

SUMMARY
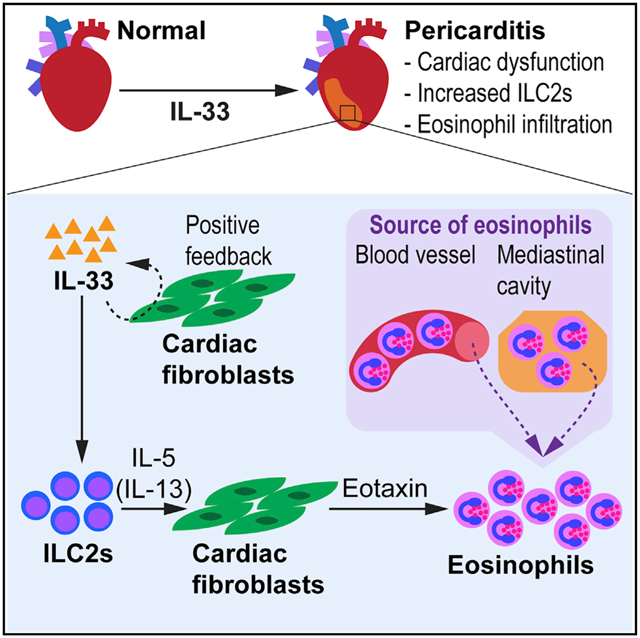
We find that cardiac group 2 innate lymphoid cells (ILC2s) are essential for the development of IL-33-induced eosinophilic pericarditis. We show a pathogenic role for ILC2s in cardiac inflammation, in which ILC2s activated by IL-33 drive the development of eosinophilic pericarditis in collaboration with cardiac fibroblasts. ILCs, not T and B cells, are required for the development of pericarditis. ILC2s transferred to the heart of Rag2−/−Il2rg−/− mice restore their susceptibility to eosinophil infiltration. Moreover, ILC2s direct cardiac fibroblasts to produce eotaxin-1. We also find that eosinophils reside in the mediastinal cavity and that eosinophils transferred to the mediastinal cavity of eosinophil-deficient ΔdblGATA1 mice following IL-33 treatment migrate to the heart. Thus, the serous cavities may serve as a reservoir of cardiac-infiltrating eosinophils. In humans, patients with pericarditis show higher amounts of ILCs in pericardial fluid than do healthy controls and patients with other cardiac diseases. We demonstrate that ILCs play a critical role in pericarditis.
Graphical Abstract
In Brief
Choi et al. show a pathogenic role for innate lymphoid cells (ILCs) in IL-33-induced eosinophilic pericarditis. ILCs are required for the development of pericarditis, and group 2 ILCs (ILC2s) promote the expression of eotaxin by cardiac fibroblasts. In humans, ILCs are found higher in the pericardial fluid of patients with pericarditis compared to others.
INTRODUCTION
Innate lymphoid cells (ILCs) are recently identified innate immune cells that serve important roles in lymphoid tissue formation, repair of damaged tissues, and homeostasis, as well as in immunity against infectious microorganisms (Spits and Cupedo, 2012; Eberl et al., 2015; Klose and Artis, 2016). ILCs are categorized into three groups based on their unique expression of transcription factors, surface markers, and the production of effector cytokines (Spits et al., 2013; Walker et al., 2013). ILC1s are characterized by interferon gamma (IFNγ) production and ILC2s by interleukin-5 (IL-5) and IL-13, while ILC3s are characterized by IL-17 and IL-22. Natural killer (NK) cells are considered a cytotoxic subset of ILC1s (Spits et al., 2013). Distinct from adaptive immune cells such as T cells and B cells, ILCs do not possess antigen-specific receptors (Spits and Cupedo, 2012). ILCs appear to be tissue resident cells and are maintained mostly by selfrenewal in homeostatic settings (Gasteiger et al., 2015; Moro et al., 2016). We recently found that cardiac ILCs are also a resident population that are type 2-skewed in both human and mouse hearts (Bracamonte-Baran et al., 2019). However, what roles cardiac ILCs play in inflammation are poorly understood.
ILC2s are found in many organs at steady state, including lung and mucosal tissues, and can be activated by IL-33, IL-25, and thymic stromal lymphopoietin (TSLP) (Kim and Artis, 2015; Klose and Artis, 2016; Moro et al., 2010; Neill et al., 2010; Price et al., 2010; Kim et al., 2013). After activation, ILC2s produce T helper 2 cell (Th2)-associated cytokines, such as IL-5 and IL-13 (Moro et al., 2010; Neill et al., 2010; Price et al., 2010). These Th2 cytokines have been implicated in eosinophil proliferation, recruitment, and homeostasis (Nussbaum et al., 2013; Price et al., 2010). The role of ILC2s in infection and inflammation has been investigated mostly in mucosa-associated tissues and skin. ILC2s are beneficial in promoting immunity to parasite infection with Nippostrongylus brasiliensis (Moro et al., 2010; Neill et al., 2010). In this helminth infection model, IL-25 and IL-33 were shown to be critical for ILC2 activation, and IL-13 produced by ILC2s mediated worm expulsion (Neill et al., 2010). In contrast to the protective immunity provided by ILC2s, IL-33-activated ILC2s are also involved in the pathogenesis of asthma and atopic dermatitis promoting inflammation in the lungs and skin, respectively (Halim et al., 2012; Salimi et al., 2013; Imai et al., 2013). Moreover, the enrichment of ILC2s was reported in biopsy samples from patients with eosinophilic esophagitis and atopic dermatitis (Doherty et al., 2015; Salimi et al., 2013).
IL-33 is a cytokine of the IL-1 family and functions as an alarmin, which is released from cells upon tissue damage or cellular stress (Schmitz et al., 2005; Cayrol and Girard, 2014; Molofsky et al., 2015). IL-33 is usually expressed and released from epithelial and endothelial cells, and it is also shown to be expressed in stromal cells such as fibroblastic reticular cells in the lymph nodes (Moussion et al., 2008; Pichery et al., 2012). The profiling of IL-33 mRNA expression in mouse tissues revealed that the heart had a relatively low expression of IL-33 compared to mucosal and barrier-related tissues such as lung and skin (Schmitz et al., 2005). The role of IL-33 in the heart has been shown to be beneficial in some mouse models of cardiovascular diseases. IL-33 treatment reduced cardiac hypertrophy in a mouse model of transverse aortic constriction and improved cardiac function and survival in a myocardial infarction mouse model (Sanada et al., 2007; Chen et al., 2015; Seki et al., 2009). However, the administration of IL-33 induced pericarditis in mice with an increased proportion of eosinophils in the heart of naive and coxsackievirus B3 (CVB3)-infected mice, demonstrating that IL-33 can also play a pathogenic role in the heart (Abston et al., 2012). IL-33 has been shown to activate and expand ILC2s (Moro et al., 2010; Neill et al., 2010; von Moltke and Locksley, 2014; Kim and Artis, 2015). This suggests that the described pathogenic effect of IL-33 in the heart could be mediated by the activation of ILC2s.
Here, we show that ILCs in the heart play a pathogenic role in promoting cardiac inflammation. Cardiac ILC2s activated by IL-33 caused eosinophilic pericarditis. The IL-33-ST2 signaling axis was important for the expansion of cardiac ILC2s and induction of pericarditis. We identified that ILCs, not T cells and B cells, were required for IL-33-induced pericarditis. Rag2−/−Il2rg−/− mice deficient in lymphocytes, including ILCs, were protected from pericarditis; however, ILC2 transfer to the heart restored the susceptibility of Rag2−/−Il2rg−/− mice to eosinophil infiltration to the heart. We also found that ILC2s induced cardiac fibroblasts to produce eotaxin-1 to direct eosinophil trafficking to the heart. Moreover, we discovered that eosinophils resided in the mediastinal cavity of naive mice, which serves as a reservoir of eosinophils. We documented the recruitment of eosinophils from the mediastinal cavity, which could be an alternate route in addition to the conventional vascular trafficking of eosinophils to the heart. Finally, we showed that, in humans, ILCs are found more frequently in pericardial fluid from pericarditis patients compared to individuals without cardiac diseases and patients with other types of cardiac diseases.
RESULTS
Number of ILC2s Increases in the Heart after IL-33 Treatment
ILC2s express the IL-33 receptor, which is also known as ST2, and become activated and proliferative upon IL-33 stimulation (Spits et al., 2013; Walker et al., 2013; Klose and Artis, 2016). To determine whether IL-33 induces an increase in ILC2s in the heart in vivo, we used the IL-33-induced pericarditis model in which mice are treated with IL-33 intraperitoneally every other day for 9 days (Abston et al., 2012). IL-33-treated mice developed severe pericarditis compared to PBS-treated mice (Figure 1A). The inflammation mostly affected the pericardium; however, in some cases, the adjacent myocardium was also inflamed (Figure 1A). We also found that IL-33-treated mice showed an increased infiltration of immune cells in other organs including esophagus and lungs (Figures S1A and S1B). We assessed cardiac function of the heart by performing M-mode and Doppler echocardiography. A typical pattern of ventricular dilation was present in IL-33-treated mice examined by M-mode echocardiography (Figure 1B). IL-33 treatment resulted in reduced ejection fraction (EF) and fractional shortening (FS) (Figures 1C and 1D). Other parameters were comparable; however, the left ventricular posterior wall at end-systole was thinner in IL-33-treated mice (Figures S1C-S1E). In addition, IL-33-treated mice showed significantly longer isovolumetric relaxation time (IVRT), an indicator of diastolic function, and higher myocardial performance index (MPI), a useful predictor of global cardiac function, than PBS-treated mice (Figures 1E and 1F). Prolonged IVRT indicates poor myocardial relaxation related to pericardial constriction, and high MPI represents both systolic and diastolic dysfunction. These data suggest that pericarditis induced by IL-33 resulted in abnormal cardiac function. We analyzed the number of heart-infiltrating CD45+ leukocytes by flow cytometry and found that it was significantly increased in IL-33-treated mice compared to PBS-treated mice, confirming that inflammation occurred in the heart as a result of IL-33 treatment (Figure 1G). We identified ILC2s in the heart as CD45+Lin−CD90+KLRG1+ST2+ (Figure S2A). Lin included CD3ε, TCRβ, CD19, B220, CD11b, CD11c, Gr-1, Ter119, FcεRIα, and NKp46, and was used to exclude cell populations that may contaminate ILC2s. Both the number and frequency of ILC2s in the heart were dramatically increased upon IL-33 treatment compared to PBS treatment (Figures 1H and S2B). We also analyzed other heart-infiltrating immune cells and found that the number and frequency of CD11b+SiglecF+ eosinophils were increased in the heart of IL-33-treated mice and that they were the most abundant cells among heart infiltrates (Figures 1I and S2C). The number and frequency of other immune cells known to express ST2, including FcεRIα+CD117(c-Kit)+ mast cells, remained unchanged (Figure 1J). To visualize the distribution of ILC2s and eosinophils in pericarditis, we used a tissue-clearing technique with the right ventricle of the heart from mice treated with IL-33. We found that ILC2s and eosinophils were located in the right ventricle near the pericardium and in the atria, which is consistent with the finding that inflammation was mostly observed in the right ventricle in the histological analysis (Figure 1K; Video S1). In addition, the number of CD11b+Ly6G+ neutrophils in the heart was decreased in IL-33-treated mice; however, we did not find significant changes in numbers of macrophages, monocytes, T cells, B cells, and NK cells (Figures S2D-S1I). We found that the level of anti-myosin immunoglobulin M (IgM) antibody in serum was significantly higher in IL-33-treated mice than in PBS-treated mice on day 9 (Figure S2J). Anti-myosin IgG and IgE were found at a negligible level in the sera of both PBS- and IL-33-treated mice (data not shown). These results indicate that IL-33-induced pericarditis is characterized by impaired cardiac function and an increased number of ILC2s and eosinophilsin the heart.
