

SUMMARY
Most monogenic disorders have a primary clinical presentation. Inherited ISG15 deficiency, however, has manifested with two distinct presentations to date: susceptibility to mycobacterial disease and intracranial calcifications from hypomorphic interferon-II (IFN-II) production and excessive IFN-I response, respectively. Accordingly, these patients were managed for their infectious and neurologic complications. Herein, we describe five new patients with six novel ISG15 mutations presenting with skin lesions who were managed for dermatologic disease. Cellularly, we denote striking specificity to the IFN-I response, which was previously assumed to be universal. In peripheral blood, myeloid cells display the most robust IFN-I signatures. In the affected skin, IFN-I signaling is observed in the keratinocytes of the epidermis, endothelia, and the monocytes and macrophages of the dermis. These findings define the specific cells causing circulating and dermatologic inflammation and expand the clinical spectrum of ISG15 deficiency to dermatologic presentations as a third phenotype co-dominant to the infectious and neurologic manifestations.
In Brief
Martin-Fernandez et al. report on five patients with inherited ISG15 deficiency, a recently discovered syndrome of type I IFN autoinflammation and mycobacterial susceptibility. This study defines an expanded clinical spectrum that now includes dermatologic disease and pinpoints the specific cell types driving inflammation.
Graphical Abstract
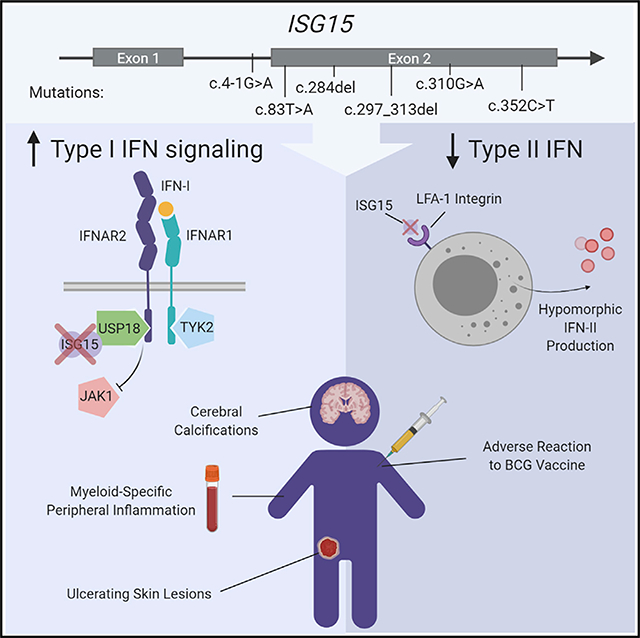
INTRODUCTION
Patients with monogenic disorders first raise clinical suspicion by the presence of a predominant disease manifestation. For example, Mendelian susceptibility to mycobacterial disease (MSMD) leads to susceptibility to mildly virulent environmental or vaccine strain (BCG) mycobacteria, which presents in infancy or early childhood as localized or disseminated lymphadenopathy. MSMD thus presents to pediatrics and pediatric infectious disease specialists. The genetic etiologies underlying MSMD center on mutations in genes encoding proteins of the IL-12/interferon-γ axis (e.g., IFNGR1, IFNGR2, IL12B, IL12RB1, STAT1, IRF8, NEMO) (Bustamante, 2020). Another example is the group of Mendelian disorders termed type I interferonopathies, which predominantly present to pediatric neurology with psychomotor delays and basal ganglia calcifications upon computed tomographic (CT) scan. Mutations of genes encoding proteins involved in nucleic acid metabolism or recognition, such as TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR1 (Crow, 2011, 2013, 2015; Crow and Rehwinkel, 2009; Rice et al., 2013; Rodero and Crow, 2016), underlie excessive interferon-I (IFN-α/β or IFN-I) production, a potent antiviral cytokine that acts developmentally as a neurotoxin. Individuals harboring mutations in these genes present with encephalopathy early in life and varying levels of neurologic dysfunction, including cognitive and motor disabilities. Secondary phenotypes may also develop, including chilblains cutaneous lesions on the distal extremities, autoantibodies, and systemic lupus erythematosus (SLE)-like clinical features.
To date, ISG15 deficiency has presented with two distinct clinical phenotypes, infectious and neurologic. ISG15 deficiency is a mixed syndrome of MSMD and monogenic type I interferonopathy. In the initial reports, MSMD was the primary phenotype in a family from Turkey and a family from Iran with ISG15 deficiencies (Bogunovic et al., 2012). MSMD was caused by lack of extracellular secreted ISG15, which normally engages the LFA-1 receptor on T and natural killer (NK) cells (Swaim et al., 2017), thus leading to hypomorphic induction of IFN-γ, akin to other genetic deficiencies leading to complete or partial loss of response to or production of IFN-γ (Bogunovic et al., 2012). In contrast, in a family from China with complete ISG15 deficiency, the primary clinical presentation was intermittent seizures stemming from intracranial calcifications (Zhang et al., 2015). Biochemically, lack of intracellular ISG15 leads to unstable levels of USP18 (Francois-Newton et al., 2012; François-Newton et al., 2011; Zhang et al., 2015), a potent negative regulator of IFN-I receptor, which results in continual downstream JAK-STAT signaling (Stark and Darnell, 2012) and augmented levels of interferon-stimulated genes (ISGs) in the blood. Importantly, the patients from Turkey and Iran previously described also had intracranial calcifications and high ISG expression in their blood, albeit clinically silent.
Here we report dermatological presentations as a third primary clinical phenotype in ISG15 deficiency.
RESULTS
Four Families, Five Patients, Six New ISG15 Alleles
We studied four families from the United States, Saudi Arabia, Spain, and Argentina. All five patients from these families presented with recurrent episodes of severe skin inflammation, and two of three vaccinated with BCG also presented with mycobacterial disease (Figures 1A and 1B, compared with previously reported patients listed in Tables S1 and S2 and Case Reports in STAR Methods). Targeted panel sequencing of primary immunodeficiency (PID) genes revealed that patient 1 (P1) harbored a compound heterozygous variant (c.310G>A and c.352C>T) in ISG15, resulting in the p.V104M substitution and a premature stop codon (p.Q118*), respectively. Whole-exome sequencing (WES) was used to identify the ISG15 variant in P2 and P3, who were found to be homozygous for an acceptor splice-site variant of ISG15 (c.4–1G>A) predicted to cause the skipping of exon 2. By targeted sequencing of PID genes, P4 was found to be compound heterozygous for the c.83T>A and c.284del variants, resulting in the p.L28Q substitution and a frameshift generating a premature stop codon, respectively. Finally, WES revealed that P5 was compound heterozygous for the c.284del variant (also found in P4) and a microdeletion c.297_313del (Figures 1C and 1D; Figure S1). These variants differed from those previously reported in ISG15-deficient patients (Bogunovic et al., 2012; Zhang et al., 2015). No rare variants of MSMD or type I interferonopathy genes were detected (Tangye et al., 2020). The familial segregation of ISG15 genotypes in these four families was consistent with an autosomal-recessive (AR) pattern of inheritance. In silico analysis revealed high CADD scores for all the alleles identified. Each of the alleles was either exceedingly rare (minor allele frequency [MAF] < 1:10,000, no homozygotes reported) or absent from public databases (Table S3). Together, these data suggest that these ISG15 genotypes may underlie additional forms of ISG15 deficiency.
Figure 1. Identification of Five Patients and Six Different Mutations of ISG15.

(A) Familial segregations of the ISG15 alleles. Family 1 includes one affected child (P1) carrying compound heterozygous variants (c.310G>A and c.352C>T) and one unaffected sibling. Family 2 includes two affected children (P2 and P3) with a splice-site variant (c.4–1G>A). Family 3 includes one affected child (P4) with compound heterozygous variants (c.83T>A and c.284del). Family 4 includes one affected child (P5) with compound heterozygous variants (c.284del and c.297_313del). The corresponding genotypes are indicated.
(B) Skin lesions from P1 (a and b) and P4 (c and d).
(C) Schematic localization of the ISG15 variants in the genomic DNA, indicated by the red arrows.
(D) Predicted localization of the four amino acid substitutions in the three-dimensional (3D) model of ISG15.
Complete and Partial Loss of Expression for Mutant ISG15 Alleles
We first investigated the effect of the compound heterozygous mutations identified in P1, P4, and P5. We transiently transfected HEK293T cells with the wild-type (WT) or mutant (c.352C>T [P1], c.310G>A [P1], c.284del [P4 and P5], c.83T>A [P4], and c.297–313del [P5]) ISG15 alleles to assess the corresponding levels of mRNA and protein. None of the variants overtly affected mRNA levels, with the exception of c.297–313del, for which the degradation of the mRNA by non-stop decay was predicted (Klauer and van Hoof, 2012) (Figure 2A). Transfection with c.352C>T, c.284del, and c.297_313del did not lead to the production of a detectable ISG15 protein. The expression of c.310G>A resulted in the production of a detectable protein, albeit at levels much lower than for WT ISG15, whereas c.83T>A was only weakly expressed, leading to the production of a protein that was smaller than the WT protein (Figures 2B–2D). We then investigated the effect of the fifth genetic variant, the homozygous essential splicing site c.4–1G>A mutation (in P2 and P3). ISG15 mRNA levels were lower in both patients with this variant than in controls (Figure S2A). We then performed 3′ RACE (rapid amplification of cDNA ends) analysis to detect splicing variants. We detected three different splicing variants, all of which were predicted to be defective (Figures 2E and 2F). Finally, western blotting of lysates from dermal hTERT-immortalized fibroblasts isolated from P2 and P3 stimulated with IFN-I revealed a complete lack of ISG15 protein production (Figure 2G). This resulted in impaired negative regulation of ISG expression, which was rescued with the WT allele (but not with a vehicle control) in the patient fibroblasts (Figures S2B and S2C). Collectively, these data suggest a complete absence of protein production from the four alleles identified in P1 (c.352C>T), P2, P3 (c.4–1G>A), P4 (c.284del), and P5 (c.297_313del) and extremely low levels of expression for the other two alleles: c.310G>A in P1 and c.83T>A in P4.
Figure 2. Allele Characterization.

(A) HEK293T cells were transfected with a plasmid encoding the various ISG15 variants: luciferase (Luc), wild-type ISG15 (WT), c.310G>A, c.352C>T, c.83T>A, c.284del ISG15, and c.297_313del. Relative mRNA levels for ISG15 assessed by qRT-PCR, performed three times for each variant, with technical triplicates; the data for a representative experiment (n = 3) are shown.
