

Abstract
Elucidating the physiological binding partners of histone post-translational modifications (hPTMs) is key to understanding fundamental epigenetic regulatory pathways. Determining such interactomes will enable the study of how perturbations of these interactions affect disease. Here we use a synthetic biology approach to set a series of hPTM-controlled photo-affinity traps in native chromatin. Using quantitative proteomics, the local interactomes of these chemically customized chromatin landscapes are determined. We show that the approach captures transiently interacting factors such methyltransferases and demethylases, as well as previously reported and novel hPTM reader proteins. We also apply this in situ proteomics approach to a recently disclosed cancer-associated histone mutation, H3K4M, revealing a number of perturbed interactions with the mutated tail. Collectively our studies demonstrate that modifying and interrogating native chromatin with chemical precision is a powerful tool for exploring epigenetic regulation and dysregulation at the molecular level.
Introduction
Protein-protein interactions mediate critical nuclear functions including transcription, chromatin remodeling, and heterochromatin establishment and maintenance. These processes are subject to precise and complex regulatory mechanisms, often involving reversible post-translational modification of the proteins involved.1 Understanding these regulatory pathways at the molecular level not only illuminates fundamental biology, but also reveals how perturbation of these interactions can lead to disease. Key to this endeavor is the determination of the set of physiological binding partners - the interactome - of post-translationally modified proteins, requiring the use of robust methodologies for trapping weak and transient protein-protein interactions in the complex nuclear environment.2
A canonical example of this regulation is the post-translational modification of histone proteins (hPTMs) associated with the eukaryotic genome. Efficient genomic packaging is achieved by wrapping approximately ≈150 base pairs of DNA around an octameric core of histones to afford a nucleosome, the fundamental repeating unit of chromatin. This assembly, in concert with myriad other factors including chromatin remodeling complexes, chromatin-associating proteins, and RNA, facilitates the exquisite temporal control of DNA-templated processes such as transcription.3 hPTMs including mono-, di-, and tri-methylation, acetylation, and ubiquitylation modulate downstream effects by binding to chromatin-associating proteins (‘readers’) or eliciting a direct biophysical effect.4 Dysregulation of epigenetic processes, often involving perturbation of normal hPTM homeostasis, is linked to a range of pathologies including cancer.5 Indeed, mutations to histone proteins themselves has recently emerged as a driver of oncogenesis,6 a paradigm that is set to broaden in terms of its clinical importance due to the recent disclosure of a vastly expanded set of these so-called “oncohistone” mutations.7,8
Current methods to determine the interactomes of hPTMs can be broadly separated into those that function in an in vitro setting (i.e. a test tube) and those that operate in cells. In general, the former employ a reductionist strategy that allows precise chemical control of the system, but at the expense of interrogating the native chromatin environment. Conversely, in-cell experiments ensure physiological relevance, but lack chemical precision and so are often correlative in nature. In vitro studies have largely deployed peptide probes bearing a hPTM, in some cases with a proximal crosslinking moiety.9–11 These can be readily synthesized, and when added to cell/nuclear lysate can identify potential interacting proteins. The chromatin context of hPTMs, however, is eliminated in these experiments. This can be especially consequential since nuclear factors are typically large multi-subunit protein complexes with the potential to engage multiple discontinuous binding epitopes on the chromatin substrate, including hPTMs and associated DNA.12,13 This issue has led to the use of reconstituted mononucleosome or oligonucleosome probes in analogous experiments, often employing quantitative proteomics workflows to identify binders.14–16 Nonetheless, these reconstituted chromatin substrates, while clearly superior to simple peptide probes, still require the use of cellular extracts as a source of potential binding factors, and as such are removed from a native chromatin context.
Progress has been made using various chemical proteomics strategies to characterize hPTM interactomes on native chromatin. In the chromatin immunoprecipitation mass-spectrometry (ChIP-MS) approach, broadly reactive crosslinking reagents (e.g. formaldehyde and BS3) are first used to covalently capture chromatin-associating proteins. A ChIP workflow is then used to enrich for an hPTM of interest, followed by proteomics analysis of the associated crosslinked proteins.17,18 While operationally straightforward, this approach is inherently limited by antibody cross-reactivity, potentially leading to false positives,19 and by epitope occlusion issues stemming from bound reader proteins or the presence of adjacent PTMs, both of which lead to false negatives. Photocrosslinking offers an alternative approach to capturing protein-protein interactions in living cells. This can be achieved either by non-specific incorporation of photo-crosslinkable amino acids into the proteome,20 or by harnessing amber suppression technology to install a probe at a specific site in a protein of interest. The latter strategy is illustrated by the work of Kapoor and co-workers who used genetic code expansion to incorporate a diazirine-bearing lysine derivative into the N-terminal tails of histones H3 and H4 in mammalian cells.21 When combined with quantitative proteomics and stable isotope labeling by amino acids in cell culture (SILAC), this approach allowed chromatin interactomes to be determined as a function of cell state. This chemo-proteomics strategy does not, however, allow direct interrogation of interactomes as a function of specific hPTMs. In principle, such information might be available were it possible to incorporate both a hPTM and a proximal photo-crosslinker into native chromatin. While progress has been made in incorporating multiple non-canonical amino acids by genetic code expansion, for example Chatterjee and co-workers have incorporated acetyl-lysine and p-benzoylphenylalanine residues into recombinant histone H3,22 such methods are largely limited to bacterially expressed proteins for use in in vitro studies.23 Moreover, key hPTMs including tri-methyllysine, are currently inaccessible by this method. Thus, there remains a need for strategies that allow more general interrogation of hPTM-specific interactomes in the context of native chromatin.
Here, we present a synthetic approach to assemble chemically modified chromatin in situ. The use of ultra-fast split inteins facilitates the installation of chemically defined hPTMs and crosslinkers with both spatial and temporal control. One portion of the split intein is fused to a truncated histone and incorporated into chromatin. The split-intein partner protein is fused to the delivery cargo, and upon protein trans-splicing (PTS) in isolated nuclei, a precisely modified histone is assembled on native chromatin. We show that multiple hPTMs can be installed, along with a proximal diazirine photo-crosslinking moiety. Once in place, UV irradiation crosslinks the modified histone tail to interacting proteins, trapping transient protein-protein interactions. This synthetic precision is combined with SILAC-based proteomics to quantitatively determine the interactome of hPTMs. The interactome of a novel “oncohistone” mutation – H3K4M – is also determined, revealing perturbations to the chromatin landscape in the presence of this cancer-associated mutation.
