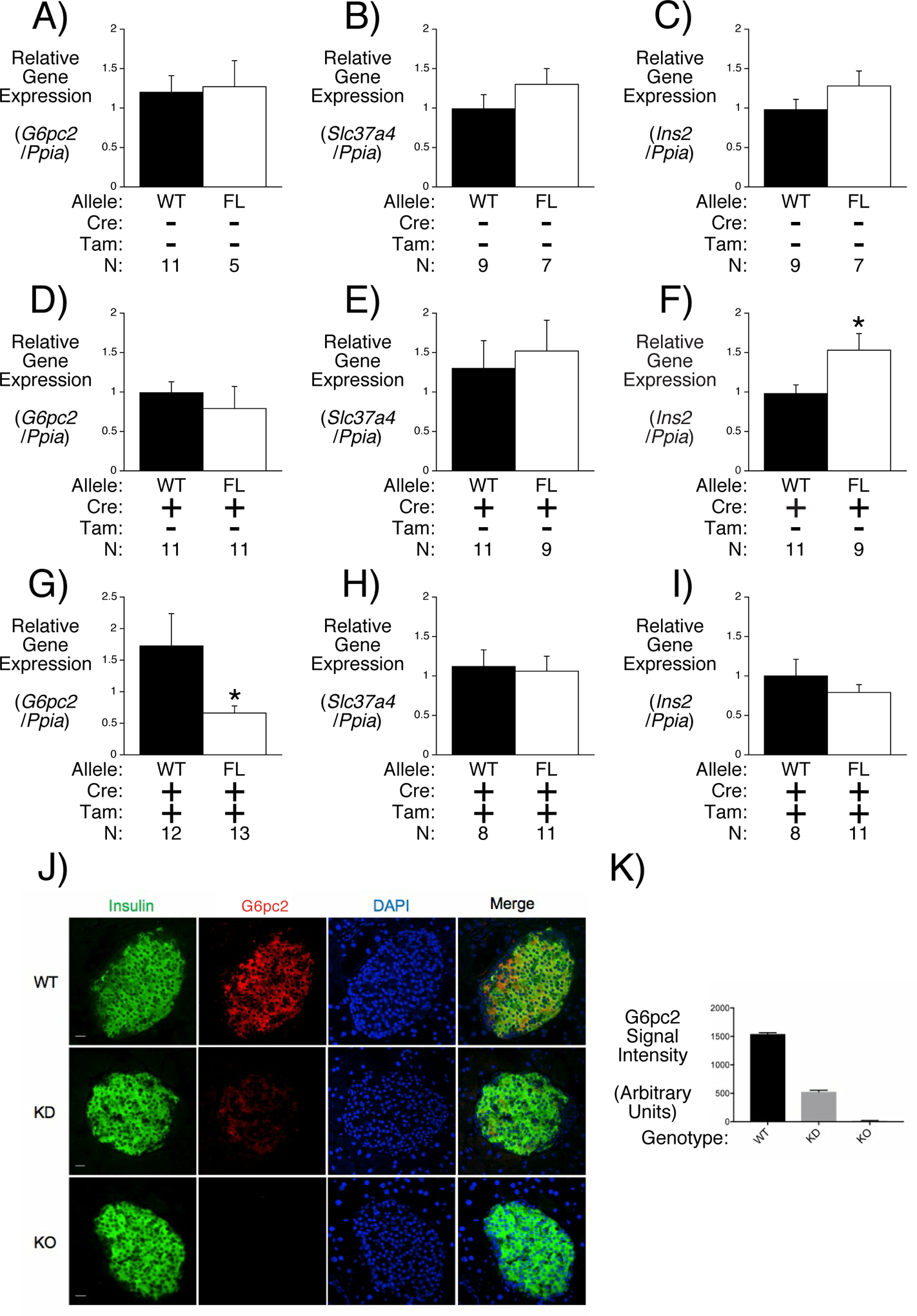
Comparison of pancreatic G6pc2 (Panels A, D & G), Slc37a4 (Panels B, E & H) and Ins2 (Panels C, F & I) expression in 16 week old 6 hr fasted male mice in the presence or absence of the MIP1-CreERT2 allele and in the presence or absence of tamoxifen (Tam) treatment. Pancreatic G6pc2, Ins2 and Slc37a4 expression were quantitated relative to Ppia (cyclophilin A) and then expressed relative to that in WT mice. Results represent the mean data ± S.E.M. derived from tissues isolated from the indicated number of male mice. Comparison of insulin and G6PC2 protein expression in representative sections of pancreas isolated from WT, germline and BCS G6pc2 KO mice (Panel J). Quantitation of G6PC2 protein expression (Panel K). Data represent the mean ± S.E.M. from 100 cells for each group. FL, floxed; KD, knockdown.
Neither the presence of LoxP sites in the G6pc2 gene nor the presence of the MIP1-CreERT2 gene prior to tamoxifen treatment affected FBG and FPI (Figs. 4A–D). In contrast, tamoxifen treatment led to a clear reduction in FBG in floxed mice (Fig. 4E) with no change in FPI (Fig. 4F) indicating that the loss of G6PC2 selectively in beta cells is sufficient to regulate FBG. As expected, the difference in FBG between WT and floxed mice (~15 mg/dl; WT n=14; floxed n=17) (Fig. 4E) is slightly less than that observed in germline G6pc2 KO mice (~21 mg/dl; WT n=29; KO n=16) (Pound et al. 2013; Wang et al. 2007), consistent with the difference between a partial reduction in G6pc2 expression in BCS G6pc2 KO mice compared to a complete loss of G6pc2 expression in germline KO mice. These data also suggest that developmental compensation, a common feature in germline KO mouse studies, has not affected the phenotype of germline G6pc2 KO mice, consistent with the absence of evidence for compensatory changes in pancreatic, islet and liver gene expression comparing WT and G6pc2 KO mice (Supplemental Figure 3).
Figure 4. Analysis FBG and FPI in Floxed G6pc2 Male Mice.