(B) HEK293T cells were transfected with a plasmid encoding the various ISG15 variants: Luciferase (Luc), wild-type ISG15 (ISG15), c.310G>A, or c.352C>T ISG15. Cell lysates were analyzed by western blotting for ISG15; a representative experiment is shown.
(C) HEK293T cells were transfected with a plasmid encoding the various ISG15 variants: Luciferase (Luc), wild-type ISG15 (ISG15), c.83T>A, or c.284del ISG15. Cell lysates were analyzed by western blotting for ISG15; a representative experiment is shown.
(D) HEK293T cells were transfected with a plasmid encoding the various ISG15 variants: Luciferase (Luc), wild-type ISG15 (ISG15), or c.297_313del ISG15. Cell lysates were analyzed by western blotting for ISG15; a representative experiment is shown.
(E) RNA was isolated from P2 and P3 and subjected to 3’RACE analysis. Schematic diagram of the ISG15 amplicons obtained on 3’RACE RT-PCR.
(F) 3′ RACE amplicons were inserted into a TOPO vector for the quantification of each splicing variant. In total, 26 colonies per patient were grown and sequenced. The graph shows the percentage of each of the three splicing variants identified.
(G) hTert-immortalized fibroblasts from a control donor (Control), P2, or P3 were treated with 1,000 U/mL of IFN-α2b for 24 h. Cell lysates were analyzed by western blotting with the indicated antibodies; data for a representative experiment are shown.
(H) HEK293T cells were transfected with a constant amount of wild-type USP18 (WT) together with various amounts of the different variants of ISG15 (WT or c.310G>A). Cell lysates were analyzed by western blotting with the indicated antibodies.
(I) HEK293T cells were transfected with a constant amount of wild-type USP18 (WT) together with various amounts of the different variants of ISG15 (WT, c.284del, or c.83T>A). Cell lysates were analyzed by western blotting with the indicated antibodies.
(J) Epstein-Barr virus (EBV)-transformed B cells from a control donor or P1 were stimulated with 1,000 U/mL IFN-α2b for 24 h. Cell lysates were analyzed by western blotting with the indicated antibodies; data from a representative experiment are shown.
(K) hTert-immortalized fibroblasts from a control or an ISG15-deficient patient were transduced with luciferase, WT ISG15, or c.310G>A ISG15 and sorted. These fibroblasts were treated with 1,000 U/mL IFN-α2bfor 12 h, washed with PBS, and left to rest for 36 h, after which relative mRNA levels were determined for MX1, three times for each individual, with technical triplicates; a representative experiment is shown. Bars represent the mean ± SD.
Partial as Opposed to Complete ISG15 Deficiency
ISG15 downregulates the IFN-I signaling pathway by stabilizing USP18, a potent negative regulator (Zhang et al., 2015). USP18 acts to prevent JAK1 from binding to IFN-I receptor and hence arrests phosphorylation of STAT1 and STAT2, essential components of the transcription factor complex that induces ISGs. ISG15 is also involved in ISGylation, a process in which ISG15 is covalently linked to newly synthesized proteins in a ubiquitin-like fashion (Hermann and Bogunovic, 2017). We first assessed the effects on USP18 stabilization of the alleles generating low levels of protein (c.310G>A or c.83T>A). We transiently transfected cells with WT USP18 and various amounts of WT, c.310G>A, or c.83T>A ISG15 (or c.284del ISG15 as a negative control). USP18 was stabilized by the c.310G>A ISG15 variant of P1, but much larger amounts of plasmid for this variant than for the WT ISG15 were required to achieve the same effect, presumably because of the very small amounts of mutant protein produced (Figure 2H). However, the c.83T>A allele of P4 was unable to stabilize USP18, suggesting a total loss of this function (Figure 2I). We then assessed the functionality of the c.310G>A allele of P1 for ISGylation. We transiently transfected HEK293T cells with constructs encoding the WT ISG15 or the c.310G>A variant, together with all the components of the ISGylation machinery (UbE1L, UBCH8, and HERC5). Again, presumably because of the smaller amounts of protein produced, the ISGylation capacity of the c.310G>A variant of P1 was significantly lower than that of WT ISG15, this phenotype being rescued by larger amounts of protein (Figure S2D). We then stimulated Epstein-Barr virus-transformed B cells (EBV-B cells) from P1 with IFN-α2b for 24 h and analyzed the levels of ISG15 and USP18 proteins. Low levels of ISG15 production were detected, and the stability of USP18 was low (Figure 2J). Furthermore, expression of the c.310G>A allele in an ISG15-null background did not rescue IFN-I-mediated inflammation, whereas expression of the WT allele restored normal regulation (Figure 2K). Overall, these results demonstrate that the protein encoded by the c.310G>A allele of P1 can stabilize USP18 and undergo ISGylation to other proteins, but it is severely hypomorphic because of the small amounts of protein produced. Thus, the autoinflammatory phenotype of P1 results from the production of insufficient quantities of ISG15 due to a premature stop codon (c.352C>T) and severe hypomorphism (~1%−5% of normal levels; c.310G>A). P1 is thus the first individual presenting with disease in which ISG15 deficiency is partial, as opposed to complete. However, this partial deficiency is so severely hypomorphic that it is clinically indistinguishable from complete ISG15 deficiency.
Myeloid Cells Display Enhanced IFN-I Activity
In the absence of free intracellular ISG15, USP18 is unstable and the downregulation of IFN-I signaling is impaired, resulting in persistent ISG expression. By contrast to STING-associated vasculopathy with onset in infancy (SAVI), but as in most interferonopathies tested to date, IFN-α levels were no higher in cells from P1 than in those of healthy donors, whereas extracellular IFN-α levels were higher in the plasma of P1, P3, and P5 than in plasma from healthy controls (peripheral blood mononuclear cells [PBMCs] from other patients and plasma from P2 and P4 were not available) (Figure S3). We also analyzed ex vivo mRNA levels for the five standard ISGs (IFIT1, MX1, IFI27, RSAD2, and IFI44I) in whole blood. P1, P2, P3, and P5 had higher steady-state levels of ISG expression than healthy controls (no blood was available for P4) (Figure 3A), suggesting that they did indeed have a classical type I interferonopathy. However, the relative contributions of specific cell types to this whole-blood phenotype were unknown. We bridged this gap in our knowledge by incorporating SIGLEC1 (CD169), a transmembrane ISG surface protein, into a 40-marker CyTOF immunophenotyping panel. The distributions of classical cell types were largely similar in P1 and a healthy control (Table S4), but t-distributed stochastic neighbor embedding (t-SNE) analysis revealed the strong expression of SIGLEC1 by a different cell population in the patient (Figure 3B). Further analyses of this cell population revealed that all monocyte subsets (CD14+CD16−, CD14+CD16+, and CD14−CD16+) and myeloid dendritic cells (Lin−CD11c+, including CD1c+ [DC2s] and CD141+ [DC1s]) had significantly higher levels of SIGLEC1 expression (Figures 3C and 3D) than the corresponding control cells. We further investigated the relative sensitivity of all whole-blood cells to circulating IFN-I by measuring the levels of STAT1, a putative marker of IFN-I exposure; phospho-STAT1, a marker of active canonical IFN-I signaling; and phospho-STAT3, a marker of non-canonical IFN-I signaling. Basal levels of STAT1, an ISG, were higher in all cell subsets (plasmacytoid dendritic cells, myeloid dendritic cells [DC1s and DC2s], classical monocytes, non-classical monocytes, CD4+ T cells, CD8+ T cells, eosinophils, basophils, neutrophils, and B cells) from P1 than in control cells (Figure 3E), indicating, a priori, a response to IFN-I. However, phospho-STAT1, a marker of active canonical IFN-I signaling, was most abundant in non-classical CD14−CD16+monocytes and CD1c+ dendritic cells (DC2) and only mildly upregulated in all other cell types, with lower levels in basophils. Furthermore, evaluations of pSTAT3, pSTAT5, pSTAT6, pp38, pMAPKAP2, pERK, and pS6 levels revealed that non-canonical IFN-I signaling was active in granulocytes and in neutrophils in particular (Figure 3F), despite all cells’ encountering IFN-I. Together, these data suggest that non-classical monocytes and DC2 cells were the principal cells involved in canonical IFN-I-induced inflammation, whereas granulocytes displayed pronounced non-canonical IFN-I-induced inflammation, in ISG15 deficiency.
Figure 3. Monocytes and Dendritic Cells in the Blood Drive Type I Interferonopathy in Patients with ISG15 Mutations.

(A) Relative mRNA levels for IFIT1, MX1, RSAD2, IFI44L, and IFI27 in peripheral blood from a healthy control, P1,P2, P3, and P5, as assessed by qRT-qPCR. Bars represent the mean ± SD.
(B) PBMCs from patients and healthy controls were immunophenotyped by CyTOF technology with a 40-marker panel. t-Stochastic neighbor embedding (t-SNE) plot of PBMCs from P1 and three healthy controls, showing SIGLEC1 (CD169) expression in the various immune populations. The monocyte compartment displays high levels of SIGLEC1 (CD169) expression in P1.
(C) SIGLEC1 (CD169) expression in the various subtypes of monocytes (CD14+CD16−, CD14+CD16+, and CD14−CD16+).
(D) SIGLEC1 (CD169) expression in dendritic cells.
(E) PBMCs from P1 and a control were analyzed by CyTOF. with a panel including several activated signaling markers. Heatmaps show the median expression levels of STAT1 and STAT3 in P1 relative to a healthy control, for the various immune cell subtypes.
(F) PBMCs from P1 and a control were analyzed by CyTOF. with a panel including several activated signaling markers. Heatmaps show the median expression levels of pSTAT1, pSTAT3, pSTAT5, pSTAT6, pp38, pMAPKAP2, pERK, and pS6 in P1 relative to a healthy control, for the various immune cell subtypes.