Results
Semi-synthesis of modified histones for in-situ quantitative proteomics
We envisioned a chemo-proteomics strategy that harnesses split intein-mediated PTS to modify chromatin in isolated nuclei.24 In principle, this protein semi-synthesis approach should allow the concomitant introduction of one or more hPTMs, a proximal photo-crosslinker, and a biotin affinity handle into a histone of choice, in the process creating a hPTM-controlled photo-affinity trap in native chromatin (Fig. 1).
Figure 1. Schematic for the in-situ chromatin interactomics approach.
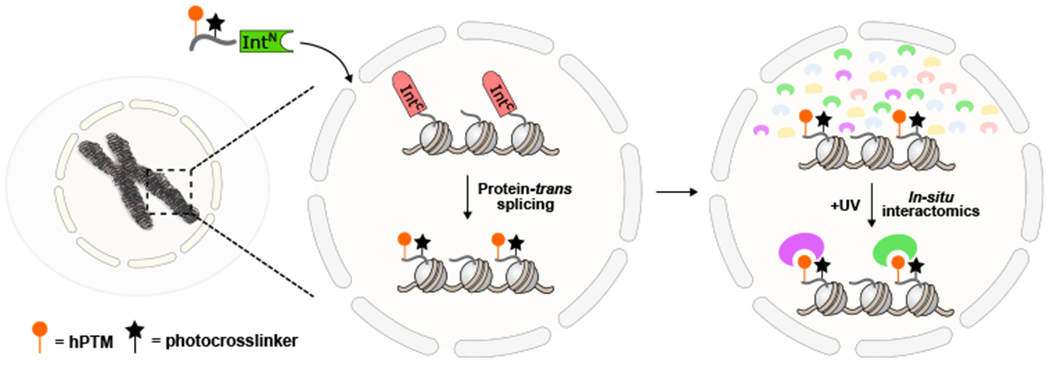
Protein-trans splicing between a chromatinized C-intein and an N-intein construct bearing hPTM(s) and a diazirine crosslinker introduces photoaffinity traps onto native chromatin. Upon UV-irradiation the chromatin-relevant hPTM interactome is determined.
Investigations began by identifying a suitable splice junction in the N-terminal disordered tail of histone H3, a region rich in PTMs and linked to numerous epigenetic processes. As part of our design, we elected to use the Cfa split intein, an engineered variant of the Nostoc punctiforme DnaE intein, which splices with a half-life of minutes, largely independent of flanking extein residues.25,26 These properties allowed us to select a splice junction between H3A31 and H3T32, thereby granting synthetic access to the majority of the H3 tail. Upon PTS, a single cysteine insertion at position 32 is present in the H3 tail; in an essentially traceless process that does not affect cell viability (Supplementary Fig. 1).
As an initial test of this in situ semi-synthesis scheme, we set out to generate a modified version of H3 containing tri-methyl-lysine at position 9 (H3K9me3), along with a minimally perturbative diazirine photo-crosslinker (photoleucine) at position 11 and an N-terminal biotin affinity handle (Fig 2a&b). This involved PTS between delivery construct 1, corresponding to residues 1-31 of histone H3 (containing the aforementioned probes) fused to the N–terminal intein fragment (CfaN), and a complementary split intein-histone fusion (FLAG-H31-28-CfaC-H332-135; construct 2) transiently transfected into HEK 293T cells. Note, the addition of H3 residues 1-28 to the N-terminus of the CfaC improved incorporation of the fusion protein into native chromatin, which we estimate at ≈15% compared to endogenous H3 (Fig. 2c; Supplementary Fig. 2). Delivery construct 1 was generated by native chemical ligation (Supplementary Fig. 3) and added to isolated nuclei expressing the CfaC-histone fusion protein. Analysis of the reaction mixture by western blotting indicated rapid formation of a new species consistent with the expected splice product 3 (Fig. 2d). The identity of the splice product was confirmed by LCMS/MS analysis of the excised band (Fig. 2e). As an initial validation of the crosslinking strategy, we asked whether we could capture a known reader of H3K9me3, namely heterochromatin protein 1 alpha (HP1α).27 Nuclei containing splice product 3 were washed to remove excess delivery material 1 and incubated with recombinant HP1α (Supplementary Fig. 4). After UV irradiation, the chromatin fraction containing the semi-synthetic histone was isolated and analyzed by western blotting (Fig. 2f). This revealed the generation of a band consistent with the expected H3K9me3-HP1α crosslink that importantly, was not present in a control experiment in which PTS was used to install the photoleucine crosslinker adjacent to a non-methylated version of H3K9 (Fig. 2f; Supplementary Figs. 5–7).
Figure 2. In-situ installation of hPTMs with ultra-fast split inteins.