Figure 1. ILC2s Increases in the Heart, and Cardiac Function Is Changed following IL-33 Treatment.

(A) Representative images of H&E-stained heart sections of the median mice treated with either PBS or IL-33. Areas marked by rectangles are shown as enlarged images in the right panels. Bars: 1 mm (left) and 100 μm (right).
(B) Representative M-mode pictures of animals treated with PBS or IL-33. Bars: blue, 1 mm; white, 0.1 s. LVESD, left ventricular end-systolic diameter; LVEDD, left ventricular end-diastolic diameter.
(C and D) Ejection fraction (EF) (C) and fractional shortening (FS) (D) of WT mice treated with PBS or IL-33.
(E and F) Isovolumetric relaxation time (IVRT) (E) and myocardial performance indexes (MPI) (F) of the heart from mice treated with PBS or IL-33 were assessed by Doppler echocardiography on day 9 post-PBS or IL-33 treatment.
(G) Total number of heart-infiltrating CD45+ leukocytes was determined by flow cytometry.
(H–J) Number of (H) ILC2s, (I) eosinophils, and (J) mast cells in the hearts. Absolute cell counts per heart were calculated using counting beads for flow cytometry (see Method Details).
(K) Image of the right ventricle of the heart from Rag2−/− mice treated with IL-33. Green and red dots represent KLRG1+ ILC2s (white arrows) and SiglecF+ eosinophils (red arrowheads), respectively. Scale bar: 500 μm. Data are representative of three independent experiments.
Data are displayed as the means. Unpaired t test (C–J) was used for statistical analysis. *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figures S1 and S2 and Video S1.
IL-33-ST2 Signaling Pathway Is Critical for ILC Accumulation in the Heart and the Development of Pericarditis
To determine whether the development of pericarditis is dependent on the IL-33-ST2 signaling pathway, ST2−/− (Il1rl1−/−) mice were treated with IL-33. ST2−/− mice were completely protected from the development of IL-33-induced pericarditis, whereas wild-type (WT) mice developed pericarditis (Figures 2A and 2B). The number of total leukocyte infiltrates was significantly reduced in the heart of ST2−/− mice compared to WT mice (Figure 2C). To further confirm the dependency of ILC2 accumulation in the heart on IL-33 signaling, we analyzed the Lin−CD90+KLRG1+ ILC population in the heart of ST2−/− mice after IL-33 treatment. We found that ILCs were significantly reduced in the heart of ST2−/− mice compared to WT mice (Figures 2D and 2E). The number of eosinophils was also decreased in the heart of ST2−/− mice (Figure 2F). This demonstrates that IL-33 signaling through ST2 is important for the development of pericarditis, and the IL-33-ST2 signaling axis is required for ILC2 accumulation in the heart. Next, we assessed whether exogenous IL-33 treatment is involved in a positive feedback loop that results in increased endogenous expression of IL-33. IL-33cit/+ reporter mice treated with IL-33 showed a higher expression of IL-33 compared to mice treated with PBS, which indicates the presence of a positive feedback loop of IL-33 expression (Figures 2G and S3A). In addition, the number of IL-33-expressing cells was increased after IL-33 treatment (Figure 2H). Most of the IL-33-expressing cells were CD45−CD31− stromal cells, and among them, Sca1+ fibroblasts were the major IL-33-expressing cells in the heart. These IL-33-expressing cardiac fibroblasts were increased in number after IL-33 treatment, suggesting that cardiac fibroblasts have a capacity to produce IL-33 in both the naive state and during cardiac inflammation (Figures 2I and 2J). Sca1+ cardiac fibroblasts also expressed platelet-derived growth factor receptor α (PDGFRα), a commonly used cardiac fibroblast marker (Figure 2K). Moreover, we found that IL-33 was spatially expressed by cardiac fibroblasts near and in the pericardium (Figure 2L). We found a correlation of IL-33 and ST2 expression in IL-33-induced pericarditis, indicating the existence of a positive feedback loop in which IL-33 induces greater ST2 expression and IL-33 production by fibroblasts (Figure S3B). Furthermore, we examined whether endogenous IL-33 is important to induce pericarditis. We found that the number of total CD45+ heart-infiltrating leukocytes was reduced in IL-33−/− mice compared to WT mice upon IL-33 treatment (Figure S3C). The number of myeloid cells and eosinophils was also decreased in the heart of IL-33−/− mice (Figures S3D and S3E). The lack of endogenous IL-33 affected the Lin−CD90+KLRG1+ ILC population, which was reduced in the heart of IL-33−/− mice compared to WT mice (Figure S3F). These results indicate that endogenous IL-33 is a required component to induce severe pericarditis, which was initiated by exogenous IL-33 administration. These data suggest that IL-33 signaling through ST2 is important for ILC accumulation in the heart and the development of pericarditis.
Figure 2. IL-33-ST2 Axis Is Critical for the Development of Pericarditis.

(A) Representative images of H&E-stained heart sections of WT and ST2−/− mice. Bars: 100 μm.
(B) Severity of pericarditis was scored on H&E-stained heart sections.
(C) Number of heart-infiltrating CD45+ leukocytes was determined by flow cytometry.
(D) Representative flow cytometry plots of CD45+Lin− cells. Gates show the frequency of CD90+KLRG1+ ILCs in the heart of WT and ST2−/− mice.
(E and F) Number of (E) Lin−CD90+KLRG1+ ILCs and (F) eosinophils in the hearts.
(G) Mean fluorescence intensity (MFI) of IL-33 expression in cardiac viable cells.
(H) Number of IL-33-expressing cells in the hearts.
(I) Mean frequency of different IL-33-expressing populations. The mean was quantified using values in a PBS-treated group (n = 4) and an IL-33-treated group (n = 5). Areas of the pie charts are proportional to total IL-33+ cells in the heart.
(J) Total number of IL-33+Sca1+ cardiac fibroblasts.
(K) PDGFRα expression by Sca1+ cardiac fibroblasts.
(L) Immunofluorescence imaging shows IL-33-expressing PDGFRα+ cardiac fibroblasts near the pericardium (white arrows) and in the pericardium (yellow arrows). Blue, green, and red color represent DAPI, IL-33, and PDGFRα, respectively. Bars: 50 μm.
Data are representative of two independent experiments and displayed as the mean. Mann-Whitney U test (B) or unpaired t test (C, E–H, and J) was used for statistical analysis. *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figure S3.
ILCs Are Required for Development of IL-33-Induced Pericarditis and Sufficient to Rescue ILC-Deficient Mice Susceptibility to Eosinophil Infiltration
To determine whether ILCs are required for the induction of pericarditis, we treated WT, Rag2−/−, and Rag2−/−Il2rg−/− mice with IL-33. While Rag2−/− mice deficient in T cells and B cells developed pericarditis comparable to WT mice, Rag2−/−Il2rg−/− mice, which lack all lymphocytes including ILCs, were completely protected from the development of pericarditis (Figures 3A and 3B). Thus, adaptive immune response is not necessary for the induction of pericarditis by IL-33. We assessed leukocytes in the heart and found a significantly reduced number of CD45+ cells in the heart of Rag2−/−Il2rg−/− mice compared to both WT and Rag2−/− mice (Figure 3C). ILC2s were increased in the heart of WT and Rag2−/− mice treated with IL-33, whereas ILC2s were not found in the heart of Rag2−/−Il2rg−/− mice (Figure 3D). IL-33-treated Rag2−/−Il2rg−/− mice showed a smaller number of eosinophils, but more neutrophils in the heart than WT and Rag2−/− mice (Figures 3E and S4A). These data suggest that ILCs are required for the development of pericarditis induced by IL-33, and adaptive immune cells such as T and B cells may not be involved in the pathogenesis of IL-33-induced pericarditis. To confirm that IL-33-induced pericarditis is ILC2-dependent, we investigated the ability of transferred ILC2s to reverse the resistance of Rag2−/−Il2rg−/− mice to pericarditis development. We performed fluorescence-activated cell sorting (FACS) to isolate CD45+Lin−CD90+ KLRG1+ST2+ ILC2s from the hearts of CD45.1+ WT donor mice after IL-33 treatment (Figure S4B). Intravenous transfer of ILC2s was not successful, and we were not able to detect any transferred ILC2s in the heart of recipient mice, which is consistent with the resident status of ILCs and their inability to traffic to the heart from the blood, as we showed before (Bracamonte-Baran et al., 2019). Therefore, we expanded FACS-sorted CD45.1+ ILC2s in vitro and injected them directly into the myocardium of ILC-deficient CD45.2+ Rag2−/−Il2rg−/− recipient mice followed by IL-33 treatment (Figure S4C). Recipient mice in a control group were injected with media in the myocardium followed by IL-33 treatment. We were able to detect CD45.1+ ILC2s in the hearts of ILC2-transferred CD45.2+ Rag2−/−Il2rg−/− mice on day 9, indicating that ILC2s injected into the myocardium remained for the course of the disease (Figure 3F). The number of heart-infiltrating CD45.2+ leukocytes was increased in Rag2−/−Il2rg−/− mice injected with ILC2s compared to mice injected with media (Figure 3G). Eosinophils were significantly increased in the hearts of Rag2−/−Il2rg−/− mice injected with ILC2s compared to mice injected with media, providing evidence of a critical role for cardiac ILC2s in eosinophil recruitment to the heart during the development of pericarditis (Figures 3H and 3I). To summarize, ILCs, but not T cells and B cells, are required for the development of pericarditis, and the adoptive transfer of cardiac ILC2s directly to the heart is sufficient to rescue Rag2−/−Il2rg−/− mice susceptibility to eosinophilic cardiac inflammation induced by IL-33.
Figure 3. ILCs Are Required for the Development of Pericarditis, and ILC2s Are Sufficient to Induce Eosinophilic Infiltration to the Heart.

(A) Representative images of H&E-stained heart sections of WT (left), Rag2−/− (center), and Rag2−/− Il2rg−/− (right) mice. Bars: 100 μm.
(B) Severity of pericarditis was scored on H&E-stained heart sections.
(C) Total number of heart-infiltrating CD45+ leukocytes was determined by flow cytometry.
(D and E) Number of (D) ILC2s and (E) eosinophils in the hearts.
(F) Representative flow cytometry plots of ILC2s found in the hearts of naive Rag2−/−Il2rg−/− mice and Rag2−/−Il2rg−/− mice injected with media or ILC2s followed by IL-33 treatment.
(G) Number of heart-infiltrating CD45.2+ leukocytes.
(H) Representative flow cytometry plots of CD45.2+ cells. Gates show the frequency of CD11b+SiglecF+ eosinophils in the hearts of naive Rag2−/−Il2rg−/− mice and Rag2−/−Il2rg−/− mice injected with media or ILC2s followed by IL-33 treatment.
(I) Number of eosinophils in the hearts.
Both male and female mice were used in each group in (B). Data are representative of two independent experiments and displayed as the mean. Kruskal-Wallis H test (B) or one-way ANOVA followed by Tukey’s post hoc test (C–E, G, and I) was used for statistical analysis. *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figure S4.