Single-Cell RNA Sequencing Reveals a Predominant ISG Signature in Myeloid Subsets
We then performed single-cell RNA sequencing (scRNA-seq) on PBMCs from P1 to analyze the impact of ISG15 deficiency on the cellular distribution and transcriptomic profiles at single-cell resolution. We performed droplet-based 3′-end massively parallel scRNA-seq on a 10X Genomics Chromium Platform, followed by unbiased clustering, with manual curation of cluster identity with known cell type markers (Figures 4A and 4B). An analysis of the genes differentially expressed between clusters revealed a high degree of enrichment in ISG expression in monocyte clusters relative to other subsets of PBMCs from the patient (CD4+ T cells, CD8+ T cells, NK cells, and B cells) (Figure 4C). These other subsets of PBMCs from the patient had similar levels of ISG expression, although more subtle differences in expression would probably be undetectable on scRNA-seq platforms, because of high mRNA dropout rates. However, eight of the ten genes most strongly upregulated in CD14+ monocytes from the patient relative to a healthy control (log fold change [FC] > 0.25 in >20% of cells) were canonical ISGs (IFITM3, IFITM2, IFI6, LY6E, IFI27, ISG15, CCL3, and LGALS3BP) (Schoggins et al., 2011). Likewise, seven of the top ten genes upregulated in non-classical monocytes were ISGs (ISG15, CCL2, IFI6, IFI27, APOBEC3A, S100A8, and LGALS2) (Figures 4D and 4E). The differences between cell types in the potency of the response to persistent IFN-I signaling probably reflect multiple layers of control. We then mined the DICE Immune Cell Atlas and found that monocytes were among the cells with the lowest basal levels of USP18, suggesting a mechanism for the differential IFN-I signatures (Figure S4). These lower initial levels of USP18 may potentiate the degradation of this molecule in the absence of ISG15, accounting for the monocytic phenotype in ISG15−/− individuals. Thus, these high-resolution analyses of PBMCs from a patient with biallelic mutations of ISG15 suggested that the type I interferonopathy was most marked in the monocyte and dendritic cell subsets.
Figure 4. ScRNA-Seq of ISG15−/− PBMCs Reveals Monocyte-Driven ISG Expression.

(A) Single-cell RNA sequencing was performed on PBMCs isolated from P1 and a healthy control, with the Chromium Platform from 10X Genomics. tSNE plots were generated by unbiased clustering and manual curation of immune cell subsets by cluster-specific genes.
(B) Bubble plots displaying the cell type markers identifying each cluster in the patient (green) and control (blue) data, with the color intensity representing scaled expression and the bubble size representing the percentage of cells expressing the transcript.
(C) Expression of representative ISGs (IFITM2, IFITM3, IFI6, LY6E, IFI27, and ISG15) in patient PBMCs, showing strong localization to monocytes.
(D) Heatmap of the top ten genes most strongly upregulated (mean cluster expression with log[fold change] > 0.25) in the patient’s classical monocytes relative to those of a healthy control.
(E) Heatmap of the top ten genes most strongly upregulated (log[fold change] > 0.25) in the patient’s non-classical monocytes relative to those of the healthy control. Columns represent expression levels in individual captured cells. Asterisk indicates well-documented interferon-stimulated genes.
Skin Lesions Display STAT Activation in Keratinocytes and Endothelial and Myeloid Cells
All five of the patients with biallelic mutations of ISG15 presented with severe skin inflammation (Figure 1B; Table 1; Table S2; Case Reports in STAR Methods). These lesions did not resemble chilblains, as in Aicardi-Goutiéres syndrome (AGS). Instead, they were ulcerative and located in the perineal, groin, scalp, neck and axillary regions, suggestive of unique patterns of skin inflammation in the absence of documented infectious disease. The lesions were largely resistant to steroid treatment, and were recurrent and necrotizing, but they resolved spontaneously, leaving post-inflammatory hyperpigmentation. Infectious complications were sometimes observed, but no common microorganism was isolated (Case Reports in STAR Methods). The combined score for clinical features placed ISG15-deficient patients (median score of 8) on par with patients with sporadic chilblain lupus and was significantly lower than that for patients with TREX1 mutations (score 18.5), although the presentations are qualitatively different (Table 1). We therefore performed further experimental investigations to determine whether the skin phenotype was intrinsic to resident skin cells, mediated by immune infiltration, or both. We first used CRISPR-Cas9 to knock out ISG15 in a human keratinocyte cell line (HaCaT) (Figure 5A). We then stimulated WT and ISG15-knockout (KO) HaCaT cells with IFN-I and measured ISG levels. ISG15-KO cells were significantly more responsive to the continuous presence of IFN-I, in terms of mRNA levels for IFIT1, MX1, OAS1, and CXCL10, suggesting that the skin epithelium can intrinsically dysregulate IFN-I responses (Figure 5B). We next generated ISG15-deficient iPSCs. Then, WT and ISG15-deficient iPSCs were differentiated into endothelial cells (Figure 5C). Following stimulation with IFN-I, ISG induction increased in ISG15-deficient cells, but not in WT endothelial cells, as shown by the levels of IFIT1, MX1, OAS1, and CXCL10 mRNA (Figure 5D). We then performed immunohistochemistry on skin sections from the lesions of P1. We monitored skin architecture, which displayed significant alterations with respect to WT skin, including a thickening of the epidermis, consistent with the clinical presentation (Figures 5Ea and 5Ee). The keratinocytes in the skin epidermis were positive for pSTAT1 in P1, but not in the control (Figures 5Eb and 5Ef). Furthermore, endothelial cells had higher pSTAT1 levels than those in control skin, whereas no such difference was observed for dermal fibroblasts (Figures 5Ec and 5Eg). The patient’s endothelial cells did not show a flat morphology, but insteada cuboidal shape suggestive of inflammation. Finally, we assessed the possible contribution of infiltrating immune cells to skin inflammation, by co-staining for CD68, a macrophage/DC marker. Skin sections from P1 had abundant CD68+ cells in the dermis, whereas this feature was not observed in control skin. The infiltrating/resident cells were also positive for pSTAT1 (Figures 5Ed and 5Eh), suggesting that IFN-I also activated myeloid cells in the skin. Thus, the inflammation of the skin in these patients is due to IFN-I dysregulation in the skin epithelium, endothelial cells, and CD68+ infiltrating or resident myeloid cells. Given the severity and depth of necrosis, the predominant etiology is likely not epithelial cell inflammation but, perhaps, severe vasculitis.
Table 1.
Severity Scoring for Skin Lesions
| Skin Lesions | |||||
|---|---|---|---|---|---|
| Patient Number | |||||
| P1 | P2 | P3 | P4 | P5 | |
| Sex | F | M | M | F | F |
| Age | 2 | 9 | 7 | 6 months | 5 |
| Mutation in ISG15 | c.310G>A and c.352C>T | c.4-1G>A | c.4-1G>A | c.83T>A and c.284del | c.284del and c.297_313del |
| Manifestation in early childhood = 2 points | 2 | 2 | 2 | 2 | 2 |
| Cold-induced skin lesions = 1 point | 0 | 0 | 0 | 0 | 0 |
| Persistence of skin lesions in summer = 1 point | 1 | 1 | 1 | 1 | 0 |
| Simultaneous skin lesions on 2 body areas = 1 point | 0 | 1 | 0 | 1 | 1 |
| Simultaneous skin lesions on 3 body areas = 2 points | 0 | 0 | 0 | 0 | 0 |
| Simultaneous skin lesions on 4 or more body areas = 3 points | 0 | 0 | 0 | 0 | 0 |
| Presence of ulcerations = 1 point | 1 | 1 | 1 | 1 | 1 |
| Necrosis, mutilations = 2 points | 2 | 2 | 0 | 2 | 2 |
| Postinflammatory hyperpigmentation = 1 point | 1 | 1 | 1 | 0 | 0 |
| Postinflammatory onychodystrophy = 1 point | 0 | 0 | 0 | 0 | 0 |
| Raynaud‘s syndrome = 1 point | 0 | 0 | 0 | 0 | 0 |
| Photosensitivity = 1 point | 0 | 0 | 0 | 0 | 0 |
| Arthritis of 1 large joint = 1 point | 0 | 0 | 0 | 0 | 0 |
| Arthritis of ≥ 2 large joints = 2 points | 0 | 0 | 0 | 0 | 0 |
| Leukopenia = 1 point | 0 | 0 | 0 | 1 | 0 |
| Lymphopenia= 1 point | 0 | 0 | 0 | 0 | 0 |
| Thrombopenia = 1 point | 0 | 0 | 0 | 0 | 0 |
| Hemoglobinemia or erythropenia = 1 point | 1 | 0 | 0 | 0 | 0 |
| Low complement C3 and/or C4 = 1 point | 0 | 0 | 0 | 0 | 0 |
| Hypergammaglobulinemia = 1 point | 0 | 0 | 0 | 0 | 0 |
| Anti-ssDNA/ anti-dsDNA antibodies = 1 point | 0 | 0 | 0 | 0 | 0 |
| Antinuclear antibodies = 1 point | 0 | 0 | 0 | 0 | 0 |
| Anti Ro/SSA; anti La/ SSB antibodies = 1 point | 0 | 0 | 0 | 0 | 0 |
| Typical lupus histology = 1 point | 0 | 0 | 0 | 0 | 0 |
| Immune complex or complement deposition at the basement membrane zone = 1 point | 0 | 0 | 0 | 0 | 0 |
| Summary | 8 | 8 | 5 | 8 | 6 |
Figure 5. IFN-I Dysregulation in Skin, Vascular Endothelial Cells, and Infiltrating Myeloid Cells.

(A) CRISPR was used to knock out ISG15 in HaCaT cells. The cells were stimulated with 1,000 U/mL IFN-α2b for 24 h. Cells were lysed, and the lysates were analyzed by western blotting with the indicated antibodies; the results of a representative experiment are shown.
(B) WT and ISG15-KO HaCaT cells were stimulated with 1,000 U/mL IFN-α2b for 12 and 24 h. Relative mRNA levels for IFIT1, MX1, OAS1, and CXCL10 were assessed by qRT-qPCR. Bars represent the mean ± standard error of the mean (SEM).
(C) Human WT and ISG15-deficient iPSCs were differentiated to develop into endothelial cells and stimulated with 1,000 U/mLIFN-α2b for 24 h. Cell lysates were analyzed by western blotting with the indicated antibodies; data from a representative experiment are shown.
(D) WT and ISG15-deficient endothelial cells were stimulated with 1,000 U/mL IFN-α2b for 12 and 24 h. Relative mRNA levels for IFIT1, MX1, OAS1, and CXCL10 were determined by qRT-qPCR. Bars represent the mean ± SEM.
(E) Representative H&E staining and staining for pSTAT1 (purple) and CD68 (brown) immunohistochemistry (IHC) results for skin biopsies from P1 and a healthy control. Original magnification: 10× (a and e), 40× (b-d, f, and g). Arrows indicate pSTAT1+ cells. Asterisks indicate CD68+ cells (macrophage infiltration).