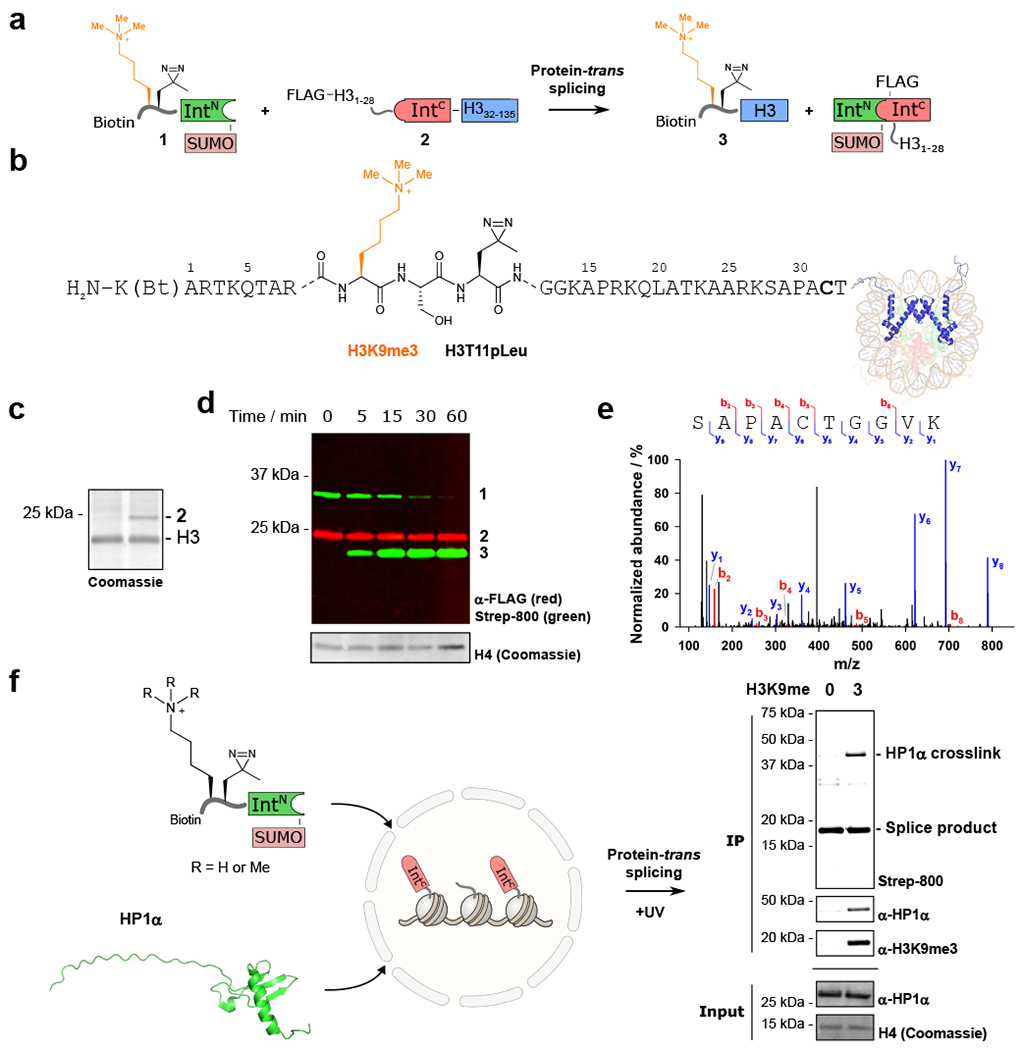
a. Protein-trans splicing between delivered material 1 and expressed construct 2 results in the semi-synthetic histone 3 bearing H3K9me3 with an adjacent photocrosslinker (H3T11pLeu). b. Expanded view of splice product 3 installing H3K9me3 and H3T11pLeu into native chromatin (Bt = biotin). c. Incorporation of 2 into chromatin is ~15%, when compared to endogenous H3. n = 2, with representative data shown. d. Time course for the trans-splicing reaction showing build-up of splice product 3. n = 2, with representative data shown. e. LCMS/MS analysis of the splice product band 3 confirms a unique peptide sequence containing the splice junction. f. An H3K9me3-dependent crosslink to HP1α is observed on addition of recombinant HP1α to nuclei bearing H3K9me3 or the wild-type tail. Right: western blot analysis displaying an H3K9me3-dependent histone-HP1α crosslink with anti-HP1α and streptavidin-800 detection. anti-HP1α and histone H4 input controls are shown below. n = 2, with representative data shown. Uncropped western blots are provided in Supplementary Fig. 26.
Encouraged by our ability to detect a known PTM-reader interaction, we next moved to a proteome-wide analysis. We implemented a SILAC-based workflow to determine the in-situ interactome of an hPTM, initially H3K9me3 (Fig. 3a). Intein-histone fusion 2 was transfected into HEK 293T cells grown in either “heavy” or “light” isotopically labeled SILAC media (Methods). Nuclei were isolated and purified, and PTS reactions were performed to install an H3K9me3-bearing tail, or the wild-type H3 tail to native chromatin (Fig. 3b). After washing the excess delivery construct from the system (Supplementary Fig. 8), the nuclei were irradiated with UV light to crosslink interacting proteins. Clear differences between the H3K9me3 and wild-type samples were observed on a streptavidin blot after UV irradiation (Fig. 3c). The nuclei were then mixed in both the forward and reversed replicates, and chromatin was sheared to approximately mono- and di-nucleosomes by sonication (Supplementary Fig. 9). A biotin-IP was performed to isolate the modified H3 proteins as well as crosslinked proteins, before on-bead digestion with trypsin, desalting, and LC-MS/MS analysis.
Figure 3. Determining the in-situ interactome of H3K9me3.