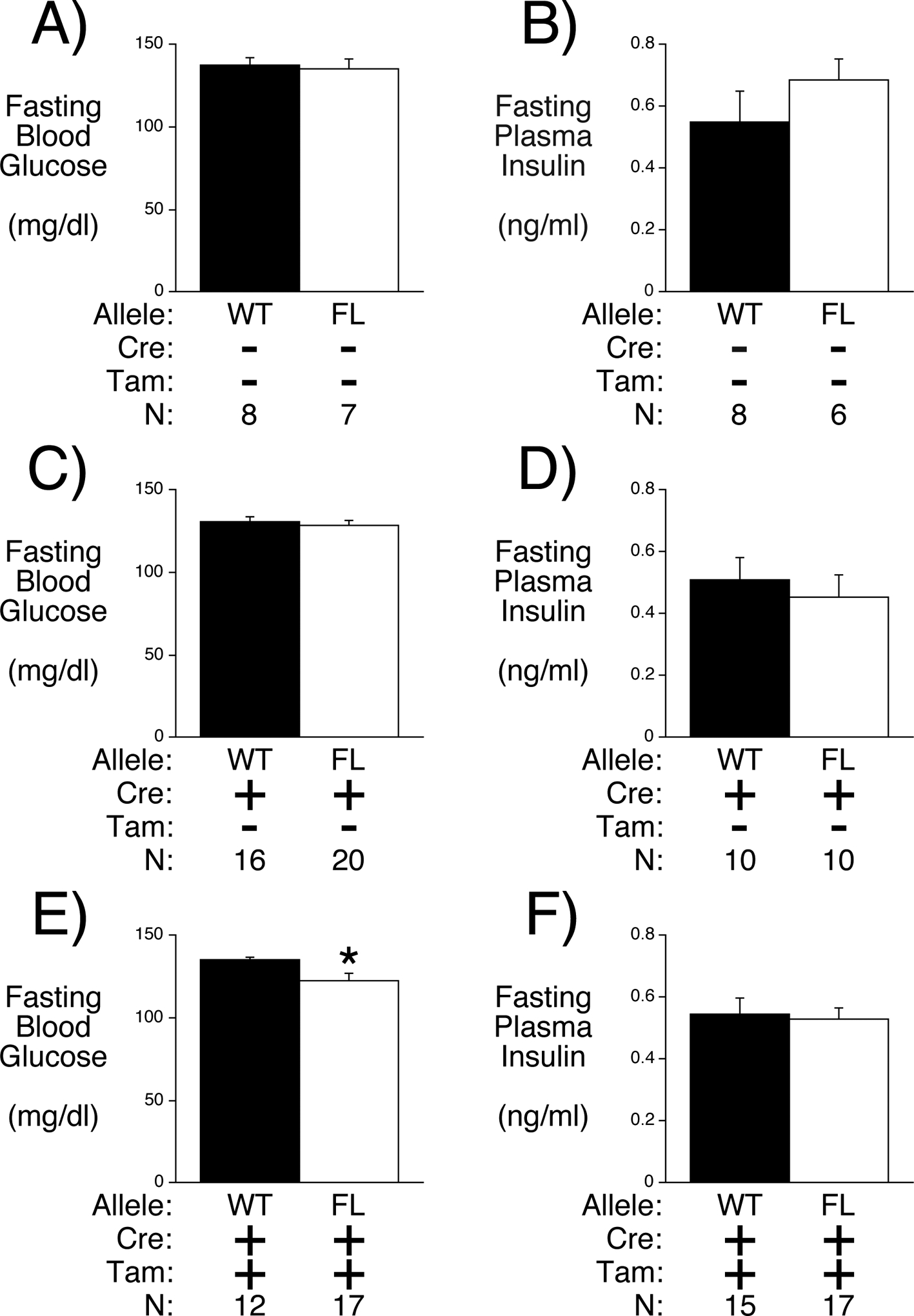
Comparison of FBG (Panels A, C & E) and FPI (Panels B, D & F) in 16 week old 6 hr fasted male mice in the presence or absence of the MIP1-CreERT2 allele and in the presence or absence of tamoxifen (Tam) treatment. Results represent the mean data ± S.E.M. derived from the indicated number of male mice. *p < 0.05 vs WT.
Human Biobank Studies Find No Evidence for Extra-Pancreatic Actions of G6PC2
To complement our mouse studies we searched for evidence for potential extra-pancreatic functions of G6PC2 in humans using Vanderbilt University’s Medical Center (VUMC) BioVU Biobank, a DNA biobank linked to a de-identified version of the Vanderbilt electronic health records, called the Synthetic Derivative (SD) (Pulley et al. 2010; Roden et al. 2008). Systematic and efficient approaches have been developed that involve screening the SD with specific SNPs to identify both novel phenotype-variant associations and plasma hormone/metabolite associations, referred to as PheWAS and LabWAS analyses, respectively (Denny et al. 2010; Denny, et al. 2011; Denny et al. 2013; Ritchie, et al. 2013; Shameer, et al. 2014). For these analyses we used the G6PC2 rs560887 SNP that has been shown to affect G6PC2 RNA splicing (Baerenwald et al. 2013) and has been linked by GWAS to variations in FBG (Bouatia-Naji et al. 2008; Chen et al. 2008) and hemoglobin A1c (HbA1c) (Soranzo, et al. 2010). PheWAS analyses showed that the G6PC2 rs560887 ‘A’ allele is associated with increased risk for acute pancreatitis in non-diabetic European Americans (EAs) (G6PC2 OR = 1.39, CI = 1.20 – 1.62, p = 2.05E-05) after Bonferroni correction for the 1212 phenotypes tested (Table 1). Similar, though less statistically significant, trends were observed in the AA sample and in the total (multi-ancestry) population (Table 1). The association with acute pancreatitis was not observed in EAs with type 2 diabetes (T2D) (Table 1) or in African Americans with T2D (AAs; data not shown). Because G6PC2 is not expressed in pancreatic exocrine or ductal tissue (Hutton and Eisenbarth 2003), this suggests that reduced islet G6PC2 expression can influence non-islet pancreatic tissue function. G6PC2 was not associated with T2D in the total population (Table 1). In contrast, BioVU analyses using the established rs13266634 T2D-associated SNP in the SLC30A8 gene (Yaghootkar and Frayling 2013) did detect an association with T2D (Table 1).
Table 1. Analysis of the Association Between G6PC2 SNP rs560887 with Disease Phenotypes Using Electronic Health Record (EHR)-Derived Analyses.
Data show an association between G6PC2 SNP rs560887 and acute pancreatitis in European Americans (EAs) without T2D and no association with T2D, in contrast to SLC30A8 SNP rs13266634. ‘All’ refers to the total EHR population. ‘Phecodes’ refer to the numerical abbreviation for individual phenotypes in the Vanderbilt EHRs. All associations were adjusted for the covariates median age of record, sex, and EHR-reported race using linear regression. SE, standard error; OR, odds ratio; N_no_SNP, number of records with a missing predictor; FDR, false discovery rate; LCI, lower confidence interval; UCI, upper confidence interval.
