

Abstract
The neonatal mammalian heart is capable of regeneration for a brief window of time after birth. However, this regenerative capacity is lost within the first week of life, which coincides with a postnatal shift from anaerobic glycolysis to mitochondrial oxidative phosphorylation, particularly towards fatty-acid utilization. Despite the energy advantage of fatty-acid beta-oxidation, cardiac mitochondria produce elevated rates of reactive oxygen species when utilizing fatty acids, which is thought to play a role in cardiomyocyte cell-cycle arrest through induction of DNA damage and activation of DNA-damage response (DDR) pathway. Here we show that inhibiting fatty-acid utilization promotes cardiomyocyte proliferation in the postnatatal heart. First, neonatal mice fed fatty-acid deficient milk showed prolongation of the postnatal cardiomyocyte proliferative window, however cell cycle arrest eventually ensued. Next, we generated a tamoxifen-inducible cardiomyocyte-specific, pyruvate dehydrogenase kinase 4 (PDK4) knockout mouse model to selectively enhance oxidation of glycolytically derived pyruvate in cardiomyocytes. Conditional PDK4 deletion resulted in an increase in pyruvate dehydrogenase activity and consequently an increase in glucose relative to fatty-acid oxidation. Loss of PDK4 also resulted in decreased cardiomyocyte size, decreased DNA damage and expression of DDR markers and an increase in cardiomyocyte proliferation. Following myocardial infarction, inducible deletion of PDK4 improved left ventricular function and decreased remodelling. Collectively, inhibition of fatty-acid utilization in cardiomyocytes promotes proliferation, and may be a viable target for cardiac regenerative therapies.
Introduction
The pathophysiological underpinning of heart failure is the inability of the adult heart to regenerate lost or damaged myocardium. Although limited myocyte turnover does occur in the adult heart, it is insufficient for restoration of contractile dysfunction1. In contrast, the neonatal mammalian heart is capable of substantial regeneration following injury through cardiomyocyte proliferation2,3, not unlike urodele amphibians4–7 or teleost fish8,9. However, this regenerative capacity is lost by postnatal day (P) 72,3 which coincides with cardiomyocyte binucleation and cell cycle arrest10. After this postnatal cell cycle arrest, cardiomyocyte growth occurs through hypertrophic increases in cardiomyocyte size11,12. Although several regulators of cardiomyocyte cell cycle arrest postnatally have been identified2,13–18, the upstream signals that cause permanent cell cycle arrest of most cardiomyocytes remain unknown.
In contrast to lower vertebrates8,9,19, mammalian cardiomyogenesis is very limited1,20–23. However, for a brief window of time after birth, the mammalian neonatal heart is capable of mounting a robust regenerative response following injury. Recent studies have demonstrated that removal of up to 15% of the left ventricular apex of postnatal day 1 (P1) mice results in regeneration by P213,24. This regenerative response is mediated by proliferation of preexisting cardiomyocytes and is lost when cardiomyocytes permanently exit the cell cycle shortly after birth3,25. Similar results were obtained using an ischemic myocardial infarction (MI) model in neonatal mice26 and large animals27,28. Given these results in neonatal mammals, significant interest has focused on mechanisms of cell cycle arrest in the early postnatal period and whether targeting these mechanisms can reverse cell cycle arrest in the adult. As expected, numerous pathways that regulate cardiomyocyte cell cycle arrest postnatally have been identified. Our lab has been interested in understanding the role of mitochondrial metabolism in regulation of cell cycle arrest postnatally. We recently demonstrated that mitochondrial derived reactive oxygen species (ROS) induce DNA damage and activate DDR in the early postnatal heart resulting in cell cycle arrest29. Importantly, during embryonic development, the heart utilizes anaerobic glycolysis as a main source of energy30,31, whereas adult cardiomyocytes utilize oxygen-dependent mitochondrial oxidative phosphorylation as the primary energy source owing to its substantial energy advantage32,33. In addition to the shift from cytoplasmic glycolysis to mitochondrial oxidative phosphorylation with developmental age, postnatal cardiomyocytes exhibit a shift in energetic substrate utilization, from pyruvate to fatty acids29,31,34.
Fatty acid utilization is energetically favourable, and it also becomes highly abundant to neonatal mammals immediately after birth because of the high fat content in breast milk, which is as high as 30%35. In the western world, such a high level of dietary fatty acid is maintained throughout life. However, whether the increase in fatty acid after birth plays a role in cardiac energy substrate utilization is not known. Importantly, increased fatty acid oxidation perpetuates dependence on fatty acid utilization by inhibiting glucose oxidation via the Randle Cycle, in which, acetyl-CoA generated from fatty acid oxidation inhibits the mitochondrial enzyme Pyruvate Dehydrogenase (PDH)36. Cardiac PDH activity is regulated by various isoforms of pyruvate dehydrogenase kinases (PDK1, 2 and 4) and phosphatases (PDP1 and 2), with phosphorylation resulting in enzyme inhibition37–39. Of the PDK isoforms, PDK4 is largely responsible for inhibiting PDH in the presence of fatty acids and increasing the reliance of the heart on fatty acid oxidation for energy production37,39–41. Intriguingly, our group and others have shown that mitochondria produce H2O2 at an elevated rate when using fatty acids rather than pyruvate as a respiratory substrate42–44.
Collectively, current evidence suggests that the postnatal increased availability of fatty acids and oxygen corresponds with a shift towards beta oxidation-dependent mitochondrial oxidative phosphorylation with an induction of oxidative DNA damage and cell cycle arrest of cardiomyocytes. Therefore, we set out to determine the role of fatty acid utilization on oxidative DNA damage and postnatal cell cycle arrest of cardiomyocytes.
Results
Feeding pups with milk deficient in fatty acids prolongs the proliferative window in young mouse hearts
To generate mice which produce milk deficient in fatty acids, we utilized a genetic model to knockout a key lipogenic enzyme involved in the production and storage of lipids specifically in mammary glands. We crossed PERKf/f mice45 with MMTV-Cre mice, a transgenic line expressing Cre recombinase predominantly in secretory cell types in the mammary epithelium under the mouse mammary tumor virus promoter (MMTV)46 (Fig. 1a). PERK has been reported to be required for proper development and maturation of mammary epithelial cells and the deletion of PERK in mammary glands inhibits key lipogenic enzymes involved in production and storage of lipids and consequently decreases lipid content in milk45.
Fig. 1: Dietary fatty acid deficiency from birth results in a pronounced prolongation of the postnatal window of cardiomyocyte proliferation.
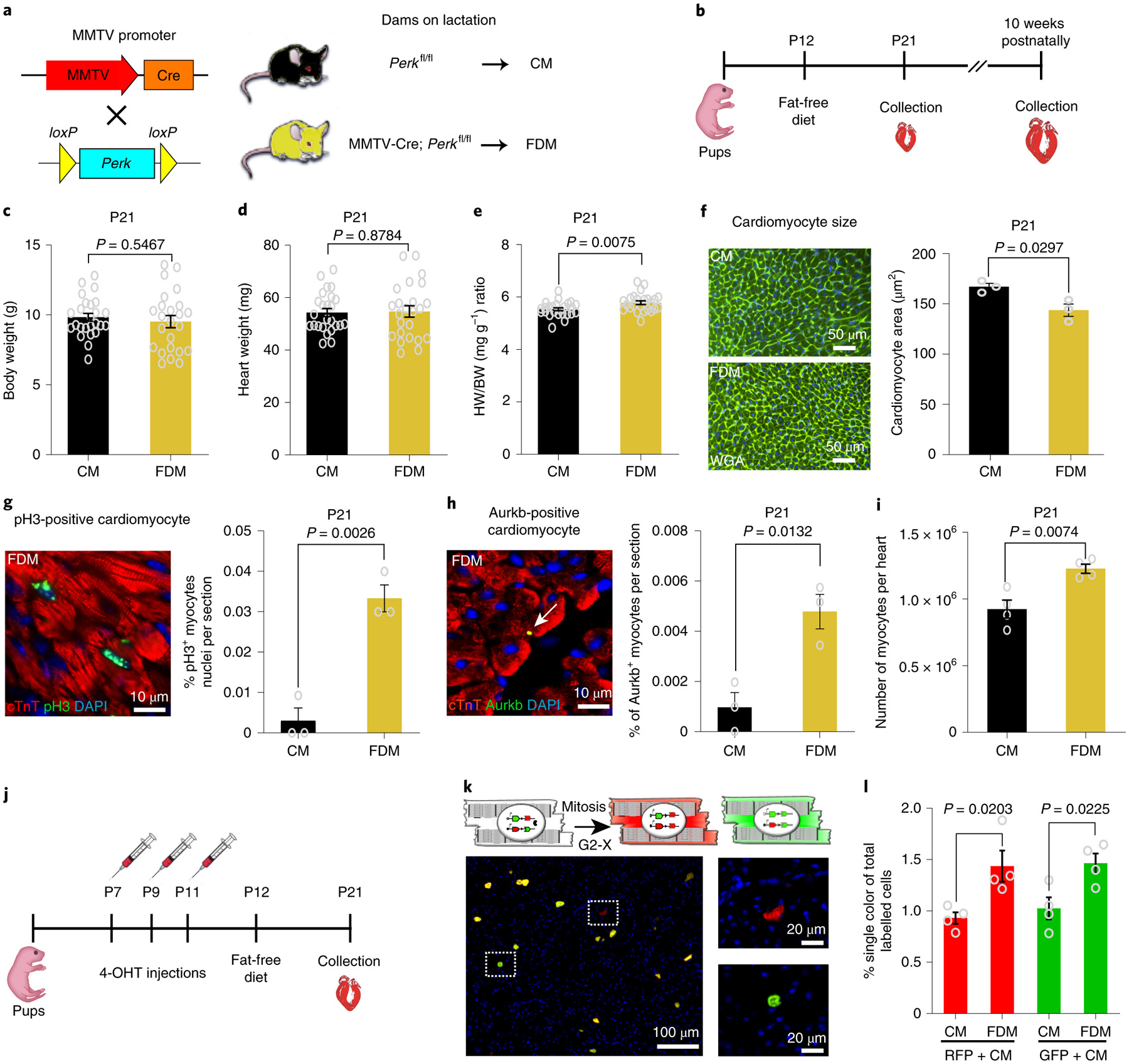
a, Schematic view of the genetic mouse model of PERK deletion specifically in mammary epithelial cells. The Cre+/PERKf/f mice produce milk deficient in fatty acids. As a control, we used the Cre−/PERKf/f mice, which produce milk with normal levels of fatty acids. b, At 12 days postnatally, beyond lactation, pups were exposed to a regular diet or Fat Free Diet. Animals were sacrificed at 21 days or 10 weeks postnatally. c, Body weight (BW) measurements show no significant difference between FDM (n=24 biologically independent mice) and CM (n=25 biologically independent mice). d, Heart weight (HW) measurements showing no difference between groups; n=24 biologically independent mice for FDM group and n=25 biologically independent mice for CM group. e, Heart weight to body weight (HW/BW) ratio shows a significant increase in FDM (n=24 biologically independent mice) compared to CM (n=25 biologically independent mice). f, WGA staining shows a significant decrease in cardiomyocyte cell size measurement in FDM compare to CM (n=3 biologically independent mice per group). g, Anti-pH3 and anti-cTnT co-immunostaining showing a significant increase in the cardiomyocyte mitosis marker in the FDM group (n=3 biologically independent mice per group). h, Anti-Aurora B kinase and anti-cTnT co-immunostaining shows a significant increase in the cardiomyocyte cytokinesis marker in the FDM compared to the CM group (n=3 biologically independent mice per group). i, A complete dissociation of cardiomyocytes by collagenases indicated a significant increase in the total number of cardiomyocytes in FDM group compared to CM (n = 4 biologically independent mice). j, Schematic view of 4-hydroxytamoxifen (4-OHT) administration and timeline experiment in MADM; MCM mice. k, (Upper) Schematic representation of MADM; MCM recombination in a parent cardiomyocyte leading to RFP+ and GFP+ single-labeled daughter cardiomyocytes. (Lower) Example of RFP+ and GFP+ single-labeled cardiomyocyte. l, Frequency of single-labeled cardiomyocytes per heart section are higher in the FDM group compared with the CM group (n=4 biologically independent mice per group). Data in bar graphs are presented as mean±s.e.m. Statistical analysis was performed using a two-tailed Student’s t-test: NS, not significant CM, control milk group; FDM, fat deficient milk group.