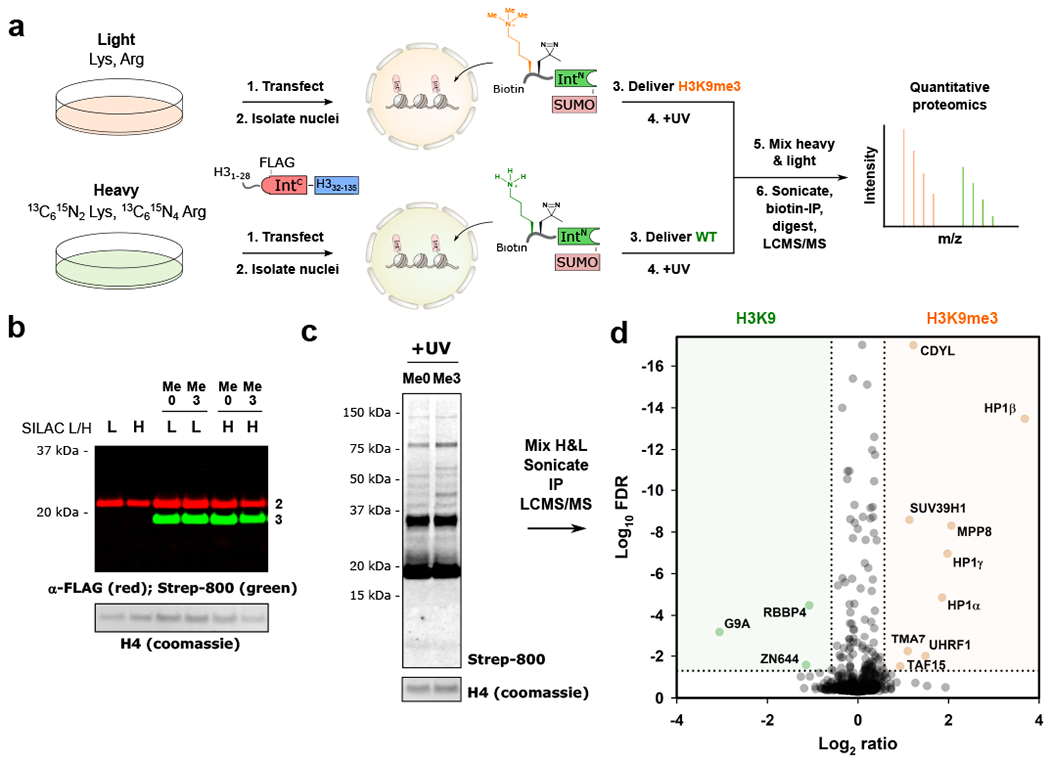
a. Schematic for the SILAC-based quantitative proteomics workflow to determine the interactome of H3K9me3, compared to the wild-type H3 tail. b. Installation of wild type (Me0) and H3K9me3-bearing tails (Me3) to endogenous chromatin in heavy (H) and light (L) labeled nuclei. Splice product (green) is observed by western blot, with excess delivered material (≈ 32 kDa) removed after the reaction. Histone H4 levels served as a loading control. c. Immunoblot analysis of UV-treated samples displaying crosslinked bands from H3K9me0- and H3K9me3-containing chromatin. Histone H4 levels served as a loading control. d. Volcano plot displaying proteins enriched in H3K9me3 (orange) and H3K9me0 samples (green); FDR < 0.05, >1.5-fold change, with tryptic peptides observed in both biological replicates. FDR values were calculated using the Benjamini-Hochberg procedure, as described in the Methods. For panels b-d experiments were performed as “forward” and “reversed” biological replicates (n = 2). Uncropped western blots are provided in Supplementary Fig. 27.
Analysis of the proteomics data (Methods) returned a number of proteins that met selected cutoffs (protein observed in both “forward” and “reversed” directions, fold change >1.5, false-discovery rate <0.05). All three HP1 isoforms were enriched in the H3K9me3 sample, as well as a number of other well-characterized reader proteins for the hPTM, including CDYL,10 MPP8,28 and UHRF1 (Fig. 3d; Supplementary Table 1).29 SUV39H1, an H3K9 di- and tri-methyltransferase was also observed. SUV39H1 dictates the spreading of heterochromatic regions through the recognition of its own reaction product by an N-terminal chromodomain.30 Potential novel interactors included the TATA-binding protein TAF15, and the putative coiled-coil domain containing protein TMA7.
G9A, an H3K9 mono- and di-methyltransferase, along with the G9A associating and zinc-finger containing protein ZN644,31 were enriched in the wild-type sample. Also enriched in the unmodified sample was RBBP4 – a constituent of PRC2 known to engage the unmodified H3 tail by its WD40 propeller domain.32 Taken together, these data confirm that the experimental workflow allows the quantitative in situ determination of hPTM interactors.
The interactome of hPTMs at H3K4
Next, we exploited the modularity of the in-situ crosslinking approach by asking how the chromatin interactome varies as a function of different modifications at a specific site. We focused on H3K4, a key hub of epigenetic regulation. Methylation of H3K4 is a hallmark of active transcription; tri-methylation of the residue (H3K4me3) marks promoters and is associated with actively transcribed genes,4 whereas mono-methylation (H3Kme1) is the most abundant modification state of H3K4, and correlates with active and primed enhancer regions.33 Moreover, the H3K4me1 mark is often found in conjunction with acetylation of H3K27 (H3K27ac), with the dual modification thought to play a key role in the activation of enhancers.
Recent analyses of patient tumor samples have highlighted a large number of mutations in canonical and variant histone proteins.7,8 These mutations are found throughout the histone primary sequences and include the established oncohistones, H3K27M and K36M, which are known to drive pediatric cancers through aberrant engagement with histone methyltransferases.34 Intriguingly, the newly expanded mutational collection includes an analogous lysine-to-methionine mutation at lysine 4 of histone 3 (H3K4M). Ectopic expression of H3K4M has recently been shown to destabilize the enhancer-specific H3K4 methyltransferases, MLL3/MLL4.35 However, the mechanism of this disruption is poorly understood, as is the broader impact of the mutation on epigenetic processes. We imagined that combining the in-situ interactome of the novel oncohistone mutation H3K4M with the aforementioned hPTM datasets would allow quantitative determination of the perturbed chromatin interactome.
We generated a series of photo-affinity traps to probe the distinct interactomes of H3K4me3, H3K4me1, a dually modified H3K4me1-H3K27ac histone, and H3K4norleucine (a non-oxidizable methionine mimic; Supplementary Figs. 10–14). Photoleucine was positioned in place of H3T6 in each of the constructs, and the biotin affinity handle was moved to H3K23, such that key binding interactions with the free N-terminus were not affected (Fig. 4a). PTS was again used to generate chromatin containing the modified H3 proteins in nuclei harboring labeled proteomes suitable for forward and reversed SILAC analyses (Supplementary Figs. 15–17&20). The presence of the installed hPTMs throughout the experimental workflow (up to 2 h) was confirmed by western blotting using modification-specific antibodies (Fig. 4b; Supplementary Fig. 19). Four proteomics datasets were collected, comparing H3K4me3 vs. wild type, H3K4me1 vs. wild type, H3K4me1 vs. H3K4me1-H3K27ac, and H3K4norleucine vs. wild type (Fig. 4c–e; Supplementary Tables 2–5).
Figure 4. Determining the interactome of H3K4 as a function of hPTMs.