DISCUSSION
We have identified 11 individuals, with nine biallelic mutations of ISG15, to date. These patients belong to seven kindreds from seven countries (Turkey, Iran, China, the United States, Saudi Arabia, Spain, and Argentina). In nine individuals, these inborn errors of ISG15 result in a complete loss of protein production and function. In one individual, some protein is produced, but the loss of function is nevertheless essentially complete. In the remaining patient, the defect is partial, defining a new form of inherited ISG15 deficiency. This c.310G>A ISG15 allele results in very low protein levels (estimated at about 5% of WT ISG15 levels). Despite the retention of all the functions of the WT ISG15 (ISGylation, deISGylation, and stabilization of USP18), this allele is rendered severely hypomorphic by its very low levels of expression, even in overexpression assays. The defect is so profound that individuals carrying it are clinically indistinguishable from those with a complete loss of function. The search for ISG15 mutations in patients with milder mycobacterial disease and/or type I interferonopathies may reveal other hypomorphic mutations. The functional characterization of four human ISG15 non-synonymous and essential splice variants found in the homozygous state in the general population would also be useful. Together, these two approaches may define the threshold of ISG15 activity that is pathogenic. These studies may also identify variants dissociating the different activities of ISG15.
Intracranial calcifications were observed in all the patients who underwent brain CT scans. Similarly, ISG products were present at high levels in the blood of all patients in which they were determined. ISG15 deficiency may therefore be considered a bona fide type I interferonopathy. The only clinical feature not considered in previous reports of ISG15 deficiency was skin inflammation, which is not infrequent in patients with type I interferonopathies (Peschke et al., 2014; Rodero and Crow, 2016). However, none of ISG15-deficient patients suffered from cold-induced inflammatory conditions affecting the skin of the distal extremities, as observed in familial AGS. Both these skin conditions have an early onset and lead to ulceration and necrosis, but the lesions of ISG15-deficient patients had a different distribution over the body, being located in the perineal, groin, scalp, neck, and axillary regions, indicating a unique pattern of skin inflammation (Crow, 2011, 2013, 2015; Crow and Rehwinkel, 2009; Rice et al., 2013; Rodero and Crow, 2016; Taft and Bogunovic, 2018). Furthermore, none of the ISG15-deficient patients presented with arthritis, hemoglobinemia, SLE, Raynaud’s syndrome, or post-inflammatory onychodystrophy resembling that observed in AGS patients with the TREX1 D18N mutation.
The pathophysiology of the skin lesions observed in ISG15-deficient patients is probably complex. Our in vitro and ex vivo data suggest that keratinocytes have high levels of pSTAT1 in the absence of ISG15. Endothelial cells in culture and in the tissue require ISG15 to curtail IFN-I-mediated inflammation. Furthermore, the in situ staining of CD68+pSTAT1+ macrophages within the lesions indicated that myeloid cells contributed to the pathological skin features. Together, our data suggest that IFN-I-mediated inflammation involves complex cooperation among keratinocytes, endothelial, and myeloid cells. This cooperation between different cell types in the skin may also provide clues to the elusive pathogenesis of the intracranial calcifications localized in the basal ganglia. Neurons, astrocytes, oligodendrocytes, microglial cells, and infiltrating leukocytes in the globus pallidus area and the putamen may cooperate to induce cell-specific IFN-I-mediated local inflammation. For diagnosis, it is therefore essential to recognize the syndromic features of ISG15 deficiency. ISG15-deficient patients have been identified on the basis of several different presentations: a reaction to BCG vaccination, intermittent seizures, or, as in the patients described here, dermatological features. Specialists in pediatric infectious diseases, neurology, and dermatology alike should therefore be aware of the phenotypic diversity with which ISG15 deficiency (and other type I interferonopathies) can present. JAK inhibitors are probably the best option for the treatment of severe cases, although the dermatologic lesions reported here resolved spontaneously or with corticosteroid therapy.
One key unanswered question in studies of autoinflammation concerns the specific identity of the cells predominantly involved in or responding to the inflammatory processes (Broderick, 2019). The production of IFN-I was recently shown to occur in pDCs and monocytes in STING-mutated patients, but no particular cell type has been identified as the main producer of IFN-I in patients with other monogenic causes of AGS, juvenile-onset dermatomyositis, or SLE (Rodero et al., 2017). The response to IFN-I across cell types has not been characterized in type I interferonopathies. We show here that myeloid cells (monocytes and dendritic cells) are the primary responders to canonical IFN-I-mediated inflammation in the peripheral blood of patients with ISG15 deficiency. This finding adds to the existing evidence suggesting a major role for myeloid cells in type I IFN inflammation (Broderick, 2019; Lavin et al., 2015; Uccellini and Garcia-Sastre, 2018). The current gold standard for the diagnosis of type I interferonopathies is the determination of bulk RNA levels in peripheral blood/PBMCs. Given our findings, indicating that not all cell types contribute equally, a study of specific populations may be warranted.
In conclusion, ISG15 deficiency is not only functionally different from other genetic forms of type I interferonopathy, except for USP18 and STAT2 deficiencies (Alsohime et al., 2020; Gruber et al., 2020; Meuwissen et al., 2016), but it is also clinically unique, as it is distinguished by MSMD, intracranial calcifications, and specific patterns of dermatological lesions, which appear to be independently sufficient for diagnosis and are co-dominant.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to the Lead Contact, Dusan Bogunovic (dusan.bogunovic@mssm.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients
At the initiation of this study, when genetic analysis was performed, the demographics of the patients were characterized as follows: P1, female age 2; P2, male age 8; P3 male age 7; P4 female age 7; P5 female age 6. The study of human subjects was approved by the IRB of Icahn School of Medicine (New York, USA) (IF2349568), INSERM and Necker Hospital for Sick Children (Paris, France), and Rockefeller University (New York, USA). P1 was enrolled in a NIAID IRB-approved protocol and provided written informed consent for study participation in accordance with the Helsinki Declaration.
Cell lines
B cells from P1 were transformed by infection with Epstein-Barr virus (EBV). In brief, human peripheral blood mononuclear cells (5–10×106 cells in 2.0 mL of HEPES [20 mM, pH 7.2–7.5]-buffered RPMI-1640 media supplemented with [final concentrations] L-glutamine [2 mM], penicillin [100 U/ml], streptomycin [100 U/ml], gentamicin [100 mg/ml], and fetal bovine serum [30%]) were cultured in T25 flasks at 37°C with 5% CO2 after the addition an equal volume of Epstein-Barr virus (EBV)-containing, sterile-filtered culture supernatant fluid from B95.8 cells (ATCC # CRL 1612; B95.8 is an immortalized cell line derived from marmoset blood leukocytes infected with EBV that sheds active EBV). Cyclosporin A [1 mg/ml], an immunosuppressive agent, was added to the cultures to inhibit the growth of T-lymphocytes and prevent killing of infected B cells. When small aggregates of growing cells were observed, the cell culture was then transferred to a T75 flask for expansion. The cell culture was replenished with fresh RPMI-1640 media containing the same supplements with reduced fetal bovine serum [20%] as needed until a daily feeding was required and the EBV-transformed cell line appeared stable.
Dermal fibroblasts from P2, P3 and healthy controls were immortalized by stable transduction with hTERT. Patient and control cells were stably complemented with WT, c.310G > A, or c.352C > T ISG15 or luciferase, by lentiviral transduction. Transduced fibroblasts were sorted on the basis of RFP expression (BD FACS Aria II). HEK293T cells (CRL-11268; ATCC) and hTERT-immortalized fibroblasts were cultured in DMEM (Dulbecco’s modified Eagle medium) supplemented with 10% fetal calf serum, 1% penicillin-streptomycin and 1% L-glutamine. All cells were cultured at 37°C, under an atmosphere containing 10% CO2. All cells were tested for mycoplasma contamination with the MycoAlert(tm) PLUS Mycoplasma Detection Kit (Lonza) according to the manufacturer’s instructions.
The HaCaT human immortal keratinocyte cell line (Kindly provided by Prof. Thomas Werfel, Department of Immunodermatology and Allergology, Hannover Medical School, Hannover, Germany) was cultured in RPMI 1640 medium (GIBCO Life Technologies) supplemented with FCS (30%), penicillin/streptomycin (100 μg/ml), gentamycin (500 μg/ml), Glutamax (1%) and NEA (1%).
For iPSC cells, we used the HSC_iso4_ADCF_SeV-iPS2 (Phoenix) cell line (Haase et al., 2017), registered as MHHi001-A (https://hpscreg.eu/). Human iPSCs were maintained under feeder-free conditions in mTeSR1, on Geltrex®-coated tissue culture flasks. The cells were cultured as single-cell monolayers at a seeding density of 3.6×104 cells/cm2. They were passaged every 3–4 days with Accutase. Endothelial cell (EC) differentiation was induced by treating the hiPSC cultures with 25 ng/ml BMP4 and the WNT pathway activator CHIR 90221 (7.5 μM) in N2B27 medium for 2 days without replacing the medium. From day (D) 3 to D7, cultures were maintained in StemPro-34 medium supplemented with 260 ng/ml rhVEGF-A165 and 2 μM For skolin with daily replacement of the medium. On D7 of differentiation the amount of ECs was determined using flow cytometric analysis for CD31. CD31 positive cells were purified by magnetic-activated cell sorting (MACS) utilizing CD31 MicroBead Kit. Human iPSC-derived ECs were cultivated in EGM-2 medium (2% FCS) on fibronectin-coated plates and were split every 3–4 days by Accutase.
METHOD DETAILS
Case reports
P1 is a girl born in 2015 to non-consanguineous parents from the USA. She was not vaccinated with the BCG vaccine. At the age of two years, she presented a right inguinal skin lesion consisting of a hypopigmented linear streak with distinct areas of circular erythema and underlying induration. Over the following year, the lesions ulcerated and spread to the suprapubic and lower abdominal regions. Biopsy revealed subcutaneous perivascular lymphocytic inflammation without vasculitis. Paniculitis, with neutrophil involvement, was noted in the underlying fat. No necrosis was observed and no fungal microorganisms were detected. A second biopsy of the abdominal lesions showed areas of normal skin and areas of necrotic skin with abundant Aspergillus hyphae invading the subcutaneous tissue. Previous treatment with topical steroids had worsened the lesions. Chest CT scans showed no signs of infection, and blood tests (beta-D-glucan and galactomannan determinations) were negative, indicating an absence of invasive fungal infection. The wound was excised and, approximately two years after their initial detection, the lesions resolved with scarring. Whole-blood activation assays showed that P1, like all ISG15-deficient patients tested to date, had a mild impairment of type II IFN production, but not of IL-12 production, in response to BCG (Figure S5A), consistent with her severely hypomorphic ISG15 allele expression. These data support previous findings suggesting a role for ISG15 in controlling type II IFN production (Bogunovic et al., 2012; Zhang et al., 2015).
