

Abstract
The microbiome is an assemblage of microorganisms living in association with a multicellular host. Numerous studies have identified a role for the microbiome in host physiology, development, immunity, and behaviour. The generation of axenic (germ-free) and gnotobiotic model systems has been vital to dissecting the role of the microbiome in host biology. We have previously reported the generation of axenic Aedes aegypti mosquitoes, the primary vector of several human pathogenic viruses, including dengue virus and Zika virus. In order to better understand the influence of the microbiome on mosquitoes, we examined the transcriptomes of axenic and conventionally reared Ae. aegypti before and after a blood meal. Our results suggest that the microbiome has a much lower effect on the mosquito’s gene expression than previously thought with only 170 genes influenced by the axenic state, while in contrast, blood meal status influenced 809 genes. The pattern of expression influenced by the microbiome is consistent with transient changes similar to infection rather than sweeping physiological changes. While the microbiome does seem to affect some pathways such as immune function and metabolism, our data suggest the microbiome is primarily serving a nutritional role in development with only minor effects in the adult.
Subject terms: Transcriptomics, Microbiome
Introduction
There is an increasing recognition that most multicellular organisms harbour microbiota, or a microbiome, that can affect their development, biology, and health. This idea has led to the concept of a “holobiont”, which suggests that the biology of an organism cannot be separated from the mutualistic, commensal, or pathogenic organisms that may stably or transiently exist with that organism1,2. Thus, there is an “extended phenotype” for the host that arises from the interactions with its microbiome3. Consequently, it can be difficult differentiating the contributions of either the host or its associated microbiome to specific phenotypic outcomes. One of the tools that can be employed to examine the microbiome’s effect on host biology is the availability of microbial free, or axenic hosts4,5. Numerous groups have developed axenic systems to study host-microbiome interactions, including Mus musculus (mice)6, Sus scrofa (pigs)7, Caenorhabditis elegans8, fruit fly (Drosophila melanogaster)9 and Gallus gallus (chickens)10. By combining these axenic systems with transcriptomics, metabolomics and epigenomics, researchers can now systematically examine the role of the microbiome in host biology4,11–14.
As with most complex organisms, mosquitoes possess a taxonomically diverse assemblage of microbes, including bacteria, viruses, fungi and protists15,16. While the mosquito microbiome can inhabit multiple tissues within the mosquito, including the germline, malpighian tubules and salivary glands, the majority of microbes reside in the gut17–19. Previous studies have found that, compared to vertebrate hosts, the mosquito microbiome is composed of relatively few community members (~ 10–70 Operational Taxonomic Units (OTUs)) which are primarily acquired from the aquatic environment as larvae16,20–22. As the aquatic environment has a large influence on the mosquito microbiome, resource use by the mosquitoes at the larval stage is likely to influence the adult core microbiome23. The composition of the mosquito microbiome reflects the habitat of the individual with the microbiome varying between sites and individuals from different species at the same location sharing similar microbiomes24. Microbiome composition transitions between the larval and adult stages; this shift reflects not only a change in their environment but also a change in their diet, with blood feeding mosquitoes possessing a different core microbiome to their sugar feeding counterparts. Blood meal acquisition results in a decrease in the number of OTUs but an increase in abundance of particular bacterial genera (Chryseobacterium and Delftia in lab reared Ae. aegypti)24.
Earlier studies reported that larvae failed to develop under sterile conditions and concluded that the microbiome played an indispensable role during larval development. This was thought to occur through a microbiome-induced hypoxic state, which triggered larval molting and growth25. However, the generation of axenic mosquitoes revealed that, with the proper nutritional supplementation, the presence of a microbiome was not essential for development, challenging this notion26,27. Transcriptomic analysis of axenic larvae revealed an enrichment of genes associated with energy metabolism similar to axenic C. elegans and indicative of dietary restrictions or starvation28,29. These findings are consistent with the delayed development time observed while rearing axenic larvae26. Together, these data suggest that the microbiome primarily serves a nutritional role during larval development26,27. In adult mosquitoes, numerous studies have observed potential effects of the microbiome on pathogen acquisition and transmission30, as well as mosquito sensitivity to insecticides31. One major example of this is the endosymbiont Wolbachia which has been shown to affect viral transmission by mosquitoes, it has also been shown that other members of the microbiome can influence vertical transmission of Wolbachia32. The capacity of the adult mosquito to transmit infections to humans could be directly influenced by their microbiome at multiple life stages23. However, these phenotypes are difficult to verify without an axenic model to systematically test the effects of manipulating the microbiome. Thus, the effects of the microbiome on adult mosquito physiology and behaviour are still largely unknown.
Because mosquitoes represent a sustained and significant public health threat33, characterising the precise role of the microbiome on mosquito physiology, development, nutrition, behaviour, and vector competency could aid the development of novel control strategies. The recent development of an axenic mosquito model now allows us to characterise microbiome–host interactions and the effects of removing the native microbiota. In order to address this, we performed transcriptomic analyses on conventionally reared (mosquitoes that possessed a native microbiota) and axenic adult mosquitoes before and after a bloodmeal to look at the effect of the microbiome on host gene expression. We found the total number of differentially expressed genes influenced by the microbiome is more consistent with transient changes akin to infection rather than sweeping physiological changes such as a those associated with bloodmeal.
Results
Transcript assembly and differential expression analysis
We used Illumina NextSeq 550 sequencing to transcriptionally profile conventional and axenic adult mosquitoes. Sterility of axenic larvae and adults were tested via culturing of viable bacteria and 16S rRNA gene PCR as previously described26. Half of the mosquitoes in each group were provided with a non-infectious bloodmeal and tissues harvested 24 h post blood meal (hpbm). The remaining half were maintained on sugar water. In order to provide more tissue level resolution, midguts and carcasses were processed and analysed separately. Three biological replicates for each treatment group and two tissue sources (midguts and carcasses) resulted in a total of 24 libraries being analysed. Across the libraries, an average of 41.3 million forward and reverse reads (range: 30.0–51.8) per sample were recovered, and this was reduced to an average of 21.2 million paired reads (range: 14.7–25.3) after quality filtering (Table S1). This filtering resulted in a total of 29.3 to 50.6 million quality-filtered reads per treatment, of which an average of 88.49% (range: 86.6–90.7%) mapped to the current assembly of the Ae. aegypti genome (AaegL5.0 https://www.vectorbase.org/organisms/aedes-aegypti/lvp_agwg/aaegl5) (Table S1).