At P12 (when mice usually start eating chow in addition to milk), the pups were exposed to a Regular Diet or Fat Free Diet (supplementary table 1). The heart and liver were collected at P21 or at 10 weeks of age (Fig. 1b). We found that 21 days postnatal (P21) mice fed from fatty acid deficient milk (FDM) dams showed a significant increase in HW to BW ratio (HW/BW ratio) compared to control (Fig.1c–e). The cardiomyocyte cell size in FDM fed mice at P21 was significantly smaller than in mice fed the control milk diet (CM) (Fig. 1f). In addition, the number of cardiomyocytes that were positive for the mitosis marker phosphorylated histone H3 Ser10 (pH3) (Fig. 1g), as well as the cytokinesis marker Aurora B Kinase (Fig. 1h), were significantly increased in the FDM hearts. Moreover, the total number of cardiomyocytes in the P21 hearts were assessed by collagenase digestion and cardiomyocyte isolation. We found that FDM hearts had a significant increase in the total number of cardiomyocytes compared to the control mice (Fig. 1i). Furthermore, we assessed generation of new cardiomyocytes by lineage tracing using the Mosaic Analysis with Double Markers (MADM) mouse model crossed with αMHC-MerCreMer mice (MCM)47. The MADM; MCM model allows clear identification of new cardiomyocyte generation, because after cytokinesis the two daughter cardiomyocytes are single-labeled either RFP+ or GFP+, whereas if complete cell separation did not occur, the cells remain double labelled (Fig. 1j–k). Therefore, the quantification of single-labeling cardiomyocytes directly correlates with completion of cytokinesis. Here, we show that P21 MADM mice fed from fatty acid deficient milk foster mothers showed a significant increase in RFP+ and GFP+ single-labeled daughter cardiomyocytes compared to control (Fig. 1l). These results indicate that dietary fatty acids contribute to cell-cycle arrest during the first weeks of postnatal life, and that a fat deficient diet increases cardiomyocyte proliferation beyond the normal postnatal proliferative window. It is important to note though that the degree of proliferation in this scenario appears to be modest.
Chronic exposure to fat free diet fails to maintain cardiomyocyte proliferation in adult
In order to evaluate the effect of chronic exposure to a fat free diet on heart and liver metabolism, FDM mice remained on a fat free diet until 10 weeks of age. The body weight and heart weight of the fat free diet mice as compared to the control mice was normal (Fig. 2a–b). Similarly, no significant difference was observed in the HW/BW ratio (Fig. 2c). Histologically, the hearts stained with Hematoxylin and Eosin (H&E) or trichrome show normal morphology without pronounced fibrosis (Fig. 2d). At the cellular level, the cardiomyocytes from mice on the fat free diet are smaller compared with mice on the control diet, as assessed by WGA staining (Fig. 2e). However, no significant difference (P value= 0.9105) was found in the percentage of pH3 positive cardiomyocytes comparing regular diet (average of 0.0017% ± 0.003, n=3) compared to fat free diet (0.0020% ± 0.001, n=3).
Fig. 2: Chronic exposure to fat free diet induces liver steatosis in adult mice.
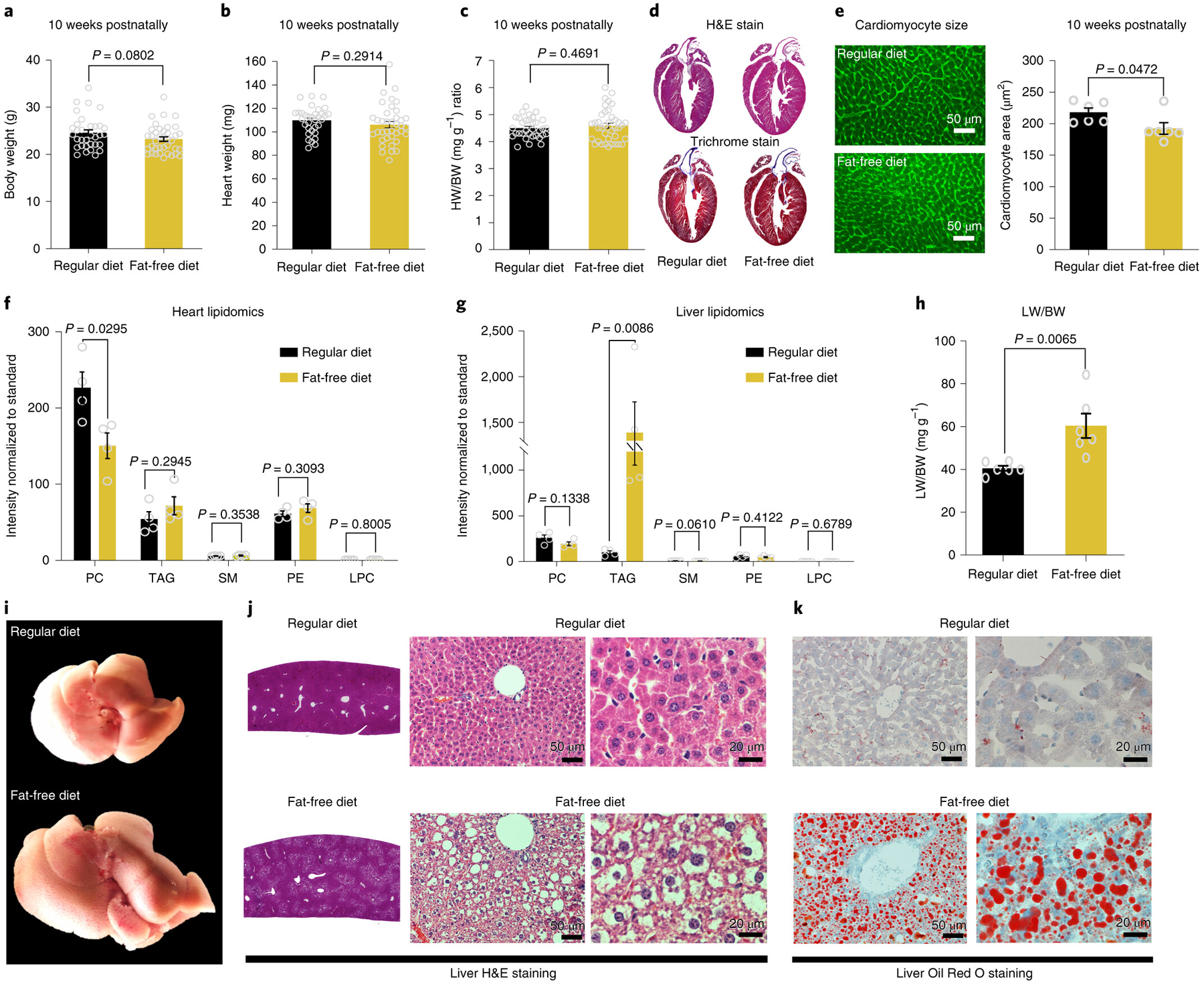
a, Body weight (BW); b, Heart weight (HW); c, HW/BW ratio measurements showing no difference between regular diet (n=33 biologically independent mice) and fat free diet (n=38 biologically independent mice). d, Heart sections stained with H&E or trichrome show no clear difference between regular diet and fat free diet. Images are representative of four independently performed experiments with similar results. e, WGA staining shows significant difference in cardiomyocyte cell size measurements in fat free diet compare to regular diet (n=6 biologically independent mice per group). f, Heart lipidomics profile shows a significant decrease in PC in fat free diet compare to regular diet group (n=4 biologically independent mice per group). g, Liver lipidomics profile shows a significant increase in TAG in fat free diet compared to the regular diet group (n=4 biologically independent mice per group). h, Liver weight to body weight (LW/BW) ratio shows a significant increase in the fat free diet liver weight compared to regular diet (n=6 biologically independent mice per group). i, Representative image of the whole liver shows a marked increase in liver size in fat free diet compared to regular diet group. j, Representative image of a histological liver section stained with H&E showing marked macrovesicular steatosis with no signal of inflammatory cell accumulation. k, Liver cryosection stained with Oil Red O showing massive positive staining lipid droplets. Data in i-k are representative of four independently performed experiments with similar results. Data in bar graphs are presented as mean±s.e.m. Statistical analysis was performed using a two-tailed Student’s t-test: NS, not significant. LPC, lysophosphatidylcholine; PC, phosphatidylcholine; PE, phosphatidylethanolamine; SM, sphingomyelin; TAG, triacylglycerol.
Next, we assessed the lipid content in the livers and hearts of fat free diet mice by performing a lipidomics profile by mass spectrometry. We found that the lipidomics profile of the fat free diet and control mice was essentially unchanged, except for the phosphatidylcholine (PC) lipid fraction, which was significantly decreased in hearts from the fat free diet group (Fig. 2f). In contrast to lipidomics profile of the hearts, we found that the total lipids in the livers of fat free diet mice were increased, mainly driven by triacylglycerol (TAG) fractions (Fig. 2g). Consistently, the liver weight to BW ratio was significantly increased in the fat free diet mice (Fig. 2h). Morphologically, these livers were enlarged with preserved shape. They were also yellowish, bulging, soft and greasy, all features consistent with fatty liver or steatosis (Fig. 2i). Under histological examination, the liver sections stained by H&E and Oil Red O showed a marked increase in fat vacuoles, increased retention of lipids and macrovesicular steatosis (Fig. 2j–k). We did not observe an increase in inflammatory cell accumulation. In addition, quantitative mass spectrometry analysis of hepatic fatty acid biosynthesis enzymes showed a significant increase in the enzymes involved in saturated fatty acid and triglyceride synthesis in fat free diet mice compared to regular diet mice (Extended Data Fig. 1). Moreover, we assessed serum lipids and blood glucose levels in mice on the fat free diet compared to mice on the regular diet. Our data showed no significant difference in blood glucose, total cholesterol, triglycerides, LDL cholesterol or HDL cholesterol between mice fed with fat free diet versus regular diet (Extended Data Fig. 2). Overall, these results indicate that mice fed with a fat deficient diet from birth develop a marked compensatory increase in fatty acid synthesis, which culminates in the development of liver steatosis, and cessation of the early cardiomyocyte proliferation phenotype by 10 weeks of age.
Loss of PDK4 shifts myocardial substrate utilization in vivo
Next, we examined the effect of inhibition of fatty acid utilization by cardiomyocyte mitochondria on cardiomyocyte proliferation. Mitochondrial Pyruvate Dehydrogenase (PDH) is a central enzyme regulating the utilization of glucose relative to fatty acids for energy homeostasis. More specifically, cardiac PDH activity is largely inhibited by Pyruvate Dehydrogenase Kinase 4 (PDK4) in the presence of fatty acids37,39–41. In order to enhance the oxidation of glycolytically-derived pyruvate, we generated an inducible cardiomyocyte-specific, PDK4 knockout (KO) mouse. To generate these mice, mice with tamoxifen-inducible Cre recombinase under control of αMHC (αMHC-MerCreMer) were crossed with PDK4loxP mice in which loxP sites flank exons 3–7. Recombination in cardiomyocytes was achieved by administering 0.5 mg of tamoxifen 3 times every other day (over 5 days) through IP injections in 10–12 weeks-old PDK4 KO mice (Fig. 3a). The hearts were harvested 2 weeks later. The efficiency of the tamoxifen administration and consequently the Cre-Lox recombination was assessed by Western blot (Fig. 3b).
Fig. 3: Conditional PDK4 deletion results in a marked shift in myocardial substrate utilization, decreased DNA damage, and increased proliferation in adult cardiomyocytes.