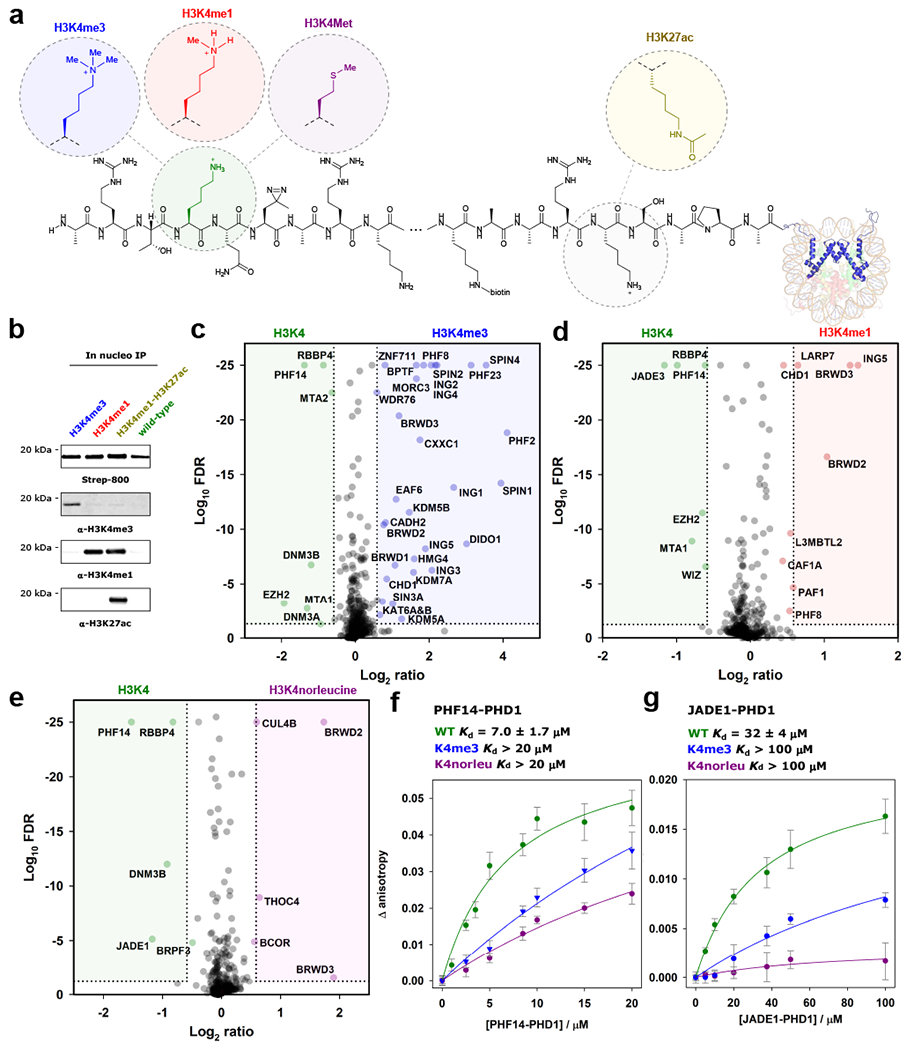
a. hPTMs and mutations are incorporated at H3K4 and H3K27, along with photoleucine at T6. b. Enrichment of the splice product shows installed methylation and acetylation PTMs are present throughout the in-situ crosslinking workflow. Immunoblots were probed with indicated antibodies. n = 2 independent experiments, representative data shown. Uncropped western blots are provided in Supplementary Fig. 28. c. Volcano plot displaying interactors enriched by H3K4me3 (blue) and H3K4me0 (green). Cutoffs as for Fig. 2d. FDR values were calculated using the Benjamini-Hochberg procedure, as described in the Methods. Proteins with Log10 FDR <25 were set to 25 for ease of visualization. d. Volcano plot displaying interactors enriched by H3K4me1 (red) and H3K4me0 (green). Cutoffs as for Fig. 2d. e. Volcano plot displaying interactors enriched by H3K4norleucine (purple) and H3K4me0 (green). Cutoffs as for Fig. 2d. For panels c-e experiments were performed as “forward” and “reversed” biological replicates (n = 2). f. Fluorescence anisotropy binding experiments for H3K4me0 (green), H3K4me3 (blue), and H3K4norleucine (purple) peptides binding to PHF14 PHD finger 1. Errors are reported as SEM (n = 3 independent experiments). g. Fluorescence anisotropy experiments for H3K4me0 (green), H3K4me3 (blue), and H3K4norleucine (purple) peptides binding to JADE1 PHD finger 1. Errors are reported as SEM (n = 3 independent experiments).
Established H3K4me3 and H3K4me1 reader proteins were identified in the respective datasets (see Supplementary Tables 2&3), including spindlin 1, 2 & 436 and BPTF for H3K4me3,37,38 and L3MBTL2 and PAF1 for H3K4me1.39,40 As expected, we observed the inhibitor of growth proteins (ING1-5) in the H3K4me3 sample.41,42 BRWD2&3, recently reported to contain a “cryptotudor” domain that recognizes modified H3K4 independent of the methylation state, were observed in both the H3K4me3 and H3K4me1 datasets.43 Both proteins were also enriched in the H3K4norleucine dataset, suggesting that H3K4M also engages the “cryptotudor” domain.
Importantly, we also observed enzymes responsible for the removal of H3K4me3, namely KDM5A and B, in the proteomics datasets (Fig. 4c). The detection of lysine demethylases using this method precludes the requirement to “trap” Fe2+-dependent demethylases through the addition of Mn2+,44 and highlights the advantage of the photo-affinity trapping method for capturing transient-type interactions on hPTM-containing chromatin.
In addition to the large number of known H3K4me3 and H3K4me1 readers detected in the SILAC datasets, we also identified several proteins that have not previously been shown to interact directly with the modifications. Among these putative novel readers are HMG4 and ZNF711 for H3K4me3 (Supplementary Table 2), and the transcriptional regulators WIZ, LARP7, and CAF1A for H3K4me1 (Supplementary Table 3). Hits enriched by the dually modified H3K4me1-H3K27ac construct over H3K4me1 included MCM5, YLPM1, and SAFB1 (Supplementary Fig. 18; Supplementary Table 4). Although observed in our datasets, we did not observe any differential engagement of MLL3/MLL4 with H3K4norleucine, however the E3 ligase CUL4B and the nuclear export factor THOC4 were enriched by the mutation (Supplementary Table 5).