| Gene | SNP | Population | Phecode | Description | Beta | SE | OR | P | N_total | N_cases | N_controls | N_no_snp | LCI | UCI | Bonferroni | FDR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G6PC2 | rs560887 | Total | 577.1 | Acute pancreatitis | 0.13 | 0.05 | 1.14 | 0.00709997 | 37343 | 411 | 36932 | 10 | 1.04 | 1.26 | FALSE | FALSE |
| G6PC2 | rs560887 | All EA | 577.1 | Acute pancreatitis | 0.15 | 0.05 | 1.16 | 0.00606234 | 31878 | 339 | 31539 | 6 | 1.04 | 1.28 | FALSE | FALSE |
| G6PC2 | rs560887 | EA Non-T2D | 577.1 | Acute pancreatitis | 0.33 | 0.08 | 1.39 | 2.05E-05 | 21357 | 145 | 21212 | 5 | 1.20 | 1.62 | TRUE | TRUE |
| G6PC2 | rs560887 | EAT2D | 577.1 | Acute pancreatitis | 2.5E-03 | 0.09 | 1.00 | 0.97696475 | 6155 | 140 | 6015 | 0 | 0.85 | 1.19 | FALSE | FALSE |
| G6PC2 | rs560887 | Total | 250.2 | Type 2 diabetes | 0.01 | 0.01 | 1.01 | 0.43847308 | 32759 | 7026 | 25733 | 9 | 0.98 | 1.04 | FALSE | FALSE |
| SLC30A8 | rs13266634 | Total | 250.2 | Type 2 diabetes | −0.06 | 0.01 | 0.94 | 3.56E-05 | 32782 | 7035 | 25747 | 16 | 0.92 | 0.97 | TRUE | TRUE |
LabWAS analyses showed that the G6PC2 rs560887 ‘A’ allele was associated with reduced blood glucose (Effect estimate = −0.05, SE = 0.01, p = 6.56E-24) in the total population. Similar trends were observed in the total EA population and in non-diabetic EAs but not EAs with T2D (Table 2) or in AAs (data not shown). Given the differences in sample sizes we cannot rule out the possibility that the lack of association in these groups is simply related to power. The G6PC2 rs560887 ‘A’ allele was also associated with reduced hemoglobin A1c (HbA1c) but only in non-diabetic EAs (Table 2). These results are consistent with GWAS data (Bouatia-Naji et al. 2008; Chen et al. 2008; Soranzo et al. 2010) and suggest that the influence of G6PC2 on blood glucose and HbA1c is lost under diabetic conditions. Surprisingly, the G6PC2 rs560887 ‘A’ allele was associated with increased taurine levels in the total population (Effect estimate = 0.32, SE = 0.08, p = 7.28E-05) with similar trends in the total EA and non-diabetic EA populations (Table 2). It is well established that insulin regulates amino acid metabolism (Rooyackers and Nair 1997), so the simplest hypothesis is that reduced islet G6PC2 expression influences the kinetics of insulin secretion which then affects amino acid metabolism. Overall these PheWAS and LabWAS results complement the conclusions of the mouse studies by not finding evidence for extra-pancreatic functions of G6PC2.
Table 2. Analysis of the Association Between G6PC2 SNP rs560887 with Laboratory Analytes Using Electronic Health Record (EHR)-Derived Analyses.
Data show an association between G6PC2 SNP rs560887 and blood glucose, hemoglobin A1c (HbA1c) and blood taurine in either the total EHR population (All) or specific European American (EA) sub-populations. No associations were observed in the African American (AA) population or separately in AAs with or without T2D (data not shown). SE, standard error; FDR, false discovery rate. For each laboratory value, we tested for associations between the median of all lab values for an individual against the number of minor alleles for that individual. All associations were adjusted for covariates median age of record, sex, and EHR-reported race using linear regression. The units for all measurements in any given lab were consistent within the lab (e.g. all blood glucose values were reported in mg/dL).
| Gene | SNP | Population | Analyte | Effect Estimate | SE | OR | P | N | LCI | UCI | Bonferroni | FDR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G6PC2 | rs560887 | Total | GLUCOSE BLOOD | −0.05 | 0.01 | 0.95 | 6.56E-24 | 38301 | 0.94 | 0.96 | TRUE | TRUE |
| G6PC2 | rs560887 | Total | HGB A1C GLYCATED | −0.01 | 0.01 | 0.99 | 0.21060335 | 14328 | 0.97 | 1.01 | FALSE | FALSE |
| G6PC2 | rs560887 | Total | TAURINE | 0.32 | 0.08 | 1.38 | 7.28E-05 | 158 | 1.22 | 1.53 | TRUE | TRUE |
| G6PC2 | rs560887 | All EA | GLUCOSE BLOOD | −0.05 | 0.01 | 0.95 | 2.08E-22 | 32686 | 0.94 | 0.96 | TRUE | TRUE |
| G6PC2 | rs560887 | All EA | HGB A1C GLYCATED | −0.01 | 0.01 | 0.99 | 0.15780832 | 12271 | 0.97 | 1.00 | FALSE | FALSE |
| G6PC2 | rs560887 | All EA | TAURINE | 0.27 | 0.08 | 1.31 | 0.00171088 | 132 | 1.15 | 1.47 | FALSE | FALSE |
| G6PC2 | rs560887 | Non T2D EA | GLUCOSE BLOOD | −0.07 | 0.01 | 0.93 | 3.97E-38 | 21728 | 0.92 | 0.94 | TRUE | TRUE |
| G6PC2 | rs560887 | Non T2D EA | HGB A1C GLYCATED | −0.05 | 0.01 | 0.96 | 3.83E-05 | 4353 | 0.93 | 0.98 | TRUE | TRUE |
| G6PC2 | rs560887 | Non T2D EA | TAURINE | 0.29 | 0.09 | 1.34 | 0.00243233 | 114 | 1.16 | 1.53 | FALSE | FALSE |
| G6PC2 | rs560887 | T2D EA | GLUCOSE BLOOD | 0.01 | 0.01 | 1.01 | 0.18306124 | 6444 | 0.99 | 1.04 | FALSE | FALSE |
| G6PC2 | rs560887 | T2D EA | HGB A1C GLYCATED | 0.02 | 0.01 | 1.02 | 0.18513367 | 5159 | 0.99 | 1.04 | FALSE | FALSE |
| G6PC2 | rs560887 | T2D EA | TAURINE | 0.30 | 0.51 | 1.35 | 0.58240028 | 12 | 0.36 | 2.35 | FALSE | FALSE |
To follow up on these observations from human population studies, we examined whether germline deletion of G6pc2 results in pancreatitis in mice. Pancreatic sections from ~8 month old 6 hour fasted mice showed no evidence of pancreatitis, with no necrosis, edema, hemorrhage, steatosis, or fibrosis within the pancreatic parenchyma and no increase in inflammatory infiltrate compared to WT controls. Furthermore, acinar cell morphology appeared normal with no evidence of degranulation, vacuolization, or luminal dilation (Fig. 5A). However, expression of Prss1, which encodes trypsinogen, was elevated in 16 week old 6 hour fasted germline G6pc2 KO mice (Fig. 5B). The expression of Ctrb1 (chymotrypsinogen), Amy2a2 (amylase) and Cpa1 (carboxypeptidase A1) were unchanged (Fig. 5B) as were Ins2, Slc30a8 (ZnT8) and Slc37a4, though Slc2a2 expression was slightly reduced (Fig. 5C). Surprisingly the opposite result was observed in 16 week old 6 hour fasted BCS G6pc2 KO mice in which expression of Prss1 was reduced (Fig. 5D).
Figure 5. Analysis Pancreatitis and Acinar Gene Expression in Germline G6pc2 KO and Floxed G6pc2 Male Mice.