Using canonical analysis of principal coordinates (CAP), the data were analysed to identify significant scale differences within the sequence datasets (Fig. 1). It was thought that CAP would confirm that the axenic state would have a significant effect on transcript abundance. However, the first component, which explained 65.9% of the variation in the data (p-value = 0.001), separated the samples by blood-fed status (Fig. 1). The second component, which explained 15.3% of the variation in the data (p-value = 0.001), separated samples by tissue type (Fig. 1). Not only did the microbial status of the mosquito explain much less of the variation between individuals (Fig. 1) but that variation was also not statistically significant (p-value = 0.077). Therefore, due to the low explanatory power in the axenic state, the decision was made to separate the data by individual treatments [midgut blood-fed (MBF), midgut sugar-fed (MSF), carcass blood-fed (CBF), carcass sugar-fed (CSF)] and to make comparison without the confounding factors of diet and tissue type.
Figure 1.
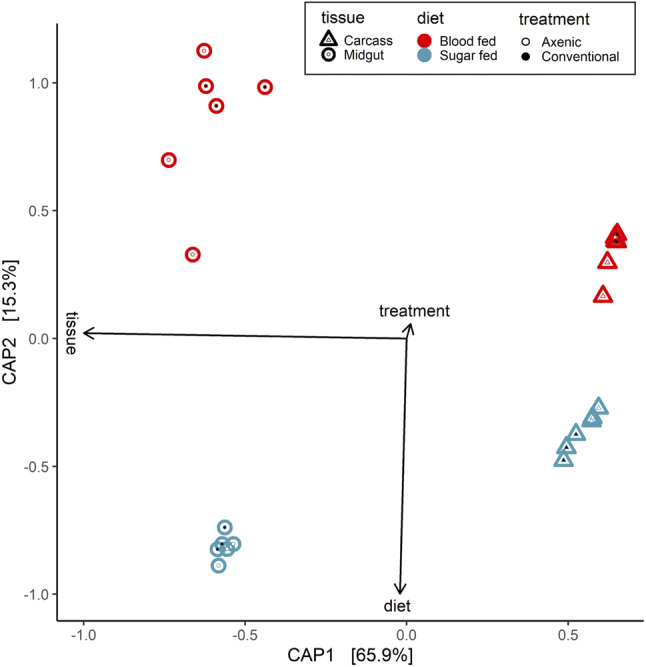
Canonical Analysis of Principal Coordinates (CAP) analysis of transcript abundance by mosquito pool. Constrained Analysis of Principal Coordinates (CAP) ordination for the transcriptomes of the mosquito pools sequenced, based on Jaccard dissimilarity index. CAP axes 1 and 2 explain 65.9 and 15.3% of the variance, respectively. Explanatory variables are indicated by arrows, while diet is indicated by the colours, tissue type by the shape and treatment by the internal dot colour.
We employed two different software packages to identify differentially expressed (DE) transcripts, DESeq234 and Sleuth35. Additionally, we applied an effect size threshold (twofold change between conditions) to focus on differences that were most likely to be biologically meaningful. DESeq2 consistently identified more DE transcripts than Sleuth (Table 1). The DESeq2 analyses identified 1,165 DE transcripts whose abundance varied significantly (adjusted p-value < 0.05) in the presence of the microbiome by at least a twofold relative to axenic mosquitoes (Table 1). The Sleuth analyses identified 181 DE transcripts whose abundance varied significantly (qval < 0.05) in the presence of the microbiome by at least a twofold change relative to axenic mosquitoes (Table 1). The overlap in DE transcripts found by both programs only accounted for on average 74% (range: 23.1–97.4%) of the total number of transcripts identified by Sleuth. Thus, Sleuth appeared to be more conservative in identifying DE transcripts and primarily overlapped with the DESeq2 data. Therefore, the Sleuth results were used for further analyses.
Table 1.
Total transcriptome abundance from both DESeq2 and Sleuth.
| MBF | MSF | CBF | CSF | Total | |
|---|---|---|---|---|---|
| DESeq2 | 537 | 189 | 261 | 292 | 1,165 |
| Sleuth | 96 | 31 | 39 | 39 | 181 |
| Shared | 75 | 30 | 38 | 9 | 152 |
| Unique DESeq2 | 462 | 159 | 223 | 283 | 1,013 |
| Unique Sleuth | 21 | 1 | 1 | 30 | 29 |
Transcriptional response to the axenic state
The DE transcripts were visualised by treatment type in MA plots (Bland–Altman plot) (Fig. 2). In all treatment groups, more transcripts were depleted in the axenic state; for the CBF, CSF and MSF treatment groups there was very little difference between the number of DE transcripts either enriched or depleted in the axenic state (Fig. 2). However, there were large differences between the number of enriched and depleted transcripts for the MBF treatment group, with 58 more transcripts showing a decreased abundance in the axenic state. The majority (89.50%) of the DE transcripts were unique to one of the four comparisons (Fig. 3), regardless of whether they were enriched or depleted in the axenic state. Only 15 (8.30%) of the DE transcripts were shared between two comparisons, four (2.21%) of the DE transcripts were shared between three comparisons and only one (0.55%) DE transcript (nesprin-1-like, AAEL025410-RA) was shared between all four comparisons. Additionally, one transcript (UDP-glucuronosyltransferase, AAEL021590 –RA) was enriched in one state (MBF) and depleted in another (MSF) (Fig. 3).