P2 is a boy born in 2009 to consanguineous parents from Saudi Arabia. He was admitted to the neonatal intensive care unit after birth for transient tachypnea and indirect hyperbilirubinemia requiring a single session of phototherapy. He was observed to have a small area of alopecia at the left occipitoparietal area of the scalp, with no cutis aplasia. At the age of one month, he was vaccinated with BCG, to which he displayed an exaggerated response, with right inguinal lymphadenopathy and erythema of the overlying skin. One month later, he developed left axillary lymphadenopathy, with extensive skin lesions, abscess formation and purulent discharge. He was repeatedly admitted for the scalp lesions, with erythema and microabscess formation, which responded partially to antibiotics. In total, the patient was admitted on five occasions for infected skin lesions. Escherichia coli (MRO) and then Staphylococcus aureus (MRSA) were cultured from the lesions. During the patient’s last admission, deep ulcerated necrotic lesions of heterogeneous size, surrounded by dusky erythema, were noted on the skin of the neck and inguinal region. Previous depressed scars were observed on the occiput. This patient has since been symptom-free.
P3 is a boy born in 2011 to consanguineous parents. He is the cousin of P2. At the age of one month, he was vaccinated with BCG. He was identified as having ISG15 deficiency during genotyping of the relatives of P2. A review of P3′s medical history revealed normal developmental milestones, subjective reporting of recurrent uncomplicated upper respiratory infections, and a history of intermittent, self-resolving skin lesions in the right axilla between the ages of six months and four years. However, these complications were never considered severe enough to seek medical attention.
P4 is a girl born in 2011 to non-consanguineous parents from Spain. She initially presented at three months of age, with nonspecific right chorioretinitis followed by severe recurrent skin ulcerations of the leg and groin at the age of six months. Histological analyses of the lesions revealed necrosis extending to the upper hypodermis, and imaging results revealed necrotizing fasciitis. Several gram-positive and gram-negative bacteria were cultured from the ulcer. Lesions continued to develop over the years, with ulcers involving the vulva and perianal region, together with mouth ulcers and erythematous papules and pustules on the abdomen and arms. Febrile flare-ups and high levels of acute-phase reactants were consistently noted. Given this history, a diagnosis of probable early-onset Behçet’s disease was made. The patient responded well to oral steroids, but she ultimately displayed resistance or intolerance to standard therapies for Behçet’s disease, including colchicine, etanercept, anakinra, adalimumab with concomitant methotrexate and infliximab. Tocilizumab treatment was then initiated, in combination with short-course prednisolone, and a complete remission of the systemic features of the disease was achieved. Since the initiation of tocilizumab at the age of five years, she has experienced one severe episode of ulceration and several subtle lesions, all of which resolved following short courses of oral corticosteroids. Genetic testing revealed ISG15 mutations, and a CT scan was performed (Figure S5B), which showed asymptomatic cerebellar and basal ganglial calcifications. The patient is currently doing well.
P5 is a girl born in 2013 to non-consanguineous parents from Argentina. She was vaccinated with BCG vaccine and displayed a local reaction at the site of vaccination without treatment. At the age of six months, she was admitted to hospital for ulcerated skin lesions, which were treated with antibiotics. At the age of one year, P5 presented ulcers of the inguinal region and vulva. A biopsy revealed necrosis of the hypodermis; cultures and direct staining for bacteria (including mycobacteria) and fungi were negative. The biopsy showed vasculitis. This patient frequently suffers cutaneous lesions and also has a history of recurrent pneumonia requiring hospitalization. She has basal cerebral calcifications. WES revealed a compound heterozygous mutation of the ISG15 gene, with both alterations affecting exon 2. Sanger sequencing confirmed this compound heterozygosity. One of the patient’s parents is heterozygous for one of the mutations and her brother is wild-type. A more extensive clinical description of this patient has been submitted to another journal as a case report.
Genetic analysis
We performed next-generation sequencing (NGS) on 320 genes associated with primary immunodeficiency disorders, as previously described (Kuehn et al., 2017), for patient 1. We captured the target regions (including coding exons and the 50 bases immediately flanking the genes) with reagents from a custom-designed HaloPlex Target Enrichment kit (Agilent Technologies, Catalog # G9912B). Briefly, we digested genomic DNA (gDNA) with restriction enzymes and hybridized fragments to probes with ends complementary to the target. We then captured the target DNA with streptavidin beads and by ligating circularized fragments, and performed polymerase chain reaction (PCR) amplification on the captured target libraries. Quality control was performed, for all libraries, with an Agilent Bioanalyzer equipped with a high-sensitivity chip. Template dilutions were calculated after the standardization of library concentrations to ~100 pM with the Ion Library Equalizer kit (Life Technologies, Catalog # 4482298). Library templates were clonally amplified and enriched with the Ion Chef system (Life Technologies, Ion PI HiQ Chef Kit, Catalog # A27198), according to the manufacturer’s protocol. Samples were processed with the standard Ion Proton PI chip v3 (Life Technologies, Catalog # A26770). Read mapping and variant calling were performed with Ion Torrent Suite software v4.4.2, as previously described (Kuehn et al., 2017). Sanger sequencing of ISG15 was performed to confirm the identity of the targeted variants detected by NGS in P1 and both parents.
For patients 2 and 3, Genomic DNA was extracted from whole blood samples with The iPrep ™ Pure Link® kit (Invitrogen™ IS10005) and iPrep Instruments from Life Technologies. The iPrep ™ Pure Link® kit contains: 1 iPrep™ PureLink® gDNA Blood cartridge, iPrep ™ Sample and elution tubes and bag iPrep™ tips and tip holders. WES was performed with 3 μg of DNA from P2, from Family 2. The gDNA was sheared with Covaris S2 Ultrasonicator (Covaris). An adaptor-ligated library was prepared with the Paired-End Genomic DNA Sample Prep kit V1 (Illumina). Exome capture was performed with the 71 Mb SureSelect Human All Exon kit (71 Mb version-Agilent Technologies). Sanger sequencing was performed to confirm the mutation in P2 and familial segregation was performed, leading to the identification of P3. The exon 2 and its flanking intron regions were amplified from gDNA extracted of whole blood with the 5′-CGGGATGTAGAGGACAGACA-3′ and 5′-ACCCTTATCCCTTCACTTGG-3′ primers. PCR was carried out with Taq polymerase (10342020, Invitrogen), the products were purified by centrifugation through Sephadex G-50 Superfine resin (GE Healthcare) and then sequenced using BigDye Terminator v3.1 (ThermoFisher). Sequencing products were purified by centrifugation through Sephadex G-50 Superfine resin, and sequences were analyzed with an ABI Prism 3700 apparatus (Applied Biosystems).
An NGS targeted panel was used for P4. Briefly, DNA extracted from peripheral blood was screened for mutations with a customized NGS gene panel containing 453 genes associated with immunopathologies. This panel was designed with NimbleDesign software. For each sample, paired-end libraries were created with the KAPA HTP Library Preparation Kit for Illumina platforms (Roche Nimblegen), SeqCap EZ Library SR (Roche Nimblegen) and the NEXTflex-96 PreCapture Combo Kit for indexing (Bioscientific). Sequencing was conducted on a MiSeq system (Illumina), according to the standard operating protocol. NGS sequencing reads were mapped to the published human genome reference (UCSC hg19) using Bowtie2 aligner. Following best practices proposed by GATK, indel realignment was done to determine suspicious intervals likely to need realignment (GATK RealignerTarget-Creator function), followed by local realignment of reads to correct misalignments due to indels (GATK IndelRealigner function). Finally, a base quality score recalibration was done (GATK BaseRecalibrator function). For variant determination, GATK Unified Genotyper and Haplotype Caller functions were applied. Various resources were used during the functional annotation, including annovar, snpEff, dbSNP, 1000G, ExAC, and ESP6500 for population frequencies, PhyloP, gerp2, and phastCons for conservation prediction, and ClinVar, CADD, Sift, PolyPhen2, MutAssesor, Fathmm, and Vest for pathogenicity prediction.
Patient 5 was subjected to WES. Her gDNA was extracted from blood samples with the QIAamp DNA Mini Kit (QIAGEN). Exon capture was performed with Agilent SureSelect Human All Exon V5 kit and the exons were sequenced on an Illumina platform with a read length of 100 base pairs and a main depth coverage of 100X. Paired-end reads were mapped to the human reference genome GRCh37, with Burrows-Wheeler Aligner (BWA) and subsequent analyses were performed in accordance with the Broad Institute guidelines, with the GATK. Duplicated reads were marked with Picard tools and variants were called with “Haplotype Caller” from GATK (100341 variants were obtained). We annotated the final VCF file with SnpEff/SnpSift. We used the dbSNP, ExAC, 1000 Genomes, and ClinVar databases, and the prediction tools PolyPhen2, SIFT and Mutation Taster. Variants were prioritized with the web-based server B_Platform (Bitgenia, https://www.bitgenia.com/). Candidate variants were analyzed in terms of their presence in the set of candidate genes (a panel of 55 autoinflammatory genes), population frequency, possible impact on protein sequence, and previous clinical reports in databases.
Plasmids and transfection
HEK293T cells were transiently transfected in the presence of Lipofectamine 2000 (Invitrogen). The variants were introduced with the QuikChange II XL site-directed mutagenesis kit, using the pDONOR221-ISG15 vector. The variants were then transferred into a pTRIP expression vector with the Gateway LR Clonase II Enzyme mix. We also used the following constructs: pCAGGS-UbE1l, pCS2+-Herc5, pFlagCMV2-UbcH8. In cotransfection experiments, empty vector was added to keep the total amount of DNA constant.