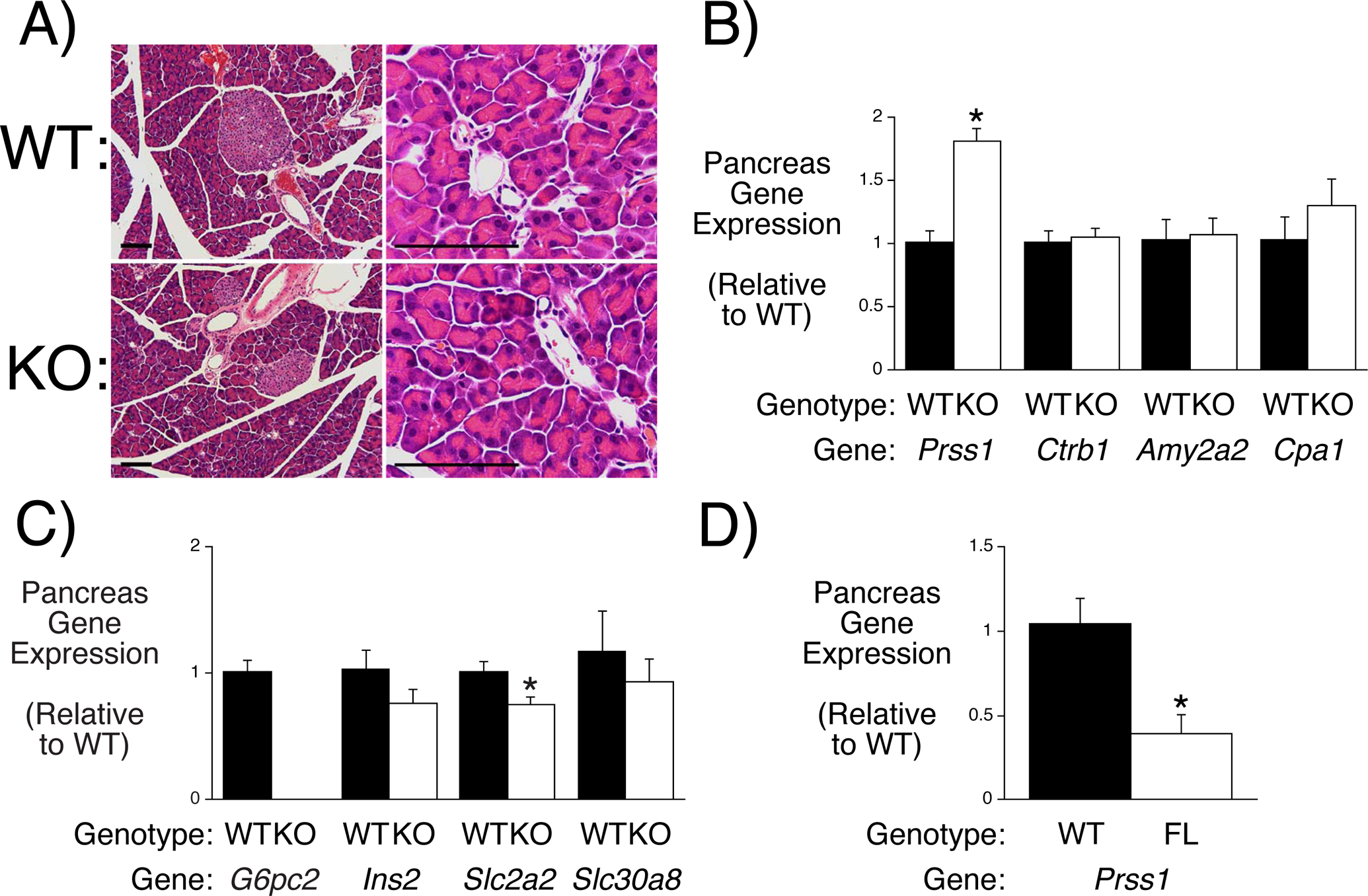
Panel A: Comparison of pancreatic tissue in WT and germline G6pc2 KO mice. Results are representative of sections analyzed from 3 WT and 3 KO mice. Size bars represent 100 μm. Panels B & C: Comparison of pancreatic acinar gene expression in 6 hr fasted 16 week old male WT and germline G6pc2 KO mice. Gene expression was quantitated relative to Ppia (cyclophilin A) and then expressed relative to that in WT mice. Results represent the mean data ± S.E.M. derived from pancreata isolated from 5 mice. *p < 0.05 vs WT. Panel D: Comparison of pancreatic gene expression in 6 hr fasted 16 week old male WT and BCS G6pc2 KO mice following tamoxifen treatment. The RNA samples were a subset of those used in Figs. 5G-I. Gene expression was quantitated relative to Ppia (cyclophilin A) and then expressed relative to that in WT mice. Results represent the mean data ± S.E.M. derived from pancreata isolated from 5 mice. *p < 0.05 vs WT.
Glycolysis is Enhanced in Germline G6pc2 KO Relative to WT Islets
Our model predicts that deletion of G6pc2 will increase glycolytic flux in islets. While glucose cycling analyses can quantitate G6P production (Wall et al. 2015) such experiments do not quantitate the rate of glycolysis. It is important to address this key difference because glycolysis, rather than the rate of G6P production, is the determinant of the magnitude of GSIS (Iynedjian 2009; Matschinsky and Wilson 2019). This distinction arises because G6P has multiple alternate metabolic fates, including conversion to fructose-6-phosphate, glucose-1-phosphate, and inositol-3-phosphate. The well-established glycolytic assay that measures the generation of 3H2O from D-[5-3H]glucose (Ashcroft et al. 1972; Tamarit-Rodriguez et al. 1998; Wang and Iynedjian 1997) was used to compare glycolytic rates in WT and germline G6pc2 KO islets. Consistent with our model, glycolysis was elevated in KO islets at 5.6 mM glucose (Fig. 6).
Figure 6. Comparison of Glycolytic Rates in WT and Germline G6pc2 KO Islets.