Figure 2.

MA plots of axenic versus conventionally reared mosquito genes. The beta value in Sleuth indicates the estimated fold change for each gene. Each dot is representative of one gene; grey dots represent no significant differences between axenic and conventionally raised groups. Red dots represent genes that were either enriched (above the x-axis) or depleted (below the x-axis) and were considered biologically significant in abundance. The four treatment groups investigated were midguts blood-fed (MBF) (a), midguts sugar-fed (MSF) (b), carcasses blood-fed (CBF) (c) and carcasses sugar-fed (CSF) (d).
Figure 3.

Upset plot of biologically significant genes expressed in the four treatment groups investigated. The plot shows the number of shared genes between the four treatment groups. The set size is the total number of biologically significant genes identified. The interaction size shows how many genes are found either only in one treatment group or in multiple treatment groups. The four treatment groups investigated were midguts blood-fed (MBF), midguts sugar-fed (MSF), carcasses blood-fed (CBF), and carcasses sugar-fed (CSF).
Annotation of differentially expressed transcripts
The DE transcripts were annotated using both NCBI and VectorBase. Due to splice variants, 170 genes were identified in the dataset (compared to 181 transcripts), 34 of which were uncharacterised to a functional annotation (Table S2). Using VectorBase36, the global classification of gene ontology (GO) terms were extracted for the DE transcripts (Table S2). There were more GO annotations than genes with many genes having multiple GO annotations. However, 47 genes had no known GO annotation. All GO annotations identified could be assigned to one of the three main domains; biological process (30.43%), cellular component (29.89%), and molecular function (39.67%). Analysis of the Level 2 GO annotations revealed that of the 14 identified, only three were unique to a single treatment type (Fig. 4). Developmental process was unique to CSF, and immune system and reproduction processes were both unique to MBF. Four annotations, response to stimulus, metabolic process, binding and catalytic activity were found in all four treatment groups. However, as with the genes, at the deepest level, a large proportion of the GO annotations were unique to each treatment (Fig. S1). However, there were more GO annotations shared between treatments with eight GO annotations shared across all four treatment groups compared to the single gene that was shared between all groups. Splitting the GO annotations into those enriched versus those that were depleted in the axenic state, more GO annotations in total were depleted with only two (protein binding and metal ion binding) being conserved among all four treatments (Fig. S1). Of the transcripts enriched in the axenic state, a single GO annotation (protein binding) was common to the four treatments. Taken together, these observations suggest that there was not a strong conserved biological response to the axenic state that was common to the different treatments.
Figure 4.

Hierarchical classification of the biologically significant GO terms identified. The treemap shows the comparative abundance of Level 2 GO terms by treatment type, midguts blood-fed (MBF) (a), midguts sugar-fed (MSF) (b), carcasses blood-fed (CBF) (c) and carcasses sugar-fed (CSF) (d).
Comparison of differential expression in axenic Aedes aegypti and Drosophila melanogaster
Using orthologues from FlyBase37 and VectorBase36 DE transcripts from axenic Ae. aegypti larvae29 and axenic Drosophila melanogaster4 were compared to those identified in this study. In total, four transcripts identified as differentially expressed due to the axenic state from a study into axenic D. melanogaster (Table 2) were also found in this study. All of the genes (defensin-A, defensin-A-like, defensin-C, lysozyme-like) are involved in immune function. Sixteen transcripts identified as differentially expressed due to the axenic state in the larvae were also identified in this study (Table 2). A quarter of the genes shared between adult, and larval axenic mosquitoes were also uncharacterised, and all but two of the remaining genes (cystathionine beta-synthase, phosphoenolpyruvate carboxykinase, purine nucleoside phosphorylase, transferrin, ATP-binding cassette sub-family A member 3, voltage-dependent calcium channel type A subunit 12, phospholipase B1, membrane-associated 1, phospholipase B1, membrane-associated 2, phosphotriesterase-related protein) were involved in nutrient metabolism, catalysis or transport. The remaining two genes were involved in cell death and inflammation (flocculation protein FLO11) toxin receptors (membrane-bound alkaline phosphatase), and wound healing (rho guanine nucleotide exchange factor 11 2).
Table 2.
Mosquito transcript ID and gene name of transcripts that were shared between axenically raised Ae. aegypti and Drosophila melanogaster.
| Transcript | Name |
|---|---|
| Larvae/adult mosquito | |
| XM_021848997.1 | Membrane-bound alkaline phosphatase |
| XM_001654437.2 | Phospholipase B1, membrane-associated X1 |
| XM_021843337.1 | Phospholipase B1, membrane-associated X2 |
| XM_021848596.1 | Phosphotriesterase-related protein |
| XM_011494827.2 | Flocculation protein FLO11 |
| XM_021853819.1 | Rho guanine nucleotide exchange factor 11 X2 |
| XM_021850833.1 | Cystathionine beta-synthase |
| XM_001647886.2 | Phosphoenolpyruvate carboxykinase |
| XM_001661117.2 | Purine nucleoside phosphorylase |
| XM_001647669.2 | Transferrin |
| XM_021841557.1 | ATP-binding cassette sub-family A member 3 |
| XM_021854675.1 | Voltage-dependent calcium channel type A subunit X12 |
| XM_001662723.2 | Uncharacterised |
| XM_001662962.2 | Uncharacterised |
| XM_001664030.2 | Uncharacterised |
| XM_021842633.1 | Uncharacterised |
| Fruit fly/adult mosquito | |
| XM_001657243.3 | Defensin-A |
| XM_001657239.3 | Defensin-A-like |
| XM_001657238.3 | Defensin-C |
| XM_021843602.1 | Lysozyme-like |
Validation of results
Quantitative PCR (qPCR) was used to validate the RNA-seq results; six genes were chosen based on their abundance profile in the dataset and the ability to design qPCR primers, specifically targeting the relevant transcripts. qPCR was performed on individual mosquitoes rather than pooled mosquitoes, producing a more robust picture of gene expression at an individual level. The genes were only tested on the conditions (i.e. tissue types and blood feed status) that showed significantly different abundance in the sequence-based analysis. Of the seven combinations tested, four comparisons came back statistically significant (Fig. 5). Thus, the qPCR data verifying transcriptional differences due to the lack of microbiome are limited.