ILC2s Drive Cardiac Fibroblasts to Upregulate CCL11/Eotaxin-1 Gene Expression
Eotaxin production is known to induce eosinophil trafficking into organs following a chemoattractant gradient (Rothenberg, 1999; Borchers et al., 2002). We previously found that the eotaxin-CCR3 pathway is the main mechanism for eosinophil trafficking to the heart (Diny et al., 2016). We found that the expression of Ccl11, a gene encoding eotaxin-1, was upregulated in the heart of IL-33-treated mice compared to PBS-treated mice, while the expression of Ccl24 encoding eotaxin-2 was comparable between groups (Figures 4A and S5A). Our previous work showed that cardiac fibroblasts produce a diverse set of cytokines and chemokines in response to different Th environments (Chen et al., 2018). Specifically, in a Th2 environment during eosinophilic cardiac inflammation, cardiac fibroblasts are the main source of eotaxin-1 (Diny et al., 2016). To investigate whether ILC2s and cardiac fibroblasts cooperate to attract eosinophils to the heart in IL-33-induced pericarditis, we devised a co-culture system in which cardiac fibroblasts are co-cultured with ILC2s separated by 0.4-μm transwells (Figure 4B). This enabled ILC2s to interact with cardiac fibroblasts through soluble factors such as cytokines, but not through direct contact. We found that cardiac fibroblasts significantly upregulated Ccl11 expression when co-cultured with ILC2s in the presence of IL-33, compared to cardiac fibroblasts cultured without ILC2s in the absence or presence of IL-33 (Figure 4C). Ccl24 expression by cardiac fibroblasts did not significantly differ (Figure S5B). Eotaxin-1 concentration in the cell culture supernatant was also significantly increased in the co-culture condition in which cardiac fibroblasts are cultured with ILC2s in the presence of IL-33 (Figure 4D). To determine whether ILC2s are required for increased eotaxin expression in vivo, we examined expression levels of genes related to eosinophil trafficking and ILC2 effector cytokines in the heart of WT, Rag2−/−, and Rag2−/−Il2rg−/− mice treated with IL-33. We found that the expression of both Ccl11 and Ccl24 was significantly decreased in Rag2−/−Il2rg−/− mice compared to WT mice (Figures 4E and 4F). The expression levels of Il5 and Il13, encoding ILC2 effector cytokines, were also downregulated in Rag2−/−Il2rg−/− mice (Figures 4G and 4H). These data suggest that ILCs may be required for eotaxin expression in vivo, possibly in an ILC2 effector cytokines-dependent way. We also examined the spatial expression of eotaxin-1 in the heart and found that eotaxin-1 was highly produced near the pericardium, where pericarditis occurs (Figure 4I). In summary, ILC2s are able to stimulate cardiac fibroblasts to produce eotaxin-1 via soluble factors, suggesting a role for ILC2s and cardiac fibroblasts in the recruitment of eosinophils into the heart.
Figure4. Cardiac Fibroblasts Increase CCL11/Eotaxin-1 Expression and Production When Co-cultured with IL-33-Stimulated ILC2s, and the Expression of Eotaxins and Type 2 Cytokines Is Dependent on ILCs.

(A) Expression of the Ccl11 gene encoding eotaxin-1 in heart homogenates was analyzed by qPCR.
(B) Schematic description of cardiac fibroblasts co-culture with ILC2s separated by 0.4-μm transwell.
(C) Expression of Ccl11 in cardiac fibroblasts was analyzed by qPCR.
(D) Eotaxin-1 concentrations in cell culture supernatants were measured by ELISA. IL-2 and IL-7 were included in culture media and IL-33 was added where indicated (C and D).
(E–H) Expression of genes in heart homogenates from WT, Rag2−/−, and Rag2−/−Il2rg−/− mice treated with IL-33 was analyzed by qPCR. Ccl11 (E), Ccl24 (F), Il5 (G), and Il13 (H).
(I) Immunohistochemistry staining for CCL11 of heart sections from WT mice treated with PBS or IL-33. Bars: 50 μm.
Data are representative of two independent experiments and displayed as the mean with SD. Unpaired t test (A) or one-way ANOVA followed by Tukey’s post hoc test (C–H) was used for statistical analysis. *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figure S5.
ILC2-Derived IL-5 Affects the Development of IL-33-Induced Pericarditis
We assessed cytokine production by cardiac ILC2s on day 9 post-IL-33 treatment using flow cytometry. We found that ILC2s in the heart produced both IL-5 and IL-13 after IL-33 treatment (Figure 5A). Eighty percent of ILC2s found in the heart produced both IL-5 and IL-13 (Figure 5B). To examine the role of IL-5 in the development of pericarditis, we depleted IL-5 in IL-33-treated mice using an anti-IL-5 monoclonal antibody (mAb). We found that mice injected with IL-5-neutralizing mAb showed a trend of less severe pericarditis by histology after IL-33 administration compared to those injected with isotype control (Figure 5C). In addition, we found that the total number of leukocytes in the heart was significantly decreased in anti-IL-5-treated mice compared to isotype-treated controls (Figure 5D). Eosinophils were also significantly reduced in the heart of anti-IL-5-treated mice compared to isotype-treated mice (Figure 5E). However, the number of ILC2s was similar between groups (Figure 5F). These data show that IL-5 produced by ILC2s drives eosinophil infiltration to the heart and contributes to the severity of pericarditis. We also examined whether IL-13 plays a role in pericarditis development. IL-13−/− mice showed pericarditis similar to WT mice (Figure 5G). We found the number of total leukocytes and eosinophils were analogous between WT and IL-13−/− mice (Figures 5H and 5I). ILC2s did not differ in number between WT and IL-13−/− mice with pericarditis (Figure 5J). These data suggest that IL-5 derived from ILC2s affects the development of pericarditis and cardiac infiltrating immune cells.
Figure 5. IL-5 Produced by ILC2s Play a Role during the Development of Pericarditis.

(A) Representative flow cytometry plot of intracellular staining of IL-5 and IL-13 in cardiac ILC2s from IL-33-treated mice.
(B) Frequency of ILC2s producing IL-5 and/or IL-13 in the hearts of IL-33-treated mice.
(C) Representative images of H&E-stained heart sections of IL-33-treated mice with isotype or anti-IL-5 administration. Bars: 100 μm.
(D) Total number of heart-infiltrating CD45+ leukocytes.
(E and F) Number of (E) eosinophils and (F) ILC2s in the hearts of IL-33-treated WT mice with isotype or anti-IL-5 administration.
(G) Representative images of H&E-stained heart sections of IL-33-treated WT and IL-13−/− mice. Bars: 100 μm.
(H) Total number of heart-infiltrating CD45+ leukocytes.
(I and J) Number of (I) eosinophils and (J) ILC2s in the hearts of IL-33-treated WT and IL-13−/− mice. Data are representative of two independent experiments and displayed as the mean. One-way ANOVA followed by Tukey’s post hoc test (B) or unpaired t test (D–F and H–J) was used for statistical analysis. **p < 0.01; ***p < 0.001.
Eosinophils Are Present in the Mediastinal Cavity and Can Migrate to the Heart
Previously, we found that the eotaxin-CCR3 pathway is critical for eosinophil migration to the heart during eosinophilic myocarditis (Diny et al., 2016). We also showed that ILC2s stimulated by IL-33 promote eotaxin-1 production by cardiac fibroblasts (Figures 4C and 4D). However, given that IL-33-induced cardiac inflammation has a unique disease pattern affecting predominantly the pericardium and myopericardium, we wanted to explore whether eosinophils could migrate from a non-vascular source such as the mediastinal cavity, a neighboring serosal cavity. In mice, there are pericardial pores between the pericardial, pleural, and mediastinal cavities (Fukuo et al., 1988). We found that eosinophils resided in the mediastinal cavity of naive WT mice at a frequency comparable to that of eosinophils in the heart (Figures 6A and 6B). To examine whether eosinophils would be present in the mediastinal cavity in higher numbers in hypereosinophilia, we examined eosinophils in the mediastinal cavity of naive IL-5 transgenic (IL-5Tg) mice, which spontaneously develop tissue and blood eosinophilia (Lee et al., 1997). In IL-5Tg mice, we found eosinophils at a high frequency in the mediastinal cavity, as well as in the heart and blood (Figures 6C and 6D). The frequency of neutrophils in the mediastinal cavity of both WT and IL-5Tg mice was significantly lower compared to the frequency in the blood of these mice, indicating that eosinophils found in the mediastinal cavity were not from blood contamination (Figures S6Aand 6B). We found that PBS-treated WT mice had a frequency of eosinophils and neutrophils in the heart, mediastinal cavity, and blood similar to that of naive WT mice (Figures 6B, S6A, S6C, and S6E). Neutrophils in the blood of IL-33-treated WT mice were found to be higher than in the heart and mediastinal cavity, as shown similarly in naive IL-5Tg mice (Figures S6B and S6F). However, in IL-33-treated mice, eosinophils were present at a higher frequency in the heart and mediastinal cavity than in the blood (Figure S6D). To determine whether eosinophils can migrate to the heart from the mediastinal cavity in addition to the vasculature, we transferred the same number of eosinophils either to the mediastinal cavity or intravenously (i.v.) to IL-33-treated ΔdblGATA1 mice lacking eosinophils (Figure 6E). Both mediastinal cavity and i.v. transfers were performed simultaneously to the same animal. The mediastinal cavity transfer was conducted by the direct injection of eosinophils into the mediastinum without an open-chest surgery. Eosinophils injected into the mediastinal cavity and i.v. were labeled with different fluorescent cell-tracking dyes, CellTrace Violet (CTV) and CellTrace Far Red (CTFR), respectively (Figures 6E and 6F). Eosinophils transferred to the mediastinal cavity were found to remain mostly in the cavity, confirming that the transfer to the cavity was correctly performed (Figure S6G). We found eosinophils transferred through both routes in the heart of ΔdblGATA1 mice deficient in eosinophils (Figures 6G and 6H). Eosinophils transferred to the mediastinal cavity were found at a significantly higher frequency in the heart of ΔdblGATA1 mice compared to eosinophils transferred intravenously (Figures 6H and 6I). This indicates that eosinophils can migrate from the mediastinal cavity to the heart in addition to traditional vascular routes. Thus, we demonstrated that eosinophils are present in the mediastinal cavity and increased in the cavity of IL-33-treated mice or hypereosinophilic mice. We also showed that eosinophils can migrate to the heart from the neighboring serosal cavities. In addition to direct eosinophil supply from the circulation, the mediastinal cavity seems to serve as an eosinophil reservoir, leading to non-vascular eosinophil trafficking to the heart in the IL-33-induced pericarditis model.
Figure 6. Eosinophils Reside in the Mediastinal Cavity from Which These Cells Can Traffic to the Heart.

(A) Representative flow cytometry plot of CD45+CD11b+ cells. Gates show CD11b+SiglecF+ eosinophils and CD11b+Ly6G+ neutrophils in the heart, mediastinal cavity, and blood of WT naive mice.
(B) Frequency of eosinophils in the heart, mediastinal cavity, and blood of WT naive mice.
(C) Representative flow cytometry plot of CD45+CD11b+ cells. Gates show CD11b+SiglecF+ eosinophils and CD11b+Ly6G+ neutrophils in the heart, mediastinal cavity, and blood of IL-5Tg naive mice.
(D) Frequency of eosinophils in the heart, mediastinal cavity, and blood of IL-5Tg naive mice.
(E) Schematic description of eosinophil transfer to eosinophil-deficient ΔdblGATA1 mice.
(F) Flow cytometry plots of CTV- or CTFR-labeled eosinophils.
(G) Flow cytometry plot of CD45+ cells in the heart. Gate shows CD11b+SiglecF+ eosinophils found in the heart of ΔdblGATA1 mice treated with IL-33 after eosinophil transfer in the mediastinal cavity and i.v.
(H) CTV- or CTFR-labeled CD11b+SiglecF+ eosinophils found in the heart of ΔdblGATA1 mice treated with IL-33 after eosinophil transfer.
(I) Frequency of CTV- or CTFR-labeled eosinophils found in the heart of ΔdblGATA1 mice treated with IL-33.
Concatenated samples (n = 7) are shown in (G) and (H). Both male and female mice were used in (G)–(I). Data are representative of two independent experiments and displayed as the mean (B and D) or the mean with SD (I). One-way ANOVA followed by Tukey’s post hoc test (B and D) or unpaired t test (I) was used for statistical analysis. **p < 0.01.
See also Figure S6.