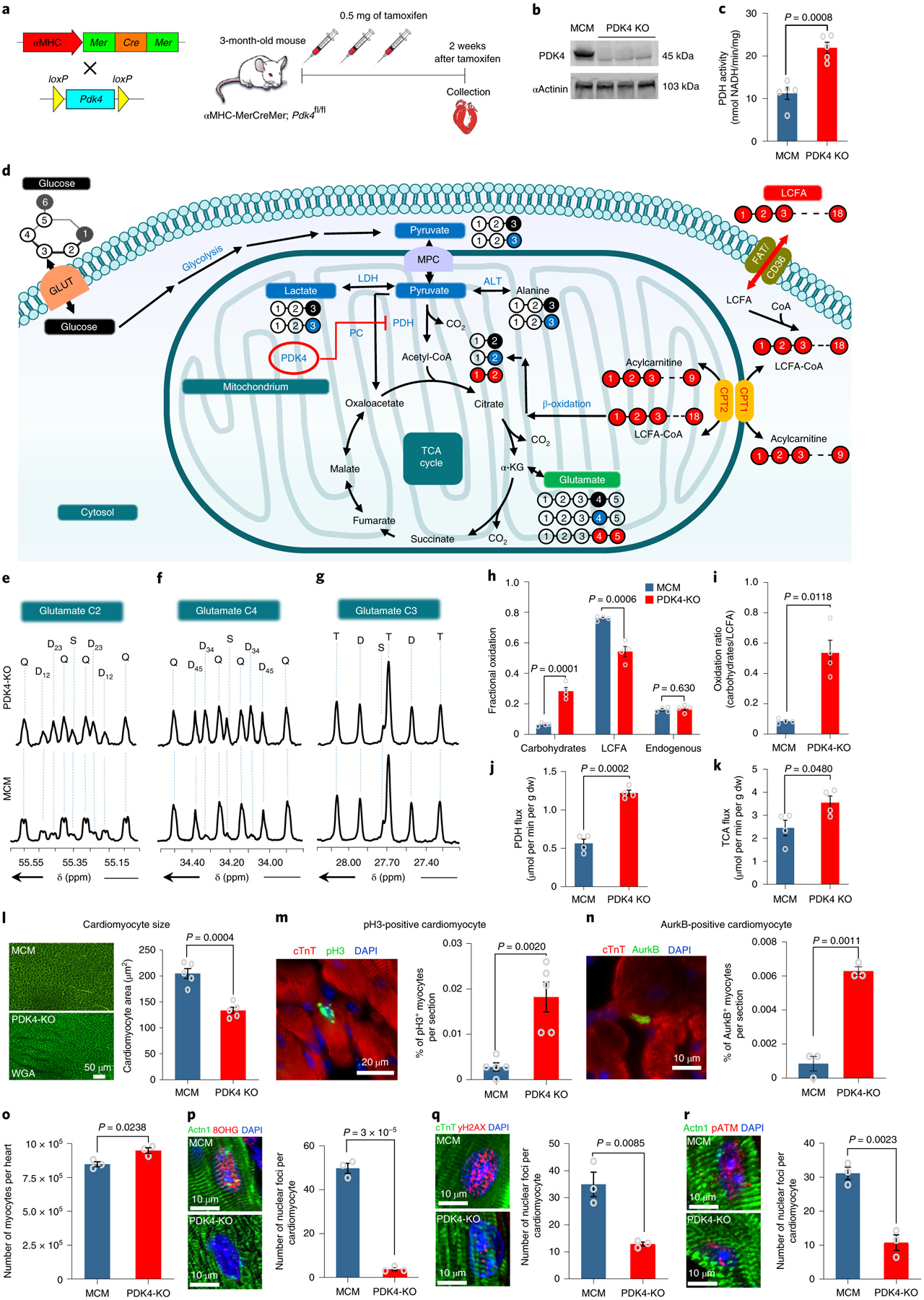
a, Schematic view of the inducible knockout αMHC-MerCreMer cross with PDK4f/f . b, Western Blot for the PDK4 protein shows noticeable depletion in the PDK4 protein expression in the PDK4 KO group compared to the control αMHC-MerCreMer (MCM) hearts. The experiment was repeated twice, with similar results. c, Pyruvate Dehydrogenase (PDH) activity (n=5 biologically independent mice per group). d, A scheme showing the generation of glutamate multiplets from glucose, lactate-pyruvate, and LCFA. Note that 13C labeling for glucose, lactate-pyruvate, and LCFA is indicated as black, blue and red balls, respectively. e, Glutamate C-2 (55.35 ppm) f, Glutamate C-4 (34.20 ppm) and g, Glutamate C-3 (27.60 ppm) spectra. The letters S, D, T, and Q refer to a singlet, doublet (with the relevant J-coupled spins), triplet (a degenerate doublet of doublets) or quartet (or doublet of doublets), respectively. h, Fractional oxidation (n=4 biologically independent mice per group). i, Carbohydrates to LCFA oxidation ratios (n=4 biologically independent mice per group). j, PDH flux and k. TCA flux (n=4 biologically independent mice per group). l, WGA staining shows a significant decrease in cardiomyocyte cell size measurement in PDK4 KO mice compared to the control (n=5 biologically independent mice per group). m, Anti-pH3 and anti-cTnT co-immunostaining shows a significant increase in the cardiomyocyte mitosis marker in cardiomyocyte-specific PDK4 KO mice (n=5 biologically independent mice per group). n, Anti-Aurora B kinase and anti-cTnT co-immunostaining shows a significant increase in cardiomyocyte cytokinesis marker in PDK4 KO compared to the control group (n=3 biologically independent mice per group). o, Quantification of total number of isolated cardiomyocytes by collagenase digestion showing a significant increase in PDK4 KO group at 4 weeks after the first tamoxifen injection (n=3 biologically independent mice per group). Data in p, q and r represent the co-immunostaining with anti-8-hydroxyguanosine (8OHG), anti-γH2AX and anti-pATM antibodies, respectively. There are significant decreases in oxidative DNA damage and the DNA damage response pathway in cardiomyocytes from PDK4 KO hearts (n=3 biologically independent mice per group). Data are presented as the mean±s.e.m. Statistical analysis was performed with two-tailed Student’s t-test: NS, not significant. Actn1, Alpha actinin-1, dw, dry weight; LCFA, long chain fatty acids; MCM: αMHC-MerCreMer mice.
Following PDK4 deletion, we found that the PDH activity in the PDK4 KO mice was significantly upregulated (Fig. 3c) with an increase in glucose relative to fatty acid oxidation. Substrate utilization was assessed by analyzing the 13C NMR multiplet ratios of glutamate isotopomers in PDK4 knockout hearts supplied with [1,6-13C2]glucose, [3-13C]lactate, [3-13C]pyruvate, and [U-13C]LCFA during perfusion. Fig. 3d illustrates the metabolism of supplied 13C-enriched substrates to produce different glutamate isotopomers. The 13C multiplets of glutamate C-2 (Fig. 3e), C-4 (Fig. 3f), and C-3 (Fig. 3g) were analyzed to evaluate the fractional substrate oxidation. The results revealed an increase in carbohydrate oxidation and a decrease in LCFA oxidation in the PDK4 KO hearts, demonstrating that deletion of PDK4 increases carbohydrate utilization for energy production compared to the control αMHC-MerCreMer (MCM) hearts (Fig. 3h–i). Higher PDH flux (Fig. 3j) was observed in the PDK4 KO hearts (1.22 ± 0.03 vs 0.56 ± 0.05μmol/min/g dw). PDK4 KO hearts also increased TCA cycle flux compared with control hearts (Fig. 3k). TCA flux was 3.55 ± 0.28 μmol/min/g dw and 2.45 ± 0.34 μmol/min/g dw for PDK4 KO and control MCM hearts, respectively. These results indicate that PDK4 has an essential role in the control of PDH activity and consequently in substrate utilization by the cardiac muscle.
Cardiac specific deletion of PDK4 promotes cardiomyocyte cell cycle progression in adult
Next, we examined the effect of the increase in glucose oxidation and decrease in fatty acid oxidation on cardiomyocyte cell size, cell proliferation and oxidative DNA damage in the adult heart. Overall, no significant difference (P value= 0.654) was found in HW/BW ratio in PDK4 KO mice (5.28 ± 0.25 n=17) compared to control MCM mice (5.12 ± 0.22 n=18). However, cardiomyocyte cell size was significantly smaller in PDK4 KO mice (Fig. 3l). Moreover, we quantified the number of cardiomyocytes that were positive for pH3 and aurora B kinase, and found a significant increase in both pH3-positive cardiomyocytes and aurora B kinase-positive cardiomyocytes in PDK4 KO mice compare to the controls (Fig. 3m–n). Also, we found that PDK4 KO hearts (at 4 weeks post tamoxifen) had a significantly higher number of cardiomyocytes following dissociation of the heart by collagenase digestion, compared to wild type hearts (Fig. 3o).
Additionally, the oxidative DNA damage caused by ROS, which was assessed and quantified by oxidative base modification of DNA 8-hydroxyguanosine (8-OHG), was significantly decreased in PDK4 KO mice compared to the control MCM mice (Fig. 3p). Furthermore, the marker for DNA double-strand breaks γH2AX was also significantly decreased in the PDK4 KO hearts (Fig. 3q). In concert, the DNA Damage Response (DDR) pathway was significantly decreased in the PDK4 KO mice, as indicated by the decreased nuclear foci of phosphorylated ataxia telangiectasia mutated (pATM) (Fig. 3r). Together these results indicate that the increase in glucose utilization leads to decreased oxidative DNA damage plays an important role in cardiomyocyte cell cycle re-entry.
RNA-sequencing analysis of PDK4 KO hearts
In order to gain insights into the pathways dysregulated in the PDK4 KO hearts, we performed an RNA-Seq analysis. RNA-seq analysis demonstrated 687 differentially expressed genes (267 downregulated and 420 upregulated) in the PDK4 KO hearts compared to the control hearts (Extended Data Fig. 3a–c). Gene Ontology analysis of the dysregulated genes showed enrichment in several pathways including DNA replication, cell cycle progression, cell growth, carbohydrates and lipid metabolic process (Extended Data Fig. 3d). Critical cell cycle inhibitors like Cdkn1a and Cdkn1c were downregulated in PDK4 KO hearts as compared to controls. In contrast, cell cycle activators like E2f1, Cdc6 and Ccnd1 were upregulated in the PDK4 KO hearts (Extended Data Fig. 3e). These results suggest that mitochondria substrate utilization influences pathways involved in cell cycle re-entry and cell growth.
Cardiac specific PDK4 KO mice show significant improvement in heart function following MI
To assess the effect of the modulation of cardiac fatty acid oxidation as a therapeutic approach to improve cardiac function and promote heart regeneration, we induced MI in adult mice. The injury was induced by permanent ligation of the proximal left anterior descending coronary artery. One week after injury, we specifically induced deletion of PDK4 in cardiomyocytes by intraperitoneal (IP) injection of tamoxifen. Deletion of PDK4 1 week following induction of myocardial infarction was specifically designed to bypass any protective effect of PDK4 deletion on ischemic cardiomyocyte death, and thus any effect of PDK4 deletion would fall in the remodeling and regeneration timeframes. The western blot of PDK4 shows elimination of the protein as early as 4 days following the first tamoxifen injection (Extended Data Fig. 4). Left ventricular systolic function was assessed by echocardiography 1 week after surgery, 1 week after tamoxifen administration and every month thereafter until 6 months post-surgery (Fig. 4a). Representative images from the echocardiography studies performed at 1 week post-MI and 6 months post-MI are presented in Fig. 4b. By 6 months post-surgery, the left ventricular ejection fraction (LVEF) of the PDK4 KO mice showed a significant improvement compared to the 1 week post-MI measurement and to the control mice (Fig. 4c). Similar improvements were seen in the other echocardiography parameters such as left ventricular internal dimension in diastole and systole consistent with reduced left ventricular dilatation (Extended Data Fig. 5 a–b). A decrease in LV dilatation and remodeling of the PDK4 KO hearts as compared to the control hearts was also seen by Trichrome staining at 6 months post-MI with significantly smaller fibrotic scars (Fig. 4d–e). By WGA staining, there was a significant decrease in cell size at the border zone of the PDK4 KO mice as compared to the control mice at 6 months post-MI (Fig. 4f). Moreover, the uninjured PDK4 KO mice showed similar decrease in cardiomyocyte cell size, compared to the uninjured control MCM mice (Fig. 4f). No significant difference was observed related to LVIDd, LVIDs between control and PDK4 KO mice 6 months post tamoxifen injection (Extended Data Fig. 5c–d). Corresponding trichrome stained short axis views are displayed below (Extended Data Fig. 5e). In addition, we observed a significant increase in the percentage of cardiomyocytes positive for pH3 and aurora B kinase in the PDK4 KO group, at 2 weeks post-MI (Fig. 4g–h). Collectively, these results indicate that deletion of PDK4 has a significant effect on the adverse cardiac remodeling and reduction in ejection fraction that occurs after induction of MI. It is important to note here that we believe that both effects are occurring simultaneously, and that the slow but gradual improvement of left ventricular systolic function over a 6 months period is indicative of a modest pro-regenerative response.
Fig. 4: PDK4 KO hearts show a higher LVEF accompanied by a remarkable decrease in LV dilatation and remodeling, as well as increase cardiomyocyte proliferation compared to control hearts.