Figure 5.

The relative expression of the six genes used to validate transcriptome results. Stars indicate statistical significance; *** < 0.0001 and * < 0.01. Gene codes used are all accepted standard abbreviations and are found in Table 2.
Finally, to confirm that the low number of transcripts discovered was not an artefact of our analysis pipeline, treatment groups were also processed via blood meal status. The two groups were separated by colonisation status or feeding status, and only the midguts were examined. As expected, there was a much larger difference in transcript expression between blood meal status than how the mosquitoes were raised (Fig. 6). There was also a much higher number of DE transcripts with 988 DE transcripts (Fig. 6). The low effect of the axenic gene transcription is further supported in that 54% of the DE transcripts were identical between the axenic and conventionally reared blood-fed groups.
Figure 6.

MA plots of significantly expressed mosquito genes during a blood or sugar meal. The beta value in Sleuth indicates the estimated fold change for each gene. Each dot is representative of one gene; grey dots represent no significant differences between axenic and conventionally raised groups. Red dots represent genes that were either enriched (above the x-axis) or depleted (below the x-axis) and were considered biologically significant. The two groups investigated were midguts conventionally raised (a), and midguts axenically raised (b).
Discussion
Alterations of transcript abundance identified in association with the axenic state in this study were relatively low, particularly in regard to other transcriptome studies of mosquitoes. For example, microarray studies on whole mosquitoes by Dissanayake et al.38 discovered 5,081 DE transcripts between male and female mosquitoes and a further 4,773 DE transcripts between blood-fed and non-blood fed females. Similarly, an Illumina-based deep sequencing study identified 5,969 DE transcripts between blood-fed and non-blood fed mosquitoes; however, a detailed examination shows that a majority of the transcripts identified showed less than a twofold change in expression39. Here we only identified 170 DE transcripts due to the axenic state. These numbers are more in line with investigations on the effect of viral and bacterial infections on the host mosquito. Studies on arbovirus infections in Ae. aegypti identified 20340 and 39741 DE genes, and a study on a native Wolbachia infection in Ae. fluviatilis found 25742 DE genes. This suggests that the transcriptional effect of removing the microbiome in adult mosquitoes might be more similar to that of a transient change rather than that of a large physiological change.
An additional explanation for the low recovery of DE transcripts may be methodological. The DESeq2 software package identified a higher number of differentially expressed transcripts (1,165 vs. 181 discovered using Sleuth Table 1). We chose the more conservative analysis as it had the highest congruence between the two methods. It may be that our estimates of DE transcripts are more conservative than in other studies. Additionally, earlier studies used microarray analysis38,40 and 45443 sequencing, whereas this and more recent studies used Illumina sequencing39,42,44,45. Looking at just the RNA Seq studies that used Illumina, including this study, four different versions of the Ae. aegypti reference were used, and no two studies employed the same bioinformatics pipeline. Earlier versions of the Ae. aegypti genome and less conservative programs to determine differential expression coupled with lower thresholds for determining differential expression might account for some of the differences found in this study. The minimal effect of the axenic state on transcript changes in mosquitoes is supported both by the qPCR and CAP results and the fact that our method still shows large scale changes associated with blood meal acquisition (Fig. 6).
A previous study has documented the transcriptional changes in larval mosquitoes in response to the axenic state and found significantly more DE genes (1,328) than we document here29. However, these studies differ in a critical factor. The larvae in the Vogel et al., study could not be reared past the L1 life stage and were only 22 h old. In comparison, we have shown that larvae can be reared in the absence of a microbiome, with the proper nutrition, and there is little detrimental effect on the mosquito26. Thus, it is likely that the axenic larvae previously described were in a stressed state due to starvation. This could account for the report of increased transcription of genes involved in amino acid transport, insulin and TOR signalling, fatty acid oxidation, and a decrease in expression of some peptidases. This pattern has widely been identified as a pattern seen in animals undergoing starvation stress, including mosquitoes29,46–49.
When trying to identify a core set of genes that were affected by the presence of a microbiome in flies, there were no genes that were identified as being shared between the adult and larval axenic Ae. aegypti and D. melanogaster, under our selection criteria. This required all genes to have been DE in all three groups with a log abundance of at least twofold (biologically significant) while also being statistically significant. There were several genes; however, that were DE but were not considered biologically significant in all three groups or the orthologue was only found in one of the two species (Table S3). The majority of the genes identified had unknown functions while those shared across species were involved in immune function and within Ae. aegypti nutrient metabolism, which supports the idea that the microbiome has a large role in supplying essential nutrients to the mosquito and the microbiome is directly interacting with the host immune system.
Mosquitoes possess a number of molecular and cellular based innate immune mechanisms50. Those most commonly associated with defending against bacteria include the Toll, Immune deficiency (Imd) and JAK-STAT signalling pathways which induce the production of antimicrobial peptides50. Not surprisingly, evidence suggests that the gut microbiome plays a role in the development and functionality of the mosquito’s innate immune response51,52. Anti-microbial peptides are regulated in mosquitoes mainly by the Imd pathway; all of the anti-microbial peptides identified in this study were down-regulated in the axenic state and were all expressed in at least two tissue types. Defensin anti-microbial peptides work against gram-positive bacteria. In this study DEFC (AAEL003832), DEFD (AAEL003857), and two of the three isoforms of DEFA (AAEL003841, AAEL027792) were differentially expressed in the carcass regardless of blood meal status, and DEFA was also differentially expressed in the blood-fed midgut. Another anti-microbial peptide, gambicin (GAM1; AAEL004522), which is regulated by the Toll, Imd, and JAK-STAT signalling pathways in combination, was differentially expressed in the midgut regardless of blood meal status. One immunity-related gene, TLR6 (AAEL023577), was upregulated in the blood-fed midgut. This may be the mosquito attempting to compensate for the decreased expression of the anti-microbial peptides as TLR6 plays a fundamental role in pathogen recognition and innate immune activation.