ILCs Are Increased in the Pericardial Fluid of Human Pericarditis
To investigate the pathophysiological relevance of ILCs in human pericarditis, we examined whether ILCs are increased in the pericardial fluid from pericarditis patients. We collected pericardial fluid from patients with diverse heart diseases, including pericarditis, and comprehensively profiled immune cells from the pericardial space using high-dimensional mass cytometry (Cy-TOF). Pericardial fluid samples were obtained from patients with arrhythmogenic right ventricular cardiomyopathy (ARVC), ischemic heart disease (IHD), valve disease, or pericarditis during surgical procedures or pericardiocentesis. Pericardial fluid specimens obtained from the rapid autopsy of deceased patients with no cardiovascular diseases served as a control. Most of the alive single cells that were isolated from the human pericardial fluid were CD45+ leukocytes (Figure 7A). To identify ILCs in the pericardial fluid, we excluded myeloid cells and other lymphoid cells such as granulocytes, macrophages, monocytes, dendritic cells, T cells, B cells, and NK cells using cell-specific markers such as CD66b, CD11b, CD68, CD3, CD19, HLA-DR, and CD56 (Figure 7A). ILCs were then defined as CD127+ cells (Figure 7A). We found that, whereas ILCs were a rare population in the pericardial fluid of autopsy controls, the frequency of pericardial ILCs was highly increased in patients with pericarditis compared to controls (Figures 7B and 7C). Although other cardiac disease groups such as ARVC, IHD, and valve disease showed a slightly increasing trend of pericardial ILCs compared to controls, the frequency of ILCs in pericarditis was significantly higher than that in other cardiac diseases (Figures 7B and 7C). Using conventional flow cytometry, we confirmed that Lin−CD127+ ILCs were increased in the pericardial fluid from pericarditis patients compared to controls (Figure 7D). These findings suggest that ILCs are a specific population that is increased in the pericardium of patients with pericarditis as we observed in the pericarditis mouse model and that the increase in pericardial fluid ILCs is a unique feature in pericarditis, which is differentiated from other cardiac diseases.
Figure 7. ILCs Are Increased in the Pericardial Fluid from Patients with Pericarditis.

(A) Gating strategy of ILCs in human pericardial fluid samples by CyTOF. L/D, live/dead.
(B) Representative CyTOF plots showing ILCs in the pericardial fluid from controls and patients with cardiac diseases. ARVC, arrhythmogenic right ventricular cardiomyopathy; IHD, ischemic heart disease.
(C) Frequency of ILCs in the pericardial fluid.
(D) Flow cytometry plots showing Lin−CD127+ ILCs in the pericardial fluid from controls and patients with pericarditis. Plots show a CD45+CD11b−CD3−CD19−CD56− population. Lin included CD1a, CD11c, CD14, CD34, CD123, CD303a(BDCA2), TCRαβ, and FcεRIα.
Concatenated samples (control, n = 3; pericarditis, n = 2) are shown in (D). One-way ANOVA followed by Tukey’s post hoc test (C) was used for statistical analysis. ***p < 0.001.
DISCUSSION
We demonstrated that ILC2s accumulate in the heart following IL-33 treatment in mice using a previously described model of IL-33-induced pericarditis (Abston et al., 2012). We report a pathogenic role for ILC2s in the cardiac inflammation, which implies that cardiac ILC2s activated by IL-33 drive eosinophilic pericarditis in collaboration with cardiac fibroblasts. With advanced visualizing tools using the tissue-clearing technique and light sheet microscopy, we revealed that cardiac ILC2s are located in the ventricles near the pericardium where eosinophilic pericarditis is observed after IL-33 treatment, which supports their role in the development of pericarditis. We identified that ILCs are required for the development of IL-33-induced pericarditis. Given that Rag2−/− mice develop comparable cardiac inflammation and pathology to WT mice following IL-33 treatment, we excluded adaptive lymphocytes such as T cells and B cells from being drivers of the development of pericarditis. The necessity of ILC2s in the induction of inflammation has been shown in other organs such as allergen-induced lung inflammation models (Halim et al., 2012; Kim et al., 2012). Papain causes asthma-like symptoms in Rag1−/− mice but not in Rag2−/−Il2rg−/− mice, and ILC-deficient Rag2−/−Il2rg−/− mice reconstituted with ILC2s develop airway inflammation (Halim et al., 2012). While Rag2−/− mice lack T cells and B cells, Rag2−/−Il2rg−/− mice are deficient in ILCs in addition to T cells and B cells. Unlike Rag2−/− mice, Rag2−/−Il2rg−/− mice were protected from IL-33-induced pericarditis, demonstrating that ILCs are required for pericarditis development. Our adoptive transfer experiments showed that cardiac ILC2s transferred directly to the heart of pericarditis-resistant Rag2−/−Il2rg−/− mice elicited cardiac eosinophil infiltration. These findings support the notion that ILCs are tissue resident cells even during acute inflammation as described by us in the heart and others in different organs (Gasteiger et al., 2015; Moro et al., 2016; Bracamonte-Baran et al., 2019). IL-33-treated mice had increased anti-myosin IgM antibody found in their sera, which suggests a possible pathogenic role for ILCs in myopericarditis. ILC2s isolated from the lungs promoted the production of immunoglobulin including IgM by B1 and B2 cells in vitro (Drake et al., 2016).
The IL-33-induced pericarditis model allowed us to investigate the pathogenic role of the IL-33-ST2 axis in ILC2 activation during cardiac inflammation. IL-33 mediates its effects through binding to its receptor ST2 as we demonstrated in cardiac ILC2 activation (Schmitz et al., 2005). ST2 is known to be expressed not only on ILC2s but also on Th2 cells, B cells, basophils, eosinophils, dendritic cells, mast cells, and NK T cells (Griesenauer and Paczesny, 2017). However, using an adoptive transfer experiment, we showed that ILC2s are essential for IL-33-induced cardiac inflammation. It was reported that IL-33 expression increases during cardiac inflammation or after cardiac injury, representing an association between IL-33 and its pathological outcome in cardiac inflammation (Chen et al., 2015; Sánchez-Más et al., 2014). ST2 is not only expressed as a membrane-bound form but also produced as a soluble form (soluble ST2 [sST2]) that is released into the circulation. sST2 has been proposed as a prognostic marker for chronic and acute heart failure and aortic stenosis (McCarthy and Januzzi, 2018; Friões et al., 2015; Coronado et al., 2019; Lindman et al., 2015). This is consistent with our findings that the IL-33-ST2 axis plays a key pro-inflammatory role in the heart by stimulating cardiac ILC2s under certain conditions related to increased IL-33 production. It should be noted that IL-33 signaling via ST2 could play a beneficial role in different types of heart disease such as pressure overload and myocardial infarction by providing improved cardiac function and survival (Sanada et al., 2007; Seki et al., 2009).
We showed that IL-33 is expressed in naive heart, and its expression is increased in pericarditis, suggesting its pathogenic role in cardiac inflammation. When mice were treated with exogenous IL-33, its endogenous expression was increased in the heart, thus offering evidence for the presence of a positive feedback loop in the heart. Moreover, IL-33-deficient mice developed less severe inflammation with smaller number of infiltrating leukocytes, including eosinophils compared to WT mice after exogenous IL-33 treatment, which indicates the importance of endogenous IL-33 as a component of the positive feedback loop in the development of pericarditis. Thus, the IL-33-induced pericarditis model does not rely solely on exogenous IL-33 administration, but endogenous IL-33 may contribute to an increased IL-33 level in the heart leading to the development of disease. We identified that Sca1+ cardiac fibroblasts are major producers of IL-33 in the heart, both in steady state and during pericarditis. We previously found that cardiac fibroblasts are a versatile stromal cell type capable of producing different sets of cytokines and chemokines in response to changes in the microenvironment (Chen et al., 2018; Diny et al., 2016). The IL-33-expressing cardiac fibroblasts are located near and in the pericardium, supporting the possible involvement of cardiac fibroblasts in ILC2 activation and pericarditis development through their IL-33 expression. Although it has been reported that endothelial cells or cardiomyocytes could be an important source for IL-33, we did not find mouse endothelial cells to be a major producer of IL-33 in the heart (Pichery et al., 2012; Moussion et al., 2008; Küchler et al., 2008; Demyanets et al., 2013). In our model, we found that cardiac fibroblasts are potent IL-33 producers and increase IL-33 production in response to systemic IL-33 administration, suggesting the existence of a feed-forward loop.
ILC2s produce Th2 cytokines such as IL-5 and IL-13 upon activation with IL-33. We were able to show that IL-33-activated ILC2s induce eotaxin-1 (CCL11) production by cardiac fibroblasts. We previously found that cardiac fibroblasts are the major CCL11 producers in eosinophilic myocarditis mouse model, and CCL11 expression is induced in cardiac fibroblasts in vitro in response to IL-4 and IL-13 (Diny et al., 2016). It has been reported that IL-13 potently induces eotaxin expression in lung epithelial cells, esophagus, and other organs (Li et al., 1999; Matsukura et al., 2001; Zuo et al., 2010; Moore et al., 2002; Takahashi et al., 2013; Blanchard et al., 2007). We found, in vivo, that cardiac ILC2s are potent producers of IL-13 upon IL-33 activation. Furthermore, cardiac ILC2s are capable of inducing the production of CCL11 by cardiac fibroblast in vitro. The expression of eotaxin and type 2 cytokines in the heart was downregulated in ILC-deficient Rag2−/−Il2rg−/− mice compared to WT and Rag2−/− mice, suggesting that eotaxin expression, and subsequent eosinophil recruitment to the heart, may occur in an ILC-dependent way in vivo. In addition, eotaxin expression was spatially concentrated in the pericardium and interstitial space, which could explain the specific inflammation pattern of eosinophilic pericarditis induced by IL-33.
We also found that cardiac ILC2s produce IL-5, which plays an important role in developing IL-33-induced pericarditis and that blocking of IL-5 protected mice from the development of eosinophilic pericarditis. In response to IL-5, eosinophils are released into the circulation, which may be hindered by blocking IL-5. Several anti-IL-5 agents, including mepolizumab, reslizumab, and benralizumab, which block IL-5 signaling via IL-5 receptor and reduce blood eosinophils, have been developed, approved, and used to treat asthma in clinical practice (Farne et al., 2017). In addition, a phase II clinical trial (NCT02130882) is evaluating the safety and efficacy of benralizumap, an anti-IL-5 receptor biologic, in decreasing eosinophils in patients with hypereosinophilic syndrome. We found that anti-IL-5 treatment had a profound effect on reducing eosinophils infiltrating to the heart during pericarditis, suggesting that anti-IL-5/IL-5R agents could be used as a potential treatment for eosinophilic pericarditis.