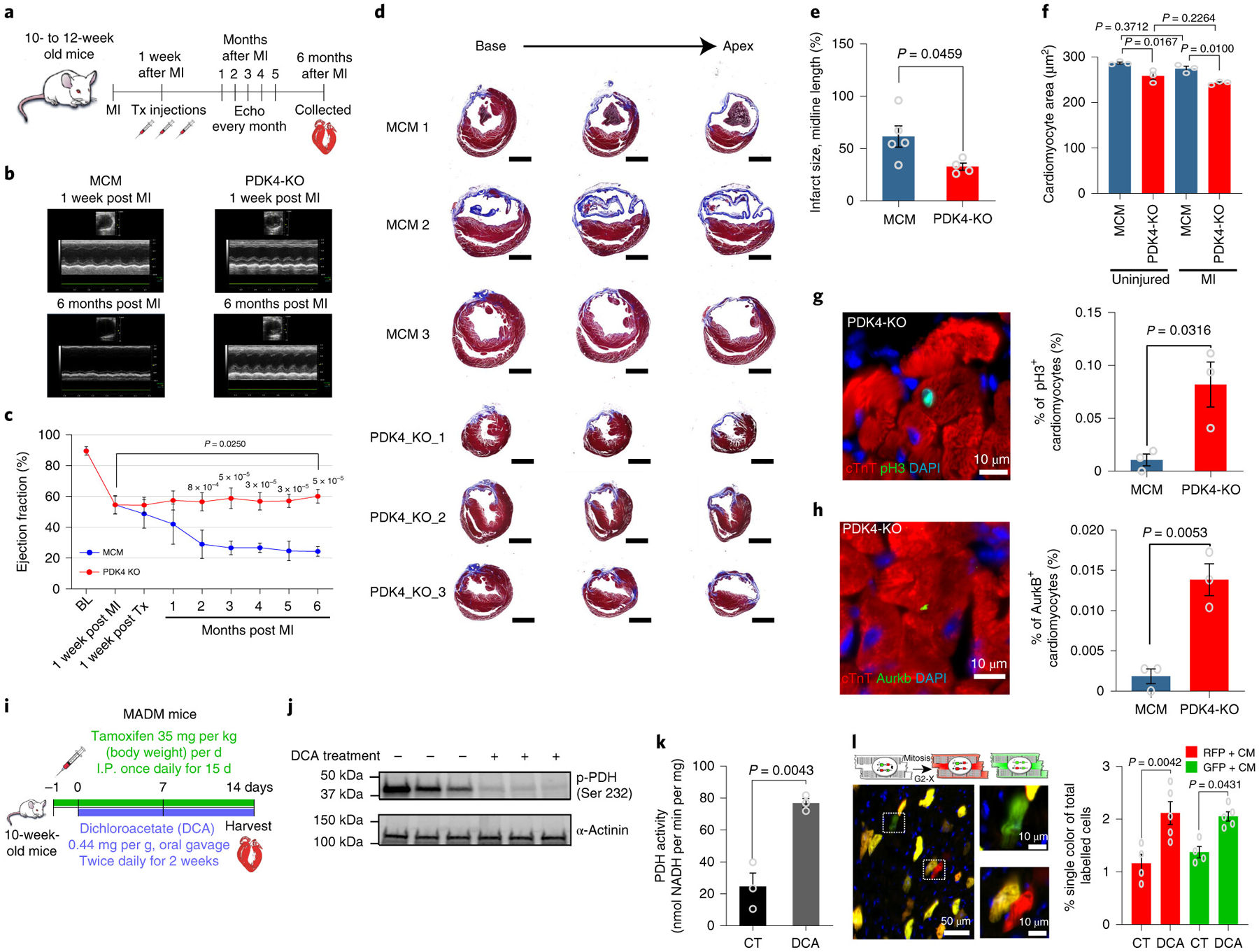
a, One week after MI induced by LAD ligation, mice were injected with Tamoxifen. The cardiac function was assessed every month by echocardiography. Hearts were harvested 6 months after MI and submitted to histology analysis. b, Representative echocardiography images comparing control MCM and PDK4 KO hearts 1 week and 6 months post-MI. c, Echocardiography analysis of LVEF showing a higher LVEF post-MI in PDK4 KO (n=5 biologically independent mice) compared to control MCM (n=5 biologically independent mice). Data are represented as the mean±s.d. Analyses were performed by unpaired two-tailed Student’s t-test. Additionally, we observed significant improvement of the LVEF 6 months post-MI compared to 1 week post-MI. Statistical analysis was performed using paired two-tailed Student’s t-test. d, Trichrome staining of hearts, 6 months post-MI, shows a marked decrease in LV dilatation and remodeling of PDK4 KO compared to control MCM hearts. The experiment was repeated four times, with similar results e, Infarct size quantification showing significantly smaller fibrotic scars in PDK4 KO group (n=4 biologically independent mice) compared to control MCM (n=5 biologically independent mice). f, WGA staining shows a significant decrease in cardiomyocyte cell size measurement in PDK4 KO compared to controls (n=3 biologically independent mice per group). g, Anti-pH3 and anti-cTnT co-immunostaining showing a significant increase in the percentage of pH3+ cardiomyocytes in the PDK4 KO group compared to control MCM (n=3 biologically independent mice per group). h, Anti-Aurora B kinase and anti-cTnT co-immunostaining shows a significant increase in the cardiomyocyte cytokinesis marker in the PDK4 KO compared to control MCM group (n=3 biologically independent mice per group). i, Schematic view of the tamoxifen and DCA administration in 10 weeks old MADM; MCM mice. j, Western blot for phopho-PDH Ser232 showing clear depletion of the phosphorylated PDH in the mice group treated with DCA (n=3 biologically independent mice per group). k, Pyruvate dehydrogenase (PDH) activity (n=3 biologically independent mice per group). l, Frequency of single-labeled cardiomyocytes per heart section, showing significant increase in DCA treated mice (n=5 biologically independent mice) compared to control (n=4 biologically independent mice). Data present in e-l are represented as the mean±s.e.m. Statistical analysis was performed using a two-tailed Student’s t-test: NS, not significant, BL, baseline; LVEF, left ventricular ejection fraction; MI, Myocardial infarction; DCA: Dichloroacetate; PDH: Pyruvate Dehydrogenase. CM: cardiomyocyte; CT: control; RFP: red fluorescent protein; GFP: green fluorescent protein; MCM: αMHC-MerCreMer.
Pharmacological inhibition of PDK4 promotes cardiomyocyte proliferation in adult mice.
In order to determine the effect of pharmacological inhibition of PDK4 in cardiomyocyte proliferation, MADM; MCM mice were used to assess genetic evidence of cardiomyocyte expansion as outlined in the Methods. We administered the known PDK inhibitor, Dichloroacetate (DCA) by gavage every 12 hours for 14 days in adult MADM; MCM mice. Concomitantly, Cre-Lox recombination was induced by tamoxifen injection for 15 days starting 1 day before DCA treatment (Fig. 4i). Hearts were harvested after 14 days of DCA treatment to assess PDH activity and total number of GFP+ and RFP+ single labeled cardiomyocytes per heart section. To confirm that DCA treatment did in fact inhibit PDH activity, we show here that DCA treatment significantly decreased phospho-PDH Ser232 (Fig. 4j) and increased PDH activity (Fig. 4k). In addition, DCA treated MADM/MCM mice displayed a significant increase in proportion of both RFP exclusive cardiomyocytes and GFP exclusive cardiomyocytes confirming a pro-proliferative effect on cardiomyocytes (Fig. 4l). These results demonstrate that an increase in PDH activity is associated with a significant increase in the number of cardiomyocytes that complete cytokinesis and results in new cardiomyocyte formation.
Discussion
Shortly after birth, cardiomyocytes exhibit a shift in energetic substrate utilization, from pyruvate to fatty acids29,31,34. The premise of the current manuscript is that we have previously found that the postnatal metabolic shift coincides with increased DNA damage, expression of DNA damage markers, and cell cycle arrest of cardiomyocytes29. Therefore, the current studies were designed to specifically determine whether modulating substrate utilization would in fact affect DNA damage and promote cell cycle re-entry in cardiomyocytes. Here, we utilized two different mouse models to assess the role of fatty acid utilization on postnatal cardiomyocyte cell cycle arrest. First, we used a combination of genetic PERK KO in mammary epithelial cells and dietary modification to induce dietary fatty acid deficiency from birth. This resulted in a pronounced prolongation of the postnatal window of cardiomyocyte proliferation. Surprisingly, after the animals were maintained on the same fat free diet for 10 weeks, we found no evidence of increased cardiomyocyte proliferation. This was associated with marked hepatomegaly and steatosis as a result of increased fatty acid synthesis, which was confirmed by lipidomics and mass spectrometry analysis of hepatic fatty acid synthesis pathways. In contrast, livers at P21 were not enlarged and that there was no fatty liver (data not shown). This indicates that the cessation of cardiomyocyte proliferation at 10 weeks coincided with enhanced FA synthesis by the liver. Previous studies have also shown this liver phenotype with exposure of adult mice to low fat diet, although these diets also contained very high sucrose content, rather than normal sucrose content in our study48. Analysis of liver lipidomics demonstrated over a 10 fold increase in TAG, which was accompanied by upregulation of several regulatory fatty acid biosynthesis enzymes including the rate limiting Acetyl CoA-carboxylase enzyme which catalyzes the irreversible carboxylation of acetyl-CoA to produce malonyl-CoA. These results suggest that impaired dietary supply of fatty acids can delay but not prevent postnatal cardiomyocyte cell cycle arrest due to a compensatory increase in hepatic fatty acid biosynthesis.
Next, we conducted a set of studies to examine the role of inhibiting fatty acid utilization by cardiomyocyte mitochondria on cardiomyocyte cell cycle. We generated a new conditional PDK4 KO mouse model, and we found that conditional PDK4 deletion in adult cardiomyocytes results in a marked shift in myocardial substrate utilization resulting in decreased fatty acid and enhanced glucose-derived pyruvate utilization. This was accompanied by a significant decrease in DNA damage, both base oxidation and double strand breaks, as well as decreased expression of pATM, an upstream DNA damage response marker. We have previously demonstrated that these pathways are activated in postnatal cardiomyocytes which coincide with cell cycle arrest29. We also noted a significant increase in cardiomyocyte mitosis and cytokinesis in response to PDK4 deletion in the adult mouse heart. It is important to note here that these results were confirmed in the MADM mouse model which allows conclusive genetic fate mapping of cardiomyocyte mitosis following pharmacological inhibition of PDK4. In support of this phenotype, RNA-seq analysis demonstrated an induction of DNA replication and cell cycle progression pathways in the PDK4 KO mice.
Intriguingly, the PDK4 KO hearts showed a remarkable decrease in LV dilatation and remodeling compared to control hearts, which was accompanied by a markedly higher LVEF compared to control hearts, and modest, but significant increase in LVEF compared to the immediate post-injury values. This was also accompanied by a decrease in MI induced cardiomyocyte hypertrophy. A number of previous elegant studies reported that activation of PDH through administration of dichloroacetate or using global PDK4 KO mice (PDK4−/−) to enhance glucose utilization is cardioprotective with regards to infarct size and contractile dysfunction following ischemia/reperfusion injury49,50. Similarly, inhibition of fatty acid utilization (malonyl coenzyme A decarboxylase KO mice) during ischemia/reperfusion injury has been found to minimize structural and functional damage50,51. However, the possibility that these interventions may influence cardiomyocyte proliferation and induce heart regeneration has not been studied. It is important to note here that although we induced Pdk4 deletion 1 week following MI to abrogate any effect on myocardial salvage in response to ischemia, the positive effect on LV function and dilatation probably reflects a combination of a reverse remodeling effect in conjunction with the modest increase in cardiomyocyte proliferation.
The possibility exists that PDK4 exerts effects not solely dependent on interaction with the PDH complex. For example, evidence indicates that PDK4 binds to and stabilizes the cAMP-response element-binding (CREB) protein, resulting in mTORC1 activation52. In addition, a recent report indicates that in mouse skeletal muscle, PDK4 interacts with and stabilizes the IP3R1-GRP75-VDAC1 protein complex at the mitochondrial-associated ER membrane, an interaction that appears to contribute to insulin resistance53. While the potential of non PDH-dependent mechanisms has not been tested in the current study, given the significant phenotype of the PDK4 mutants it is important for future experiments to consider the potential contributions of alternative roles for PDK4.
In summary, the findings in the current report indicate that fatty acid utilization by cardiomyocytes is an important contributor to postnatal cardiomyocyte cell cycle arrest, at least in part through modulation of DNA damage and the DNA damage response pathway. Our results also suggest that PDK4 may be a viable therapeutic target for prevention of myocardial remodeling and induction of cardiomyocyte proliferation in the adult heart.
Methods
Animals:
All protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of UTSW. All experiments were performed on age and sex matched mice. The following mice were obtained from the Jackson Laboratory: αMHC-MerCreMer [(Tg(Myh6-Cre/Esr1*)1Jmk) (stock number 005650)]; MADM-11GT/TG [(Igs2tm1(ACTB-EGFP,-tdTomato)Zng/Igs2 tm2(ACTB-tdTomato,-EGFP)Zng/J) (stock number 030578)]; PERKloxP [(Eif2ak3tm1.2Drc) (stock number 023066)] and MMTV-cre mice [Tg(MMTV-cre)4Mam (stock number 003553)].
Generation and genotyping of the PDK4 knockout mice:
The PDK4tm1a(KOMP)Wtsi embryonic stem cells (ESCs) (cell clone EPD0180_2_B01) was purchased from the Knockout Mouse Project (KOMP) Repository. The ESC clones were injected into the blastocysts of C57BL/6 mice in the Transgenic Core Facility at the University of Texas Southwestern Medical Center, Dallas (UTSW). Mice were genotyped using PDK4 sense (5’-TTCCATGAGAAGAGCCCAGAAGACC-3’) and PDK4 antisense (5’-AGAAGAGGCAGCTTCATGGAAGG-3’) primers. The wild type allele was identified as a 553bp long PCR product and the PDK4f/f mice were determined by a 745bp long PCR product on a 1.2% agarose gel. Inducible cardiomyocyte-specific deletion of PDK4 (PDK4 KO) was achieved by crossing PDK4f/f mice with αMHC-MerCreMer mice54 [(tamoxifen-inducible Cre recombinase under control of a cardiomyocyte-specific alpha-myosin heavy chain promoter (αMHC)]. Homologous recombination in cardiomyocytes was achieved by administering 0.5 mg of tamoxifen 3 times every other day (over 5 days) through intraperitoneal (IP) injections in 8–12 week-old PDK4 KO mice. The hearts were harvested 2–4 weeks later. αMHC-MerCreMer mice were genotyped using αMHC-MerCreMer sense (5’- GATTTCCGTCTCTGGTGTAGCTGATGATCC-3) and αMHC-MerCreMer antisense (5’-GCCAGGTATCTCTGACCAGAGTCATCC-3) primers. Age and sex matched αMHC-MerCreMer were used as controls and received the same dose of tamoxifen.