Vitellogenesis (yolk deposition) is vital for successful reproduction in mosquitoes. The process is regulated by the juvenile hormone (JH) pathway53–55. Previous studies have shown that nutritionally deprived mosquito larvae have lower levels of JH resulting in the production of fewer eggs as adults39. The influence of the gut microbiome on nutritional status has been observed in multiple studies56,57. For example, there is a significant delay in the developmental time of axenically reared Ae. aegypti compared to their conventionally reared counterparts26,58. In this study, we found an enrichment of juvenile hormone epoxide hydrolase (AAEL011313) in blood-fed midguts of axenic adults. This enzyme is a negative regulator of JH and its enrichment may indicate an active effort on behalf of the axenic mosquito to conserve nutritional resources potentially resulting in lower egg production. Similarly, we observed a reduction in forkhead A2 (AAEL003173) transcripts in the axenic blood-fed midgut. Forkhead box (FOX) proteins are transcription factors that regulate the expression of genes associated with cell growth, proliferation and differentiation51. In fact, suppression of fat body associated FOX family proteins have been shown to reduce egg production in Ae. aegypti; however, midgut associated FOX family members have not been assessed for their role in oogenesis59. While the axenic mosquitoes tested in Correa et al.26 did not show a statistical difference in the number of eggs laid, there are still potentially lifelong effects from an axenic state during development. Experiments using gnotobiotic Ae. aegypti and Aedes atropalpus mosquitoes demonstrated that multiple different bacterial species supported oogenesis in Ae. aegypti while only one bacterial species supported normal egg production in Ae. atropalpus60. When investigated the differences between treatments within Ae. atropalpus nutrient levels of stored lipids appeared to make the difference in egg production60. Additionally, a study examining the role of the gut microbiome and blood digestion demonstrated that bacteria plays a key role in the digestion of proteins and that the lack of bacteria could deprive the mosquito of essential nutrients and affect oocyte maturation in Ae. aegypti61.
In other axenic species, there has been evidence of intestinal remodelling, with the presence of a microbiome required for normal development4,5,62,63. In both vertebrates and invertebrates studied, the absences of a gut microbiome affected gut morphology in a number of ways, including epithelial cell renewal, cell composition and spacing, and a decrease in total intestinal size4,64. Several genes were upregulated in the midgut that could relate to intestinal remodelling and function including genes involved in muscle formation (AAEL018288), mucus production (AAEL019619, AAEL023490), microtubule formation (AAEL019567), and ciliary formation (AAEL018303). Additionally, genes that are involved in the regulation of intestinal homeostasis (AAEL014566, AAEL007658) were downregulated in the axenic state, which might suggest that it is more difficult for axenic mosquitoes to recover from insults.
In summary, while this transcriptional analysis has identified several differentially expressed genes that could point to specific gene expression or metabolic pathways that may be remodelled in the absence of a microbiome, the overall effect of the axenic state on the adult mosquito is relatively small. This raises interesting questions as to how mosquitoes acquire and regulate their internal bacterial load to avoid the potential negative consequences of unchecked bacterial growth and potential phenotypic outcomes associated with altered microbiome compositions, such as vector competence.
Materials and methods
Aedes aegypti rearing
Sterilisation of the Aedes aegypti eggs was carried out as previously described26,65. Briefly Ae. aegypti eggs were collected from colony mosquitoes, and in a sterile hood the eggs were serially rinsed for five minutes in 70% ethanol, followed by a five-minute wash in a 3% bleach and 0.2% ROCCAL-D (Pfizer) solution, and then again for five minutes in fresh 70% ethanol. The sterilised eggs were then rinsed three times in autoclaved DI water and placed in a Petri dish filled with phosphate-buffered saline (PBS). Eggs were hatched in a vacuum oven (Precision Scientific Model 29) at 25 Hz for 15 min at room temperature. The sterility of larvae and mosquitoes was tested via culturing viable bacteria and 16S rRNA gene PCR as previously described26. Conventionally reared mosquitoes were established by colonising axenic mosquitoes with bacteria from a homogenized colony mosquito.
Larvae were transferred from the Petri dish to individual wells of six-well tissue culture plates; each well contained 5 ml of sterilised DI water and a 0.6 g plug of liver yeast agar18. After pupation, the mosquitoes were transferred to autoclaved mosquito emergence chambers. All the mosquitoes were fed on filter sterilised 10% sucrose solution. Approximately a week after the first emergence, half the mosquitoes were blood-fed using a circulating water bath and membrane feeder. Mosquitoes were fed sterile defibrinated sheep blood (Hemostat Laboratories), and axenic Swiss Webster mouse pelts (provided by Dr Andrew Goodman, Yale University) were used as the membrane in lieu of parafilm. All feeds were carried out in a biosafety cabinet under sterile conditions.
RNA extractions and library preparation
Midguts were dissected from sugar-fed and 24 hpbm groups in sterile PBS and pools of five midguts or carcasses/group were placed in mirVana lysis buffer. There were three pooled replicates of each group. RNA was extracted using the mirVana miRNA Isolation kit according to the manufacturer’s instructions (Ambion). Total RNA was quantified using the BR RNA assay on the Qubit (Thermo Fisher Scientific) and quality assessed using an RNA 6,000 chip on an Agilent 2,100 Bioanalyzer (Agilent Technologies). Library construction and sequencing were performed at the Next Generation Sequencing Core at the University of Texas Medical Branch. 1 µg of total RNA was used to construct libraries using the NEBNext Ultra II RNA Library kit according to the manufacturer’s recommendations (New England Biolabs). The resultant DNA libraries with nonhomologous 5′ and 3′ ends were analysed by qPCR to determine the template concentration of each library. The 24 samples were pooled and sequenced on an Illumina NextSeq550 using a high-output flow-cell and paired-end 75 base reads following the manufacturer's protocol.