IL-33-induced cardiac inflammation has a unique pattern affecting mostly pericardium and myopericardium; therefore, we explored whether eosinophils could migrate from a non-vascular source such as the mediastinal cavity, a neighboring serosal cavity. We identified that eosinophils are present in the mediastinal cavity of naive mice and that adoptively transferred eosinophils to the mediastinal cavity of IL-33-treated eosinophil-deficient mice are able to traffic to the heart. This suggests that the serous cavities may serve as a reservoir of cardiac-infiltrating eosinophils. Immune cells have been previously identified in serous cavities such as the peritoneum and pleural cavity (Ha et al., 2006; Shimotakahara et al., 2007; Wang and Kubes, 2016; Weber et al., 2014; Deniset et al., 2019). Macrophages residing in the peritoneal cavity are rapidly recruited to the injured liver through a non-vascular route and display tissue-reparative phenotypes (Wang and Kubes, 2016). Macrophages found in mouse pericardial fluid played a reparative role after myocardial infarction (Deniset et al., 2019). The pleural space is also shown to possess B1a B cells, which can migrate to the lung and produce protective IgM in response to bacterial infection (Weber et al., 2014). We found that eosinophils residing in the mediastinal cavity can migrate to the heart. There are pericardial pores between pericardial, pleural, and mediastinal cavities (Fukuo et al., 1988). Therefore, all of these cavities should be considered a possible source of cells infiltrating to the heart. This does not exclude the contribution of circulating eosinophils to the development of pericarditis; however, it is noteworthy that the mediastinal cavity can serve as an alternative reservoir of eosinophils, which may lead to rapid infiltration during inflammation in close proximity to the heart. IL-33 treatment also induces inflammation in lungs and esophagus, as we demonstrated here and shown before by others (Travers et al., 2017; Judd et al., 2016). After treating mice with IL-33, we found pericardial, subserosal, and peribronchial eosinophil infiltrates in the heart, esophagus, and lungs, respectively. IL-33-induced esophagitis has a subserosal inflammation pattern that is similar to IL-33-induced pericarditis, with the inflammation mostly confined to the subserosa rather than to the mucosa. Based on these observations, we speculate that the chest serosal cavities could also be a source of eosinophils for other organs located in the mediastinal cavity. This could be important in patients with hypereosinophilia, since we showed that hypereosinophilic mice have more eosinophils in their mediastinum than do WT mice. Our findings suggest that ILC2s may direct eosinophils to infiltrate organs, not only from the blood but also from the neighboring serosal cavity where these cells reside locally.
Serosal fluid in the pericardium contains immune cells and can become a source of infiltrating leukocytes under certain insults such as exposure following cardiac surgery and myocardial infarction (Butts et al., 2017; Horckmans et al., 2018). We found that ILCs were present at a higher frequency in the pericardial fluid of patients with pericarditis, whereas healthy controls and patients with other types of cardiac diseases such as cardiomyopathy showed a much lower frequency of ILCs in the pericardial fluid. The number of patients tested was limited; therefore, further clinical investigation will be needed. However, this result suggests that ILCs may also be involved in the pathogenesis of pericarditis in humans. It is unclear whether the increase in ILCs results from other underlying causes such as infection and whether ILCs directly induce human pericarditis, as shown in the IL-33-induced pericarditis mouse model.
Although many pericarditis case reports have been published, the etiology is still largely unknown. Contemporary treatments for pericarditis mostly aim to resolve symptoms by administrating anti-inflammatory agents such as nonsteroidal anti-inflammatory drugs (NSAIDs, especially indomethacine), colchicine, and corticosteroids (Imazio et al., 2013, 2015). A phase III clinical trial (ClinicalTrials.gov: NCT03737110) is assessing the IL-1 blocker in patients with recurrent pericarditis. Another treatment option for severe cases is a pericardiocentesis or pericardiectomy to remove the inflamed pericardium. These symptomatic therapies, however, do not address a specific cause of pericarditis. One of the problems in developing new targeted therapies is a lack of classification on subtypes of pericarditis. There are several case reports of eosinophilic pericarditis in humans (Li et al., 2002; Spiegel et al., 2004; Herry et al., 1997; AbdullGaffar and Almulla, 2015; Van den Bosch et al., 1986). In this study, using the IL-33-induced eosinophilic pericarditis mouse model, we identified cardiac ILC2s playing a critical role in the pathogenesis of eosinophilic pericarditis. We also showed limited human data that suggest an increased number of ILCs in the pericardial fluid of pericarditis patients. These findings augment our understanding of how ILC2s contribute to cardiac inflammation and provide insights into targeted therapy for eosinophilic pericarditis. In a setting where there is no specific way to target ILC2s, it may be helpful to alleviate eosinophilic pericarditis by blocking the IL-33-ST2-ILC2s axis.
Our findings suggest a pathogenic role for ILCs in cardiac inflammation. ILCs play a critical role in the development of pericarditis induced by IL-33, as their presence and activation are essential components to inducing pericarditis. Our study provides insights into the role of cardiac ILCs in inflammation that can be helpful to developing therapeutic targets for pericarditis.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniela Čiháková (cihakova@jhmi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples
Human pericardial fluid samples were obtained from patients with pericarditis, ARVC, IHD or valve disease. Samples were collected while these patients underwent coronary artery bypass grafting, valve replacement surgery or pericardiocentesis at the Institute for Clinical and Experimental Medicine (IKEM) in Czech Republic. Informed consent was obtained from subjects and the study protocol was approved by the IKEM Ethics Committee (G-18-60). Control pericardial fluid specimens were obtained from rapid autopsy of patients who had no cardiovascular diseases at the Johns Hopkins University. Informed consent was obtained from subjects and the study protocol was approved by the Committee for the Protection of Human Subjects (IRB NA_00036610, PI: Jody Hooper). Sex of human sample donors is indicated below: Controls, 1 male and 2 females; ARVC, 1 male and 2 females; IHD, 11 males and 4 females; Valve disease, 2 males and 2 females; Pericarditis, 1 male and 1 female. Pericardial fluid cells were isolated by centrifugation, resuspended in fetal bovine serum (FBS) supplemented with 10% DMSO, and stored in a liquid nitrogen tank. Frozen samples were shipped on dry ice. Patients’ information of all samples was processed with random non-linked code labeling in a database at the time of preservation. For each sample, a separate random non-linked code was assigned for the analysis process.
Animals
Wild-type BALB/cJ (JAX 651), Rag2−/− (JAX 8448), Rag2−/−γc−/− (JAX 14593), CD45.1/cByJ (JAX 6584), ΔdblGATA1 (JAX 5653) mice were obtained from the Jackson Laboratory (Bar Harbor, ME). ST2−/−, IL-13−/− and IL-33cit/+ mice were kindly provided by Andrew N.J. McKenzie (Medical Research Council, Cambridge, UK). IL-33cit/cit mice were used as IL-33−/− mice. IL-5Tg mice were kindly provided by James Lee (Mayo Clinic, Scottsdale, AZ). Mice used were 6 to 12-week-old male mice on the BALB/c background unless otherwise mentioned in the figure legends. Mice were maintained at the Johns Hopkins University School of Medicine specific pathogen-free animal facility. Experiments were performed with age-matched mice in accordance with the guidelines set forth in the Guide for the Care and Use of Laboratory Animals. All methods and protocols were approved by the Animal Care and Use Committee of Johns Hopkins University.
Isolation of primary adult mouse cardiac fibroblasts
Hearts were isolated from 6-12-week-old WT BALB/c mice pretreated i.p. with PBS with 50U/ml Heparin. Hearts were cannulated through aorta and perfused for 3 min at 37°C with calcium-free perfusion buffer (7.03 g/L NaCl, 1.1 g/L KCl, 0.082 g/L KH2PO4, 0.085 g/L Na2HPO4, 0.144 g/L MgSO4, 2.38 g/L HEPES, 0.39 g/L NaHCO3, 1 g/L glucose, 3.74 g/L Taurine, 1 g/L 2,3-Butanedione monoxime; all Sigma-Aldrich) and for 8 min with the addition of Collagenase II (Worthington Biochemical Corporation) and Protease XIV (Sigma-Aldrich) and 0.03M CaCl2. The heart was cut into pieces and digested by gentle pipetting for 3 min or until large pieces are fully digested. After filtering through a 100 μm strainer and washing with DMEM (GIBCO), cardiac fibroblasts in cell suspensions were separated from cardiac myocytes that precipitated rapidly and spontaneously. Cardiac fibroblasts were cultured at 37°C in DMEM with 20% FBS (GE Healthcare Life Siences), Non-essential amino-acids (Sigma Aldrich), Penicillin-Streptomycin, 2 mM L-Glutamine, and 25 mM HEPES (all Quality Biological). Nonadherent cells were washed off after 1 h. Culture media was changed every day for 5 days until cardiac fibroblasts are confluent. Cardiac fibroblasts were passaged twice before co-culture.
METHOD DETAILS
Induction of experimental pericarditis
Mice were injected with 1 μg of recombinant mouse IL-33 (BioLegend) in 100 μL PBS intraperitoneally on days 0, 2, 4, 6 and 8. On day 9 or 10, hearts were harvested for further analysis detailed below.
Assessment of pericarditis severity
Hearts were cut transversely, fixed in SafeFix (Thermo Fisher Scientific), embedded in paraffin, cut into 5 μm-thick sections and stained with H&E (Histoserv, MD). The severity of pericarditis was assessed by scoring infiltration of the area of pericardium around right ventricle (RV) on H&E-stained sections based on histopathology score from 0 to 4 using the following criteria for hematopoietic infiltrates: grade 0, no inflammation; grade 1, < 20% of RV is involved and/or mild inflammation; grade 2, 20%–50% of RV is involved and/or intermediate inflammation; grade 3, 50%–80% of RV is involved and/or severe inflammation; grade 4, >80% of RV is involved and/or severe inflammation with adjacent myocardial infiltrates. Scoring was performed by two blinded investigators and averaged.
Light microscopy
Images on H&E-stained and immunostained sections were acquired on the BX43 microscope (Olympus) with the DS-Fi3 camera (Nikon) using NIS-Elements D Software (v. 5.10.01, Nikon).
Tissue clearing with immunolabeling and light sheet microscopy
Hearts were perfused with PBS for 3 min followed by 4% paraformaldehyde in PBS for 3 min for fixation. After perfusion, hearts were removed from mice and the right ventricle was obtained and immersed in 4% paraformaldehyde in PBS for 2 h at 4°C to complete fixation. After washing with PBS, the sample was permeabilized, washed, penetrated, and blocked using Visikol HISTO reagents (Visikol) according to manufacturer’s protocols. Primary and secondary antibody staining were performed using 10 μg/ml anti-mouse SiglecF (BD) and KLRG1 (ProSci) antibodies and 20 μg/ml anti-rat and anti-hamster IgG conjugated with fluorophores (Thermo Fisher Scientific), respectively, in Visikol HISTO antibody buffer (Visikol) according to manufacturer’s protocols. After immunolabeling, tissue clearing process was conducted using Visikol HISTO-1 and −2 (Visikol) following manufacturer’s instruction. Images of cleared tissue with immunolabeling were acquired using a light sheet microscope, Ultra Microscope II (LaVision Biotec). Processing of images was carried out with Imaris (Bitplane).
Immunofluorescence staining and confocal microscopy
Mouse heart was perfused with PBS for 3 min followed by 4% paraformaldehyde in PBS for 3 min for fixation. After perfusion, heart was removed from mice and immersed in 4% paraformaldehyde in PBS for 2 h at 4°C to complete fixation. After fixation, the heart was placed in 30% sucrose in PBS overnight or until tissue sinks at 4°C before embedding in OCT. Cryostat sectioning was performed to obtain heart sections in a 100 μm-thickness. Sections were washed in PBS, permeabilized in 0.5% Triton X-100 in PBS and blocked in 10% normal goat serum and 0.3% Triton X-100 in PBS. After blocking, sections were incubated with Rabbit anti-PDGFRα antibody (Abcam). Goat anti-Rabbit IgG conjugated with Alexa Fluor 555 (Thermo Fisher) was used as secondary antibody and DAPI was used for counterstaining. Images were acquired using Zeiss LSM 700 microscope (Zeiss) at 20X and 40X magnification with the ZEN Black software (Zeiss). ImageJ/Fiji was used to process images.
Immunohistochemistry staining
Paraffin-embedded hearts were cut into 4-μm-thick sections. After deparaffinization, heat-induced antigen retrieval and blocking, sections were stained with 5 μg/ml Polyclonal Goat Anti-Mouse CCL11 antibody (R&D Systems) and Anti-Goat HRP-DAB Cell & Tissue Staining Kit (R&D Systems) and counter stained with Hematoxylin.