Fatty acid deficient milk mouse model:
The Protein Kinase R (PKR)-like endoplasmic reticulum kinase (PERK) or Eukaryotic translation initiation factor 2 alpha-subunit kinase 3 (Eif2ak3) plays a significant role in the unfolded protein response55,56. PERK is required for proper development and maturation of mammary epithelial cells. Deletion of PERK in mammary glands inhibits key lipogenic enzymes involved in lipid production and storage (Fatty acid synthase, ATP citrate lyase and stearyl-CoA desaturase-1) decreasing the lipid content in milk45. To generate mice which produce milk deficient in fatty acids, PERKloxP mice with loxP sites which flank exons 3–557 were mated with Mouse mammary tumor virus (MMTV)-Cre transgenic mice46. MMTV-Cre transgenic mouse lines express Cre recombinase predominantly in secretory cell types in the mammary epithelium46. Female mice homozygous for PERKloxP and MMTV-Cre produce mammary glands in which the milk is deficient in fatty acids45. Here, only male mice were used in our studies to avoid variations between groups. The PERKloxP and MMTV-cre mice were obtained from Jackson Laboratory (Eif2ak3tm1.2Drc stock number 023066); and Tg(MMTV-cre)4Mam - stock number 003553), respectively.
MADM (Mosaic Analysis with Double Markers) lineage tracing:
To corroborate that completed cytokinesis has occurred in cardiomyocytes, MADM-11GT/TG; αMHC-MerCreMer (MADM; MCM) transgenic mice were used to assess cytokinesis events in cardiomyocytes by lineage tracing47. In short, tamoxifen injections in MADM; MCM mice induce allelic recombination of green fluorescent protein (GFP) and red fluorescent protein (RFP) in cardiomyocytes, so that daughter cells which do not complete cytokinesis remain yellow (GFP+ and RFP+) and daughter cells which complete cytokinesis will be GFP+ or RFP+. It is important to note here that although the MADM system represents a highly rigorous method of assessment of cellular proliferation, it underestimates the degree of proliferation by at least 50%47.
In the fat deficient milk model, MADM; MCM neonatal mice at P0 were transferred to a Fat deficient milk foster mother (MMTV-Cre; PERKf/f) and fat free diet introduce from P12. The control group consisted of MADM; MCM with its own mother on the regular diet. 4-hydroxytamoxifen (4-OHT) was used to induce recombination from P7 (following postnatal cell cycle arrest that normally occurs around P7). MADM; MCM mice were injected with 4-OHT (200μg/pup) at P7, P9, and P11, and harvested at P21 and assessed for GFP+ (green) and RFP+ (red) exclusive cells relative to the total number of GFP+/RFP+ (yellow) recombined cardiomyocytes per section.
To assess the effect of increased PDH activity, MADM; MCM mice were treated with Dichloroacetate (DCA). DCA (0.44mg/g of bodyweight) by oral gavage inhibits phospho-PDH (increases PDH activity) for approximately 12 hours58, we therefore administer DCA (0.44mg/g of bodyweight) by gavage, every 12 hours in MADM; MCM mice for 14 days. Adult (10 weeks old) MADM; MCM mice were injected with tamoxifen (35mg/kg) once a day for 15 days beginning 1 day prior to DCA treatment. Hearts were harvested after 14 days of DCA treatment and assessed for GFP+ and RFP+ exclusive cells relative to the total number of GFP+/RFP+ recombined cardiomyocytes per section. The MADM-11GT/TG (Igs2tm1(ACTB-EGFP,-tdTomato)Zng/Igs2 tm2(ACTB-tdTomato,-EGFP)Zng/J) mice were obtained from the Jackson Laboratory (stock number 030578).
Cardiomyocyte isolation:
Hearts were freshly harvested and fixed in 4% PFA at 4°C overnight. The ventricles were minced to smaller pieces and subsequently incubated with collagenase D (2.4 mg mL-1, Roche) and B (1.8 mg mL-1, Roche) for 12 hours at 37°C using an end-over-end shaker. The supernatants were collected via 160 μm nylon mesh filter, and the procedure was repeated until no more cardiomyocytes were dissociated from the tissue. A portion of the isolated cardiomyocytes were co-stained with Connexin 43 antibody (IHCWORLD IW-PA1026) and DAPI (Sigma D9542) for quantification.
13C NMR isotopomer analysis:
All experimental procedures were approved by IACUC at UTSW. Substrate competition between glucose and fatty acid was evaluated using 13C-NMR isotopomer analysis in isolated Langendorff perfused hearts from control and PDK4 KO mice. The hearts were quickly excised, arrested, and cannulated via the aorta following cervical dislocation of the mouse. The cannulated heart was then connected to a perfusion column apparatus maintained at 37°C using a temperature controlled bath. Hearts were perfused retrograde for 30 minutes at 80 cm H2O pressure with a modified Krebs-Henseleit buffer containing 8 mM [1,6-13C]glucose, 1.2 mM [3-13C]lactate, 0.12 mM [3-13C]pyruvate, 0.4 mM [U-13C]long chain fatty acids (LCFAs), 0.75% bovine serum albumin (BSA). The LCFA is a physiological mix of oleic, palmitic, stearic, palmitoleic and linolenic acids. The non-recirculating buffer was oxygenated with a thin-film oxygenator with a 95:5 mixture of O2:CO2. The heart rate was continually monitored throughout perfusion with a fluid-filled catheter in the left ventricle. Subsequently, hearts were snap-frozen, pulverized in liquid nitrogen and extracted with 4% perchloric acid. The perchloric extracts were then neutralized and reconstituted in D2O containing 1 mM ethylenediaminetetraacetic acid (EDTA) and 0.5 mM 2,2-dimethyl-2-silapentane-5-sulfonate (DSS). Proton-decoupled 13C-NMR spectra of heart extracts were acquired with a 14.1 T spectrometer (Bruker, USA) equipped with 5-mm cryoprobe. 13C NMR resonances from glutamate multiplets were deconvoluted using ACD/SpecManager (ACD Labs, Canada). Multiplet ratios were calculated to determine the relative oxidation of carbohydrates (glucose, lactate, pyruvate), LCFAs, and unlabeled endogenous substrates (e.g., triglycerides and glycogen). The labeling patterns from [1,6-13C]glucose, [3-13C]lactate, and [3-13C]pyruvate eventually generate the same labeling pattern in Ac-CoA, so these were collectively termed carbohydrates. Multiplet ratios were submitted as input in tcaCALC v.2.07 for isotopomer analysis. Data is presented as the mean±s.e.m. (n=4 per group) and analyzed with Welch’s t-test for statistical significance.
Immunostaining:
Tissues were fixed in 4% paraformaldehyde in PBS overnight at 4°C and incubated in 30% sucrose in PBS at 4°C overnight. Tissues were embedded in tissue freezing medium and cut 8 μm thicknesses. For antigen retrieval, either 1 mM EDTA with 0.05% Tween 20 in boiling water or epitope retrieval solution (IHC World) in a steamer (IHC-Tek Epitope Retrieval Streamer Set) were used, then sections were blocked with 10% serum from the host animal of secondary antibodies, and incubated with primary anti-bodies overnight at 4 °C. The sections were subsequently washed with PBS and incubated with corresponding secondary antibodies conjugated to Alexa Fluor 488 or 555 (Invitrogen). The slides were mounted in antifade mounting medium (Vector Laboratories, Burlingame, California). Primary antibodies used are as follows: anti-phospho histone H3 Ser10 (Millipore 06–570, 1:100), anti-aurora B (Sigma A5102, 1:100), anti-troponin T, cardiac isoform Ab-1, clone 13–11 (Thermo Scientific MS-295-P1, 1:100), anti-sarcomeric α-actinin (Abcam, ab68167, 1:100), anti-8 hydroxyguanosine (Abcam ab62623, 1:50), anti-phosphorylated ATM (Santa Cruz Biotechnology sc-47739, 1:100), Anti-phospho-Histone gamma H2AX (Millipore-Sigma 05–636). DAPI was used for the nuclear staining.
Drug administration:
Tamoxifen (Sigma) was dissolved in 90% sesame oil/10% ethanol and stored at −20°C. Prior to IP injection (0.5mg/day/mouse), tamoxifen solution was heated at 55°C for 10 min.
Fat free diet:
The modified diet was purchased from Research Diets, Inc (Product number D18012302). Details of diet composition can be accessed in the Supplementary Table 1.
Blood collection and plasma lipid analyses:
Blood was extracted by puncturing capillary tube through mouse orbital sinus. Plasma was collected by centrifuging at 8000 g for 10 min. Total cholesterol and triglyceride concentrations were determined by colorimetric enzymatic assay (Infinity, Thermo Scientific). HDL levels were measured by HDL cholesterol Kit (Wako) after precipitation of apoB-containing lipoproteins using phosphotungstate-magnesium. LDL cholesterol was calculated by Friedewald equation59.
Wheat Germ Agglutinin (WGA) Staining and Cell Size Quantification:
For antigen retrieval, the slides were boiled in citrate buffer (BioGenex) for 20 min, followed by cooling down for 30 min in a water container then washed three times with PBS. The slides were permeabilized with 0.3% Triton X, washed with PBS three times, and incubated with wheat germ agglutinin (WGA) conjugated to Alexa Fluor 488 (50 mg/ml, Life Technologies) for 1 h at room temperature. The slides were washed with PBS three times and mounted in antifade mounting medium (Vector Laboratories, Burlingame, California). The cell size quantification was performed in 3 independent samples per group with 3 different fields and positions, each from left and right ventricles, and septum were captured at 60 x magnifications. For post-MI group, 6 images per section were capture (3 images per side of the border zone). ImageJ software (National Institutes of Health) was used to quantify the size of each cell.
Proteomics Analysis:
Quantitative proteomics was used to determine changes in the expression of the hepatic enzymes involved in the fatty acid biosynthesis. For each liver homogenate, an aliquot containing 100 μg of total protein was taken for analysis, mixed with 8 pmol Bovine Serum Albumin (BSA) as a non-endogenous internal standard, and heated to equilibrate. The proteins were precipitated with acetone. The dried protein pellet was reconstituted in 100 μL Laemmli sample buffer and 20 μL (containing 20 μg) used to run a short (1.5 cm) SDS-PAGE gel. The gel was fixed and stained with Coomassie blue. Each sample was cut from the gel as the entire lane and divided into smaller pieces. The gel pieces were washed to remove the Coomassie blue, then reduced, alkylated, and digested with trypsin. The mixture of peptides was extracted from the gel, evaporated to dryness in a SpeedVac, and reconstituted in 200 μL 1% acetic acid for analysis. The analyses were carried out on a TSQ Quantiva triple quadrupole mass spectrometry system. The HPLC was an Ultimate 3000 nanoflow system with a 10 cm x 75 μm i.d. C18 reversed phase capillary column. 5 μL aliquots were injected and the peptide eluted with a 60 min gradient of acetonitrile in 0.1% formic acid. The mass spectrometer was operated in the selected reaction monitoring mode. For each protein assayed, the method measured 2 to 3 ideal peptides. The program Skyline integrated the peak areas of the appropriate chromatographic peaks. The response for each protein was calculated as the geometric mean of the peak areas for those 2 to 3 peptides. These values were normalized to the response for the BSA standard and expressed as pmol/100 μg total protein.