Cytokine Stimulation
Transduced hTERT fibroblasts were primed by incubation with 0, 10, 100, or 1,000 IU ml−1 IFN-α2b (Merck IntronA) in normal growth medium for 12 h.The IFN-α2b was then eliminated by thorough washing with PBS and the cells were allowed to rest for 36 h in normal medium. HaCaT cells and hiPSC-ECs were used to seed 24-well plates at a density of 2 × 105 cells per well. The following day, the medium was removed and the cells were washed twice with PBS, and then treated with IFN-α (1000 IU/ml, Peprotech) for 12 or 24 h.
ISG expression analysis
RNA was extracted from fibroblasts (QIAGEN RNeasy) or whole blood (PAXgene Blood RNA Kit) and reverse-transcribed (ABI High Capacity Reverse Transcriptase) according to the instructions provided by the manufacturer. Levels of ISG expression (IFIT1, MX1, RSAD2, IFI27, IFI44L, ISG15, CXCL10, OAS1) relative to the 18SRNA housekeeping gene were analyzed by Taqman quantitative real-time PCR (TaqMan Universal Master Mix II w/ UNG, Roche LightCycler 480 II). The relative expression levels of the ISGs were calculated by the ΔΔCT method, with comparison to the mean value for mock-treated controls.
3′ RACE PCR
Total RNA was extracted from the patient’s hTERT-immortalized fibroblasts. We performed 3′ RACE PCR with the 3′ RACE System for the Rapid Amplification of cDNA ends (Invitrogen), according to the manufacturer’s instructions. We used the 3′ RACE Universal Amplification Primer (UAP) supplied in the kit, which was complementary to the adaptor primer, and a gene-specific primer that bound to ISG15. PCR products were cloned by TOPO-TA cloning (Thermo Fisher Scientific). Twenty-five colonies were grown. The plasmids isolated were subjected to Sanger sequencing to quantify the different splicing variants.
ISG15 knockout in hiPSC and HaCaT cells
For the ISG15 knockout we applied the CRISPR/Cas9 plasmid transfection system. We selected two guide RNAs resulting in the deletion of a 492 bp sequence comprising parts of exon 1 and exon 2. Each single guide RNA was cloned into the PX458_pSpCas9–2A-GFP plasmid. We used Lipofectamine 3000 for the transfection of iPSCs and Lipofectamine LTX for HaCaT cells, using 2.75 μg and 1 ug of each plasmid, respectively. After 1–3 days the cells were sorted for eGFP expression. iPSC cells were seeded in low density on irradiated mouse embryonic fibroblasts in iPSC medium (knockout-DMEM supplemented with 20% knockout serum replacement, 1mM L-glutamine, 0.1mM β-mercaptoethanol, 1% nonessential amino acid stock, 10ng/ml b-FGF) supplemented with 10μM Rock Inhibitor Y27632. HaCaT cells were plated as single cells into 96-well plates. Arising clones were picked manually and further cultivated clonally. PCR analysis for the presence of the deletion as well as the absence of the 5′ and 3′ junction of a wild-type allele, confirmed the homozygous knockout in two hiPSC clones. Sanger sequencing of the targeted region and western blotting confirmed ISG15 knockout.
Protein analyses
For the experiment shown in Figure 2, cells were lysed in RIPA buffer (Thermo Fisher Scientific), DTT and a protease/phosphatase inhibitor cocktail (Cell Signaling Technology) and subjected to western blotting. For the experiment shown in Figure 5, cells were harvested in RA1 lysis buffer (Macherey-Nagel, Düren, Germany) containing 1% β-mercaptoethanol. The resulting cell lysates were stored at −20°C until RNA extraction and qPCR analysis. The antibodies used were directed against STAT1 (Santa Cruz Biotechnology), STAT2 (Millipore), phospho-Tyr 701 STAT1 (Cell Signaling Technology), phospho-Tyr 689 STAT2 (Millipore), USP18 (Cell Signaling Technology), ISG15 (Santa Cruz Biotechnology), β-actin (Cell Signaling Technology), and IFIT1 (Cell Signaling Technology). An enhanced chemiluminescence detection reagent was used for detection (Pierce ECL Western Blotting Substrate, Thermo Fisher Scientific).
Mass cytometry
Whole blood from P1 and healthy controls was centrifuged on a Ficoll gradient to collect the mononuclear cell layer. PBMCs were stained and analyzed by mass cytometry (CyTOF) at the Human Immune Monitoring Center of the Icahn School of Medicine at Mt. Sinai. The barcoded cells were combined and stained by incubation with antibodies against selected surface markers for 30 minutes on ice. Cells were then washed and fixed, resuspended in diH2O containing EQ Four Element Calibration Beads (Fluidigm) and acquired on a CyTOF2 mass cytometer (Fluidigm). Data files were normalized with a bead-based normalization algorithm (CyTOF software, Fluidigm) and debarcoded with CD45 gating. The gated populations were visualized in lower dimensions with viSNE in Cytobank (https://www.cytobank.org/).
Immunohistochemistry
Immunohistochemistry staining was performed using a Discovery Ultra Ventana instrument (Roche), with the staining module of RUO Discovery Multimer V2 (V0.00.0083). Slides were first incubated in blocking agent containing 2% BSA PBS, and Discovery Ultra antibody block (Cat # 760–4204) (Roche). Slides were incubated with primary antibodies for 60 minutes at 37°C. The following primary antibodies were used: anti-phospho-STAT1 (Tyr701) 58D6 (Cell Signaling) and anti-CD68 Clone Kp1 (Abcam) at recommended dilution in blocking agent. The slides were then incubated with Omni Map anti-rabbit HRP-conjugated secondary antibody (Multimer HRP) (Cat # 760–4311) (Roche) for 32 minutes. Positive signals were detected with the Discovery ChromoMap DAB Kit (Cat #760–159) (Roche).
Single-cell RNA sequencing
PBMCs were isolated by centrifugation on a Ficoll gradient from fresh heparin-stabilized whole blood. PBMCs from all individuals were then immediately processed together on the 10X Genomics Chromium platform for droplet generation and reverse transcription. In accordance with the protocol, approximately 12,000 PBMCs were loaded into the Chromium Single Cell 3′ v2 assay with a planned targeted cell recovery of 7,000 cells. Libraries were prepared and subjected to quality control according to the manufacturer’s protocol. Final libraries were sequenced on an Illumina HiSeq 4000 to a targeted depth of 20,000 reads per cell. The sequencing data were processed with the Cell Ranger pipeline. Gene-cell matrices were then imported into the SEURAT V2.3 R toolkit (https://satijalab.org/seurat/) for quality control, clustering and data analysis. The standard workflow was followed, with the filtering out of clear outliers of UMIs (presumed debris and doublets) and filtering according to the percentage of mitochondrial genes (dying cells), data normalization according to total expression, scaling by a factor of 1E4 and log-transformation, calculation of genes with variable expression, which are used for canonical correlation analysis for common sources of variation between datasets, the selection and alignment of statistically significant and relevant canonical correlation vectors for graph-based clustering, non-linear dimensionality reduction by tSNE for visualization, the definition of conserved biomarkers unique to each cluster via differential expression analysis, the manual curation of cluster identity with cluster-specific gene enrichments that are well-documented markers of cell type and the exploration of genes displaying differential expression between individuals.
Single molecule array (SiMoA)
Digital ELISA for type I IFN was performed with the single molecular array (SiMoA), using the IFNα Assay Kit (Quanterix, 100860), according to the manufacturer’s instructions, on a Simoa HD1 Analyzer. Plasma samples were obtained from the patients and three healthy donors by Ficoll gradient centrifugation and further clarified by high-speed centrifugation. Cellular cytokines were analyzed in healthy control and patient PBMCs after the overnight treatment of these cells with brefeldin-A. Cells in RIPA lysis buffer were then sorted by FACS, as follows: lymphocytes (CD3+, CD19+ or CD56+), monocytes (CD3−CD19−CD56−HLADR+CD14+), myeloid DCs (CD3−CD19−CD56−HLADR+CD14−CD123−) or plasmacytoid DCs (CD3−CD19−CD56−HLADR−CD14−CD123+).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as the mean ± standard deviation (SD) or standard error of the mean (SEM), as indicated in the legend for each figure. Statistical significance between sample groups was evaluated using the Student’s t test, based upon the assumption that values exhibit a Gaussian distribution. A p value < 0.05 was considered significant. Statistical parameters including the exact values of n, identity of replicates, definitions of center and dispersion and statistical significance are reported in the Figure Legends when necessary. All statistical tests were calculated with GraphPad PRISM.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-STAT1 Clone C-111 | Santa Cruz Biotechnology | Cat No. sc417; RRID:AB_675902 |
| Rabbit anti-STAT2 | Millipore Sigma | Cat No. 06502; RRID:AB_31014 |
| Rabbit anti-phospho-Tyr 701-STAT1 Clone 58D6 | Cell Signaling Technology | Cat No. 9167; RRID:AB_561284 |
| Rabbit anti-phospho-Tyr-689-STAT2 Clone D3P2P | Cell Signaling Technology | Cat No. 88410; RRID:AB_2800123 |
| Rabbit anti-USP18 Clone D4E7 | Cell Signaling Technology | Cat No. 4813; RRID:AB_10614342 |