Echocardiography
Conscious mice were held in a supine position and M-mode or Doppler echocardiography was performed using Vevo 2100 with a MS400 or MS550D transducer (FUJIFILM VisualSonics, Ontario, Canada). In M-mode, the heart was imaged in the parasternal short axis view to measure parameters such as left ventricular internal dimension (LVID) and ejection fraction (EF). To measure the Myocardial Performance Index (MPI), tissue Doppler imaging of the mitral annulus was obtained. Cardiac time intervals, isovolumetric contraction time (IVCT), isovolumetric relaxation time (IVRT) and ejection time (ET), were measured and MPI was calculated based on these parameters (Gao et al., 2011).
High-dimensional mass cytometry (CyTOF)
Frozen cells were thawed at 37°C and washed with PBS twice. Single cell suspensions were stained with Cell-ID Cisplatin (Fluidigm), blocked with Fc Receptor Binding Inhibitor (Thermo Fisher), and stained with heavy metal-conjugated antibodies (Fluidigm) for surface staining. Cells were fixed and permeabilized with FoxP3 Transcription Factor Staining Buffer Set (Thermo Fisher) followed by intracellular staining with metal-conjugated antibodies (Fluidigm). Cells were fixed in methanol-free formaldehyde (Thermo Fisher) and kept in Cell-ID Intercalator-Ir solution (Fluidigm). Stained cells were shipped on ice to the CyTOF Facility at the University of Pennsylvania and acquired on Helios (Fluidigm). CyTOF data were normalized in the CyTOF software (Fluidigm) and analyzed using Cytobank (Cytobank Inc).
Flow cytometry and cell sorting
Hearts were perfused with PBS through ventricles, cut into small pieces and digested in gentleMACS C Tubes (Miltenyi Biotec) with 3000U Collagenase II and 300U DNase I (Worthington Biochemical Corporation) in HBSS for 30 min at 37°C. To generate single cell suspensions, hearts were mechanically dissociated using GentleMACS dissociator (Miltenyi Biotec) following manufacturer’s protocols. Cells in the mediastinal cavity were harvested by lavage with PBS. Blood was collected in PBS with 100U/ml Heparin and overlaid on Histopaque-1119 (Sigma Aldrich) to remove red blood cells. Remaining red blood cells were lysed using ACK buffer (Quality Biological). Cells from heart samples were strained through a 40 μm filter, and cells from other organs were strained through a 70 μm filter. For intracellular cytokine staining, cells were stimulated with 50 ng/ml PMA, 750 ng/ml Ionomycin (Sigma-Aldrich), GolgiStop and GolgiPlug (BD Biosciences) for 4 h at 37°C before staining. Single cell suspensions were stained with LIVE/DEAD Fixable Aqua (Thermo Fisher Scientific). FcγRII/III was blocked with anti-mouse CD16/CD32 (eBioscience), and markers of interest were stained with flourochrome-conjugated antibodies (BD, BioLegend and eBioscience). Lineage (Lin) used for identifying mouse ILC2s included CD3ε, TCRβ, CD19, B220, CD11b, CD11c, Gr-1, Ter119, FcεRIα and NKp46. To quantify absolute number of cells, viable cells were counted using CountBright Absolute Counting Beads (Thermo Fisher Scientific). Samples were acquired on BD LSR II or Fortessa (BD Biosciences) and data were analyzed using FlowJo V10 (Tree Star). For cardiac ILC2 sorting, after enzymatic and mechanical dissociation of hearts, single cell suspensions were obtained using Histopaque-1077 (Sigma-Aldrich) according to manufacturer’s instructions. Collected mononuclear cells were stained and sorted on BD FACSAria II (BD Biosciences).
Isolation and in vitro expansion of cardiac ILC2
Cardiac ILC2s were isolated using FACSAria II (BD Biosciences) from the hearts of IL-33-treated mice and cultured at 37°C in RPMI 1640 (GIBCO) with 10% FBS (GE Healthcare Life Sciences), 1% Penicillin-Streptomycin, 2 mM L-Glutamine, 10 mM HEPES, 1 mM Sodium Pyruvate (all Quality Biological), 0.1 mM Non-essential amino acids (Sigma Aldrich), 0.05 mM 2-Mercaptoethanol (GIBCO). Cytokines, IL-2, IL-7 and IL-33, at a concentration of 25 ng/ml were added in media to expand ILC2s in vitro.
Cardiac injection of ILC2
CD45.2+ Rag2−/−γc−/− mice were anesthetized with 3.5% isoflurane (Baxter) and tracheal intubation was performed immediately. While on intubation, mice were provided with oxygen with 2% isoflurane by mechanical ventilation system (Model 845, Harvard Apparatus). Pre-operational analgesics (0.05 mg/kg Buprenorphine, Reckitt Benckiser) and paralytics (1 mg/kg Succinylcholine, Henry Schein) were treated before operation. Thoracotomy was done to access the chest cavity and expose the ventricles of the heart. 5x105 CD45.1+ cardiac ILC2s were injected into the myocardium on three ventricular locations using a 29G ½ insulin syringe (BD). Post-operational analgesics (0.05 mg/kg Buprenorphine) were administered during recovery.
In vitro co-culture of cardiac fibroblasts and ILC2
ILC2s were FACS-sorted from the hearts of I L-33-treated mice and expanded in vitro as described above. 1x105 ILC2s were placed on Transwell with 0.4 μm pore polyester membrane insert (Corning). Cardiac fibroblasts from second passage were co-cultured with ILC2s located on Transwell at 37° C in ILC2 culture media for 24 h. 25 ng/ml IL-2 and 25 ng/ml IL-7 were included in co-culture and 25 ng/ml IL-33 was added in certain conditions indicated. Cells and supernatants were harvested after co-culture and used for further analysis.
In vivo IL-5 neutralization
To block IL-5 in vivo, mice were treated i.p. with 300 μg of anti-IL-5 monoclonal antibody (Clone: TRFK5, BioXCell) or isotype antibody (Clone: HRPN, BioXCell) every three days starting from a day before pericarditis induction.
Eosinophil isolation, labeling, and transfer to the mediastinal cavity or intravenously
Blood from donor IL-5Tg mice was collected in PBS with 100U/ml Heparin and overlaid on Histopaque-1119 (Sigma Aldrich) to remove red blood cells. Anti-CD90.2 and anti-CD45R(B220) microbeads (Miltenyi Biotec) were used to deplete lymphocytes and enrich eosinophils. Eosinophil purity (78%–95%) was determined by flow cytometry. For labeling, cells were stained with either CellTrace™ Violet (CTV) or CellTrace™ Far Red (CTFR) according to manufacturer’s instructions (Thermo Fisher). Recipient mice were anesthetized with 500 μL avertin or isoflurane and 1-8x106 cells were injected to the mediastinal cavity or by retro-orbital injection using a 29G ½ insulin syringe (BD).
Quantitative PCR
RNA was extracted in TRIzol (Invitrogen) and cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) and executed on MyiQ2 thermal cycler (Bio-Rad) using iQ5 optical system software (Bio-Rad). Acquired data were analyzed by the 2−ΔΔCt method (Livak and Schmittgen, 2001). Threshold cycles were normalized to the expression of Gapdh and then to controls. Primers for genes (Ccl11: 5′-GAATCACCAACAA CAGATGCAC-3′ and 5′-ATCCTGGACCCACTTCTTCTT-3′, Ccl24: 5′-TCTTAGGGCCCTTCTTGGTG-3′ and 5′- AATTCCAGAAAAC CGAGTGG-3′, Il5: 5′-CTCTGTTGACAAGCAATGAGACG-3′ and 5′-TCTTCAGTATGTCTAGCCCCTG-3′, Il13: 5′-CCTGGCTCTTG CTTGCCTT-3′ and 5′-GGTCTTGTGTGATGTTGCTCA-3′, Gapdh: 5′-TTGATGGCAACAATCTCCAC-3′ and 5′-CGTCCCGTAGACA AAATGGT-3′) were commercially synthesized (Integrated DNA Technologies).
ELISA
Supernatants from cardiac fibroblasts co-culture with ILC2s or sera from mice were stored at −80°C prior to ELISA. Eotaxin-1 was determined by quantitative sandwich ELISA using Mouse CCL11/Eotaxin Quantikine ELISA Kit (R&D Systems) according to manufacturer’s protocols. For anti-myosin IgM ELISA, plates were coated with 0.5 μg/well myosin heavy chain α peptide MyHCα614-62g (Ac-SLKLMATLFSTYASAD; Genscript) and Alkaline Phosphatase AffiniPure goat anti-mouse IgM, μ chain specific (Jackson ImmunoResearch) at a 1:4000 dilution was used to detect anti-myosin IgM in sera.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Two group comparisons were performed using Student’s t test for normally distributed data. Multiple group comparisons were performed using one-way ANOVA followed by Tukey’s post hoc test. Mann-Whitney U test or Kruskal-Wallis H test for two groups or multiple groups, respectively, was used for nonparametric data. Pearson correlation coefficient, r, was calculated using correlation analysis in Prism 6 (GraphPad Software Inc.). Statistical analysis was conducted in Prism 6. Statistically significant comparisons were represented by asterisks: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Statistical analysis details are also described in the figure legends.