Lipidomics analysis:
Approximately 100 mg of tissue (liver and heart) in 1mL of methanol/dichloromethane (1:2, v/v) were homogenized with a Bead Ruptor (Omni International) for 50s (5.5 mps, 3 cycles, 10 sec/cycle, 5 s dwell time). The homogenates were transferred to glass tubes and diluted to a final concentration of 20 mg/mL using methanol/dichloromethane (1:2, v/v). The lipid liquid extractions were performed at room temperature (including centrifugation). Aliquots equivalent to 0.5mg of homogenized tissue, were transferred to fresh glass tubes where 1mL of methanol, dichloromethane and water were added. The mixture was vortexed for 5 seconds and centrifuged at 2671×g for 5 min. The organic phase was collected to a fresh glass tube with a Pasteur pipette and dried under N2. The dried extracts were reconstituted in 600μL of dichloromethane/methanol/isopropanol (2:1:1, v/v/v) containing 8mM ammonium fluoride (NH4F) and 33 μL/mL of 3:50 diluted SPLASH LipidoMix™ internal standards mixture (Supplementary Table 2). The samples were direct-infused into a SCIEX quadrupole time-of-flight (QTOF) TripleTOF 6600+ mass spectrometer (Framingham, MA, USA) via a custom configured LEAP InfusePAL HTS-xt autosampler (Morrisville, NC, USA). Samples were infused for 3 min at a flow rate of 10μL/min. Electrospray ionization (ESI) source parameters were, GS1 25, GS2 55, curtain gas (Cur) 20, source temperature 300°C and ion spray voltage 5500V in positive ionization mode. GS1 and GS2 were zero-grade air while Cur and CAD gas were nitrogen. Optimal declustering potential and collision energy settings were 120V and 40eV. MS/MSALL 60 analysis was performed by collecting product-ion spectra at each unit mass from 200–1200 Da with an accumulation time of 0.3s per mass. Analyst® TF 1.7.1 software (SCIEX) was used for MS/MSALL data acquisition. Chronos XT® software from Axel Semrau (Srockhovel, Germany) was used to control the InfusePAL system. Data analysis was performed by MarkerView (Sciex), followed by lipid species identification using LipPy, an in-house script.
Myocardial Infarction Model:
For the experiment, 10–12 weeks old, sex matched αMHC-MerCreMer/PDK4f/f or control αMHC-MerCreMer mice received an IP injection of ketamine/xylazine cocktail (100mg/kg, 5mg/kg) to facilitate surgical prep and endotracheal intubation to maintain the airway. The mice were placed in the supine position on a heated (37°C) platform. Anesthesia was maintained throughout the procedure with 1.0% isoflurane. After performing a thoracotomy between the fourth and fifth ribs, myocardial infarction (MI) was induced by ligation of the left anterior descending (LAD) coronary artery. An irreversible ligature was placed at a level ≈1 mm below the left atrium by tightening a suture loop around the vessel. Successful coronary artery occlusion was determined by observed regional myocardial surface cyanosis distal to the suture. Age and sex matched αMHC-MerCreMer mice were used as controls and received the same dose of tamoxifen.
RNA sequencing:
Snap frozen heart tissues from 5 control and 4 PDK4 KO samples were pulverized using mortar and pestle under liquid nitrogen. Pulverized samples were resuspended in TRIzol™ reagent (Thermo Fisher Scientific, Cat#:15596018) and total RNA was prepared using Direct-zol™ RNA miniprep (Zymo Research, Cat#:R2061) according to the manufacturer’s protocol. RNA integrity was assessed using the Agilent bioanalyzer and only samples with RIN > 6 were used for the library prep. Paired-end libraries were constructed using Tru-seq kits (Illumina) and the libraries were sequenced on the Hiseq4000, generating 2×151-bp paired-end reads. Reads were mapped against the mouse genome using Tophat version 2.0.11 with default parameters. Gene count was computed using htseq-count. Differential gene expression analysis was performed using Edge R.A gene and was considered to be differentially expressed between control and PDK4 KO heart samples if the FDR-adjusted p-value was less than 0.05. Gene expression heat map was generated using R (version 3.1.2).
PDH activity:
Isolated cardiac mitochondria were diluted to 0.05 mg/ml in a buffer containing 25 mM MOPS and 0.05% Triton X-100 at pH 7.4. Solubilization of mitochondria with 0.05% Triton X-100 inhibits complex I of the respiratory chain, preventing consumption of NADH. PDH activity was measured spectrophotometrically (Agilent 8452A) as the rate of NAD+ reduction to NADH (340 nm, ε= 6,200 M−1cm−1) upon addition of 2.5 mM pyruvate, 0.1 mM CoASH, 0.2 mM thiamine pyrophosphate, 1.0 mM NAD+, and 5.0 mM MgCl2 at pH 7.4.
Echocardiography:
Assessment of in vivo heart function was performed on conscious, non-sedated mice performed using a Vevo2100 micro-ultrasound system (VisualSonics). Measurements were obtained at baseline, 1 week after MI, 1 week after tamoxifen induced gene deletion and monthly after MI until 6 months. M-mode images were obtained from a parasternal short-axis view at the level of the papillary muscles. All echocardiography measurements were performed and analyzed in a blinded manner.
Statistical Analysis:
All statistical analyses were performed using GraphPad Prism 8 software and a P value of <0.05 was considered statistically significant. Sample size, data and statistical analyses are present and described in all legends. Unpaired two-tailed Student’s t-test was used to determine statistical significance between two independent groups. Paired two-tailed Student’s t-test was used to compare paired mice at two points in time.
Reporting summary:
Further information on research design is available in the Reporting Summary linked to this paper.
Supplementary Material
Extended Data
Extended Data Fig. 1. Quantitative mass spectrometry analysis of fatty acids biosynthesis liver enzymes.
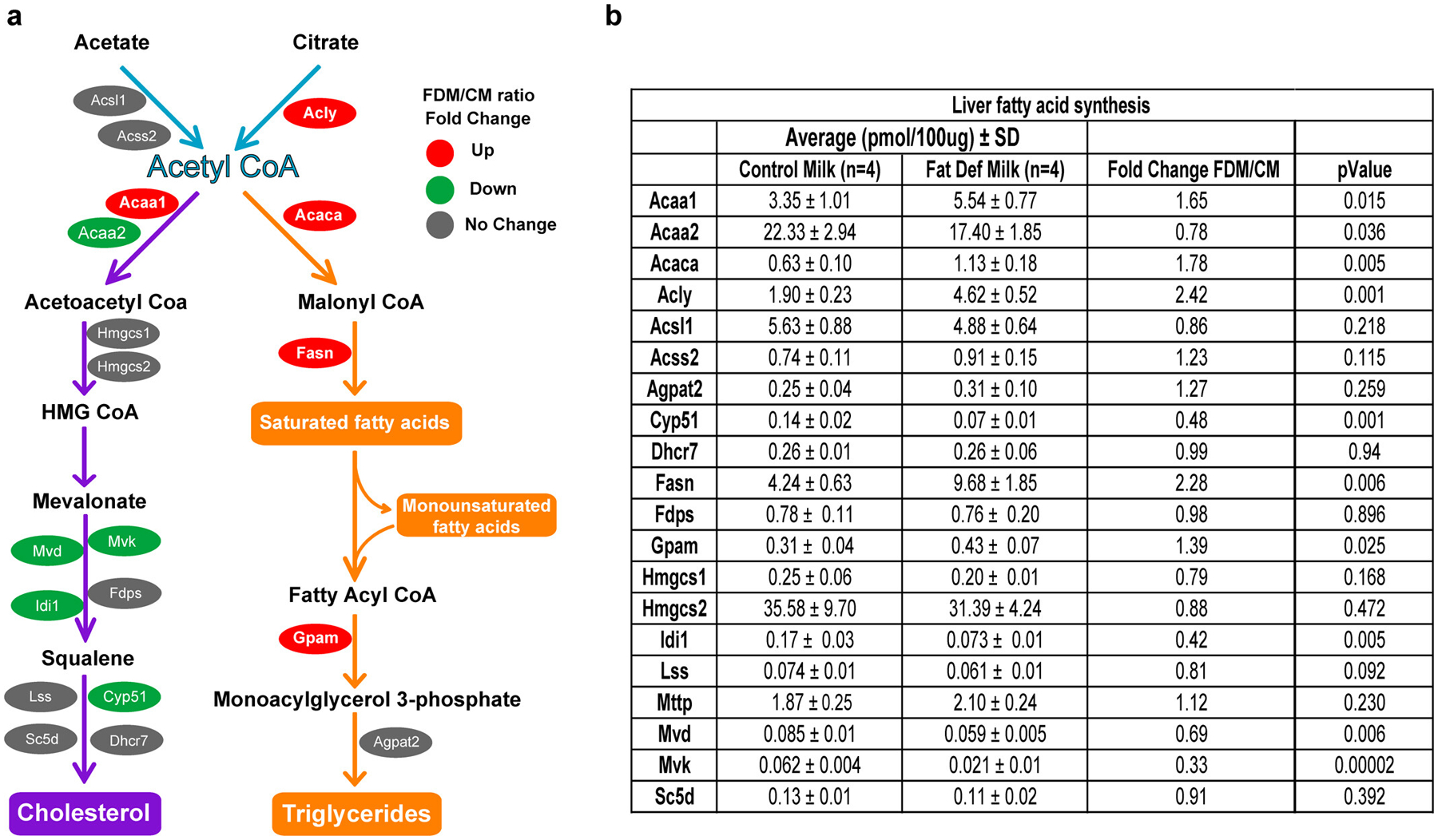
a, Schematic view of cholesterol and triglyceride biosynthesis pathways. Red, green and gray colors represent the upregulated, downregulated and unchanged protein expression, respectively. The analysis shows a significant increase in the enzymes involved in the synthesis of saturated fatty acids and triglycerides in FDM compare to CM mice. Enzymes involved in cholesterol biosynthesis were downregulated in the FDM liver. b, Table showing the average, fold change and p-value of the hepatic enzymes involved in fatty acid biosynthesis, identified by quantitative mass spectrometry between FDM and CM mice (n=4 biologically independent mice per group). CM, control milk group; FDM, fat deficient milk group. Statistical analysis was performed with two-tailed Student’s t-test.
Extended Data Fig. 2. Serum lipids and blood glucose measurements.
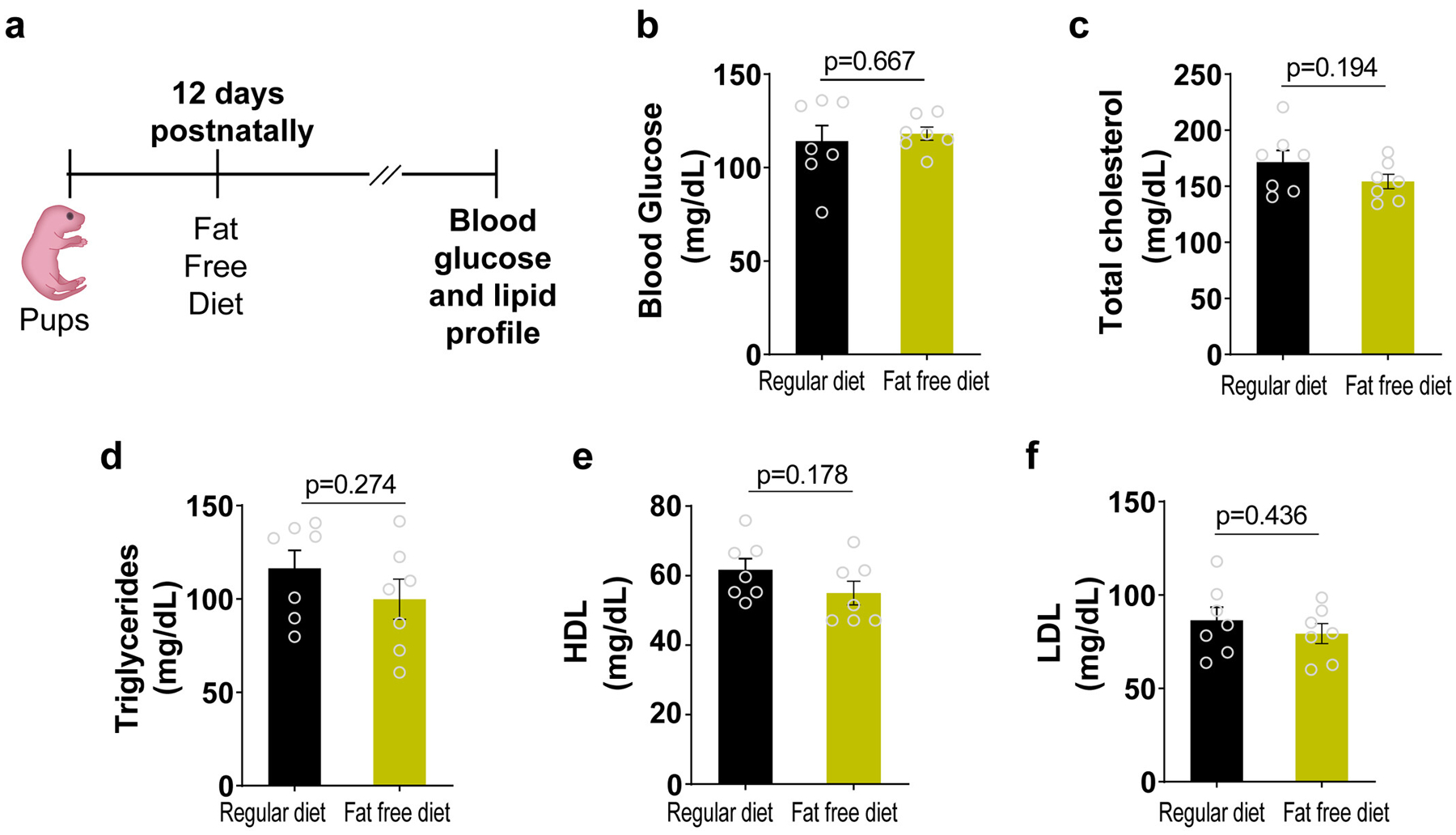
a, At 12 days postnatally, beyond lactation, pups were exposed to a regular diet or Fat Free Diet. Samples were collected at 10 weeks postnatally. Data in b-f represent the blood glucose, total cholesterol, triglycerides, HDL cholesterol or LDL cholesterol measurements, respectively. (n=7 biologically independent mice per group) Data presented as the mean±s.e.m. Statistical analysis was analyzed with two-tailed Student’s t-test: NS, not significant
Extended Data Fig. 3. RNA-seq analysis of PDK4 KO and Control hearts.