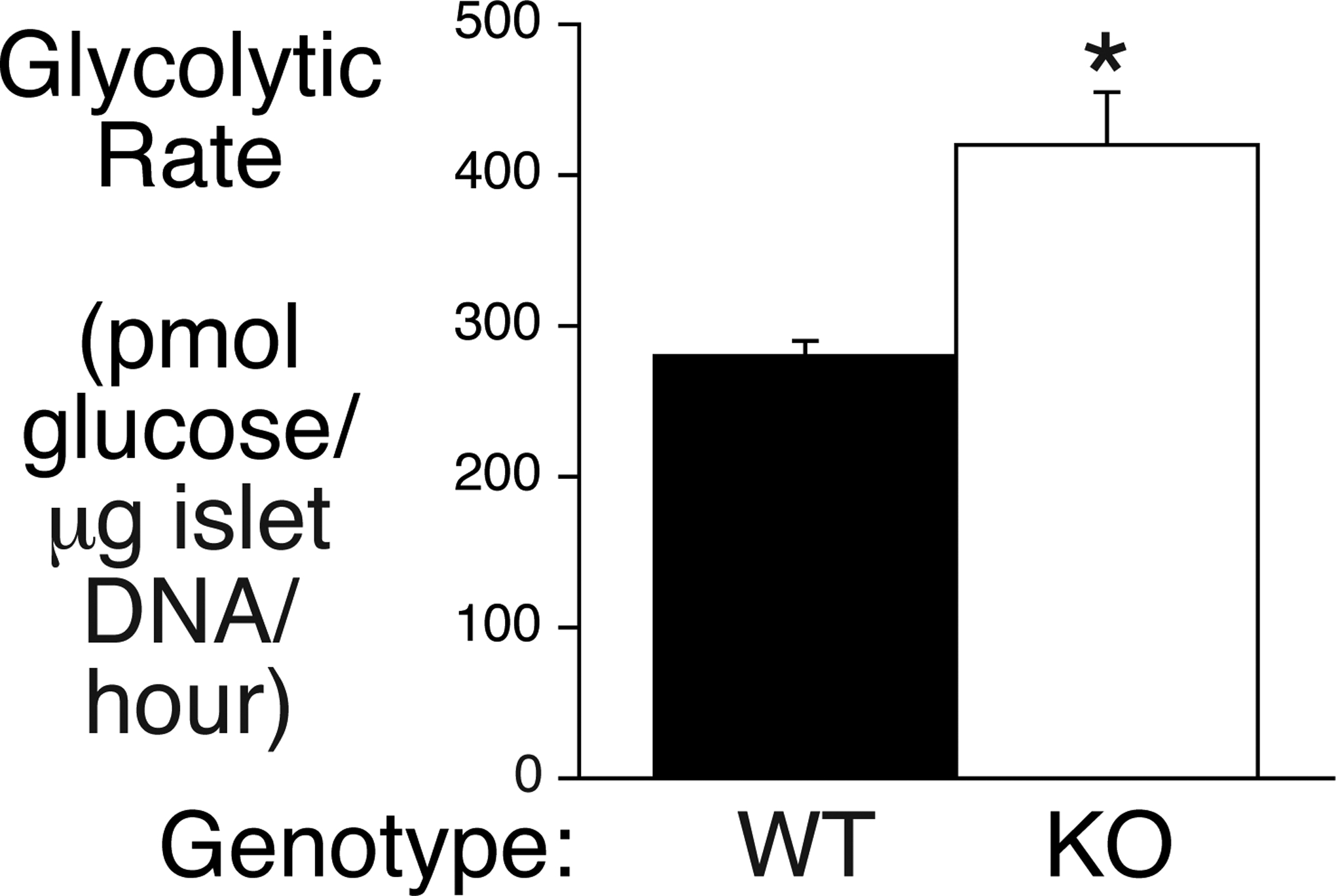
Data show the rates of glycolysis in isolated islets from WT and germline G6pc2 KO mice at 5 mM glucose and represent the mean ± S.E.M. of 5 – 7 islet samples. *p < 0.05 vs WT.
Glucose Cycling Exists in Human Islets
Using a novel stable isotope-based methodology that eliminates assumptions associated with the use of radioisotopes, we have recently shown that mouse islets exhibit significant rates of glucose cycling (Wall et al. 2015). The same methodology was used to address the key question as to whether glucose cycling also occurs in human islets. Kayton et al. (Kayton et al. 2015) have recently used islet perifusion assays to define 5 patterns of GSIS in human islets, designated as Group 1 – 5. Group 1 islets exhibit optimal responses to secretagogues whereas GSIS shows distinct abnormalities in Group 2–5 islets (Kayton et al. 2015). Glucose uptake and cycling were compared at both 5 mM and 11 mM glucose in human islet preparations from four donors that were defined as Group 1 using perifusion assays (Supplemental Figure 4). Significant rates of glucose uptake and glucose cycling were detected and were typically greater at 11 mM than 5 mM though, for reasons that are unclear, this was not uniform and the islet preparation that exhibited decreased glucose uptake at 11 mM was distinct from the preparation that exhibited decreased glucose cycling at 11 mM (Fig. 7). At 5 mM glucose the level of glucose uptake inversely correlated with the level of glucose cycling (Fig. 7). Since the rate of glucose cycling is determined by quantitating the glucose isotopomers generated by cycling that are transported out of the islets and back into the media, one possible interpretation of this observation is that in cells with high glycolytic flux, re-cycled glucose preferentially re-enters the glycolytic pathway rather than leaving the cell, leading to an underestimate of the rate of cycling. Another possible interpretation is that the rate of G6P dephosphorylation is fairly constant despite changes in glucose uptake, so glucose cycling represents a smaller percentage of glucose uptake when the glucose uptake rate is high. The mean rate of glucose cycling is clearly lower in human (Fig. 7) than mouse (Wall et al. 2015) islets. This correlates with higher basal GSIS (Conrad, et al. 2016) and lower G6PC2 expression (Xin, et al. 2016) in human versus mouse islets as well as lower FBG in healthy humans (2019) than C57BL/6J mice (Pound et al. 2013).
Figure 7. Comparison of Glucose Uptake Cycling Rates in Group 1 Human Islets.