| Mouse anti-ISG15 Clone F9 | Santa Cruz Biotechnology | Cat No. sc166755; RRID:AB_2126308 |
| Rabbit anti-β-actin Clone 13E5 | Cell Signaling Technology | Cat No. 4970; RRID:AB_2223172 |
| Rabbit anti-IFIT1 Clone D2X9Z | Cell Signaling Technology | Cat No. 14769; RRID:AB_2783869 |
| Mouse anti-CD68 Clone KP1 | Abcam | Cat No. ab955; RRID:AB_307338 |
| Goat anti-mouse IgG HRP-conjugated | Southern Biotech | Cat No. 101005; RRID:AB_2728714 |
| Goat anti-rabbit IgG HRP-conjugated | Southern Biotech | Cat No. 403005; RRID:AB_2687483 |
| Discovery OmniMap anti-rabbit HRP (RUO) | Roche | Cat No. 760-4311; RRID:AB_2811043 |
| anti-CD123 BV421-conjugated Clone 6H6 | Biolegend | Cat No. 306017; RRID:AB_10900244 |
| anti-CD11c BV711-conjugated Clone 3.9 | Biolegend | Cat No. 301629; RRID:AB_11219609 |
| anti-HLADR PE-conjugated Clone L243 | Biolegend | Cat No. 307605; RRID:AB_314683 |
| anti-CD56 APC-conjugated Clone 5.1H11 | Biolegend | Cat No. 362503; RRID:AB_2563912 |
| anti-CD19 APC-conjugated Clone HIB19 | Biolegend | Cat No. 302212; RRID:AB_314242 |
| anti-CD14 AF488-conjugated Clone M5E2 | Biolegend | Cat No. 301811; RRID:AB_493159 |
| anti-CD3 APC-conjugated Clone UCHT1 | Biolegend | Cat No. 300411; RRID:AB_314065 |
| anti-CD45 89Y-conjugated Clone HI30 | Fluidigm | Cat No.3089003B; RRID:AB_2661851 |
| anti-CD57 113In-conjugated Clone HCD57 | Biolegend | Cat No.322302; RRID:AB_2661815 |
| anti-CD11c 115In-conjugated Clone Bu15 | Biolegend | Cat No.337202; RRID:AB_1236381 |
| anti-IgD 141Pr-conjugated Clone IA6-02 | Biolegend | Cat No.348202; RRID:AB_10550095 |
| anti-CD19 142Nd-conjugated Clone HIB19 | Biolegend | Cat No.302202; RRID:AB_2661817 |
| anti-CD45RA 143Nd-conjugated Clone HI100 | Biolegend | Cat No.304102; RRID:AB_314406 |
| anti-CD141 144Nd-conjugated Clone M80 | Biolegend | Cat No.344102; RRID:AB_2661788 |
| anti-CD4 145Nd-conjugated Clone RPA-T4 | Biolegend | Cat No.300502; RRID:AB_314070 |
| anti-CD8 146Nd-conjugated Clone RPA-T8 | Biolegend | Cat No.301002; RRID:AB_2661818 |
| anti-CD20 147Sm-conjugated Clone 2H7 | Biolegend | Cat No.302302; RRID:AB_314250 |
| anti-CD16 148Nd-conjugated Clone 3G8 | Biolegend | Cat No.302014; RRID:AB_314214 |
| anti-CD127 149Sm-conjugated Clone A019D5 | Fluidigm | Cat No.3149011B; RRID:AB_2661792 |
| anti-CD1c 150Nd-conjugated Clone L161 | Biolegend | Cat No.331502; RRID:AB_2661820 |
| anti-CD123 151Eu-conjugated Clone 6H6 | Biolegend | Cat No.306002; RRID:AB_2661822 |
| anti-CD66b 152Sm-conjugated Clone G10F5 | Biolegend | Cat No.305102; RRID:AB_2661823 |
| anti-PD1 153Eu-conjugated Clone EH12.2H7 | Biolegend | Cat No.329926; RRID:AB_11147365 |
| anti-CD86 154Sm-conjugated Clone IT2.2 | Biolegend | Cat No.305410; RRID:AB_314530 |
| anti-CD27 155Gd-conjugated Clone O323 | Biolegend | Cat No.302802; RRID:AB_2661825 |
| anti-PDL1 156Gd-conjugated Clone 29E.2A3 | Biolegend | Cat No.329710; RRID:AB_2275581 |
| anti-CD33 158Gd-conjugated Clone WM53 | Biolegend | Cat No.303402; RRID:AB_314346 |
| anti-CD24 159Tb-conjugated Clone ML5 | Biolegend | Cat No.311102; RRID:AB_314851 |
| anti-CD14 160Gd-conjugated Clone M5E2 | Biolegend | Cat No.301810; RRID:AB_314192 |
| anti-CD56 161Dy-conjugated Clone B159 | BD Biosciences | Cat No.555513; RRID:AB_395903 |
| anti-CD169 162Dy-conjugated Clone 7-239 | Biolegend | Cat No.346002; RRID:AB_2189031 |
| anti-CXCR5 163Dy-conjugated Clone REA103 | Miltenyi | Cat No.130-122-325; RRID:AB_2801905 |
| anti-CD69 164Dy-conjugated Clone FN50 | Biolegend | Cat No.310902; RRID:AB_314837 |
| anti-CCR6 165Ho-conjugated Clone G034E3 | Biolegend | Cat No.353402; RRID:AB_10918625 |
| anti-CD25 166Er-conjugated Clone M-A251 | Biolegend | Cat No.356102; RRID:AB_2661833 |
| anti-CCR7 167Er-conjugated Clone G043H7 | Biolegend | Cat No.353256; RRID:AB_2814291 |
| anti-CD3 168Er-conjugated Clone UCHT1 | Biolegend | Cat No.300402; RRID:AB_2661835 |
| anti-CX3CR1 169Tm-conjugated Clone 2A9-1 | Biolegend | Cat No.341602; RRID:AB_1595422 |
| anti-CD38 170Er-conjugated Clone HB-7 | Biolegend | Cat No.356602; RRID:AB_2661836 |
| anti-CD161 171Yb-conjugated Clone HP-3G10 | Biolegend | Cat No.339902; RRID:AB_2661837 |
| anti-CD209 172Yb-conjugated Clone 9E9A8 | Biolegend | Cat No.330102; RRID:AB_1134253 |
| anti-CXCR3 173Yb-conjugated Clone REA232 | Miltenyi | Cat No.130-108-022; RRID:AB_2655743 |
| anti-HLADR 174Yb-conjugated Clone L243 | Biolegend | Cat No.307602; RRID:AB_314680 |
| anti-Axl 175Lu-conjugated Clone 108724 | R&D Systems | Cat No.MAB154; RRID:AB_2062558 |
| anti-CCR4 176Yb-conjugated Clone 205410 | R&D Systems | Cat No.MAB1567; RRID:AB_2074395 |
| anti-pSTAT5 147 Sm-conjugated Clone 47 | Fluidigm | Cat No.3147012A; RRID:AB_2827887 |
| anti-pSTAT6 149 Sm-conjugated Clone 18/P-Stat6 | Fluidigm | Cat No.3149004A |
| anti-pSTAT1 153 Eu-conjugated Clone 4a | Fluidigm | Cat No.3153005A; RRID:AB_2744689 |
| anti-pp38 156 Gd-conjugated Clone D3F9 | Fluidigm | Cat No.3156002A; RRID:AB_2661826 |
| anti-pSTAT3 158 Gd-conjugated Clone 4/P-Stat3 | Fluidigm | Cat No.3158005A; RRID:AB_2811100 |
| anti-pMAPKAP2 159 Tb-conjugated Clone 27B7 | Fluidigm | Cat No.3159010A; RRID:AB_2661828 |
| anti-STAT3 165 Ho-conjugated Clone 124H6 | Fluidigm | Cat No.3173003A |
| anti-STAT1 169 Tm-conjugated Clone 10C4B40 | Biolegend | Cat No.661002; RRID:AB_2563664 |
| anti-pERK 171 Yb-conjugated Clone D13.14.4E | Fluidigm | Cat No.3171010A; RRID:AB_2811250 |
| anti-pS6 175 Lu-conjugated Clone N7-548 | Fluidigm | Cat No.3175009A; RRID:AB_2811251 |
| anti-CD31 APC-conjugated | Miltenyi Biotech | Cat No. 130110808; RRID:AB_2657282 |
| Bacterial and Virus Strains | ||
| DH5-Alpha Competent E. Coli | Molecular Cloning Laboratories | Cat No. DA-196 |
| bacille Calmette-Guerin strain mycobacteria | This paper | N/A |
| Biological Samples | ||
| Human whole blood samples | Various institutions | N/A |
| Dermal punch biopsies | National Institute of Health | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Intron-A Recombinant Interferon Alpha-2b | Merck Pharmaceuticals | Cat No. NDC0085057102 |
| Proteomic Stabilizer Prot1 | SMART TUBE Inc | Cat No. 501351691 |
| Heparin | Sigma | Cat No. 201060 |
| Osmium tetroxide (99.9%) | ACROS organics | Cat No. 191180010 |
| Discovery Ultra antibody block | Roche | Cat No. 760-4204 |
| Pierce ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat No. 32106 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | Cat No. 89901 |
| Protease/Phosphatase Inhibitor Cocktail | Cell Signaling Technologies | Cat No. 5872 |
| Macherey-Nagel RNA Isolation, RA1 Lysis Buffer | Thermo Fisher Scientific | Cat No. 10335832 |
| Brefeldin A | Thermo Fisher Scientific | Cat No. B7450 |
| Histopaque 1077 | Millipore Sigma | Cat No. 10771-500 |
| Cyclosporin | Sigma Aldrich | Cat No. C3662 |
| BMP4 | R&D | Cat No. 324-BP |
| CHIR90221 | Technical Chemistry, LUH | Cat No. 252917-06-9 |
| rhVEGF-A165 | Peprotech | Cat No. 100-20 |
| Forskolin | Sigma-Aldrich | Cat No. F3917; CAS_66575-29-9 |
| Accutase | ThermoFisher | Cat No. A1110501 |
| Getrex | ThermoFisher | Cat No. A1413301 |
| mTeSR1 | StemCellTechnologies | Cat No. 85850 |
| N2 supplement | Thermo Fisher | Cat No. 17502048 |
| B27 supplement | ThermoFisher | Cat No. 0080085SA |
| StemPro-34 medium | ThermoFisher | Cat No. 10639011 |
| EGM-2 | Lonza | Cat No. CC-3163 |
| Fibronectin | Corning | Cat No. 354008 |
| Rock Inhibitor Y27632 | Tocris | Cat No. 1254; CAS_129830-38-2 |
| Critical Commercial Assays | ||
| QuikChange II XL site-directed mutagenesis kit | Agilent Technologies | Cat No. 200522 |
| Gateway LR Clonase II Enyme Mix | Thermo Fisher Scientific | Cat No. 11791100 |
| Gateway BP Clonase Enzyme Mix | Thermo Fisher Scientific | Cat No. 11789020 |
| QIAamp DNA Mini Kit | QIAGEN | Cat No. 51304 |
| RNeasy RNA Isolation Kit | QIAGEN | Cat No. 74106 |
| PAXgene Blood RNA Kit | BD Biosciences | Cat No. 762165 |
| Applied Biosystems High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat No. 4368814 |
| TaqMan Universal Master Mix II with UNG | Thermo Fisher Scientific | Cat No. 4440039 |
| 3′ Rapid Amplification of cDNA Ends Kit | Thermo Fisher Scientific | Cat No. 18373019 |
| TOPO TA Cloning Kit | Thermo Fisher Scientific | Cat No. K457540 |
| Interferon-alpha Simoa Assay Kit | Quanterix | Cat No. 100860 |
| Disc Kit for Simoa HD1 | Quanterix | Cat No. 103347 |
| Simoa System Buffer Combo Pack | Quanterix | Cat No. 100488 |
| Human Magnetic Luminex Assay | R&D | Cat No. LXSAHM |
| Cell-ID 20-Plex Pd Barcoding Kit | Fluidigm | Cat No. 201060 |
| Chromium single cell Chip Kit V2 | 10X Genomics | Cat No. 120236 |
| Chromium single cell 3′ Library and Gel Bead Kit | 10X Genomics | Cat No.120237 |
| Discovery ChromoMap DAB Kit | Roche | Cat No. 760-159 |
| MycoAlert PLUS Mycoplasma Detection Kit | Lonza | Cat No. LT07-703 |
| SureSelect Human All Exon V5 kit | Agilent | N/A |
| CD31 MicroBead Kit | Miltenyi Biotec | Cat No. 130-091-935 |
| Lipofectamine 3000 Transfection Reagent | Thermo Fisher | Cat No. L3000001 |
| Lipofectamine™ LTX Reagent | Thermo Fisher | Cat No. 15338500 |
| iPrep Pure Link kit Invitrogen | Thermo Fisher | Cat No IS10005 |
| Invitrogen Taq polymerase | Thermo Fisher | Cat No. 10342020 |
| Sephadex G-50 Superfine Resin | GE Healthcare | Cat No. 17004101 |