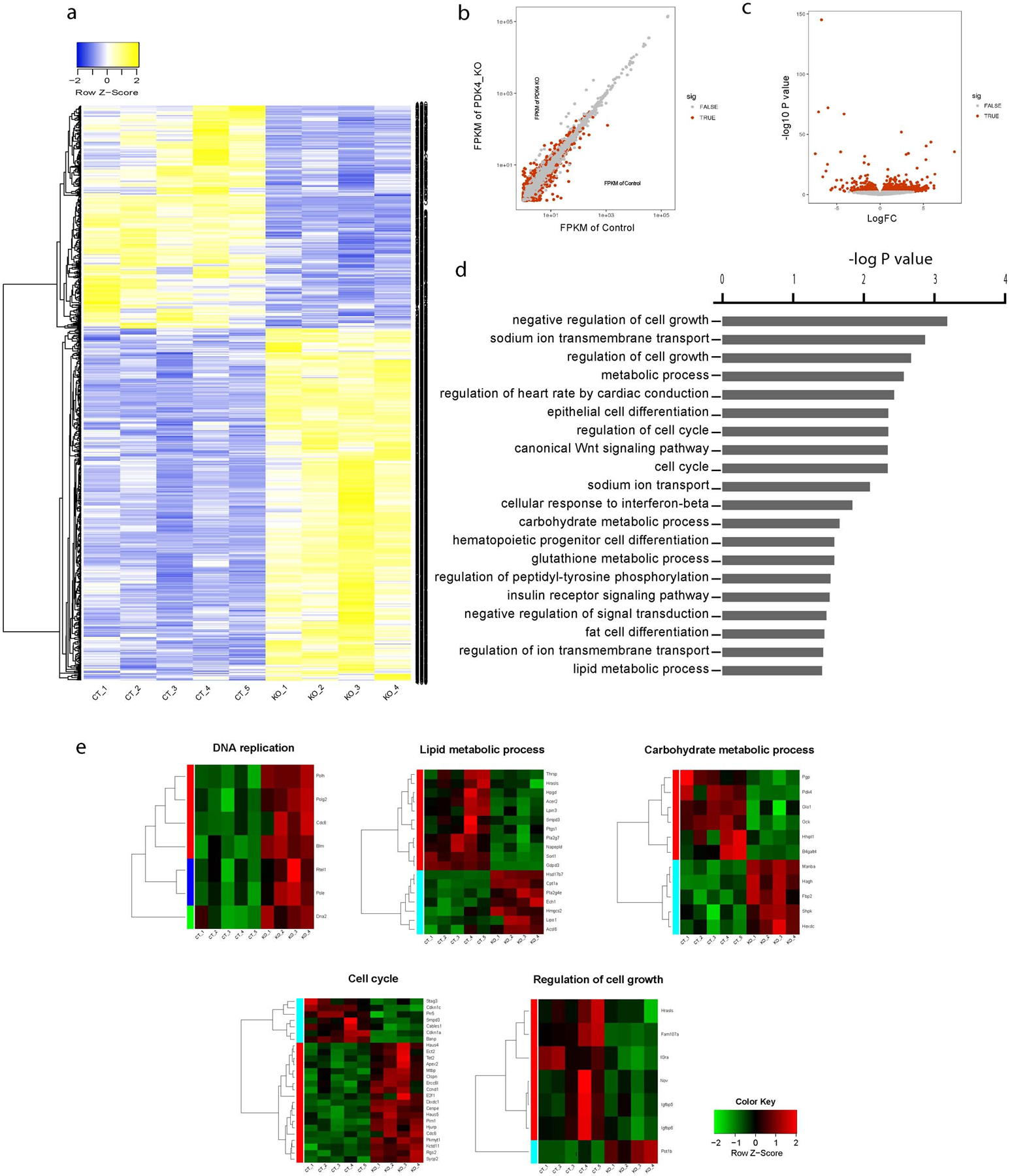
a, Heat-map of all dysregulated genes. Blue and yellow colors represent upregulated and downregulated genes, respectively (n=5 biologically independent mice for control and n=4 biologically independent mice for PDK4 KO group). b, Scatter plot showing the Fragments Per Kilobase of transcript, per Million mapped reads (FPKM) in control and PDK4 KO group (n=5 biologically independent mice for control and n=4 biologically independent mice for PDK4 KO). c, Volcano plot of the log of fold change dysregulated genes between Control and PDK4 KO group. (n=5 biologically independent mice for control and n=4 biologically independent mice for PDK4 KO). Statistic analysis was performed by two-tailed F-test for differential gene expression analysis. d, Ontology analysis performed using DAVID Functional Annotation Tool. Statistics analysis was performed by hypergeometric test. e, heat maps showing a number of dysregulated pathways, including DNA replication, lipid metabolic process, carbohydrate metabolic process, cell cycle and cell growth. Red and green colors represent upregulated and downregulated genes, respectively (n=5 biologically independent mice for control and n=4 biologically independent mice for PDK4 KO). CT: Control
Extended Data Fig. 4. Timeline of PDK4 deletion in the inducible model.
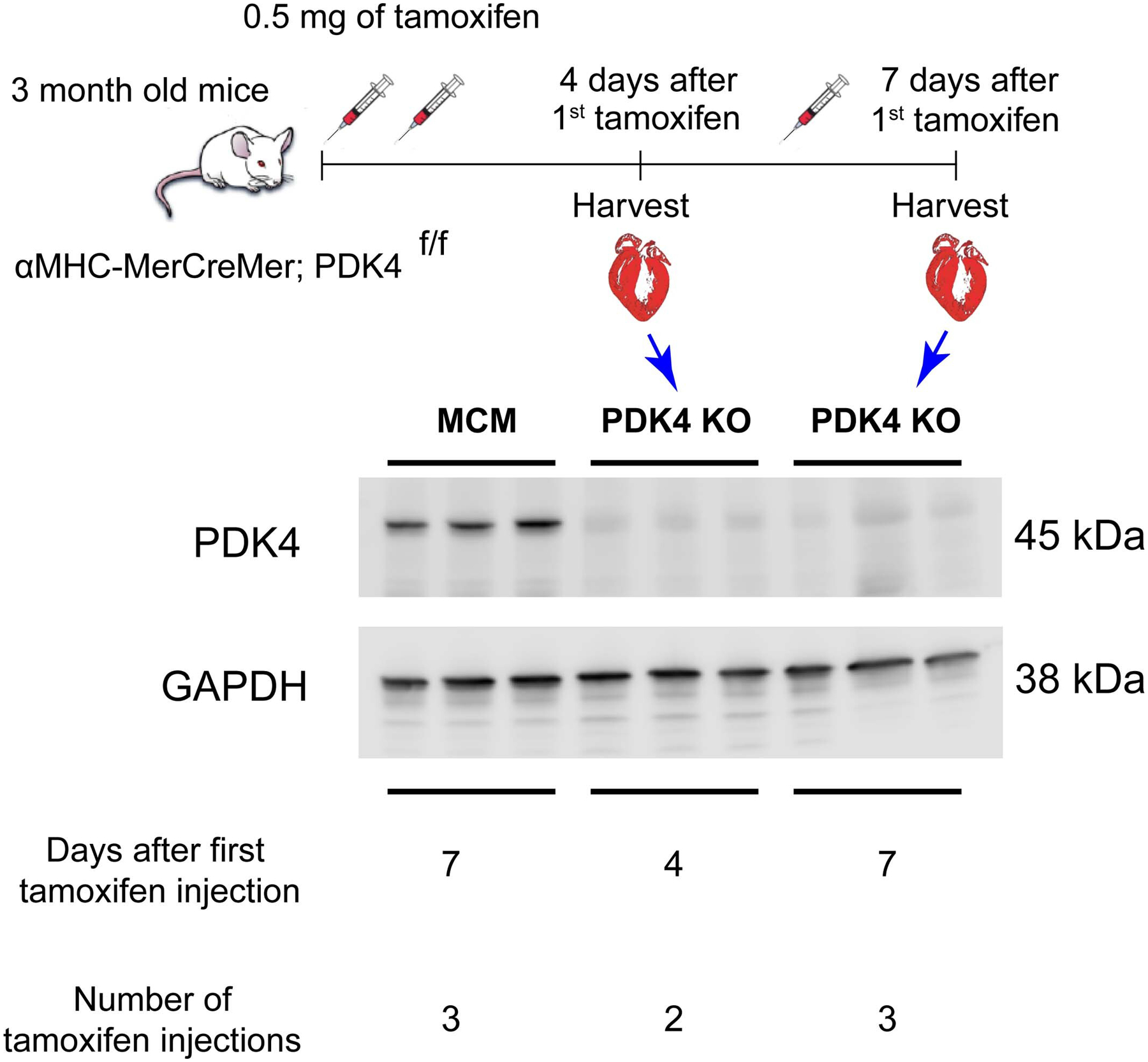
Western blot of PDK4 shows elimination of the protein as early as 4 days following the first tamoxifen injection.
Extended Data Fig. 5. Echocardiography parameters.
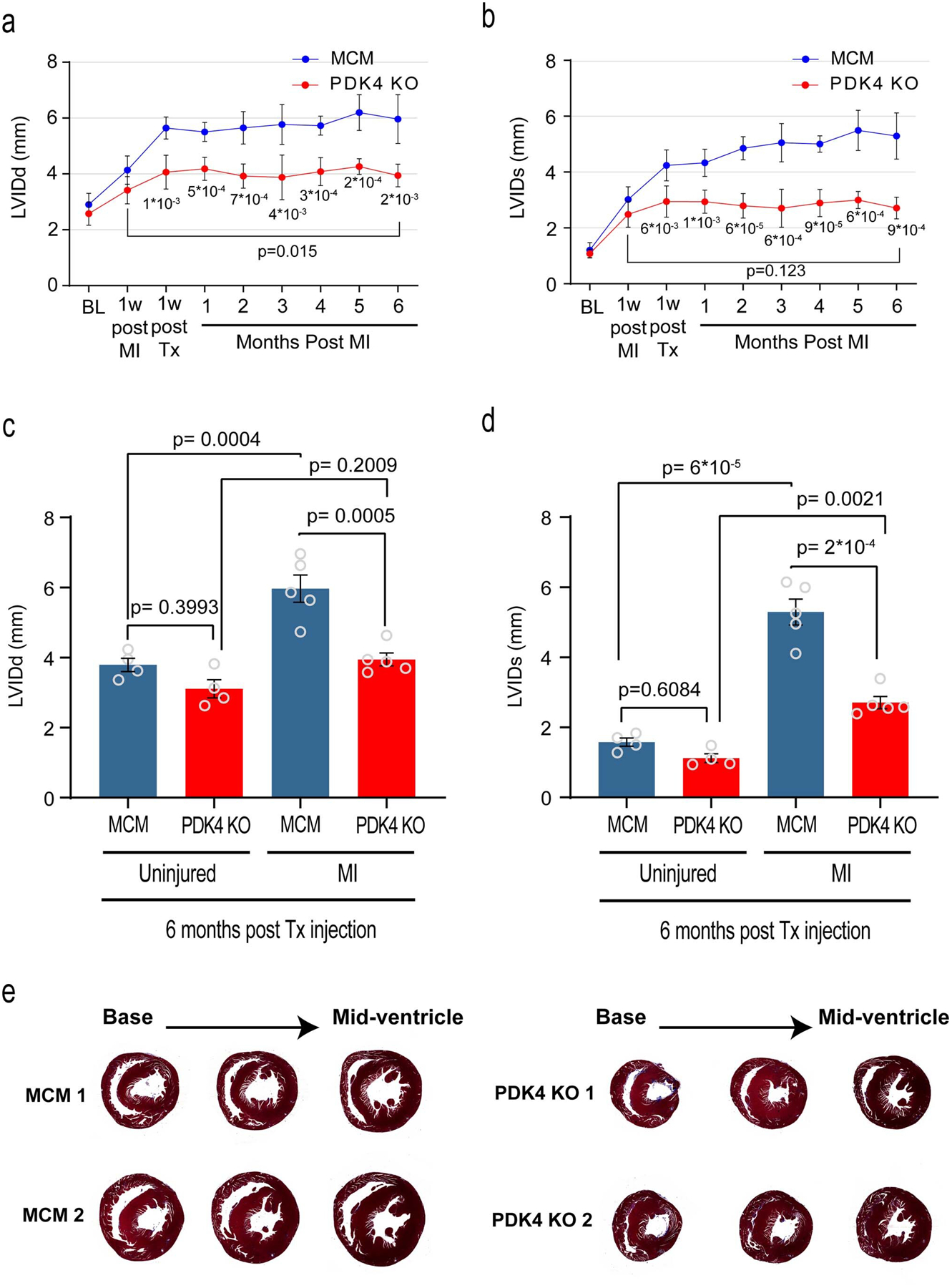
a, left ventricular internal dimension in diastole (LVIDd); b, left ventricular internal dimension in systole (LVIDs), echocardiogram measurements, comparing PDK4 KO and the control αMHC-MerCreMer (MCM) at different time points (n=5 biologically independent mice per group). Data are represented as the mean±s.d. Analyses were performed by unpaired two-tailed Student’s t-test to compare MCM vs PDK4 KO. Paired two-tailed Student’s t-test was used to compare two time points within the same mouse (6 months post MI versus 1 week post MI). c, LVDd; d, LVIDs echocardiogram measurements, comparing uninjured versus myocardial infarction (MI) MCM and PDK4 KO groups (n=4 biologically independent mice for uninjured and n=5 biologically independent mice for MI groups). Data are represented as the mean±s.e.m. Analyses were performed by Two-way ANOVA followed by Tukey’s multiple comparisons test, with individual variance computed for each comparison. ns, not significant. e, Trichrome staining of control MCM and PDK4 KO uninjured hearts, 6 months post Tamoxifen injection. Images are representative of three independently performed experiments, with similar results.