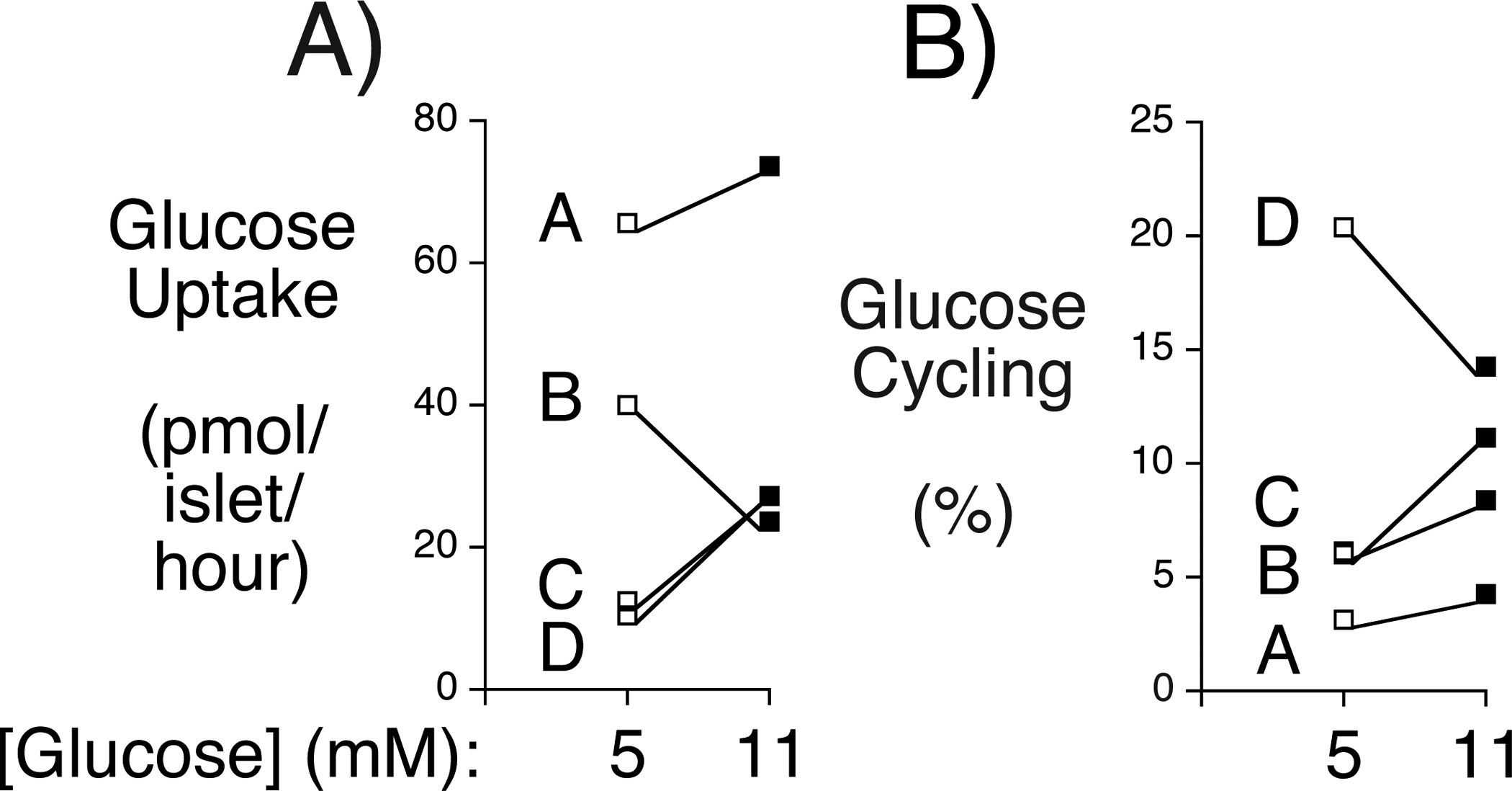
Data show the rates of glucose uptake (Panel A) and cycling (Panel B) in individual preparations of Group 1 islets at either 5 mM or 11 mM glucose from four donors (designated A-D). Information on the individual human donors is presented in Supplemental Table 1 and information of the insulin secretory profile of each islet preparation, as assessed by islet perifusion, is presented in Supplemental Figure 4.
Discussion
The experiments described here provide further support for the concept that G6PC2 activity in pancreatic islets creates a glucokinase/G6PC2 futile cycle that determines the rate of beta cell glycolytic flux and hence the sensitivity of GSIS to glucose rather than this rate being determined by glucokinase alone. Specifically, we show that deletion of G6pc2 enhances glycolysis in mouse islets (Fig. 6) and glucose cycling also exists in human islets (Fig. 7). In addition, gene expression analyses show that G6PC2 is unlikely to be important in non-islet tissues where the more active G6PC isoforms, G6PC1 and G6PC3, are more abundantly expressed (Fig. 1) and that deletion of G6pc2 specifically in beta cells is sufficient to reduce FBG (Figs. 3 & 4). Future studies will be designed to confirm that GSIS is enhanced at sub-maximal glucose in perfused pancreata and isolated islets from BCS G6pc2 KO mice as previously observed in germline G6pc2 KO mice (Pound, et al. 2013).
Several interesting observations arose from the human biobank studies. First, we observed a strong association between G6PC2 and blood glucose levels (Table 2) even though the BioVU population represents individuals who visit VUMC with a wide range of medical issues and who were not all under overnight fasting conditions when blood was isolated. This suggests that G6PC2 can influence blood glucose under non-fasting conditions, consistent with the concept that altered G6PC2 expression affects the sensitivity of GSIS to glucose over a range of glucose concentrations rather than just at fasting glucose levels (Supplemental Fig. 2). Second, the results show that altered G6PC2 expression in humans can affect the risk of acute pancreatitis (Table 1) and that deletion of G6pc2 in mice alters Prss1 expression (Fig. 5B & D). Since altered Prss1 expression may affect the risk of pancreatitis (Boulling, et al. 2016; Le Marechal, et al. 2006) this provides a potential mechanistic link between these observations. Since G6PC2 is expressed selectively in pancreatic islet cells (Hutton and Eisenbarth 2003) the mechanism by which it affects Prss1 expression and especially why the direction of the effect differs with germline and BCS deletion is unclear but will be the subject of future studies. Finally, we observed that G6PC2 was not associated with altered risk for T2D (Table 1), matching GWAS data that have failed to establish a consistent link with T2D risk (Shi, et al. 2017). It will be of considerable interest to identify individuals with mutations that have a major effect on G6PC2 function to determine whether an association with T2D risk then emerges. Elevations in FBG in the prediabetic state correlate with (Brunzell, et al. 1976) and appear sufficient to drive (Brereton, et al. 2014; Wang, et al. 2014) the decline in beta cell function, likely mediated by reactive oxygen species (ROS), that underlies the progression from prediabetes to T2D (Halban, et al. 2014; Weir and Bonner-Weir 2004). However, unless G6PC2 has unknown additional effects on beta cell metabolic flux, a therapy designed to inhibit G6PC2 and thereby lower FBG may fail to prevent this progression because the reduced FBG would be associated with unchanged glycolytic flux and therefore unchanged generation of damaging ROS. Future studies will compare the generation of ROS in WT and KO islets. While a therapy directed at G6PC2 may not affect T2D risk it would likely still be beneficial in preventing cardiovascular-associated mortality (CAM) in individuals with elevated FBG. Thus, in Europeans, an increase in FBG levels from less than 90 mg/dl to between 99–108 mg/dl is associated with 30% increased risk of CAM (Coutinho, et al. 1999), and in Asians a reduction in FBG from 99 to 90 mg/dl is associated with a 25% reduction in the risk of CAM (Lawes, et al. 2004). The risk of CAM increases still further in individuals with the high FBG levels characteristic of diabetes (Coutinho et al. 1999; DECODE 2003; Khaw, et al. 2001; Lawes et al. 2004).
In mice, G6pc2 expression is ~20 fold higher in beta than alpha cells (Xin et al. 2016), G6PC2 protein is undetectable in alpha cells (Hutton and Eisenbarth 2003), and glucagon levels are unchanged in germline G6pc2 KO mice (Wang et al. 2007). The reduction in FBG in germline G6pc2 KO mice with unchanged FPI is therefore solely due to the shift in the sensitivity of GSIS to glucose, consistent with our glycolysis and BCS G6pc2 KO mouse data. In contrast, in humans G6PC2 expression is only ~5 fold higher in beta than alpha cells (Xin et al. 2016) such that altered alpha cell glycolysis could contribute to the reduction in FBG in humans with reduced G6PC2 expression.
In summary, these studies show that islet-specific G6PC2 activity is sufficient to regulate FBG, although the possibility that G6PC2 plays some role in tissue-specific metabolism outside the pancreas cannot be excluded.