| BigDye Terminator v3.1 Cycle Sequencing Kit | Thermo Fisher | Cat No. 4337457 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | ATCC CRL-3216 |
| hTERT-immortalized dermal fibroblasts | Icahn School of Medicine at Mount Sinai | N/A |
| EBV-immortalized B cells | National Institute of Health | N/A |
| B95.8 cells | ATCC | ATCC CRL-1612 |
| HSC_iso4_ADCF_SeV-iPS2 | Haase et al., 2017 | N/A |
| HaCaT (Human Keratinocyte cell line) | Boukamp et al., 1988 | N/A |
| Oligonucleotides | ||
| MX1 mRNA TaqMan FAM Hs200895608_m1 | Thermo Fisher | Cat No. 4331182 |
| IFI27 mRNA TaqMan FAM Hs01086373_g1 | Thermo Fisher | Cat No. 4351370 |
| IFIT1 mRNA TaqMan FAM Hs03027069_s1 | Thermo Fisher | Cat No. 4331182 |
| IFI44L mRNA TaqMan FAM Hs00915292_m1 | Thermo Fisher | Cat No. 4331182 |
| RSAD2 mRNA TaqMan FAM Hs00369813_m1 | Thermo Fisher | Cat No. 4351370 |
| ISG15 CRISPR gRNA1 GCGCAGATCACCCAGAAGAT CGG | This paper. | N/A |
| ISG15 CRISPR gRNA2 GGTAAGGCAGATGTCACAGG TGG | This paper. | N/A |
| 3′ RACE ISG15 primer: GAACTCATCTTTGCCAGtACAGGAG | This paper. | N/A |
| ISG15 gDNA PCR F: CGGGATGTAGAGGACAGACA | This paper. | N/A |
| ISG15 gDNA PCR R: ACCCTTATCCCTTCACTTGG | This paper. | N/A |
| Recombinant DNA | ||
| pTRP IRES RFP PuroR | This paper | N/A |
| pDONR221 Gatweway Vector | Thermo Fisher | Cat No. 12536017 |
| pCAGGS-UbE1l, | This paper | N/A |
| pCS2+-Herc5 | This paper | N/A |
| pFlagCMV2-UbcH8 | This paper | N/A |
| pSpCas9-2A-GFP (PX458) | Addgene | Cat No. 48138 |
| Software and Algorithms | ||
| Cytobank | Beckman Coulter | N/A |
| FlowJo | Becton Dickinson Company | N/A |
| RStudio | RStudio | N/A |
| CellRanger | 10X Genomics | N/A |
| Seurat V2.3 R toolkit | https://satijalab.org/seurat/ | N/A |
| Genome Analysis Toolkit (GATK) | Broad Institute | N/A |
| B Platform | Bitgenia | N/A |
| NimbleDesign | Roche Sequencing | N/A |
| Ion Torrent Sofware V4.4.2 | Thermo Fisher | N/A |
| SNPEff | http://snpeff.sourceforge.net | |
| GraphPad Prism 7 | GraphPad Software | N/A |
Highlights.
ISG15 deficiency is identified in five new patients with six novel genetic lesions
All patients presented with dermatologic complications
ISG15 mutations lead to hyper-activated responses to type I interferon
Patient cells exhibit striking cell type specificity to the interferon response
ACKNOWLEDGMENTS
We would like to thank Ludovic Debure and Dr. Thomas Wisniewski of the New York State Center of Excellence for Alzheimer’s Disease for their technical assistance with the SiMoA assay. This research was supported in part by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH). We would like to thank Rebeca Perez de Diego for her advice. This work was supported by National Institute of Allergy and Infectious Diseases/NIH grants R01AI127372, R21AI134366, and R21AI129827, a grant from the March of Dimes to D.B., and The Helmholtz Association’s Cross-Program Initiative on Personalized Medicine, iMed. The Laboratory of Human Genetics of Infectious Diseases is supported by grants from the St. Giles Foundation, the Jeffrey Modell Foundation, The Rockefeller University Center for Clinical and Translational Science, grant UL1TR001866 from the National Center for Research Resources and the National Center for Advancing Sciences (NCATS), National Institutes of Health, the National Institute of Allergy and Infectious Diseases (5R01AI089970–02 and 5R37AI095983), grants from the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), the French Foundation for Medical Research (Fondation pour la Recherche Médicale [FRM], EQU201903007798) and the French National Research Agency (ANR) under the “Investments for the Future” program (ANR-10-IAHU-01), GENMSMD (ANR-16-CE17–0005-01), Institut National de la Santé et de la Recherche Médicale (INSERM), Paris University, and The Rockefeller University. C.G. was supported by T32 training grant 5T32HD075735–07 at the Icahn School of Medicine at Mount Sinai.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107633.
REFERENCES
- Alsohime F, Martin-Fernandez M, Temsah MH, Alabdulhafid M, Le Voyer T, Alghamdi M, Qiu X, Alotaibi N, Alkahtani A, Buta S, et al. (2020). JAK inhibitor therapy in a child with inherited USP18 deficiency. N. Engl. J. Med. 382, 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, et al. (2012). Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science 337, 1684–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, and Fusenig NE (1988). Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106, 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick L (2019). Hereditary autoinflammatory disorders: recognition and treatment. Immunol. Allergy Clin. North Am. 39, 13–29. [DOI] [PubMed] [Google Scholar]
- Bustamante J (2020). Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum. Genet 10.1007/s00439-020-02120-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ (2011). Type I interferonopathies: a novel set of inborn errors of immunity. Ann. N Y Acad. Sci. 1238, 91–98. [DOI] [PubMed] [Google Scholar]
- Crow YJ (2013). Aicardi-Goutiéres syndrome. Handb. Clin. Neurol. 113, 1629–1635. [DOI] [PubMed] [Google Scholar]
- Crow YJ (2015). Type I interferonopathies: Mendelian type I interferon upregulation. Curr. Opin. Immunol. 32, 7–12. [DOI] [PubMed] [Google Scholar]
- Crow YJ, and Rehwinkel J (2009). Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum. Mol. Genet. 18 (R2), R130–R136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, Pellegrini S, and Uzé G (2011). USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLoS ONE 6, e22200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois-Newton V, Livingstone M, Payelle-Brogard B, Uzé G, and Pellegrini S (2012). USP18 establishes the transcriptional and anti-proliferative interferon α/β differential. Biochem. J. 446, 509–516. [DOI] [PubMed] [Google Scholar]
- Gruber C, Martin-Fernandez M, Ailal F, Qiu X, Taft J, Altman J, Rosain J, Buta S, Bousfiha A, Casanova JL, et al. (2020). Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J. Exp. Med. 217, e20192319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase A, Göhring G, and Martin U (2017). Generation of non-transgenic iPS cells from human cord blood CD34+ cells under animal component-free conditions. Stem Cell Res. (Amst.) 21, 71–73. [DOI] [PubMed] [Google Scholar]
- Hermann M, and Bogunovic D (2017). ISG15: in sickness and in health. Trends Immunol. 38, 79–93. [DOI] [PubMed] [Google Scholar]
- Klauer AA, and van Hoof A (2012). Degradation of mRNAs that lack a stop codon: a decade of nonstop progress. Wiley Interdiscip. Rev. RNA 3, 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn HS, Niemela JE, Sreedhara K, Stoddard JL, Grossman J, Wysocki CA, de la Morena MT, Garofalo M, Inlora J, Snyder MP, et al. (2017). Novel nonsense gain-of-function NFKB2 mutations associated with a combined immunodeficiency phenotype. Blood 130, 1553–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Mortha A, Rahman A, and Merad M (2015). Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 15, 731–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, Li Z, van Unen L, Heijsman D, Goldmann T, et al. (2016). Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 213, 1163–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschke K, Friebe F, Zimmermann N, Wahlicht T, Schumann T, Achleitner M, Berndt N, Luksch H, Behrendt R, Lee-Kirsch MA, et al. (2014). Deregulated type I IFN response in TREX1-associated familial chilblain lupus. J. Invest. Dermatol. 134, 1456–1459. [DOI] [PubMed] [Google Scholar]
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, et al. (2013). Assessment of interferon-related biomarkers in Aicardi-Goutiéres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 12, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero MP, and Crow YJ (2016). Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J. Exp. Med. 213, 2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, McGlasson SL, Alyanakian MA, Bader-Meunier B, Barnerias C, et al. (2017). Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J. Exp. Med. 214, 1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011).A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR, and Darnell JE Jr. (2012). The JAK-STAT pathway at twenty. Immunity 36, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaim CD, Scott AF, Canadeo LA, and Huibregtse JM (2017). Extracellular ISG15 signals cytokine secretion through the LFA-1 integrin receptor. Mol. Cell 68, 581–590.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft J, and Bogunovic D (2018). The Goldilocks zone of type I IFNs: lessons from human genetics. J. Immunol. 201, 3479–3485. [DOI] [PubMed] [Google Scholar]
- Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, et al. (2020). Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 40, 24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uccellini MB, and Garcia-Sastre A (2018). ISRE-reporter mouse reveals high basal and induced type I IFN responses in inflammatory monocytes. Cell Rep. 25, 2784–2796.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, et al. (2015). Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 517, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.