Acknowledgements
Herman I. May (Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA) for the myocardial infarction surgeries in mice. H.A.S. is supported by grants from the NIH (1R01HL115275 and 5R01H2131778), National Aeronautics and Space Administration (NNX-15AE06G), American Heart Association (16EIA27740034), Cancer Prevention and Research Institute of Texas (RP160520), Hamon Center for Regenerative Science and Medicine, and Fondation Leducq. N.T.L. is supported by a Haberecht Wildhare-Idea Research Grant. A.D.S. is supported by grant from the NIH (R37-HL034557), C.R.M. is supported by grant from the NIH (P41-EB015908) and G.S. is supported by the grant from the AHA (18POST34050049). M.K. is supported by the grants from NIH (3P20GM103447 and 5P30AG050911). I.M.M is supported by Alfonso Martin Escudero Foundation Fellowship. N.U.N.N. is supported by AHA Postdoctoral Fellowship 19POST34450039.
Footnotes
Competing interests
None of the authors have competing interests
Data Availability
RNA-seq raw data have been deposited in the NCBI Sequence Read Archive (SRA) database with accession number: PRJNA593900
References
- 1.Bergmann O et al. Evidence for cardiomyocyte renewal in humans. Science 324, 98–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahmoud AI et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 497, 249–253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porrello ER et al. Transient regenerative potential of the neonatal mouse heart. Science (New York, N.Y 331, 1078–1080 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker RO, Chapin S & Sherry R Regeneration of the ventricular myocardium in amphibians. Nature 248, 145–147 (1974). [DOI] [PubMed] [Google Scholar]
- 5.Oberpriller JO & Oberpriller JC Response of the adult newt ventricle to injury. The Journal of experimental zoology 187, 249–253 (1974). [DOI] [PubMed] [Google Scholar]
- 6.Neff AW, Dent AE & Armstrong JB Heart development and regeneration in urodeles. The International journal of developmental biology 40, 719–725 (1996). [PubMed] [Google Scholar]
- 7.Flink IL Cell cycle reentry of ventricular and atrial cardiomyocytes and cells within the epicardium following amputation of the ventricular apex in the axolotl, Amblystoma mexicanum: confocal microscopic immunofluorescent image analysis of bromodeoxyuridine-labeled nuclei. Anatomy and embryology 205, 235–244 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Jopling C et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 464, 606–609, doi: 10.1038/nature08899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kikuchi K et al. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 464, 601–605, doi:nature08804[pii]10.1038/nature08804 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soonpaa MH, Kim KK, Pajak L, Franklin M & Field LJ Cardiomyocyte DNA synthesis and binucleation during murine development. The American journal of physiology 271, H2183–2189 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Nassar R, Reedy MC & Anderson PA Developmental changes in the ultrastructure and sarcomere shortening of the isolated rabbit ventricular myocyte. Circ Res 61, 465–483 (1987). [DOI] [PubMed] [Google Scholar]
- 12.Wu Y & Wu EX MR study of postnatal development of myocardial structure and left ventricular function. J Magn Reson Imaging 30, 47–53 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Sdek P et al. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. The Journal of cell biology 194, 407–423, doi: 10.1083/jcb.201012049 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xin M et al. Hippo pathway effector Yap promotes cardiac regeneration. Proceedings of the National Academy of Sciences of the United States of America 110, 13839–13844, doi: 10.1073/pnas.1313192110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J et al. mir-17–92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res 112, 1557–1566, doi: 10.1161/CIRCRESAHA.112.300658 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eulalio A et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492, 376–381, doi: 10.1038/nature11739 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Bersell K, Arab S, Haring B & Kuhn B Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138, 257–270, doi: 10.1016/j.cell.2009.04.060 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Porrello ER et al. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res 109, 670–679, doi: 10.1161/CIRCRESAHA.111.248880 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poss KD, Wilson LG & Keating MT Heart regeneration in zebrafish. Science 298, 2188–2190 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Bergmann O et al. Cardiomyocyte renewal in humans. Circ Res 110, e17–18; author reply e19–21, doi:110/1/e17[pii]10.1161/CIRCRESAHA.111.259598 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Laflamme MA, Myerson D, Saffitz JE & Murry CE Evidence for cardiomyocyte repopulation by extracardiac progenitors in transplanted human hearts. Circ Res 90, 634–640 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Nadal-Ginard B [Generation of new cardiomyocytes in the adult heart: Prospects of myocardial regeneration as an alternative to cardiac transplantation]. Revista espanola de cardiologia 54, 543–550 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Quaini F et al. Chimerism of the transplanted heart. The New England journal of medicine 346, 5–15, doi: 10.1056/NEJMoa012081 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Lam NT & Sadek HA Neonatal Heart Regeneration. Circulation 138, 412–423, doi: 10.1161/CIRCULATIONAHA.118.033648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elhelaly WM et al. C-Kit Cells Do Not Significantly Contribute to Cardiomyogenesis During Neonatal Heart Regeneration. Circulation 139, 559–561, doi: 10.1161/CIRCULATIONAHA.117.033150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porrello ER et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proceedings of the National Academy of Sciences of the United States of America 110, 187–192, doi: 10.1073/pnas.1208863110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu W et al. Regenerative Potential of Neonatal Porcine Hearts. Circulation, doi: 10.1161/CIRCULATIONAHA.118.034886 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye L et al. Early Regenerative Capacity in the Porcine Heart. Circulation, doi: 10.1161/CIRCULATIONAHA.117.031542 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Puente BN et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157, 565–579, doi: 10.1016/j.cell.2014.03.032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisher DJ, Heymann MA & Rudolph AM Myocardial oxygen and carbohydrate consumption in fetal lambs in utero and in adult sheep. The American journal of physiology 238, H399–405 (1980). [DOI] [PubMed] [Google Scholar]
- 31.Lopaschuk GD, Collins-Nakai RL & Itoi T Developmental changes in energy substrate use by the heart. Cardiovascular research 26, 1172–1180 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Wisneski JA et al. Metabolic fate of extracted glucose in normal human myocardium. The Journal of clinical investigation 76, 1819–1827, doi: 10.1172/JCI112174 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gertz EW, Wisneski JA, Stanley WC & Neese RA Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. The Journal of clinical investigation 82, 2017–2025, doi: 10.1172/JCI113822 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehman JJ & Kelly DP Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clinical and experimental pharmacology & physiology 29, 339–345 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Anderson SM, Rudolph MC, McManaman JL & Neville MC Key stages in mammary gland development. Secretory activation in the mammary gland: it’s not just about milk protein synthesis! Breast Cancer Res 9, 204, doi: 10.1186/bcr1653 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rindler PM, Crewe CL, Fernandes J, Kinter M & Szweda LI Redox regulation of insulin sensitivity due to enhanced fatty acid utilization in the mitochondria. American journal of physiology. Heart and circulatory physiology 305, H634–643, doi: 10.1152/ajpheart.00799.2012 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Bowker-Kinley MM, Davis WI, Wu P, Harris RA & Popov KM Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. The Biochemical journal 329 (Pt 1), 191–196 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rardin MJ, Wiley SE, Naviaux RK, Murphy AN & Dixon JE Monitoring phosphorylation of the pyruvate dehydrogenase complex. Analytical biochemistry 389, 157–164, doi: 10.1016/j.ab.2009.03.040 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugden MC & Holness MJ Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Archives of physiology and biochemistry 112, 139–149, doi: 10.1080/13813450600935263 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Holness MJ, Kraus A, Harris RA & Sugden MC Targeted upregulation of pyruvate dehydrogenase kinase (PDK)-4 in slow-twitch skeletal muscle underlies the stable modification of the regulatory characteristics of PDK induced by high-fat feeding. Diabetes 49, 775–781 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Holness MJ et al. Evaluation of the role of peroxisome-proliferator-activated receptor alpha in the regulation of cardiac pyruvate dehydrogenase kinase 4 protein expression in response to starvation, high-fat feeding and hyperthyroidism. The Biochemical journal 364, 687–694, doi: 10.1042/BJ20011841 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson EJ, Yamazaki H & Neufer PD Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. The Journal of biological chemistry 282, 31257–31266, doi: 10.1074/jbc.M706129200 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Rindler PM, Plafker SM, Szweda LI & Kinter M High dietary fat selectively increases catalase expression within cardiac mitochondria. J Biol Chem 288, 1979–1990, doi: 10.1074/jbc.M112.412890 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seifert EL, Estey C, Xuan JY & Harper ME Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. The Journal of biological chemistry 285, 5748–5758, doi: 10.1074/jbc.M109.026203 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bobrovnikova-Marjon E et al. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proceedings of the National Academy of Sciences 105, 16314–16319, doi: 10.1073/pnas.0808517105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner K-U, Ward T, Davis B, Wiseman R & Hennighausen L Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Research 10, 545–553, doi: 10.1023/a:1013063514007 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Ali SR et al. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proceedings of the National Academy of Sciences 111, 8850–8855, doi: 10.1073/pnas.1408233111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smagris E et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 61, 108–118, doi: 10.1002/hep.27242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lewandowski ED & White LT Pyruvate dehydrogenase influences postischemic heart function. Circulation 91, 2071–2079 (1995). [DOI] [PubMed] [Google Scholar]
- 50.Ussher JR et al. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovascular research 94, 359–369, doi: 10.1093/cvr/cvs129 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Dyck JR et al. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 114, 1721–1728, doi: 10.1161/CIRCULATIONAHA.106.642009 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Liu Z et al. PDK4 protein promotes tumorigenesis through activation of cAMP-response element-binding protein (CREB)-Ras homolog enriched in brain (RHEB)-mTORC1 signaling cascade. J Biol Chem 289, 29739–29749, doi: 10.1074/jbc.M114.584821 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thoudam T et al. PDK4 Augments ER-Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity. Diabetes, doi: 10.2337/db18-0363 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sohal DS et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89, 20–25 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Harding HP, Zhang Y & Ron D Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274, doi: 10.1038/16729 (1999). [DOI] [PubMed] [Google Scholar]
- 56.Cavener DR, Gupta S & McGrath BC PERK in beta cell biology and insulin biogenesis. Trends in Endocrinology & Metabolism 21, 714–721, doi: 10.1016/j.tem.2010.08.005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang P et al. The PERK Eukaryotic Initiation Factor 2α Kinase Is Required for the Development of the Skeletal System, Postnatal Growth, and the Function and Viability of the Pancreas. Molecular and Cellular Biology 22, 3864–3874, doi: 10.1128/mcb.22.11.3864-3874.2002 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsuhashi T et al. Activation of pyruvate dehydrogenase by dichloroacetate has the potential to induce epigenetic remodeling in the heart. Journal of Molecular and Cellular Cardiology 82, 116–124, doi: 10.1016/j.yjmcc.2015.02.021 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Friedewald WT, Levy RI & Fredrickson DS Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clinical chemistry 18, 499–502 (1972). [PubMed] [Google Scholar]
- 60.Ejsing CS et al. Automated identification and quantification of glycerophospholipid molecular species by multiple precursor ion scanning. Anal Chem 78, 6202–6214, doi: 10.1021/ac060545x (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.