A number of proteins were excluded by the presence of the hPTMs (Fig. 4c–e; Supplementary Tables 2–4). For instance, the PRC2 subunits EZH2 and RBBP4 were enriched by the unmodified H3 tail over H3K4me3 and H3K4me1. The molecular precision afforded by the approach confirmed the exclusion of the de novo DNA methyltransferases DNMT3A&B by H3K4me3, but not by H3K4me1, consistent with cytosine methylation being mutually exclusive to promoter-marking H3K4me3 regions.45 Interestingly, DNMT3B was also excluded by H3K4norleucine, suggesting a potential role for the mutation in disrupting de novo cytosine methylation. The NuRD complex members MTA1&2 were also found to be sensitive to methylation at H3K4. Methylation of the H3 tail is known to abrogate binding of NuRD to chromatin,46 and the specificity afforded by diazirine-based crosslinking suggests that MTA1&2 may play a direct role in regulating this interaction. The comparison of H3K4me1 and H3K4me1-H3K27ac interactomes revealed EZH2 as enriched by the unmodified H3K27 side chain, along with the E3 ligase PIAS1, the PAF1 subunit CTR9, and the SWI/SNF complex member SMCA1 (Supplementary Fig. 18; Supplementary Table 4).
The largely uncharacterized PHD-domain containing protein PHF14 was enriched by the wild-type tail in the H3K4me3, H3K4me1, and H3K4norleucine datasets. The first PHD domain of PHF14 has been implicated in chromatin binding,47 and was expressed as a GST-fusion protein (Supplementary Fig. 21). Low-micromolar binding of the unmodified H3 tail to PHF14-PHD1 was confirmed by fluorescence anisotropy (Kd = 7.0 ± 1.7 μM), with both H3K4me3 and H3K4norleucine perturbing this interaction (Fig. 4F; Supplementary Figs. 22–24).
Also excluded by H3K4norleucine was JADE1, a member of the HBO1 complex responsible for the majority of H4 acetylation (at H4K5, K8, and K12) in vivo.48,49 JADE1 contains two PHD fingers, and both engage H3 tails methylated at K4 to various degrees. PHD1 binds the unmodified H3 tail, and overrides the ability of PHD2 to associate with the H3 tail irrespective of the methylation state of K4. Consistent with this previous work, we found that JADE1-PHD1 binds to the wild-type H3 tail peptide with a Kd = 32 ± 4 μM, based on fluorescence anisotropy measurements (Fig. 4f; Supplementary Fig. 25). By contrast, the binding affinity of the H3 tail is largely abolished in the presence of H3K4me3 and H3K4norleucine (Kd > 100 μM).
Taken together, the quantitative comparison of H3 interactomes allows the identification of numerous factors involved in the deposition, interrogation, and removal of individual or multiple hPTMs in the complex nuclear environment. That we were able to reliably capture many of the known binding partners of the H3K4 and H3K9 methylation states, provides strong justification for future biochemical studies focusing on the novel interacting factors identified using our approach. Moreover, the confirmation of disrupted interactions between the H3 tail and PHD finger domains of chromatin-associating proteins in the presence of H3K4M presents exciting opportunities for investigation.
Conclusion
Here, we present an in-situ crosslinking approach that places selected PTMs along with an adjacent diazirine crosslinker into native chromatin. We have used the technology to generate the first interactomes of hPTMs in a physiologically relevant chromatin context. The chemical precision and flexibility afforded by split-intein mediated histone semi-synthesis, in principle, allows the in nucleo installation of a broad range of hPTMs into histone tails, i.e. beyond the methylation and acetylation marks described in the current study. Further, PTS-based semi-synthesis should be applicable to accessible PTMs in other chromatin-associating proteins. Importantly, the crosslinking approach reliably identifies enzymes responsible for the deposition and removal of the targeted PTMs (e.g. methyltransferases and demethylases), which are challenging to detect using other methods. Moreover, the sensitivity afforded by quantitative proteomics allows subtle perturbations in interactomes to be detected, as shown in the comparison of wild-type and H3K4M-samples.
The current ‘bait and trap’ workflow is performed in isolated cell nuclei, which may perturb native chromatin to some extent. However, that we were able to reliably identify established readers, writers and erasers of several hPTMs argues that this disruption is not a major limitation. Nonetheless, extension of the photo-affinity approach to living cells, perhaps employing engineered atypical or conditionally splicing split inteins, is clearly an exciting direction for future study.50,51 We envision that the in-situ crosslinking and proteomics workflow described herein will be useful for determining physiological interactomes of a range of well-established and novel hPTMs, as well as post-translationally modified nuclear proteins.
Methods
Stable isotope labeling of amino acids in cell culture (SILAC)
HEK 293T cells were plated in DMEM for SILAC media (Thermo Fisher), supplemented with dialyzed FBS (Thermo Fisher), penicillin-streptomycin, and “heavy” isoptope labeled (13C6-15N2-Lys and 13C6-15N4-Arg) or “light” Lys and Arg (Cambridge Isotope Laboratories). The cells were passaged at 90% confluency to a total of eight doublings to ensure complete isotopic labeling. The cells were expanded into 10 cm plates and transfected with a plasmid encoding FLAG-H31-28-CfaC-H332-135 using Lipofectamine 2000 following the manufacturer’s instructions. After 26 h, the plates were harvested and the cell pellets flash frozen and stored at −80 °C.
In-situ crosslinking protocol
SILAC experiments were performed as biological duplicates in a “forward” and “reversed” fashion to control for effects of metabolic incorporation of isotopic labels. Each “forward” and “reversed” experimental replicate was carried out starting from 6 ×107 cells. “Heavy” and “light” HEK 293T cells were lysed by hypotonic lysis in RSB buffer (10 mM tris, 15 mM NaCl, 1.5 mM MgCl2, Roche cOmplete EDTA-free protease inhibitors, pH 7.6) for 10 min on ice. The crude nuclei were isolated by centrifugation at 400 g for 5 min at 4 °C. The nuclei were resuspended in RSB buffer, and homogenized with ten strokes of a loose pestle Dounce homogenizer, and pelleted at 400 g for 5 min at 4 °C. The nuclei were resuspended in crosslinking buffer (20 mM HEPES, 1.5 mM MgCl2, 150 mM KCl, Roche cOmplete EDTA-free protease inhibitors, pH 7.6) and centrifuged at 400 g for 5 min at 4 °C. Finally, the nuclei were resuspended in 300 μl of crosslinking buffer per 1 ×107 cells. To the isolated nuclei was added the appropriate delivery construct bearing the selected PTMs and/or photoleucine to a final concentration of 0.3 μM. The splicing reactions were incubated at 37 °C for 30 min protected from light. The nuclei were pelleted at 400 g for 5 min at 4 °C, and washed twice with crosslinking buffer to remove excess delivered material. The nuclei were then resuspended in 200 μl crosslinking buffer per 1 ×107 cells and incubated at 37 °C for 30 min.