Supplementary Material
Acknowledgements
We thank Susan Hajizadeh, Tracy O’Brien and Brooke Olesnevich for performing insulin, glycogen and glucose cycling assays, respectively. We also thank Joshua C. Denny and Huan Mo for their insights on BioVU-related studies. This research was supported by the following grants: R.O’B., DK92589; O.P.M., DK043748 and DK078188; A.C.P., UC4 DK104211, DK106755, DK89572, and by grants from the JDRF and the Department of Veterans Affairs (BX000666); W.H.L. JDRF grant 1-SRA-2018-675-S-B; L.K.D., R56MH120736; J.D.Y DK106348. The isolation of mouse islets by the Vanderbilt Islet Procurement and Analysis Core and the measurement of plasma insulin by the Vanderbilt Hormone Assay & Analytical Services Core were supported by NIH grant P60 DK20593, to the Vanderbilt Diabetes Research Training Center. Human pancreatic islets were provided by the NIDDK-funded Integrated Islet Distribution Program at the City of Hope (NIH Grant # 2UC4 DK098085). K.J.B., S.B.G., M.L.W., and K.E.S. were supported by the Vanderbilt Molecular Endocrinology Training Program grant 5T32 DK07563.
R.O’B. is the guarantor of this work, had full access to all the data, and takes full responsibility for the integrity of data and the accuracy of data analysis.
Footnotes
Conflict of Interest Statement
There is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- 2019. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2019. Diabetes Care 42 S13–S28. [DOI] [PubMed] [Google Scholar]
- Arden SD, Zahn T, Steegers S, Webb S, Bergman B, O’Brien RM & Hutton JC 1999. Molecular cloning of a pancreatic islet-specific glucose-6-phosphatase catalytic subunit-related protein. Diabetes 48 531–542. [DOI] [PubMed] [Google Scholar]
- Ashcroft SJ, Weerasinghe LC, Bassett JM & Randle PJ 1972. The pentose cycle and insulin release in mouse pancreatic islets. Biochem J 126 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenwald DA, Bonnefond A, Bouatia-Naji N, Flemming BP, Umunakwe OC, Oeser JK, Pound LD, Conley NL, Cauchi S, Lobbens S, et al. 2013. Multiple functional polymorphisms in the G6PC2 gene contribute to the association with higher fasting plasma glucose levels. Diabetologia 56 1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouatia-Naji N, Rocheleau G, Van Lommel L, Lemaire K, Schuit F, Cavalcanti-Proenca C, Marchand M, Hartikainen AL, Sovio U, De Graeve F, et al. 2008. A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science 320 1085–1088. [DOI] [PubMed] [Google Scholar]
- Boulling A, Abrantes A, Masson E, Cooper DN, Robaszkiewicz M, Chen JM & Ferec C 2016. Discovery and Functional Annotation of PRSS1 Promoter Variants in Chronic Pancreatitis. Hum Mutat 37 1149–1152. [DOI] [PubMed] [Google Scholar]
- Boustead JN, Martin CC, Oeser JK, Svitek CA, Hunter SI, Hutton JC & O’Brien RM 2004. Identification and characterization of a cDNA and the gene encoding the mouse ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein. J Mol Endocrinol 32 33–53. [DOI] [PubMed] [Google Scholar]
- Brereton MF, Iberl M, Shimomura K, Zhang Q, Adriaenssens AE, Proks P, Spiliotis II, Dace W, Mattis KK, Ramracheya R, et al. 2014. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun 5 4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunzell JD, Robertson RP, Lerner RL, Hazzard WR, Ensinck JW, Bierman EL & Porte D Jr. 1976. Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol Metab 42 222–229. [DOI] [PubMed] [Google Scholar]
- Chen WM, Erdos MR, Jackson AU, Saxena R, Sanna S, Silver KD, Timpson NJ, Hansen T, Orru M, Grazia Piras M, et al. 2008. Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J Clin Invest 118 2620–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY & Mansfield BC 2008. Mutations in the glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen storage disease. Hum Mutat 29 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad E, Dai C, Spaeth J, Guo M, Cyphert HA, Scoville D, Carroll J, Yu WM, Goodrich LV, Harlan DM, et al. 2016. The MAFB transcription factor impacts islet alpha-cell function in rodents and represents a unique signature of primate islet beta-cells. Am J Physiol Endocrinol Metab 310 E91–E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho M, Gerstein HC, Wang Y & Yusuf S 1999. The relationship between glucose and incident cardiovascular events. A metaregression analysis of published data from 20 studies of 95,783 individuals followed for 12.4 years. Diabetes Care 22 233–240. [DOI] [PubMed] [Google Scholar]
- DECODE 2003. Is the current definition for diabetes relevant to mortality risk from all causes and cardiovascular and noncardiovascular diseases? Diabetes Care 26 688–696. [DOI] [PubMed] [Google Scholar]
- Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, Wang D, Masys DR, Roden DM & Crawford DC 2010. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26 1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny JC, Crawford DC, Ritchie MD, Bielinski SJ, Basford MA, Bradford Y, Chai HS, Bastarache L, Zuvich R, Peissig P, et al. 2011. Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome- and phenome-wide studies. Am J Hum Genet 89 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, Field JR, Pulley JM, Ramirez AH, Bowton E, et al. 2013. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 31 1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerin I, Veiga-da-Cunha M, Achouri Y, Collet JF & Van Schaftingen E 1997. Sequence of a putative glucose 6-phosphate translocase, mutated in glycogen storage disease type Ib [see comments]. FEBS Lett 419 235–238. [DOI] [PubMed] [Google Scholar]
- Halban PA, Polonsky KS, Bowden DW, Hawkins MA, Ling C, Mather KJ, Powers AC, Rhodes CJ, Sussel L & Weir GC 2014. beta-Cell Failure in Type 2 Diabetes: Postulated Mechanisms and Prospects for Prevention and Treatment. Diabetes Care 37 1751–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton JC & Eisenbarth GS 2003. A pancreatic beta-cell-specific homolog of glucose-6-phosphatase emerges as a major target of cell-mediated autoimmunity in diabetes. Proc Natl Acad Sci U S A 100 8626–8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton JC & O’Brien RM 2009. The glucose-6-phosphatase catalytic subunit gene family. J Biol Chem 284 29241–29245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iynedjian PB 2009. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 66 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayton NS, Poffenberger G, Henske J, Dai C, Thompson C, Aramandla R, Shostak A, Nicholson W, Brissova M, Bush WS, et al. 2015. Human Islet Preparations Distributed for Research Exhibit a Variety of Insulin Secretory Profiles. Am J Physiol Endocrinol Metab 308 E592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaw KT, Wareham N, Luben R, Bingham S, Oakes S, Welch A & Day N 2001. Glycated haemoglobin, diabetes, and mortality in men in Norfolk cohort of european prospective investigation of cancer and nutrition (EPIC-Norfolk). Bmj 322 15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku GM, Kim H, Vaughn IW, Hangauer MJ, Myung Oh C, German MS & McManus MT 2012. Research resource: RNA-Seq reveals unique features of the pancreatic beta-cell transcriptome. Mol Endocrinol 26 1783–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawes CM, Parag V, Bennett DA, Suh I, Lam TH, Whitlock G, Barzi F & Woodward M 2004. Blood glucose and risk of cardiovascular disease in the Asia Pacific region. Diabetes Care 27 2836–2842. [DOI] [PubMed] [Google Scholar]