Data analysis
Sequences were quality checked using FastQC66 and quality filtered using Trimmomatic 0.3667, sequencing adapters and reads with Phred-equivalent scores of < 33 were removed at this stage. Potential sequence contamination arising from phiX, human, and sheep (blood source) were removed using BBDuk 36.62 and BBMap 36.6268. Quality filtered reads then pseudoaligned to the Aedes aegypti genome (assembly AaegL5) using Kallisto 0.43.169. Data were then imported into RStudio and analysed with DESeq234, and Sleuth35 to determine differential abundance. Default parameters were used in both analyses.
Gene Ontology terms (GO) were obtained from VectorBase annotations. Statistical analyses were performed in R using the packages agricolae70 and vegan71.
Real-time quantitative PCR assays
Quantitative PCR (qPCR) assays verified patterns in the transcriptome datasets. Mosquitoes were raised, dissected, and subjected to RNA extraction as described above The quantitative PCR assays were performed using the iTaq Universal SYBR Green One-Step Kit (Bio-Rad, Hercules, California, USA) in a 20-µl reaction volume, 50 ng of RNA was added to all reactions, and all samples were tested in duplicate and NTC, and NRT controls were included. The genes were chosen if they did not have any splice variants, and primers could be designed to cover exon-exon boundaries. The primer sequences used in these assays are listed in Table S4.
Supplementary information
Acknowledgements
We would like to thank Tonya Petruff and John Shepard for their efforts maintaining the mosquito colony. We would also like to thank Andrew Goodman and Anjelica Marin from the Yale School of Medicine for providing axenic mice carcasses. Funding for this project was provided by the Louis A. Magnarelli Post-Doctoral Research Fellowship program. This publication was also supported by the Cooperative Agreement Number U01CK000509, funded by the Centers for Disease Control and Prevention. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention or the Department of Health and Human Services. GLH is supported by NIH grants (R21AI138074 and R21AI129507), the Wolfson Foundation and Royal Society (RSWF\R1\180013), the John S. Dunn Foundation Collaborative Research Award and the BBSRC (BB/T001240/1).
Author contributions
D.E.B.., B.S., and G.L.H conceived and designed the experiments. M.A.C. and J.H. performed the laboratory experiments and J.H. and B.S. processed the NGS data and performed the analysis. J.H., B.S., D.E.B., and G.L.H. wrote and edited the manuscript. B.S. and D.E.B. oversaw the project.
Data availability
Sequences and related information determined in this work can be found at GenBank accession #'s SRR11209355-SRR11209378 and Bioproject PRJNA609359.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-67811-y.
References
- 1.Bordenstein SR, Theis KR. Host biology in light of the microbiome: Ten principles of holobionts and hologenomes. PLoS Biol. 2015;13:e1002226. doi: 10.1371/journal.pbio.1002226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg E, Zilber-Rosenberg I. Symbiosis and development: The hologenome concept. Birth Defects Res. Part C. Embryo Today Rev. 2011;93:56–66. doi: 10.1002/bdrc.20196. [DOI] [PubMed] [Google Scholar]
- 3.Dawkins R. The Extended Phenotype: The Long Reach of The Gene. Oxford: Oxford University Press; 1999. [Google Scholar]
- 4.Broderick NA, Buchon N, Lemaitre B. Microbiota-induced changes in Drosophila melanogaster host gene expression and gut morphology. MBio. 2014;5:e01117-14. doi: 10.1128/mBio.01117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith, K., McCoy, K. D. & Macpherson, A. J. in Seminars in immunology. 59–69 (Elsevier, Amsterdam).
- 6.Wagner M. Determination of germfree status. Ann. N. Y. Acad. Sci. 1959;78:89–101. doi: 10.1111/j.1749-6632.1959.tb53097.x. [DOI] [Google Scholar]
- 7.Schaffer J, et al. Studies on fatal hypoglycemia in axenic (germfree) piglets. Proc. Soc. Exp. Biol. Med. 1965;118:566–570. doi: 10.3181/00379727-118-29906. [DOI] [PubMed] [Google Scholar]
- 8.Buecher EJ, Hansen EL. Mass culture of axenic nematods uising continuous aeration. J. Nematol. 1971;3:199. [PMC free article] [PubMed] [Google Scholar]
- 9.Bakula M. The persistence of a microbial flora during postembryogenesis of Drosophila melanogaster. J. Invertebr. Pathol. 1969;14:365–374. doi: 10.1016/0022-2011(69)90163-3. [DOI] [PubMed] [Google Scholar]
- 10.Reyniers J, et al. A complete life-cycle in the" germ-free’bantam chickèn. Nature. 1949;163:67–68. doi: 10.1038/163067a0. [DOI] [PubMed] [Google Scholar]
- 11.Bost A, et al. How gut transcriptional function of Drosophila melanogaster varies with the presence and composition of the gut microbiota. Mol. Ecol. 2018;27:1848–1859. doi: 10.1111/mec.14413. [DOI] [PubMed] [Google Scholar]