The nuclei were placed on ice and irradiated with 365 nm UV light for 20 min. The UV-irradiated nuclei were mixed as necessary for “forward” and “reversed” experiments and pelleted at 1,500 g for 5 min at 4 °C. The chromatin fraction was isolated by sequential resuspension and centrifugation in LB1 (50 mM HEPES, 140 mM NaCl, 1 mM EDTA, 10% v/v glycerol, 0.5% NP-40 alternative, 0.25% Triton-X100, Roche cOmplete EDTA-free protease inhibitors, pH 7.5), LB2 (10 mM tris, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, Roche cOmplete EDTA-free protease inhibitors, pH 8.0), and LB3 (10 mM tris, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% sodium lauroylsarcosine, Roche cOmplete EDTA-free protease inhibitors, pH 8.0); 1.5 ml, 1.5 ml, and 1.1 ml, respectively. Chromatin was sheared to approximately mono- and di-nucleosomes with 12 cycles of a probe sonicator at 25% amplitude, for 15 sec on and 45 sec off. 0.5% v/v Triton X-100 was added to the sheared chromatin and any insoluble material was pelleted at 12,000 g for 10 min at 4 °C. Binding buffer (25 mM tris, 150 mM NaCl, 0.5% v/v NP-40 alternative, 1 mM DTT, pH 7.6) was added to the supernatant in a ratio of 1 ml supernatant to 2.5 ml binding buffer, and added to equilibrated Sepharose magnetic streptavidin beads (GE Healthcare). The immunoprecipitation was performed at room temperature for two hours, with end-over-end rotation.
The beads were washed sequentially with 1x binding buffer, 2× 0.5% SDS in PBS, 2× 1 M NaCl in PBS, 2x tris-buffered saline, and 10× 50 mM ammonium bicarbonate at (pH 8.0). Finally, trypsin Gold (Promega; 2 μl of a 1 μg/ul stock) was added to the beads in 200 μl ammonium bicarbonate, and on-bead digestion was performed overnight at 37 °C with end-to-end rotation. A further 1 μl of trypsin was added the following morning, and the digestion incubated at 37 °C for a further 2 h. The supernatant was removed, and the beads washed with ammonium bicarbonate. The combined supernatants were dried in a SpeedVac vacuum concentrator (Thermo Fisher).
SILAC-based mass spectrometry data collection and analysis
Trypsin-digested samples were desalted using an Empore SDB-XC disc (3M) and dried in a SpeedVac vacuum concentrator (Thermo Fisher). Dried samples were resuspended in 21 μl of 0.1% formic acid (pH 3.0). 2 μl was injected per run using an Easy-nLC 1200 UPLC system. Samples were loaded directly onto a 45 cm x 75 μm nanocapillary column packed with 1.9 μm C18-AQ (Dr. Maisch, Germany) mated to a metal emitter in-line with an Orbitrap Fusion Lumos (Thermo Scientific). The column temperature was set to 45 °C and a 2 h gradient method with a flow rate of 300 nl min−1 was used. The mass spectrometer was operated in data dependent mode with a 120,000 resolution MS1 scan (positive mode, profile data type, AGC 4 ×105, Max IT 50 ms, 375-1500 m/z) in the Orbitrap, followed by HCD fragmentation in the ion trap with 35% collision energy. A dynamic exclusion list was invoked to exclude previously fragmented peptides for 60 s and maximum cycle time of 3 s was used. Peptides were isolated for fragmentation using a 1.2 Da window in the quadrupole. The ion trap was operated in Rapid mode with AGC 1 ×104, maximum IT of 54 ms, and a minimum of 5000 ions.
Data processing was performed using MaxQuant (v. 1.6.2.6)52 using default parameters unless otherwise noted. Replicates were analyzed in parallel with match between runs selected. The Uniprot human proteome reference database was used (proteome ID UP000005640) and Trypsin/P was selected as the enzyme. Met oxidation (+15.9949) and N-terminal acetylation (+42.0106) were specified as variable modifications. Homothreonine (+14.0157), a product of the reaction of photoleucine with water, was selected as a variable modification for Thr. For protein quantification, requantify was enabled and the minimum label ratio count was set to one. Only proteins quantified in both experimental replicates were reported. Mean and standard errors of protein ratios were calculated with values from experimental replicates. p-values were calculated by fitting log protein ratios to a normal distribution and calculating the probability that each protein ratio exhibited no change. These values were subsequently adjusted for multiple hypothesis testing using the Benjamini-Hochberg method.53 Protein ratios with an average H/L ratio greater than 1.5 and FDR-adjusted p-values less than 0.05 were considered significant.
Supplementary Material
Acknowledgements
The authors thank Felix Wojcik and Robert Thompson, and other members of the Allis and Muir laboratories for valuable discussions. We thank Tharan Srikumar, Saw Kyin, and Henry Shwe from the Princeton Proteomics & Mass Spectrometry Core. AJB is a Damon Runyon Fellow of the Damon Runyon Cancer Research Foundation (DRG-2283-17). JDB was funded by a postdoctoral fellowship from the U.S. National Institute of Health (GM123659). This work was supported by the U.S. National Institutes of Health (NIH grants R37-GM086868 to TWM, and PO1-CA196539 to CDA and TWM).
Footnotes
Competing Interests Statement
The authors declare no competing interests.
Data Availability Statement
All relevant data are included in the manuscript and supplementary information. Mass spectrometry data files have been uploaded to the PRIDE proteomics database (PXD017447).