DATA AND CODE AVAILABILITY
This study did not generate datasets.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Table S1 | N/A | N/A |
| Biological Samples | ||
| Pericardial fluid samples | Johns Hopkins University School of Medicine | https://www.hopkinsmedicine.org/som/ |
| Pericardial fluid samples | Institute for Clinical and Experimental Medicine, Prague, Czech Republic | https://www.ikem.cz/en/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse IL-33 (carrier-free) | BioLegend | Cat#580508 |
| Collagenase, Type 2 | Worthington | Cat#LS004177 |
| Deoxyribonuclease I | Worthington | Cat#LS002139 |
| Protease, Type XIV: Bacterial, From Streptomyces griseus | Sigma | Cat#P5147-5G |
| ACK Lysing Buffer | Quality Biological | Cat#118-156-721 |
| Bovine Serum Albumin | Sigma | Cat#A3608-100G |
| 0.5M EDTA, pH8.0 | Corning | Cat#46-034-CI |
| PMA | Sigma | Cat#P1585 |
| Ionomycin | Sigma | Cat#I9657 |
| BD GolgiPlug | BD Biosciences | Cat#555029 |
| BD GolgiStop | BD Biosciences | Cat#554724 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Thermo Fisher | Cat#L34966 |
| BV711 Streptavidin | BioLegend | Cat#405241 |
| Streptavidin Alexa Fluor 350 Conjugate | Thermo Fisher | Cat#S11249 |
| BD CytoFix Fixation Buffer | BD Biosciences | Cat#554655 |
| Fixation/Permeabilization Solution Set | BD Biosciences | Cat#554714 |
| eBioscience Foxp3/Transcription Factor Staining Buffer Set | Thermo Fisher | Cat#00-5523-00 |
| CountBright Absolute Counting Beads | Thermo Fisher | Cat#C36950 |
| SafeFix II All-Purpose Fixative | Fisher Scientific | Cat#23-042600 |
| DMEM | GIBCO | Cat#11995-065 |
| RPMI 1640 | GIBCO | Cat#11875-093 |
| Heat Inactivated Fetal Bovine Serum | Thermo Fisher | Cat#10082147 |
| L-Glutamine | Corning | Cat#25-005-CI |
| Penicillin-Streptomycin | Sigma | Cat#P4333 |
| Antibiotic-Antimycotic solution (100x) | Corning | Cat#30-004-CI |
| Sodium Pyruvate | Sigma | Cat#S8636 |
| 1M HEPES Buffer pH7.3 | Quality Biological | Cat#118-089-721 |
| MEM Non-essential Amino Acid solution (100x) | Sigma | Cat#M7145-100ML |
| 2-Mercaptoethanol | GIBCO | Cat#21985-023 |
| Recombinant Mouse IL-2 (carrier-free) | BioLegend | Cat#575404 |
| Recombinant Mouse IL-7 (carrier-free) | BioLegend | Cat#577802 |
| Heparin sodium salt from porcine intestinal mucosa | Sigma | Cat#H3393 |
| Trypsin 2.5% | Quality Biological | Cat#118-086-721 |
| Histopaque-1119 | Sigma | Cat#11191-100ML |
| Histopaque-1077 | Sigma | Cat#10771-100ML |
| CD90.2 MicroBeads, mouse | Miltenyi Biotec | Cat#130-049-101 |
| CD45R (B220) MicroBeads, mouse | Miltenyi Biotec | Cat#130-049-501 |
| TRIzol Reagent | Thermo Fisher | Cat#15596026 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat#1708891 |
| Power SYBR Green PCR Master Mix | Thermo Fisher | Cat#4367659 |
| Normal Goat Serum Blocking Solution | Vector Laboratories | Cat#S-1000-20 |
| Prolong Glass Antifade Mountant | Thermo Fisher | Cat#P36980 |
| Visikol HISTO Starter Kit | VISIKOL | Cat#HSK-1 |
| Forane Liquid for inhalation 100ml | Baxter | Cat#1001936040 |
| Buprenorphine(1mg/ml) | ZooPharm | Cat#BZ8069317 |
| Succinylcholine (20mg/ml) | Hospira, Inc. | Cat#00409662902 |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher | Cat#C34557 |
| CellTrace Far Red Cell Proliferation Kit | Thermo Fisher | Cat#C34564 |
| MyHCα614-629 (Ac-SLKLMATLFSTYASAD) | GenScript | N/A |
| Trilogy | Cell Marque | Cat#920P-04 |
| TBS with Tween (TBST), 20X Solution | Thermo Fisher | Cat#J75500-K2 |
| Dual Endogenous Enzyme Block | Dako | Cat#S2003 |
| Antibody Dilution Buffer | Ventana | Cat#ADB250 |
| Richard-Allan Scientific Signature Series Hematoxylin 1 | Thermo Fisher | Cat#7221 |
| Richard-Allan Scientific Signature Series Clear-Rite 3 | Thermo Fisher | Cat#6901 |
| Shandon Bluing Reagent | Thermo Fisher | Cat#6769001 |
| Optic Mount I Toluene | Mercedes Scientific | Cat#MER7722 |
| Maxpar PBS | Fluidigm | Cat#201058 |
| Cell-ID Intercalator-Ir | Fluidigm | Cat#201192B |
| Cell-ID Cisplatin | Fluidigm | Cat#201064 |
| Maxpar Cell Staining Buffer | Fluidigm | Cat#201068 |
| Maxpar Fix and Perm Buffer | Fluidigm | Cat#201067 |
| 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher | Cat#28906 |
| Critical Commercial Assays | ||
| Mouse CCL11/Eotaxin-1 Quantikine ELISA Kit | R&D Systems | Cat#MME00 |
| Anti-Goat HRP-DAB Cell & Tissue Staining Kit | R&D Systems | Cat#CTS008 |
| Experimental Models: Organisms/Strains | ||
| Mouse: WT: BALB/cJ | The Jackson Laboratory | Cat#000651 |
| Mouse: CD45.1+: CByJ.SJL(B6)-Ptprca/J | The Jackson Laboratory | Cat#006584 |
| Mouse: Rag2−/−: C.B6(Cg)-Rag2tm1.1Cgn/J | The Jackson Laboratory | Cat#008448 |
| Mouse: Rag2−/−Il2rg−/−: C;129S4-Rag2tm1.1Flv Il2rgtm1.1Flv/J | The Jackson Laboratory | Cat#014593 |
| Mouse: ΔdblGATA1: C.129S1(B6)-Gata1tm6Sho/J | The Jackson Laboratory | Cat#005653 |
| Mouse: ST2−/− (BALB/c) | Townsend et al., 2000 (Dr. McKenzie) | N/A |
| Mouse: IL-13−/− (BALB/c) | McKenzie et al., 1998 (Dr. McKenzie) | N/A |
| Mouse: IL-33cit/+ (BALB/c) | Wong et al., 2012 (Dr. McKenzie) | N/A |
| Mouse: IL-33−/−: IL-33cit/cit (BALB/c) | This paper | N/A |
| Mouse: IL-5Tg (BALB/c) | Lee et al., 1997 (Dr. Lee) | N/A |
| Oligonucleotides | ||
| Ccl11 forward primer: 5′-GAATCACC AACAACAGATGCAC-3′ | This paper | N/A |
| Ccl11 reverse primer: 5′-ATCCTGGAC CCACTTCTTCTT-3’ | This paper | N/A |
| Ccl24 forward primer: 5′-TCTTAGG GCCCTTCTTGGTG-3’ | This paper | N/A |
| Ccl24 reverse primer: 5′-AATTCCAG AAAACCGAGTGG-3′ | This paper | N/A |
| Il5 forward primer: 5′-CTCTGTTG ACAAGCAATGAGACG-3′ | This paper | N/A |
| Il5 reverse primer: 5′-TCTTCAGT ATGTCTAGCCCCTG-3′ | This paper | N/A |
| Il13 forward primer: 5′-CCTGGCTCTTGCTTGCCTT-3′ | This paper | N/A |
| Il13 reverse primer: 5′-GGTCTTGT GTGATGTTGCTCA-3′ | This paper | N/A |
| Gapdh forward primer: 5′-TTGATGG CAACAATCTCCAC-3′ | This paper | N/A |
| Gapdh reverse primer: 5′-CGTCCC GTAGACAAAATGGT-3′ | This paper | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo | RRID: SCR_008520 |
| PRISM | GraphPad | RRID: SCR_002798 |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Imaris | BitPlane | RRID: SCR_007370 |
| Cytobank | Cytobank | RRID: SCR_014043 |
Highlights.
ILCs are required for the development of IL-33-induced eosinophilic pericarditis
ILC2s facilitate eotaxin expression in cardiac fibroblasts
Eosinophils in the mediastinum can migrate to the heart during pericarditis
ILCs are more frequently present in the pericardial fluid of pericarditis patients
ACKNOWLEDGMENTS
The authors would like to extend their gratitude to Jobert G. Barin, Nicola L. Diny, Megan K. Wood, David Hughes, Hannah Kalinoski, and Paul Delgado for insightful discussions; Megan K. Wood and Jiyeon Lee for manuscript editing; Dr. Andrew N.J. McKenzie (Medical Research Council, Cambridge, UK) for the ST2−/−, IL-13−/− and IL-33cit/+ mice; Dr. James Lee (Mayo Clinic, Scottsdale, AZ, USA) for the IL-5Tg mice; Xiaoling Zhang (Johns Hopkins Ross Flow Cytometry Core) for cell sorting; Djahida Bedja and Nadan Wang for echocardiography; Kevin Brown (Fluidigm) for the CyTOF antibody panel design; Takuya Ohtani (Penn Institute for Immunology at the University of Pennsylvania) for operating CyTOF and acquiring samples; and the animal resources at Johns Hopkins University. This work was supported by grants from the National Institutes of Health/National Heart, Lung, and Blood Institute (R01HL118183 and R01HL136586), the American Heart Association AWRP Winter 2017 Grant-in-Aid (17GRNT33700274), the Johns Hopkins 2015 Catalyst Award, and the American Heart Association 2019 Transformational Project Award (19TPA34910007) to D.C.; the Matthew Vernon Poyner (MVP) Memorial Myocarditis Research Fund to D.C. and Nisha Gilotra; the American Heart Association Predoctoral Fellowship (16PRE31170040) to H.S.C.; the Myocarditis Foundation 2018 Rhett Lundy Memorial Research Fellowship to T.W.; the Johns Hopkins Autoimmune Disease Research Center O’Leary-Wilson Fellowship, the Johns Hopkins Bloomberg School of Public Health Richard J. and Margaret Conn Himelfarb Student Support fund, and the Katherine E. Welsh Fellowship to X.H.; the Myocarditis Foundation 2016 Research Fellowship to G.C.; and the American Heart Association Postdoctoral Fellowship (16POST31330012) and American Autoimmune-Related Diseases Association (AARDA) 2016 Young Investigator Award to W.B.-B.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.040.
REFERENCES
- AbdullGaffar B, and Almulla A (2015). Eosinophilic pericardial effusion in hypereosinophilic syndrome with restrictive cardiomyopathy. Cytopathology 26, 388–389. [DOI] [PubMed] [Google Scholar]
- Abston ED, Barin JG, Cihakova D, Bucek A, Coronado MJ, Brandt JE, Bedja D, Kim JB, Georgakopoulos D, Gabrielson KL, et al. (2012). IL-33 independently induces eosinophilic pericarditis and cardiac dilation: ST2 improves cardiac function. Circ. Heart Fail 5, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard C, Mingler MK, Vicario M, Abonia JP, Wu YY, Lu TX, Collins MH, Putnam PE, Wells SI, and Rothenberg ME (2007). IL-13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibility with glucocorticoids. J. Allergy Clin. Immunol 120, 1292–1300. [DOI] [PubMed] [Google Scholar]
- Borchers MT, Ansay T, DeSalle R, Daugherty BL, Shen H, Metzger M, Lee NA, and Lee JJ (2002). In vitro assessment of chemokine receptor-ligand interactions mediating mouse eosinophil migration. J. Leukoc. Biol 71, 1033–1041. [PubMed] [Google Scholar]
- Bracamonte-Baran W, Chen G, Hou X, Talor MV, Choi HS, Davogustto G, Taegtmeyer H, Sung J, Hackam DJ, Nauen D, and Čiháková D (2019). Non-cytotoxic Cardiac Innate Lymphoid Cells Are a Resident and Quiescent Type 2-Commited Population. Front. Immunol 10, 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butts B, Goeddel LA, George DJ, Steele C, Davies JE, Wei CC, Varagic J, George JF, Ferrario CM, Melby SJ, and Dell’Italia LJ (2017). Increased Inflammation in Pericardial Fluid Persists 48 Hours After Cardiac Surgery. Circulation 136, 2284–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrol C, and Girard JP (2014). IL-33: an alarmin cytokinewith crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol 31, 31–37. [DOI] [PubMed] [Google Scholar]
- Chen WY, Hong J, Gannon J, Kakkar R, and Lee RT (2015). Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proc. Natl. Acad. Sci. USA 112, 7249–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Bracamonte-Baran W, Diny NL, Hou X, Talor MV, Fu K, Liu Y, Davogustto G, Vasquez H, Taegtmeyer H, et al. (2018). Sca-1+ cardiac fibroblasts promote development of heart failure. Eur. J. Immunol 48, 1522–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronado MJ, Bruno KA, Blauwet LA, Tschöpe C, Cunningham MW, Pankuweit S, van Linthout S, Jeon ES, McNamara DM, Krejčí J, et al. (2019). Elevated Sera sST2 Is Associated With Heart Failure in Men ≤ 50 Years Old With Myocarditis. J. Am. Heart Assoc 8, e008968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanets S, Kaun C, Pentz R, Krychtiuk KA, Rauscher S, Pfaffenberger S, Zuckermann A, Aliabadi A, Gröger M, Maurer G, et al. (2013). Components of the interleukin-33/ST2 system are differentially expressed and regulated in human cardiac cells and in cells of the cardiac vasculature. J. Mol. Cell. Cardiol 60, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniset JF, Belke D, Lee WY, Jorch SK, Deppermann C, Hassanabad AF, Turnbull JD, Teng G, Rozich I, Hudspeth K, et al. (2019). Gata6(+) Pericardial Cavity Macrophages Relocate to the Injured Heart and Prevent Cardiac Fibrosis. Immunity 51, 131–140.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diny NL, Hou X, Barin JG, Chen G, Talor MV, Schaub J, Russell SD, Klingel K, Rose NR, and Čiháková D (2016). Macrophages and cardiac fibroblasts are the main producers of eotaxins and regulate eosinophil trafficking to the heart. Eur. J. Immunol 46, 2749–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty TA, Baum R, Newbury RO, Yang T, Dohil R, Aquino M, Doshi A, Walford HH, Kurten RC, Broide DH, and Aceves S (2015). Group 2 innate lymphocytes (ILC2) are enriched in active eosinophilic esophagitis. J. Allergy Clin. Immunol 136, 792–794.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake LY, Iijima K, Bartemes K, and Kita H (2016). Group 2 Innate Lymphoid Cells Promote an Early Antibody Response to a Respiratory Antigen in Mice. J. Immunol 197, 1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl G, Colonna M, Di Santo JP, and McKenzie AN (2015). Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science 348, aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farne HA, Wilson A, Powell C, Bax L, and Milan SJ (2017). Anti-IL5 therapies for asthma. Cochrane Database Syst. Rev 9, CD010834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friões F, Lourenço P, Laszczynska O, Almeida PB, Guimarães JT, Januzzi JL, Azevedo A, and Bettencourt P (2015). Prognostic value of sST2 added to BNP in acute heart failure with preserved or reduced ejection fraction. Clin. Res. Cardiol 104, 491–499. [DOI] [PubMed] [Google Scholar]