- 12.Larsson E, et al. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut. 2012;61:1124–1131. doi: 10.1136/gutjnl-2011-301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun J, et al. Gene expression profiles of germ-free and conventional piglets from the same litter. Sci. Rep. 2018;8:1–9. doi: 10.1038/s41598-018-29093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volf J, et al. Gene expression in the chicken caecum is dependent on microbiota composition. Vet. Res. 2017;48:85. doi: 10.1186/s13567-017-0493-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandler JA, Liu RM, Bennett SN. RNA shotgun metagenomic sequencing of northern California (USA) mosquitoes uncovers viruses, bacteria, and fungi. Front. Microbiol. 2015;6:185. doi: 10.3389/fmicb.2015.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guégan M, et al. The mosquito holobiont: Fresh insight into mosquito-microbiota interactions. Microbiome. 2018;6:49. doi: 10.1186/s40168-018-0435-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hegde S, Rasgon JL, Hughes GL. The microbiome modulates arbovirus transmission in mosquitoes. Curr. Opin. Virol. 2015;15:97–102. doi: 10.1016/j.coviro.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma P, et al. Salivary glands harbor more diverse microbial communities than gut in Anopheles culicifacies. Parasites Vectors. 2014;7:235. doi: 10.1186/1756-3305-7-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chavshin AR, et al. Malpighian tubules are important determinants of Pseudomonas transstadial transmission and longtime persistence in Anopheles stephensi. Parasites Vectors. 2015;8:36. doi: 10.1186/s13071-015-0635-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vázquez-Martínez MG, et al. Cyanobacteria associated with Anopheles albimanus (Diptera: Culicidae) larval habitats in southern Mexico. J. Med. Entomol. 2002;39:825–832. doi: 10.1603/0022-2585-39.6.825. [DOI] [PubMed] [Google Scholar]
- 21.Lindh J, Borg-Karlson A-K, Faye I. Transstadial and horizontal transfer of bacteria within a colony of Anopheles gambiae (Diptera: Culicidae) and oviposition response to bacteria-containing water. Acta Trop. 2008;107:242–250. doi: 10.1016/j.actatropica.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 22.Gimonneau G, et al. Composition of Anopheles coluzzii and Anopheles gambiae microbiota from larval to adult stages. Infect. Genet. Evol. 2014;28:715–724. doi: 10.1016/j.meegid.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 23.Bennett KL, et al. Dynamics and diversity of bacteria associated with the disease vectors Aedes aegypti and Aedes albopictus. Sci. Rep. 2019;9:1–12. doi: 10.1038/s41598-018-37186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scolari F, Casiraghi M, Bonizzoni M. Aedes spp. and their microbiota: A review. Front. Microbiol. 2019;10:2036. doi: 10.3389/fmicb.2019.02036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coon KL, et al. Bacteria-mediated hypoxia functions as a signal for mosquito development. Proc. Natl. Acad. Sci. 2017;114:E5362–E5369. doi: 10.1073/pnas.1702983114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Correa MA, Matusovsky B, Brackney DE, Steven B. Generation of axenic Aedes aegypti demonstrate live bacteria are not required for mosquito development. Nat. Commun. 2018;9:4464. doi: 10.1038/s41467-018-07014-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romoli O, Schonbeck JC, Hapfelmeier S, Gendrin M. Bacteria contribute to mosquito development via folate biosynthesis and energy storage. bioRxiv. 2019 doi: 10.1101/2019.12.31.891671. [DOI] [Google Scholar]
- 28.Castelein N, Hoogewijs D, De Vreese A, Braeckman BP, Vanfleteren JR. Dietary restriction by growth in axenic medium induces discrete changes in the transcriptional output of genes involved in energy metabolism in Caenorhabditis elegans. Biotechnol. J. Healthcare Nutr. Technol. 2008;3:803–812. doi: 10.1002/biot.200800003. [DOI] [PubMed] [Google Scholar]
- 29.Vogel KJ, Valzania L, Coon KL, Brown MR, Strand MR. Transcriptome sequencing reveals large-scale changes in axenic Aedes aegypti larvae. PLoS Negl. Trop. Dis. 2017;11:e0005273. doi: 10.1371/journal.pntd.0005273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caragata EP, Tikhe CV, Dimopoulos G. Curious entanglements: Interactions between mosquitoes, their microbiota, and arboviruses. Curr. Opin. Virol. 2019;37:26–36. doi: 10.1016/j.coviro.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dada N, Sheth M, Liebman K, Pinto J, Lenhart A. Whole metagenome sequencing reveals links between mosquito microbiota and insecticide resistance in malaria vectors. Sci. Rep. 2018;8:2084. doi: 10.1038/s41598-018-20367-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Audsley MD, Yixin HY, McGraw EA. The microbiome composition of Aedes aegypti is not critical for Wolbachia-mediated inhibition of dengue virus. PLoS Negl. Trop. Dis. 2017;11:e0005426. doi: 10.1371/journal.pntd.0005426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.33Webb, C., Doggett, S. & Russell, R. A guide to mosquitoes of Australia. (Csiro Publishing, 2016).
- 34.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods. 2017;14:687. doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 36.Giraldo-Calderón GI, et al. VectorBase: An updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res. 2014;43:D707–D713. doi: 10.1093/nar/gku1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thurmond J, et al. FlyBase 20: The next generation. Nucleic Acids Res. 2018;47:D759–D765. doi: 10.1093/nar/gky1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dissanayake SN, et al. aeGEPUCI: A database of gene expression in the dengue vector mosquito, Aedes aegypti. BMC Res. Notes. 2010;3:248. doi: 10.1186/1756-0500-3-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonizzoni M, et al. RNA-seq analyses of blood-induced changes in gene expression in the mosquito vector species, Aedes aegypti. BMC Genomics. 2011;12:82. doi: 10.1186/1471-2164-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colpitts TM, et al. Alterations in the Aedes aegypti transcriptome during infection with West Nile, dengue and yellow fever viruses. PLoS Pathog. 2011;7:e1002189. doi: 10.1371/journal.ppat.1002189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonizzoni M, et al. Complex modulation of the Aedes aegypti transcriptome in response to dengue virus infection. PLoS ONE. 2012;7:e50512. doi: 10.1371/journal.pone.0050512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caragata EP, et al. The transcriptome of the mosquito Aedes fluviatilis (Diptera: Culicidae), and transcriptional changes associated with its native Wolbachia infection. BMC Genomics. 2017;18:6. doi: 10.1186/s12864-016-3441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Price DP, et al. The fat body transcriptomes of the yellow fever mosquito Aedes aegypti, pre-and post-blood meal. PLoS ONE. 2011;6:e22573. doi: 10.1371/journal.pone.0022573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong S, Behura SK, Franz AW. The midgut transcriptome of Aedes aegypti fed with saline or protein meals containing chikungunya virus reveals genes potentially involved in viral midgut escape. BMC Genomics. 2017;18:382. doi: 10.1186/s12864-017-3775-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang X, Poelchau MF, Armbruster PA. Global transcriptional dynamics of diapause induction in non-blood-fed and blood-fed Aedes albopictus. PLoS Negl. Trop. Dis. 2015;9:e0003724. doi: 10.1371/journal.pntd.0003724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perez-Hedo M, Rivera-Perez C, Noriega FG. Starvation increases insulin sensitivity and reduces juvenile hormone synthesis in mosquitoes. PLoS ONE. 2014;9:e86183. doi: 10.1371/journal.pone.0086183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harbison ST, Chang S, Kamdar KP, Mackay TF. Quantitative genomics of starvation stress resistance in Drosophila. Genome Biol. 2005;6:R36. doi: 10.1186/gb-2005-6-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schuier AM, Wood PA. Mouse models for disorders of mitochondrial fatty acid β-oxidation. ILAR J. 2002;43:57–65. doi: 10.1093/ilar.43.2.57. [DOI] [PubMed] [Google Scholar]
- 49.Moskalev A, et al. A comparison of the transcriptome of Drosophila melanogaster in response to entomopathogenic fungus, ionizing radiation, starvation and cold shock. BMC Genomics. 2015;16:S8. doi: 10.1186/1471-2164-16-S13-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prasad AN, Brackney D, Ebel GD. The role of innate immunity in conditioning mosquito susceptibility to West Nile virus. Viruses. 2013;5:3142–3170. doi: 10.3390/v5123142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ricci I, et al. Mosquito/microbiota interactions: From complex relationships to biotechnological perspectives. Curr. Opin. Microbiol. 2012;15:278–284. doi: 10.1016/j.mib.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 52.Weiss B, Aksoy S. Microbiome influences on insect host vector competence. Trends Parasitol. 2011;27:514–522. doi: 10.1016/j.pt.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raikhel AS, et al. Molecular biology of mosquito vitellogenesis: From basic studies to genetic engineering of antipathogen immunity. Insect Biochem. Mol. Biol. 2002;32:1275–1286. doi: 10.1016/s0965-1748(02)00090-5. [DOI] [PubMed] [Google Scholar]
- 54.Raikhel AS, Dhadialla T. Accumulation of yolk proteins in insect oocytes. Annu. Rev. Entomol. 1992;37:217–251. doi: 10.1146/annurev.en.37.010192.001245. [DOI] [PubMed] [Google Scholar]
- 55.Hansen IA, Attardo GM, Rodriguez SD, Drake LL. Four-way regulation of mosquito yolk protein precursor genes by juvenile hormone-, ecdysone-, nutrient-, and insulin-like peptide signaling pathways. Front. Physiol. 2014;5:103. doi: 10.3389/fphys.2014.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Douglas AE. Lessons from studying insect symbioses. Cell Host Microbe. 2011;10:359–367. doi: 10.1016/j.chom.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minard G, Mavingui P, Moro CV. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasites Vectors. 2013;6:146. doi: 10.1186/1756-3305-6-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamada R, Deshpande SA, Bruce KD, Mak EM, William WJ. Microbes promote amino acid harvest to rescue undernutrition in Drosophila. Cell Rep. 2015;10:865–872. doi: 10.1016/j.celrep.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hansen IA, et al. Forkhead transcription factors regulate mosquito reproduction. Insect Biochem. Mol. Biol. 2007;37:985–997. doi: 10.1016/j.ibmb.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coon KL, Brown MR, Strand MR. Gut bacteria differentially affect egg production in the anautogenous mosquito Aedes aegypti and facultatively autogenous mosquito Aedes atropalpus (Diptera: Culicidae) Parasites Vectors. 2016;9:375. doi: 10.1186/s13071-016-1660-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de O Gaio A, et al. Contribution of midgut bacteria to blood digestion and egg production in Aedes aegypti (Diptera: Culicidae)(L.) Parasites Vectors. 2011;4:105. doi: 10.1186/1756-3305-4-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levenson, S. M. & Tennant, B. in Federation proceedings. 109. [PubMed]
- 63.Meslin, J., Sacquet, E. & Guenet, J. in Annales de biologie animale, biochimie, biophysique. 203. [PubMed]
- 64.Abrams GD, Bauer H, Sprinz H. Influence of the normal flora on mucosal morphology and cellular renewal in the ileum. A comparison of germ-free and conventional mice. New York: Mount Sinai Hospital; 1962. [PubMed] [Google Scholar]
- 65.66Hyde, J., Correa, M. A., Brackney, D. E. & Steven, B. Generation and rearing of axenic Aedes aegypti mosquitoes Protocol Exchange (2019).
- 66.FastQC: A quality control tool for high throughput sequence data (Babraham Bioinformatics, Babraham Institute, Cambridge, 2010).
- 67.Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bushnell B. BBMap: A fast, accurate, splice-aware aligner. Berkeley: Lawrence Berkeley National Lab. (LBNL); 2014. [Google Scholar]
- 69.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 70.agricolae: Statistical Procedures for Agricultural Research. (2019).
- 71.vegan: Community Ecology Package. R package version 2.5–2. 2018 (2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequences and related information determined in this work can be found at GenBank accession #'s SRR11209355-SRR11209378 and Bioproject PRJNA609359.