References
- 1.Bannister AJ & Kouzarides T Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aebersold R & Mann M Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Li B, Carey M & Workman JL The Role of Chromatin during Transcription. Cell 128, 707–719 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Kouzarides T Chromatin Modifications and Their Function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Dawson MA & Kouzarides T Cancer epigenetics: From mechanism to therapy. Cell 150, 12–27 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Weinberg DN, Allis CD & Lu C Oncogenic mechanisms of histone H3 mutations. Cold Spring Harb. Perspect. Med 7, a026443 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett RL et al. A Mutation in Histone H2B Represents a New Class of Oncogenic Driver. Cancer Discov. 9, 1438–1451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nacev BA et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 567, 473–478 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vermeulen M et al. Selective Anchoring of TFIID to Nucleosomes by Trimethylation of Histone H3 Lysine 4. Cell 131, 58–69 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Vermeulen M et al. Quantitative Interaction Proteomics and Genome-wide Profiling of Epigenetic Histone Marks and Their Readers. Cell 142, 967–980 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Li X et al. Quantitative chemical proteomics approach to identify post-translational modification-mediated protein-protein interactions. J. Am. Chem. Soc 134, 1982–1985 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Machida S et al. Structural Basis of Heterochromatin Formation by Human HP1. Mol. Cell 69, 385–397 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Poepsel S, Kasinath V & Nogales E Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat. Struct. Mol. Biol 25, 154–162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartke T et al. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143, 470–484 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikolov M et al. Chromatin affinity purification and quantitative mass spectrometry defining the interactome of histone modification patterns. Mol. Cell Proteomics 10, 1–16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Local A et al. Identification of H3K4me1-associated proteins at mammalian enhancers. Nat. Genet 50, 73–82 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engelen E et al. Proteins that bind regulatory regions identified by histone modification chromatin immunoprecipitations and mass spectrometry. Nat. Commun 6, 7155 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji X et al. Chromatin proteomic profiling reveals novel proteins associated with histone-marked genomic regions. Proc. Natl. Acad. Sci. U.S.A 112, 3841–3846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah RN et al. Examining the Roles of H3K4 Methylation States with Systematically Characterized Antibodies. Mol. Cell 72, 162–177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suchanek M, Radzikowska A & Thiele C Photo-leucine and photo-methionine allow identification of protein-protein interactions in living cells. Nat. Meth 2, 261–267 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Kleiner RE, Hang LE, Molloy KR, Chait BT & Kapoor TM A Chemical Proteomics Approach to Reveal Direct Protein-Protein Interactions in Living Cells. Cell Chem. Biol 25, 110–120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Y, Gilgenast MJ, Hauc S & Chatterjee A Capturing Post-Translational Modification-Triggered Protein-Protein Interactions Using Dual Noncanonical Amino Acid Mutagenesis. ACS Chem. Biol 13, 1137–1141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Y, Addy PS, Mukherjee R & Chatterjee A Defining the current scope and limitations of dual noncanonical amino acid mutagenesis in mammalian cells. Chem. Sci 8, 7211–7217 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.David Y, Vila-Perelló M, Verma S & Muir TW Chemical tagging and customizing of cellular chromatin states using ultrafast trans-splicing inteins. Nat. Chem 7, 394–402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevens AJ et al. Design of a Split Intein with Exceptional Protein Splicing Activity. J. Am. Chem. Soc 138, 2162–2165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stevens AJ et al. A promiscuous split intein with expanded protein engineering applications. Proc. Natl. Acad. Sci. U.S.A 114, 8538–8543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischle W et al. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116–1122 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Chang Y, Horton JR, Bedford MT, Zhang X & Cheng X Structural insights for MPP8 chromodomain interaction with histone h3 lysine 9: Potential effect of phosphorylation on methyl-lysine binding. J. Mol. Biol 408, 807–814 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothbart SB et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol 19, 1155–1160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lachner M, O’Carroll D, Rea S, Mechtler K & Jenuwein T Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Bian C, Chen Q & Yu X The zinc finger proteins ZNF644 and WIZ regulate the G9A/GLP complex for gene repression. Elife e05606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmitges FW et al. Histone Methylation by PRC2 Is Inhibited by Active Chromatin Marks. Mol. Cell 42, 330–341 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Creyghton MP et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A 107, 21931–21936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis PW et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jang Y et al. H3.3K4M destabilizes enhancer H3K4 methyltransferases MLL3/MLL4 and impairs adipose tissue development. Nucleic Acids Res. 47, 607–620 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W et al. Nucleolar protein Spindlin1 recognizes H3K4 methylation and stimulates the expression of rRNA genes. EMBO Rep. 12, 1160–1166 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442, 91–95 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wysocka J et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442, 86–90 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Guo Y et al. Methylation-state-specific recognition of histones by the MBT repeat protein L3MBTL2. Nucleic Acids Res. 37, 2204–2210 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen FX et al. PAF1 regulation of promoter-proximal pause release via enhancer activation. Science 357, 1294–1298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi X et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peña PV et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442, 100–103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan MAJ et al. A cryptic tudor domain links BRWD2/PHIP to COMPASS-mediated histone H3K4 methylation. Genes Dev. 31, 2003–2014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rotili D et al. A photoreactive small-molecule probe for 2-oxoglutarate oxygenases. Chem. Biol 18, 642–654 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharifi-Zarchi A et al. DNA methylation regulates discrimination of enhancers from promoters through a H3K4me1-H3K4me3 seesaw mechanism. BMC Genomics 18, 964–985 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zegerman P, Canas B, Pappin D & Kouzarides T Histone H3 lysine 4 methylation disrupts binding of nucleosome remodeling and deacetylase (NuRD) repressor complex. J. Biol. Chem 277, 11621–11624 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Huang Q et al. Depletion of PHF14, a novel histone-binding protein gene, causes neonatal lethality in mice due to respiratory failure. Acta Biochim. Biophys. Sin 45, 622–633 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Saksouk N et al. HBO1 HAT Complexes Target Chromatin throughout Gene Coding Regions via Multiple PHD Finger Interactions with Histone H3 Tail. Mol. Cell 33, 257–265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerace M et al. The scaffolding protein JADE1 physically links the acetyltransferase subunit HBO1 with its histone H3–H4 substrate. J. Biol. Chem 293, 4498–4509 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stevens AJ, Sekar G, Gramespacher JA, Cowburn D & Muir TW An Atypical Mechanism of Split Intein Molecular Recognition and Folding. J. Am. Chem. Soc 140, 11791–11799 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gramespacher JA, Burton AJ, Guerra LF & Muir TW Proximity Induced Splicing Utilizing Caged Split Inteins. J. Am. Chem. Soc 141, 13708–13712 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
References (Methods section only)
- 52.Cox J & Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.