- Le Marechal C, Masson E, Chen JM, Morel F, Ruszniewski P, Levy P & Ferec C 2006. Hereditary pancreatitis caused by triplication of the trypsinogen locus. Nat Genet 38 1372–1374. [DOI] [PubMed] [Google Scholar]
- Lei KJ, Shelly LL, Pan CJ, Sidbury JB & Chou JY 1993. Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a. Science 262 580–583. [DOI] [PubMed] [Google Scholar]
- Livak KJ & Schmittgen TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 402–408. [DOI] [PubMed] [Google Scholar]
- Martin CC, Bischof LJ, Bergman B, Hornbuckle LA, Hilliker C, Frigeri C, Wahl D, Svitek CA, Wong R, Goldman JK, et al. 2001. Cloning and Characterization of the Human and Rat Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein (IGRP) Genes. J Biol Chem 276 25197–25207. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM & Wilson DF 2019. The Central Role of Glucokinase in Glucose Homeostasis: A Perspective 50 Years After Demonstrating the Presence of the Enzyme in Islets of Langerhans. Front Physiol 10 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CR & Lazarow AL 1963. Immunoassay of insulin: two antibody system: plasma insulin of normal, subdiabetic, and diabetic rats. Am J Med Sci 257 415–419. [Google Scholar]
- Pan CJ, Chen SY, Jun HS, Lin SR, Mansfield BC & Chou JY 2011. SLC37A1 and SLC37A2 are phosphate-linked, glucose-6-phosphate antiporters. PLoS One 6 e23157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrolonis AJ, Yang Q, Tummino PJ, Fish SM, Prack AE, Jain S, Parsons TF, Li P, Dales NA, Ge L, et al. 2004. Enzymatic characterization of the pancreatic islet-specific glucose-6-phosphatase-related protein (IGRP). J Biol Chem 279 13976–13983. [DOI] [PubMed] [Google Scholar]
- Pound LD, Oeser JK, O’Brien TP, Wang Y, Faulman CJ, Dadi PK, Jacobson DA, Hutton JC, McGuinness OP, Shiota M, et al. 2013. G6PC2: A Negative Regulator of Basal Glucose-Stimulated Insulin Secretion. Diabetes 62 1547–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulley J, Clayton E, Bernard GR, Roden DM & Masys DR 2010. Principles of human subjects protections applied in an opt-out, de-identified biobank. Clin Transl Sci 3 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie MD, Denny JC, Zuvich RL, Crawford DC, Schildcrout JS, Bastarache L, Ramirez AH, Mosley JD, Pulley JM, Basford MA, et al. 2013. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation 127 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR & Masys DR 2008. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 84 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooyackers OE & Nair KS 1997. Hormonal regulation of human muscle protein metabolism. Annu Rev Nutr 17 457–485. [DOI] [PubMed] [Google Scholar]
- Shameer K, Denny JC, Ding K, Jouni H, Crosslin DR, de Andrade M, Chute CG, Peissig P, Pacheco JA, Li R, et al. 2014. A genome- and phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Hum Genet 133 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly LL, Lei KJ, Pan CJ, Sakata SF, Ruppert S, Schutz G & Chou JY 1993. Isolation of the gene for murine glucose-6-phosphatase, the enzyme deficient in glycogen storage disease type 1A. J Biol Chem 268 21482–21485. [PubMed] [Google Scholar]
- Shi Y, Li Y, Wang J, Wang C, Fan J, Zhao J, Yin L, Liu X, Zhang D & Li L 2017. Meta-analyses of the association of G6PC2 allele variants with elevated fasting glucose and type 2 diabetes. PLoS One 12 e0181232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh JJ, Pan CJ, Mansfield BC & Chou JY 2003. A glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. J Biol Chem 278 47098–47103. [DOI] [PubMed] [Google Scholar]
- Soranzo N, Sanna S, Wheeler E, Gieger C, Radke D, Dupuis J, Bouatia-Naji N, Langenberg C, Prokopenko I, Stolerman E, et al. 2010. Common variants at ten genomic loci influence hemoglobin A1C levels via glycemic and non-glycemic pathways. Diabetes 59 3229–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B & Dor Y 2015. Weaning triggers a maturation step of pancreatic beta cells. Dev Cell 32 535–545. [DOI] [PubMed] [Google Scholar]
- Syring KE, Boortz KA, Oeser JK, Ustione A, Platt KA, Shadoan MK, McGuinness OP, Piston DW, Powell DR & O’Brien RM 2016. Combined Deletion of Slc30a7 and Slc30a8 Unmasks a Critical Role for ZnT8 in Glucose-Stimulated Insulin Secretion. Endocrinology 157 4534–4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamarit-Rodriguez J, Idahl LA, Gine E, Alcazar O & Sehlin J 1998. Lactate production in pancreatic islets. Diabetes 47 1219–1223. [DOI] [PubMed] [Google Scholar]
- Thorens B, Tarussio D, Maestro MA, Rovira M, Heikkila E & Ferrer J 2015. Ins1(Cre) knock-in mice for beta cell-specific gene recombination. Diabetologia 58 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-da-Cunha M, Chevalier N, Stephenne X, Defour JP, Paczia N, Ferster A, Achouri Y, Dewulf JP, Linster CL, Bommer GT, et al. 2019. Failure to eliminate a phosphorylated glucose analog leads to neutropenia in patients with G6PT and G6PC3 deficiency. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall ML, Pound LD, Trenary I, O’Brien RM & Young JD 2015. Novel Stable Isotope Analyses Demonstrate Significant Rates of Glucose Cycling In Mouse Pancreatic Islets. Diabetes 64 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H & Iynedjian PB 1997. Modulation of glucose responsiveness of insulinoma beta-cells by graded overexpression of glucokinase. Proc Natl Acad Sci U S A 94 4372–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Martin CC, Oeser JK, Sarkar S, McGuinness OP, Hutton JC & O’Brien RM 2007. Deletion of the Gene Encoding the Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein Autoantigen Results in a Mild Metabolic Phenotype. Diabetologia 50 774–778. [DOI] [PubMed] [Google Scholar]
- Wang Z, York NW, Nichols CG & Remedi MS 2014. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab 19 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC & Bonner-Weir S 2004. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 53 Suppl 3 S16–21. [DOI] [PubMed] [Google Scholar]
- Xin Y, Kim J, Okamoto H, Ni M, Wei Y, Adler C, Murphy AJ, Yancopoulos GD, Lin C & Gromada J 2016. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab 24 608–615. [DOI] [PubMed] [Google Scholar]
- Yaghootkar H & Frayling TM 2013. Recent progress in the use of genetics to understand links between type 2 diabetes and related metabolic traits. Genome Biol 14 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.