- Fukuo Y, Nakatani T, Shinohara H, and Matsuda T (1988). The mouse pericardium: it allows passage of particulate matter from the pleural to the pericardial cavity. Anat. Rec 222, 1–5. [DOI] [PubMed] [Google Scholar]
- Gao S, Ho D, Vatner DE, and Vatner SF (2011). Echocardiography in Mice. Curr. Protoc. Mouse Biol 1, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger G, Fan X, Dikiy S, Lee SY, and Rudensky AY (2015). Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesenauer B, and Paczesny S (2017). The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol 8, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha SA, Tsuji M, Suzuki K, Meek B, Yasuda N, Kaisho T, and Fagarasan S (2006). Regulation of B1 cell migration by signals through Toll-like receptors. J. Exp. Med 203, 2541–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim TY, Krauss RH, Sun AC, and Takei F (2012). Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 36, 451–463. [DOI] [PubMed] [Google Scholar]
- Herry I, Philippe B, Hennequin C, Danel C, Lejeunne C, and Meyer G (1997). Acute life-threatening toxocaral tamponade. Chest 112, 1692–1693. [DOI] [PubMed] [Google Scholar]
- Horckmans M, Bianchini M, Santovito D, Megens RTA, Springael JY, Negri I, Vacca M, Di Eusanio M, Moschetta A, Weber C, et al. (2018). Pericardial Adipose Tissue Regulates Granulopoiesis, Fibrosis, and Cardiac Function After Myocardial Infarction. Circulation 137, 948–960. [DOI] [PubMed] [Google Scholar]
- Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, Nakanishi K, and Yamanishi K (2013). Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc. Natl. Acad. Sci. USA 110, 13921–13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imazio M, Brucato A, Cemin R, Ferrua S, Maggiolini S, Beqaraj F, Demarie D, Forno D, Ferro S, Maestroni S, et al. ; ICAP Investigators (2013). A randomized trial of colchicine for acute pericarditis. N. Engl. J. Med 369, 1522–1528. [DOI] [PubMed] [Google Scholar]
- Imazio M, Gaita F, and LeWinter M (2015). Evaluation and Treatment of Pericarditis: A Systematic Review. JAMA 314, 1498–1506. [DOI] [PubMed] [Google Scholar]
- Judd LM, Heine RG, Menheniott TR, Buzzelli J, O’Brien-Simpson N, Pavlic D, O’Connor L, Al Gazali K, Hamilton O, Scurr M, et al. (2016). Elevated IL-33 expression is associated with pediatric eosinophilic esophagitis, and exogenous IL-33 promotes eosinophilic esophagitis development in mice. Am. J. Physiol. Gastrointest. Liver Physiol 310, G13–G25. [DOI] [PubMed] [Google Scholar]
- Kim BS, and Artis D (2015). Group 2 innate lymphoid cells in health and disease. Cold Spring Harb. Perspect. Biol 7, a016337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, Hepworth MR, Van Voorhees AS, Comeau MR, and Artis D (2013). TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci. Transl. Med 5, 170ra16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, Savage PB, Mckenzie AN, Smith DE, Rottman JB, et al. (2012). Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J. Allergy Clin. Immunol 129, 216–227.e1-e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose CS, and Artis D (2016). Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol 17, 765–774. [DOI] [PubMed] [Google Scholar]
- Küchler AM, Pollheimer J, Balogh J, Sponheim J, Manley L, Sorensen DR, De Angelis PM, Scott H, and Haraldsen G (2008). Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am. J. Pathol 173, 1229–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NA, McGarry MP, Larson KA, Horton MA, Kristensen AB, and Lee JJ (1997). Expression of IL-5 in thymocytes/T cells leads to the development of a massive eosinophilia, extramedullary eosinophilopoiesis, and unique histopathologies. J. Immunol 158, 1332–1344. [PubMed] [Google Scholar]
- Li L, Xia Y, Nguyen A, Lai YH, Feng L, Mosmann TR, and Lo D (1999). Effects of Th2 cytokines on chemokine expression in the lung: IL-13 potently induces eotaxin expression by airway epithelial cells. J. Immunol 162, 2477–2487. [PubMed] [Google Scholar]
- Li Q, Gupta D, Schroth G, Loghin C, Letsou GV, and Buja LM (2002). Images in cardiovascular medicine. Eosinophilic pericarditis and myocarditis. Circulation 105, 3066. [DOI] [PubMed] [Google Scholar]
- Lindman BR, Breyley JG, Schilling JD, Vatterott AM, Zajarias A, Maniar HS, Damiano RJ Jr., Moon MR, Lawton JS, Gage BF, et al. (2015). Prognostic utility of novel biomarkers of cardiovascular stress in patients with aortic stenosis undergoing valve replacement. Heart 101, 1382–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Matsukura S, Stellato C, Georas SN, Casolaro V, Plitt JR, Miura K, Kurosawa S, Schindler U, and Schleimer RP (2001). Interleukin-13 upregulates eotaxin expression in airway epithelial cells by a STAT6-dependent mechanism. Am. J. Respir. Cell Mol. Biol 24, 755–761. [DOI] [PubMed] [Google Scholar]
- McCarthy CP, and Januzzi JL Jr. (2018). Soluble ST2 in Heart Failure. Heart Fail. Clin 14, 41–48. [DOI] [PubMed] [Google Scholar]
- McKenzie GJ, Emson CL, Bell SE, Anderson S, Fallon P, Zurawski G, Murray R, Grencis R, and McKenzie AN (1998). Impaired development of Th2 cells in IL-13-deficient mice. Immunity 9, 423–432. [DOI] [PubMed] [Google Scholar]
- Molofsky AB, Savage AK, and Locksley RM (2015). Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 42, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PE, Church TL, Chism DD, Panettieri RA Jr., and Shore SA (2002). IL-13 and IL-4 cause eotaxin release in human airway smooth muscle cells: a role for ERK. Am. J. Physiol. Lung Cell. Mol. Physiol 282, L847–L853. [DOI] [PubMed] [Google Scholar]
- Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, and Koyasu S (2010). Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544. [DOI] [PubMed] [Google Scholar]
- Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, Fukunaga K, Asano K, Betsuyaku T, and Koyasu S (2016). Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat. Immunol 17, 76–86. [DOI] [PubMed] [Google Scholar]
- Moussion C, Ortega N, and Girard JP (2008). The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One 3, e3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, et al. (2010). Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, Thornton EE, Krummel MF, Chawla A, Liang HE, and Locksley RM (2013). Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, and Girard JP (2012). Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J. Immunol 188, 3488–3495. [DOI] [PubMed] [Google Scholar]
- Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, and Locksley RM (2010). Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 107, 11489–11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg ME (1999). Eotaxin. An essential mediator of eosinophil trafficking into mucosal tissues. Am. J. Respir. Cell Mol. Biol 21, 291–295. [DOI] [PubMed] [Google Scholar]
- Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, Huang LC, Johnson D, Scanlon ST, McKenzie AN, et al. (2013). A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med 210, 2939–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, and Lee RT (2007). IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J. Clin. Invest 117, 1538–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Más J, Lax A, Asensio-López Mdel.C., Fernandez-Del Palacio MJ, Caballero L, Santarelli G, Januzzi JL, and Pascual-Figal DA (2014). Modulation of IL-33/ST2 system in postinfarction heart failure: correlation with cardiac remodelling markers. Eur. J. Clin. Invest. 44, 643–651. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, et al. (2005). IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki K, Sanada S, Kudinova AY, Steinhauser ML, Handa V, Gannon J, and Lee RT (2009). Interleukin-33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ. Heart Fail 2, 684–691. [DOI] [PubMed] [Google Scholar]
- Shimotakahara A, Kuebler JF, Vieten G, Metzelder ML, Petersen C, and Ure BM (2007). Pleural macrophages are the dominant cell population in the thoracic cavity with an inflammatory cytokine profile similar to peritoneal macrophages. Pediatr. Surg. Int 23, 447–451. [DOI] [PubMed] [Google Scholar]
- Spiegel R, Miron D, Fink D, Gavriel H, and Horovitz Y (2004). Eosinophilic pericarditis: a rare complication of idiopathic hypereosinophilic syndrome in a child. Pediatr. Cardiol 25, 690–692. [DOI] [PubMed] [Google Scholar]
- Spits H, and Cupedo T (2012). Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu. Rev. Immunol 30, 647–675. [DOI] [PubMed] [Google Scholar]
- Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, et al. (2013). Innate lymphoid cells–a proposal for uniform nomenclature. Nat. Rev. Immunol 13, 145–149. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Imaeda H, Fujimoto T, Ban H, Bamba S, Tsujikawa T, Sasaki M, Fujiyama Y, and Andoh A (2013). Regulation of eotaxin-3/CC chemokine ligand 26 expression byThelpertype 2 cytokines in human colonic myofibroblasts. Clin. Exp. Immunol 173, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, and McKenzie AN (2000). T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J. Exp. Med 191, 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers J, Rochman M, Caldwell JM, Besse JA, Miracle CE, and Rothenberg ME (2017). IL-33 is induced in undifferentiated, non-dividing esophageal epithelial cells in eosinophilic esophagitis. Sci. Rep 7, 17563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bosch JM, Wagenaar SS, and Westermann CJ (1986). Asthma, eosinophilic pleuropneumonia, and pericarditis without vasculitis. Thorax 41, 571–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, and Locksley RM (2014). I-L-C-2 it: type 2 immunity and group 2 innate lymphoid cells in homeostasis. Curr. Opin. Immunol 31,58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JA, Barlow JL, and McKenzie AN (2013). Innate lymphoid cells–how did we miss them? Nat. Rev. Immunol 13, 75–87. [DOI] [PubMed] [Google Scholar]
- Wang J, and Kubes P (2016). A Reservoir of Mature Cavity Macrophages that Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell 765, 668–678. [DOI] [PubMed] [Google Scholar]
- Weber GF, Chousterman BG, Hilgendorf I, Robbins CS, Theurl I, Gerhardt LM, Iwamoto Y, Quach TD, Ali M, Chen JW, et al. (2014). Pleural innate response activator B cells protect against pneumonia via a GM-CSF-IgM axis. J. Exp. Med 211, 1243–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, Barlow JL, Neill DR, Panova V, Koch U, et al. (2012).Transcription factor RORa is critical for nuocyte development. Nat. Immunol 13, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L, Fulkerson PC, Finkelman FD, Mingler M, Fischetti CA, Blanchard C, and Rothenberg ME (2010). IL-13 induces esophageal remodeling and gene expression by an eosinophil-independent, IL-13R alpha 2-inhibited pathway. J. Immunol 185, 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets.