

Abstract
Insect herbivory is pervasive in plant communities, but its impact on microbial plant colonizers is not well-studied in natural systems. By calibrating sequencing-based bacterial detection to absolute bacterial load, we find that the within-host abundance of most leaf microbiome (phyllosphere) taxa colonizing a native forb is amplified within leaves impacted by insect herbivory. Herbivore-associated bacterial amplification reflects community-wide compositional shifts towards lower ecological diversity, but the extent and direction of such compositional shifts can be interpreted only by quantifying absolute abundance. Experimentally eliciting anti-herbivore defenses reshaped within-host fitness ranks among Pseudomonas spp. field isolates and amplified a subset of putatively phytopathogenic P. syringae in a manner causally consistent with observed field-scale patterns. Herbivore damage was inversely correlated with plant reproductive success and was highly clustered across plants, which predicts tight co-clustering with putative phytopathogens across hosts. Insect herbivory may thus drive the epidemiology of plant-infecting bacteria as well as the structure of a native plant microbiome by generating variation in within-host bacterial fitness at multiple phylogenetic and spatial scales. This study emphasizes that “non-focal” biotic interactions between hosts and other organisms in their ecological settings can be crucial drivers of the population and community dynamics of host-associated microbiomes.
Keywords: co-infection, Pseudomonas, Brassicaceae, phyllosphere, microbiome, indirect interactions
Introduction
For many organisms, attack by multiple enemies is inevitable and often occurs sequentially during the lifetime of individual hosts. Prior attack can alter host phenotypes and change how future attacks unfold, often generating cascading effects at larger spatial and temporal scales1–4. Given the large effects of co-infection on host health and the population dynamics of their parasites, explicitly studying co-infection is becoming increasingly common4–6. But rarely has this perspective been extended to studies of diverse host-associated microbial communities (‘microbiomes’). Instead, microbiome studies tend to focus on effects of host genotype or abiotic variation on microbiome diversity patterns7–11. This has left a major gap in our understanding of how host colonization from non-microbial enemies impacts the population biology of microbiome-associated taxa.
For plants, there is tremendous interest in understanding the structure and function of the microbiome both for applied purposes, such as engineering growth promotion and disease resistance12,13, and as model systems for host-microbial symbioses more generally. Insect herbivory represents a pervasive threat to plants in wild and agricultural settings alike14. Herbivory alters plant phenotypes through tissue damage and induction of plant defenses, which can change susceptibility of plants to attack by insects15 as well as microbes16,17. Thus, factors that influence the impact of herbivores on hosts will likely affect the colonization and growth of plant-associated microbes. While insect herbivores14 and plant-associated microbes18 have clear effects on plant phenotypes and fitness, they are generally considered independently. Our study addresses this gap by explicitly considering how patterns of abundance and diversity of leaf-colonizing (endophytic) bacterial taxa are altered in the presence of insect herbivory and by exploring the associations among herbivory, bacterial infection, and plant fitness in a native forb (Cardamine cordifolia, Brassicaceae; ‘bittercress’).
We first used marker gene sequencing (16S rRNA) coupled with paired leaf culturing to establish and validate sequence-based estimates of absolute bacterial load in host tissue. By elucidating a relationship between bacterial load and the sequence counts of bacteria- versus host-derived 16S (Box 1), our approach enabled standard 16S marker gene sequencing to quantitatively reveal variation in abundance distributions of entire suites of bacterial taxa across hosts with and without prior herbivory by the specialist leaf-mining insect Scaptomyza nigrita (Drosophilidae). We then assessed the extent of co-clustering between microbial abundance and intensity of insect herbivory at the plant patch scale across our natural study populations, and we related microbe-herbivore co-aggregation to fruit set, a component of plant fitness. In parallel, we directly examined variation in sensitivity to inducible plant defenses against chewing herbivores among 12 genetically diverse, bittercress-derived isolates of Pseudomonas spp. bacteria. We did so by experimentally infecting accessions of native bittercress in which prior herbivory was simulated by exogenously pre-treating plants with the plant defense hormone jasmonic acid (JA), which induces canonical defenses against chewing herbivores in plants17, including bittercress19.
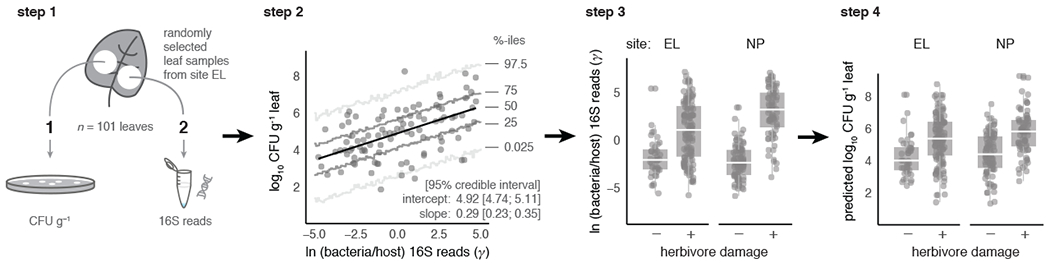
Box 1: Devising an estimator of bacterial load from 16S data.
Defining the estimator
To establish an estimator of bacterial load using 16S sequence data, we hypothesized that the composition of the sequencing data, in terms of host-versus bacteria-derived 16S reads, may provide information about the underlying density of bacteria. This occurs, we reasoned, because DNA templates of the two sources compete as targets during the amplification reaction, and biases towards one or the other will accrue exponentially. By this logic, the logarithm of the relative abundance of bacteria-to-host 16S counts captures information about the density of bacterial cells in the starting material. Accordingly, for each sample, we calculated the following estimator
where rB and rH are the read counts of bacteria- and curated host-derived 16S counts for a given sample, respectively. rB can be calculated at any taxonomic level, ranging from the single bASV to all of the bacteria present in the sample, by summing the sequence counts at the desired taxonomic scale.
Validating and deploying the estimator
Step 1: Collect paired tissue samples and enumerate bacteria independently.
We validated this estimator empirically by examining the relationship between γ and an independent measure of bacterial abundance in leaf tissues derived from bacterial culturing of a subset (n = 101) of the samples from the EL study. These samples were surface-sterilized, homogenized, and plated on non-selective King’s B media to enumerate bacterial colony forming units (CFU) per g starting leaf material, following Humphrey et al. 20. This approach is appropriate because a majority of bacterial taxa typically found to colonize leaf tissues can be cultivated in the laboratory on rich media20,43.
Step 2: Quantify relationship between 16S data and directly observed bacterial load.
We then estimated the slope and intercept of the relationship between observed log10 CFU g−1 leaf tissue (hereafter log CFU) and the predictor variable γ for this sample set using a Bayesian linear regression, which allowed us to directly incorporate uncertainty in model fit into downstream analyses. We found a clear positive association between γ and log CFU (see figure), validating our usage of γ as an estimator of absolute bacterial abundance in leaf tissues.
Step 3: Model relationship between observed γ and herbivore damage.
We then deployed the validated estimator to test whether bacterial abundance as measured by γ was elevated in insect-damaged plant tissues. To begin, we modeled how γ varied across herbivore-damaged and undamaged leaves for various bacterial taxa. The illustrating example below shows that the distributions of γ calculated for all bacteria are elevated in herbivore-damaged bittercress tissues sampled from both sites EL and NP.
Step 4: Transform results for γ into predicted bacterial load via parameters from Step 2.
Finally, we used posterior draws of parameters from the Step 2 model to predict how variation in γ translates into predicted bacterial load as expressed in log CFU. Here see that elevated γ in herbivore-damaged tissues translates into higher bacterial loads when predicted based on the relationship between γ and log CFU derived in Step 2. Further details on how we specified and estimated models, as well as how we incorporated parameter uncertainty throughout this approach, can be found in Methods: Quantifying and modeling bacterial abundance patterns.
Our experiments reveal that insect herbivory, via induction of plant defenses, can modify endophytic bacterial diversity patterns by amplifying naturally prevalent and potentially phytopathogenic bacterial taxa within a native plant host. This mechanism may be at least partly responsible for the strong positive association between herbivory and endophytic bacterial abundance within leaf microbiomes seen under field conditions. Crucially, the patterns and degree of bacterial abundance variation we found cannot be revealed by traditional compositional analysis of high-throughput marker gene sequencing, which masks the extent and direction of within-host variation in bacterial load. By linking marker gene counts to an absolute standard, our study reveals how insect herbivory associates with variation in bacterial loads at leaf and patch scales within a natural plant population. More generally, this work highlights the importance of (a) accounting for prevalent but ‘non-focal’ biotic interactions hosts have with other colonizers in their natural contexts, and (b) using detection and analytical approaches to quantify these effects on components of microbial fitness.
Results
Bacterial loads are amplified in insect-damaged leaf tissues
We devised an estimator of bacterial absolute abundance (γ) from 16S sequence data by statistically calibrating information on 16S counts from host versus bacteria to a culture-based standard of bacterial load (Box 1). Using this approach, we found that bacteria within herbivore-damaged leaves at our field sites (see Methods) exhibited local population sizes several doublings greater compared to the bacteria found in undamaged leaves (Fig. 1a,b; median ± 95% credible interval of posterior predicted additional doublings: 2.5 [2.1; 3.9] site Emerald Lake [EL]; 4.5 [3.6; 5.3] site North Pole Basin [NP]). This result, rooted in sequence data, is further validated by the parallel observation that damaged leaves showed higher bacterial loads than undamaged leaves via culturing of the n = 101 calibration set (Supplementary Figure 5; mean difference of 3.7 bacterial doublings [1.8–5.6, 95% c.i. on mean difference]; Welch’s unequal variance two-sample t-test, t = 3.86, p < 0.001), which is quantitatively consistent with a prior result from a parallel and independent culture-based study in this system20.
Figure 1: Pervasive increases in endophytic bacterial load in herbivore-damaged leaves.
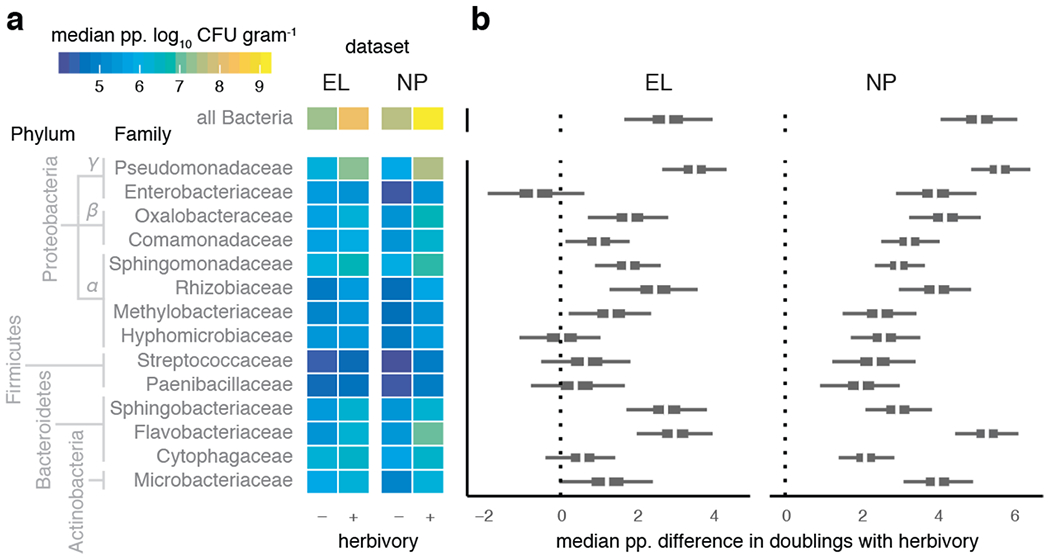
a–b. Posterior predicted (‘pp.’) infection intensity of bacterial amplicon sequence variants (bASV) from the 14 most prevalence bacterial families show variation in the extent of elevated growth in herbivore-damaged leaf tissue. a. Heatmap shows median predicted log10 bacterial abundance (colony-forming units, CFU) per g starting leaf material) from 200 posterior simulations of the best-fitting model of each bacterial family separately (see Methods). b. Boxplots showing median (white), 95% (thin), and 50% (thick) quantiles of the posterior predicted median difference in the number of bacterial cell divisions (i.e., doublings) achieved in herbivore-damaged leaves compared to undamaged leaves, for sites EL and NP separately.
Herbivore-associated bacterial amplification is both community-wide and taxon-specific
We then capitalized on the high taxonomic resolution and sampling depth afforded by amplicon sequencing to examine shifts in the abundance and distribution of diverse bacteria within the bittercress leaf microbiome. We constructed exact bacterial amplicon sequence variants (bASVs) from our 16S data and estimated how bASVs from each bacterial family varied across damaged and un-damaged leaves (see Methods). The within-host density of bacterial bASVs from several bacterial families was elevated in herbivore-damaged leaves compared to undamaged leaves (Fig. 1). For most bacterial families, the relative increase in within-host density with herbivory was greater at site NP than at site EL (Fig. 1b). This was largely because several taxa showed lower baseline loads in undamaged leaves at site NP (Extended Data 1). In contrast, bacterial loads for all families were similar for damaged leaves at both site (Extended Data 1). Pseudomonadaceae was the most abundant taxon across all leaves and also showed the greatest fold increase under herbivory (Fig. 1).
Several statistical models of abundance (γ; see Methods) at the level of bacterial family showed support for bASV-level differences in intercept and slope values (Table S2), including Pseudomonadaceae and Sphingomonadaceae. Two individual bASVs in particular drove family-level patterns in these clades (Supplementary Figure 6), which together comprised ~ 20% of all sequencing reads across the two sample sets. Within the Pseudomonadaceae, Pseudomonas3 was the most abundant bASV, which falls within the putatively phytopathogenic P. syringae clade (Supplementary Figure 7). We previously showed that P. syringae strains can be pathogenic, induce chlorosis, and reduce leaf photosynthetic function in bittercress20. Thus, a major component of the signal of elevated bacterial load in the presence of insect herbivory comes from putatively phyopathogenic genotypes within the group P. syringae.
Compositional shifts in leaf bacterial communities under herbivory
When the absolute bacterial abundance patterns described above were analyzed in a compositional framework, we detected differences in overall community structure and ecological diversity between damaged and undamaged leaves. Specifically, we found lower evenness (J′; Fig. 2b) in damaged leaves, indicating a stronger skew towards a smaller number of bacterial taxa: Pseudomonadaceae comprise an even greater proportion of the population in damaged leaves owing to their already-high average abundance in undamaged leaves and large fold-increase under herbivory. Family-level relative abundances differed in terms of Shannon-Jensen divergence (i.e., β-diversity) between damaged versus un-damaged leaves (Fig. 2c). These observations indicate that amplification of bacteria in herbivore-damaged leaves can produce community-wide signatures of reduced within-host diversity and elevated between-host diversity at broad taxonomic scales.
Figure 2: Herbivore-damaged leaves harbor compositionally diverged microbiomes with reduced ecological diversity shifted heavily towards Pseudomonadaceae.
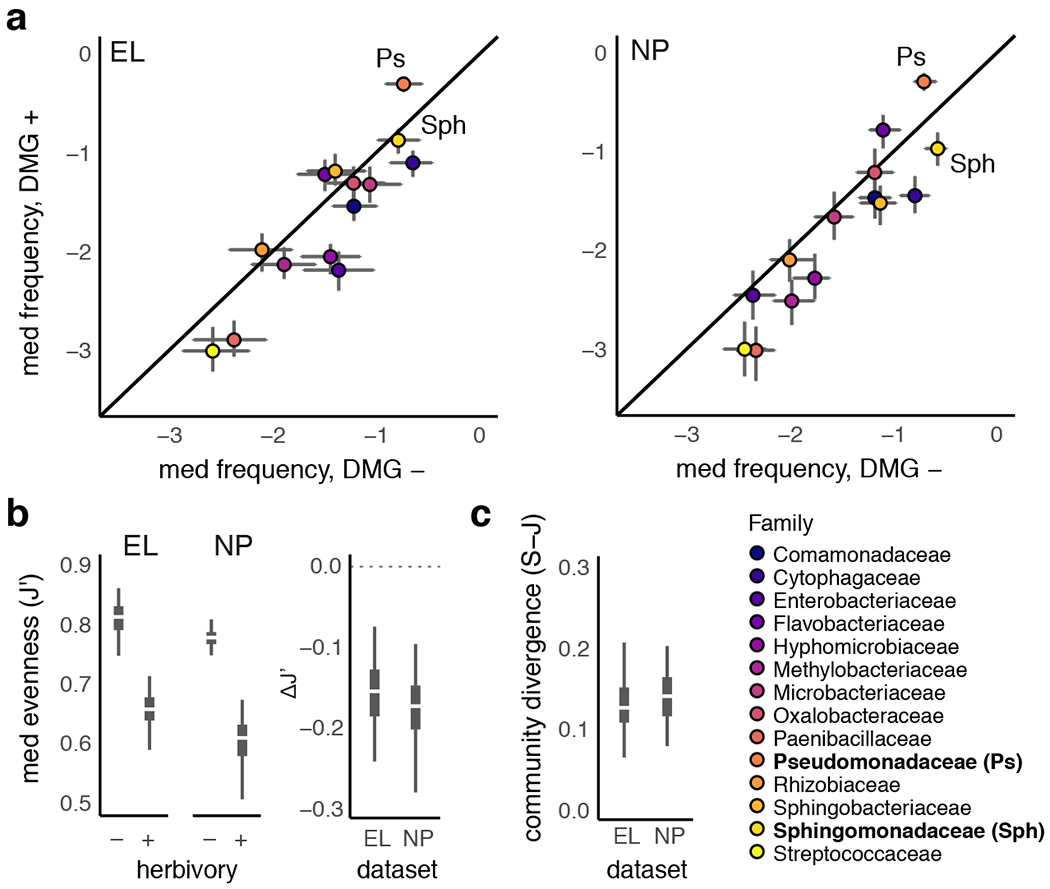
a. log10 relative abundance of each family in undamaged (x-axis) versus damaged (y-axis) samples shows skew towards Pseudomonadaceae (Ps) and relative reductions of abundance among most other taxa at both study sites, including Sphingomonas (Sph), which shows a ~ 2-fold increase in number of doublings in herbivore damaged leaves. Error bars represent 95% predicted median frequency intervals calculated from posterior simulations of bacterial abundance across damaged and undamaged leaf classes. b. Compositional changes from the amplification of already abundant taxa (e.g., Pseudomonadaceae) produces reduced community-level evenness (J′) and leads to compositional divergence (i.e., β-diversity) between damaged and undamaged leaves at both study sites (c). Boxplots represent the median (white), 95% (thin), and 50% (thick) quantiles of each statistic calculated from the posterior predicted median relative frequency data depicted in a.
Plant defenses against chewing herbivores enhance growth of putative phytopathogens in planta
We then directly addressed how inducible plant defenses against chewing herbivores impacted the within-host fitness of a suite of bittercress-derived endophytic bacteria from the family Pseudomonadaceae. Plant pre-treatment with the plant defense hormone JA caused statistically clear alterations in within-host growth of five of the twelve Pseuomonas spp. strains tested (Fig. 3a), with the most pronounced changes resulting in 2.5–5 additional doublings of two phylogenetically distinct P. syringae isolates (20A and 02A; Table S3). Increased within-host density of these two strains can account for differences in total Pseudomonas abundance, as well as differences in abundance patterns summed at the level of bacterial clade (P. syringae versus P. fluorescens; Fig. 3b). By recapitulating the elevated P. syringae and family-wide increased abundance under herbivory seen in our field studies, this greenhouse result highlights that induction of plant defenses against chewing herbivores is one potential mechanism whereby insect herbivory could lead to amplification of bacterial taxa within the bittercress leaf microbiome.
Figure 3: Eliciting plant defenses against chewing herbivores alters within-host performance of putative phytopathogens.
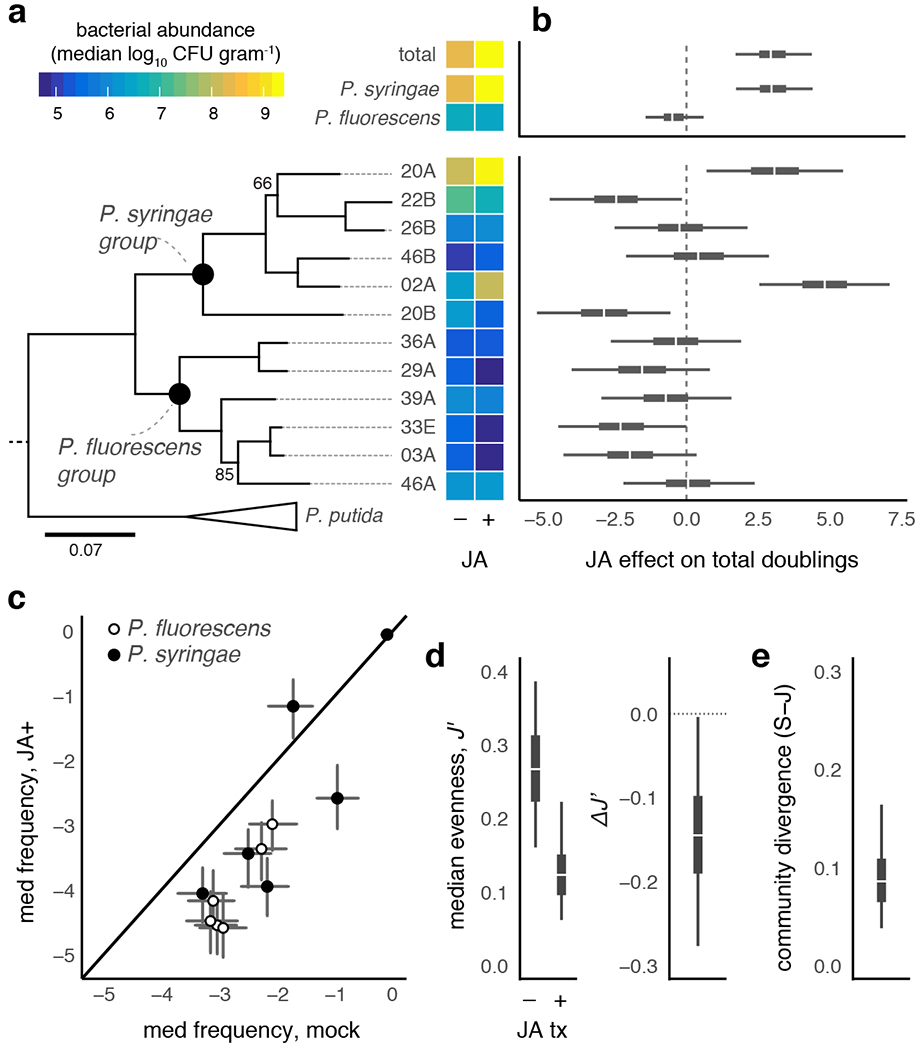
a. Experimental infections with 12 Pseudomonas spp. strains, concurrently isolated from EL study site20, reveal strain-to-strain variation in growth under mock-treated (M) and jasmonic acid (JA) induced plants. Heatmap shows median log10CFU g−1 surface sterilized plant tissue 2 d post inoculation. Maximum likelihood phylogeny of strains estimated with four housekeeping loci (2951 bp)20. b. The median (white), 95% (thin), and 50% (thick) quantiles of the posterior difference between the number of bacterial doublings attained by bacteria growing in JA-versus mock-treated leaves (see Supplemental Methods). c. Compositional analysis of relative abundances (± 95% posterior interval) calculated from (a) reflect decreased evenness (J′; d) in JA-treated plant tissues, leading to overall community-level divergence (e). Shown in d and e are boxplots of the median (white), 95% (thin), and 50% (thick) quantiles from 200 posterior simulations of abundance from the model results depicted in (b).
Notably, two strains (22B and 20B) exhibited markedly decreased within-host fitness in JA-treated compared to mock-treated leaves (Fig. 3a–b). Such herbivore-driven fitness variation among P. syringae is undetectable when only considering larger taxonomic scales of genus or family (Fig. 3), where genotypes which increase in local abundance contribute to an overall signature of elevated taxon-wide abundance as measured by lower resolution tools (e.g., 16S sequencing). Thus, induction of plant defenses against chewing herbivores leads to the amplification and numerical dominance of a narrow subset of the P. syringae community within this host population (Fig. 3c). Such changes result in compositional shifts towards decreased Shannon evenness in JA-treated leaves (Fig. 3d) and an overall community-wide divergence with mock-treated leaves (Fig. 3e).
Putative phytopathogens are aggregated in highly herbivore-damaged plant patches
We then analyzed how Pseudomonas3, a highly abundant individual bASV within the P. syringae group, varied across bittercress plant patches in relation to the level of herbivory on those plant patches. At site NP, we found a highly aggregated (i.e., right-skewed) distribution of herbivore loads across plant patches (Fig. 4a, top marginal density plot). This aggregated herbivore distribution across plant patches results in a predicted 50-fold enrichment of local density of Pseudomonas3 in the most-damaged compared to the least-damaged plant patch (Fig. 4). Analyzed in a more general framework, over half the predicted Pseudomonas3 population is harbored in just one fifth of the plant patches in the bittercress population at our study site NP (Extended Data 2).
Figure 4: Co-infection by herbivores and phytopathogenic bacteria is aggregated across plant populations and is associated with lower plant reproduction.
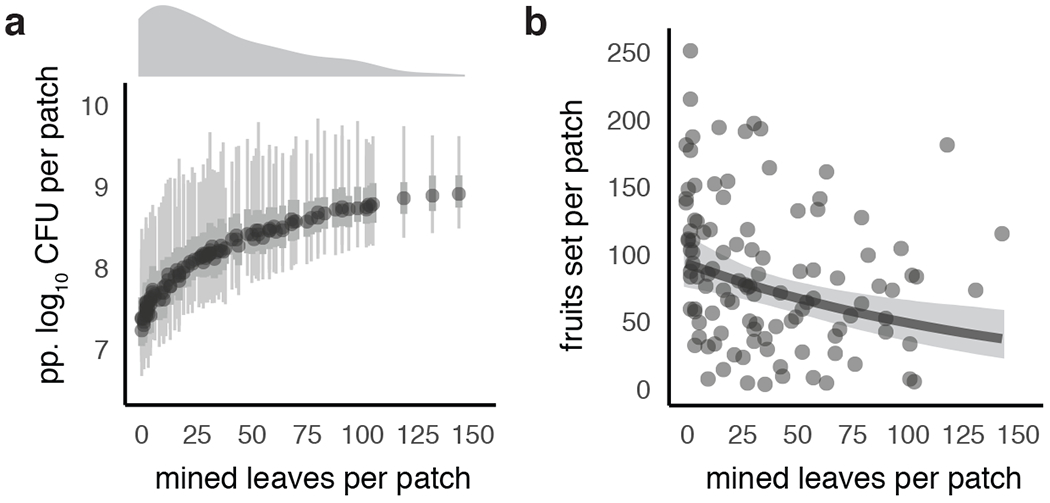
a. Median, 95%, and 50% quantiles of 200 posterior simulations of predicted (‘pp.’) bacterial load across plant patches (n = 110 at site NP; n = 16 stems sampled per patch). Density plot above x-axes exhibits right-skewed (i.e., aggregated) distribution of herbivore damage at the plant patch level. b. Patch-level herbivory (and thus co-infection intensity) is associated with decreased fruit-set in this native plant population. Plotted are raw fruit-set data summed at the patch level (n = 16 stems per patch), with marginal effects slope (and its 95% credible interval) plotted after accounting for average plant height (see Table S4).
Herbivore–bacteria co-aggregation is associated with lower plant fitness
At site NP, bittercress patches with higher herbivore intensity showed lower reproductive success, with the most damaged patches estimated to produce half as many fruits as patches with no herbivore damage (Fig. 4b, Table S4). Plants with more insect damage tend to have higher levels of bacterial infection. Thus, standing variation in fruit set is closely associated with levels of co-aggregation of these plant natural enemies across our sample within this native bittercress population.
Causes of herbivore aggregation in natural plant populations
Although not the primary focus of our study, our field experiments were also designed to test how mid-season pre-treatment with the exogenous plant defense hormones JA or salicylic acid (SA) impacted plant attack rates by S. nigrita (see Methods). JA-induced bittercress can locally deter adult S. nigrita and reduce larval feeding rates19. SA treatment canonically induces defenses against biotrophic microbial colonizers and often pleiotropically suppresses plant defenses against chewing herbivores17, including S. nigrita 20. Thus, treatment with either plant defense hormone has the potential to modify the foraging behavior of S. nigrita.
By the end of the growing season, we observed that the degree of herbivore aggregation among host plants at site NP varied extensively across plant patches at site NP (Extended Data 3a,b) and reflected little statistical signature of causation by the early-season hormone treatments with either JA or SA. Estimates for both SA and JA treatment coefficients were elevated above the mock/control condition, but the posterior distribution for both hormone effects overlapped zero (lower 4th %-ile < 0 for JA; lower 15th %-ile < 0 for SA; Table S5; Extended Data 3c). Thus, while prior plant exposure to JA, and possible also SA, may cause elevated S. nigrita herbivory at the patch scale, standing variation in S. nigrita herbivory arising stochastically or from unmeasured factors at site NP dominates over any causal effects of our population-level defense hormone manipulation (Extended Data 3). Additionally, these early-season plant defense hormone treatments showed no discernible effects on distributions of γ for overall or family-wise bacterial abundance (Extended Data 4).
Discussion
Overview
Here we show that insect herbivory is strongly associated with bacterial abundance and diversity within a native plant microbiome using field and greenhouse experiments. We provide evidence that activation of plant defenses against chewing herbivores is at least one causal mechanism whereby such within-host amplification of leaf-colonizing bacteria can occur. Specifically, the growth of a majority of bacterial taxa found in the leaf microbiome of native bittercress was amplified in plant tissues damaged by the specialist herbivore S. nigrita (Fig. 1) at two separate sub-alpine field sites. These ecological effects were only detectable by linking sequence-based bacterial quantification to an external standard of absolute abundance (Box 1), rather than relying on compositional analysis as is commonly done with studies of both plant and animal microbiomes (but see Vandeputte et al. 21).
The bacterial clades most altered under herbivory include strains from groups well-known for causing plant disease (P. syringae), and our follow-up experimental work in the greenhouse showed that inducing plant defenses against chewing herbivores in bittercress was sufficient to cause similar degrees of amplification of putatively phytopathogenic P. syringae genotypes in leaf tissues. Amplification of specific P. syringae genotypes can largely account for species- and family-level patterns seen in our field studies, which has coarser taxonomic resolution. Overall, these experiments suggest that S. nigrita herbivores may play a larger role than previously appreciated in promoting the within-host growth of particular bacterial genotypes or pathovars in bittercress, although the causal nature of the role of herbivores was not gleaned from the field portion of this study. Given that the majority of plants face herbivore attack to some degree14,22, it is possible that our results generalize across plant-microbe systems.
Herbivore-inducible plant defenses can amplify putative phytopathogens
The mechanisms governing the growth-promoting effects of insect herbivory on leaf-colonizing bacteria are potentially numerous. Leaf damage itself can release nutrients, alleviating resource constraints for bacteria while also creating routes for colonization of the leaf interior from the leaf surface23,24. Plant defenses induced by chewing herbivores could directly or indirectly alter interactions with bacteria independent of the physical effects of plant tissue damage. It is known that JA-dependent anti-herbivore defenses can suppress the subsequent activity of signaling pathways responsive to bacterial infection17, allowing bacteria (including strains of P. syringae) to reach higher densities within JA-affected leaf tissues25. S. nigrita can trigger JA-dependent host defenses in bittercress19, and this form of defense signaling is widely conserved among diverse plant groups17. Released from top-down control, a diversity of resident microbes may then proliferate as defenses are more strongly directed against herbivory, which may manifest in community-wide patterns of abundance changes as noted in our study (Fig. 1).
The hypothesis that anti-herbivore defenses pleiotropically increase bacterial growth is consistent with results from our greenhouse experiment. We found that the within-host fitness of several strains of putatively phytopathogenic P. syringae increased within bittercress leaves pre-treated with JA compared to mock-treated leaves (Fig. 2). Although our experiment is consistent with this plausible mechanism by which herbivores can facilitate bacterial growth within plants, it does not identify the proximal mechanism(s) responsible for these effects. JA induction may have instead stressed the plants, or triggered expression of traits unrelated to defense per se, reducing basal tolerance to infection by a subset of the bacterial commuity. Such net effects of JA induction on bacterial abundance are also likely influenced by underlying constitutive levels of genetic resistance and/or tolerance to herbivory, traits which often exhibit quantitative variation within and among plant populations26. The role of host genetic variation in mediating the impacts of herbivore attack on microbial plant colonization is an open avenue of future research.
Finally, several other abundant bacterial groups (e.g., Sphingomonadaceae, Flavobacteriaceae) exhibited amplified abundance under insect herbivory in our field studies (Fig. 1). Functional studies examining finer-scale variation among genotypes of these relatively less well-studied bacterial groups would be highly fruitful for establishing a more general understanding of the mechanistic basis of plant-microbe interactions in the context of inducible defenses against chewing herbivores.
Herbivore distributions can alter the spatial patterning of plant disease
The impact of insect herbivory on phyllosphere bacteria can be observed at several spatial scales. Herbivore damage was highly clustered on a subset of hosts (Fig. 4), which is a pattern consistent with other plant-herbivore systems27 as well as many host–macroparasite systems more generally28. An accompanying effect of aggregated herbivore damage is the enrichment of bacterial infection on a subset of the host population (Fig. 4; Extended Data 2), which alters the spatial structure of growth and potentially also the transmission of plant-colonizing microbes. Uncovering the temporal dynamics of how herbivore aggregation precedes or follows microbial attack—or whether the two colonizers cyclically amplify one another—will require more controlled studies that manipulate the timing and density of herbivory itself.
Regardless of the precise mechanisms resulting in such herbivore-microbe co-aggregation, plant patches with the highest levels of co-aggregation had substantially (~ 50%) lower reproductive success compared to minimally-damaged plant patches (Fig. 4b). Although we cannot identify the cause of lower plant fitness from our study, the co-aggregation of these distinct plant colonizers may at least partly explain it. Whether these plants were more stressed to begin with or achieved lower fruiting success because of their infestation with herbivores and phytopathogens cannot be resolved without future studies which isolate the causal effects of single and multiple infection on plant fitness.
What drives the highly skewed pattern of herbivory among host plants? Plant populations often display a patchwork of defensive phenotypes, influenced by plant abiotic stress, variation in the underlying defensive alleles, or by defense induction from prior herbivore or microbial attack. While plant defenses can shape herbivore attack rates in the laboratory and over wide spatial extents26, less is known about how patterns of defense induction impact the population dynamics of insect herbivores27,29. Many insects are deterred by anti-herbivore defenses, but some specialist herbivores use these same cues as attractants owing to detoxification mechanisms which confer resistance to such defenses30. S. nigrita uses anti-herbivore plant defenses to locate host but also avoids high concentrations of defensive compounds when given the option19. Thus, the joint expression of positive chemo-taxis towards JA-inducible compounds, coupled to aversion of high levels of JA responsive defensive chemistry, may influence where herbivory becomes concentrated among plants in native bittercress populations.
Results from our field hormone treatments using JA and SA both showed elevated patch-level S. nigrita leaf miner damage compared to mock-treated patches (Extended Data 3a,b). However, the statistical signature of these treatment effects was not clear enough to confidently conclude that our field trials substantially altered natural patterns of herbivory by this specialist, given the high degree of stochastic or unexplained variation in herbivore damage we observed across plant patches (Extended Data 3c; Table S5). Discovering the biotic and abiotic factors structuring herbivory patterns in natural host populations thus remains a challenge in this system31 as well as many others29, and we have not attempted to solve this problem in the present study. Nonetheless, our study suggests that predicting herbivore distributions may be key for understanding population-level distributions of plant-associated bacteria. Regardless of its causes, insect herbivore damage can be readily measured and incorporated into plant microbiome studies in order to help reveal the drivers of variation in microbiome abundance and diversity within plant communities.
Quantifying bacterial loads is crucial for understanding the ecology of the microbiome
The patterns of abundance variation among bacterial taxa across leaf types, when distilled into a community-level compositional metric, showed decreased ecological diversity (i.e., evenness) in damaged versus un-damaged leaves, resulting in overall compositional divergence between sample sets (Fig. 2–3). This results from particular taxa undergoing larger absolute changes in abundance than other taxa, which leads to stronger skews in the composition of the community calculated on the relative scale. Compositional analysis on its own would preclude inference of the direction or magnitude of changes in bacterial abundance 32 , even though this is of primary interest to researchers exploring the microbiome and its impact on host fitness21,33,34. Compositional methods are thus poorly suited to studies of the population biology of microbiome-dwelling bacterial taxa when bacterial load varies or when microbial fitness is a desired response variable.
Direct bacterial quantification21, as well as controlled DNA spike-ins35, can correct for biases and ambiguities inherent to compositional analyses. Our study provides an additional framework for enabling standard high-throughput 16S sequencing approaches to provide quantitative measures of bacterial abundance when canonical approaches (e.g., qPCR) are infeasible due to host organelle contamination. Rather than being discarded, 16S read counts derived from host organelles—once curated—can provide an internal reference population against which the proportionality of other taxa can be measured36. While we have established the usefulness of our estimator of bacterial load (γ) using a paired culture-based experiment, this need not be the only way. Bacterial culturing is an intrinsically noisy means of enumerating bacteria, due to dilution and/or counting noise. In addition, chemically-mediated antagonism or facilitation among bacterial species or strains can cause over- or under-detection of particular combinations of taxa on agar plates37. These limitations will no doubt set a lower limit to the resolution of biological effects one is capable of detecting with culture-dependent methods. Testing the generality of our approach across other plant–microbe systems, and with other means of enumerating bacteria in samples, is therefore a priority.
Conclusions
Our study emphasizes that large effects on the population biology of P. syringae, and many other lineages of leaf-colonizing bacteria, may stem from the action of insect herbivores. Biotic interactions such as herbivory are absent from the classic ‘disease triangle’ of plant pathology. The role of insect herbivores in P. syringae epidemiology—and plant-microbiome relations in general—has been under-appreciated. Variation in bacterial abundance across samples, and the implications of relative abundance changes for bacterial fitness, are not easily detectable via compositional analyses applied to 16S data, which typically do not utilize external or internal standards. Thus, studies aiming to decipher why plant microbiomes differ in structure or function should endeavor to quantify bacterial loads in order to retain this important axis of variation as a focal response variable, while also considering additional biotic interactions commonly encountered by the hosts under study.
Methods
Field studies of herbivore–bacteria co-infection
We surveyed herbivore damage arising from S. nigrita (Supplementary Figure 1f–g) in replicate 0.5 m2 plots of native bittercress along transects in sub-alpine and alpine streams near the Rocky Mountain Biological Laboratory (RMBL) in each of two years (2012, Emerald Lake [EL], n = 24 plots; 2013, site North Pole Basin [NP], n = 60 plots; Supplementary Figure 1a–d). Our analysis of the impact of hormone treatments on S. nigrita foraging patterns was previously published for site EL20, and we implemented a similar approach for site NP in this study. Full experimental design details are given in the Supplemental Methods and are depicted in the schematic in Supplementary Figure 1e.
By the end of the growing season, when herbivory and bacterial infection had run their course, we determined S. nigrita leaf-miner damage status of all leaves (both sites) as well as fruit set (site NP only) produced on each of the focal bittercress stems (stem-level sample size n = 768, site EL; n = 1920, site NP). At both sites, we collected leaf tissue in a randomized manner (see Supplemental Methods) to quantify the abundance and diversity of bacteria that had colonized the leaf interior.
Amplicon sequencing of bacteria in leaf tissues
We quantified bacterial abundance in leaf tissues using next-generation amplicon sequencing of the bacterial 16S rRNA locus using the Illumina MiSeq platform. In order to enrich our samples for endophytic bacteria, we surface-sterilized all samples prior to DNA extraction, which achieved a reliable reduction of bacterial abundance as detected by our 16S analysis approach (Supplementary Figure 2). Subsequently, we extracted DNA from the 192 leaf discs (~ 0.8 cm2) from site EL and 192 tissue pools from site NP (4 four discs per pool) and amplified bacterial 16S following published protocols38 (see Supplemental Methods). We amended this protocol by including peptic nucleic acid (PNA) PCR clamps into reaction mixtures to reduce amplification of host chloroplast- and mitochondria-derived 16S, following Lundberg et al.39. This was highly effective at reducing the proportion of host-derived 16S reads per library in our sample sets (Supplementary Figure 3).
We then used DADA240 to error-correct, trim, quality-filter, and merge the paired-end sequencing reads that passed error thresholds off the sequencer. Of the approximately 4 million raw reads, ~ 90% were retained following quality control via DADA2 (Supplementary Figure 4), and these reads were then delineated into exact amplicon sequencing variants (ASVs). 16S reads from bittercress chloroplast or mitochondria were manually curated and summed into ‘host-derived’ for comparison with bacteria-derived 16S (see Supplemental Methods).
Quantifying and modeling bacterial abundance patterns
In order to quantitatively asses how herbivore damage relates to abundance and diversity of microbial plant colonizers, we required a link between 16S counts and bacterial load. We therefore devised and validated an estimator (γ) of the abundance of bacterial ASVs within host tissues (Box 1). Using γ as an empirically validated estimator of absolute bacterial load in leaf tissues, we then constructed a two-stage modeling approach to estimate bacterial load across our complete sample set.
We first fit and compared a series of increasingly flexible Bayesian regression models to estimate how γ varies as a function of herbivore damage in leaves (see Supplemental Methods). When calculating γ, we took rB at the bASV-level for all bASVs within each of the 14 most abundant bacterial families, together comprising > 95% of total bASV counts in the datasets. We then took the candidate best stage-1 model, heuristically defined as the model with the lowest leave-one-out Bayesian information criterion (LOO-IC)41, and used it to generate n = 200 replicate sets of simulated response values (γ) predicted by the model parameters fit to the original data.
In the next stage, we used this distribution of γ as an input predictor variable to the model we fit between our observed γ and observed log CFU (Box 1). This allowed us to report bacterial abundance estimates, based initially on 16S count data, on the scale of predicted log CFU per unit leaf mass—a more directly interpretable measure of within-host fitness. Rather than point estimates, we sampled intercept, slope, and residual error parameters from their joint posterior distribution of the calibration CFU model for each data point independently. Specifically, this has the effect of incorporating uncertainty in the fit between observed log CFU and γ such that downstream predictions are not overly biased by the precise value of any regression slope estimate, which may itself arise from peculiarities in the action of PNA during amplification of 16S. Overall, this two-stage modeling approach was designed to incorporate uncertainty in the model fit for γ as well as in the relationship between observed γ and observed log CFU. The endpoint of this approach is 200 sets of posterior predicted log CFU values for each sample in the dataset, which formed the basis of downstream calculations of bacterial abundance variation, as well as ecological diversity (Shanon evenness J′) and similarity (Shannon–Jensen divergence SJ) in and between damaged and undamaged leaf sets, respectively (see Supplemental Methods).
Population-level analysis of herbivore–bacteria co-aggregation
We assessed how patch-level variation in herbivory correlated with bacterial infection intensity at the field-scale by focusing on the most highly abundant bASV at both field sites (‘Pseudomonas3’). Leaf-level abundance of this individual bASV was predicted using the approach described above. We then summed the predicted abundance (on the linear scale) of bacteria across leaves within each plant patch and used these predicted bacterial sums to calculate the cumulative proportion of the total Pseudomonas3 population harbored by plant patches with differing levels of herbivory, which portrays the extent of co-aggregation of herbivores and bacteria across the host population.
Experimental infections in planta
We directly examined how inducible plant defenses against chewing herbivores impacted within-host bacterial performance using field-derived accessions of bittercress plants and their bacterial colonizers. We randomized the selection of six focal strains from within each of the two dominant Pseudomonas clades (P. syringae, and P. fluorescens) represented in our endophytic strain collection from bittercress20. We infected each strain into a single leaf on each of n = 32 distinct bittercress clones that had been randomized to receive pre-treated 3 days prior with JA (1 mM; Sigma) or a mock solution. Bittercress clones were originally isolated as rhizomes from various sites within 2 km of the RMBL in 201242 and were re-grown in the greenhouse at University of Arizona for up to 12 months prior to use. Two days post infection, we sampled, sterilized, homogenized, and dilution-plated leaf discs onto non-selective rich King's B media, following Humphrey et al. 20. We compared bacterial abundance (log2) between treated and untreated samples using Gaussian Bayesian regression models. We subsequently used posterior predicted abundances as the basis for considering how herbivore-inducible defenses impact the composition and diversity of this Pseudomonas community at different taxonomic levels (see Supplemental Methods).
Extended Data
Extended Data Fig. 1. Correspondence between predicted bacterial abundance and herbivory effects between sites EL and NP.
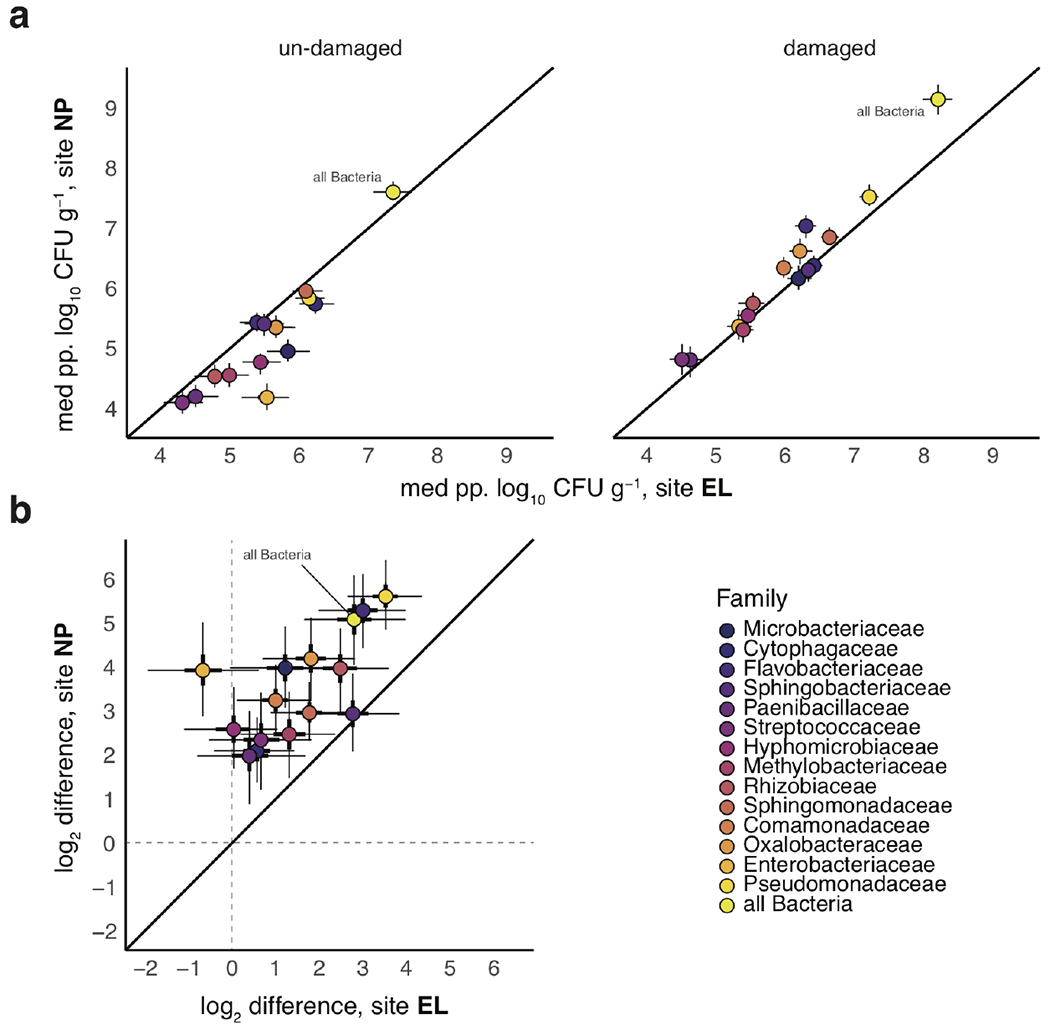
a. Plotted are median (circles) ± 95% posterior distributions of predicted abundance for bacterial bASVs summed at the family level for sites EL (x-axis) and NP (y-axis). b. Comparison between the magnitudes of log2-fold differences between damaged and undamaged leaves at sites EL (x-axis) and NP (y-axis). Middle 50%-ile and 95%-iles of median effects (circles) are depicted by thick and thin bars, respectively. On both plots, we also show data summed across all taxa in the dataset (‘all Bacteria’).
Extended Data Fig. 2. Plant patches with high herbivory harbor a disproportionate fraction of the estimated population of the most abundant P. syringae bASV.
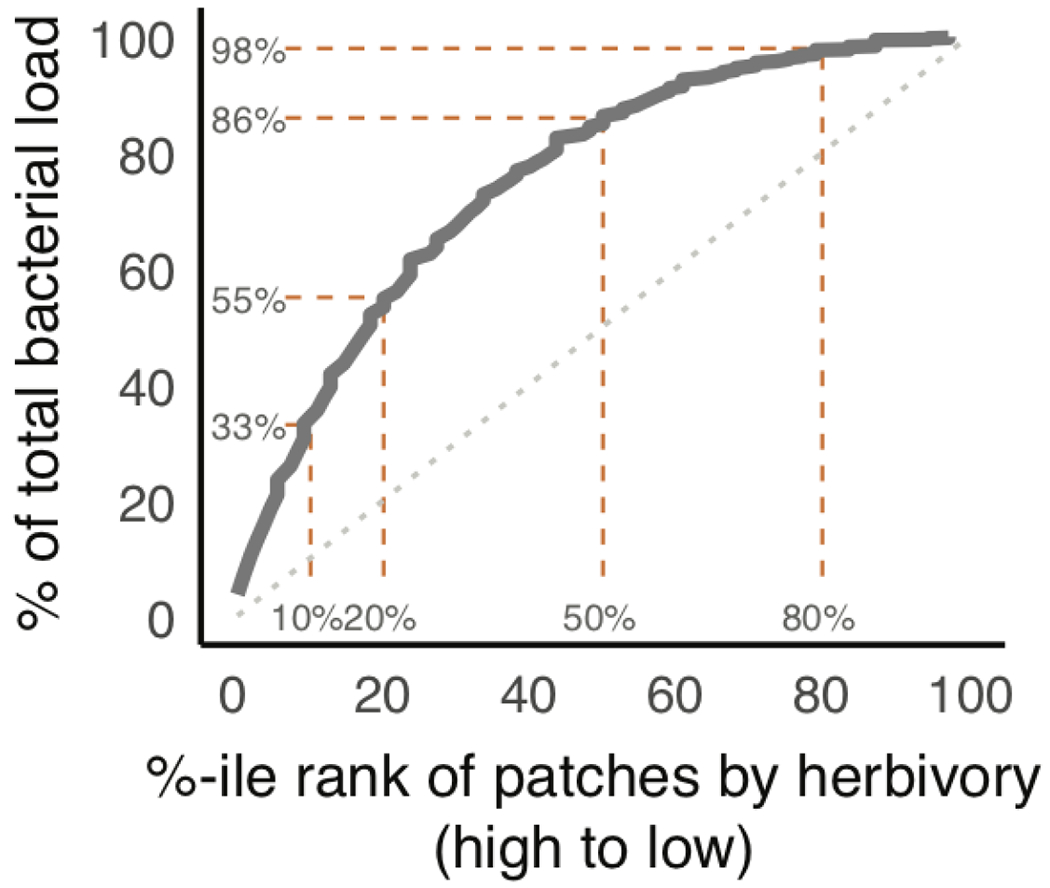
Scaling of percentile rank (high to low) of patch-level herbivore load with total population-level percentage of bacterial propagules present in the sampled patch. At site NP, the top 20% of plant patches with the most herbivory harbor >50% of bacterial propagules in the plant population.
Extended Data Fig. 3. Effects of hormone pre-treatment effects on levels of S. nigrita herbivory in bittercress populations at site NP.
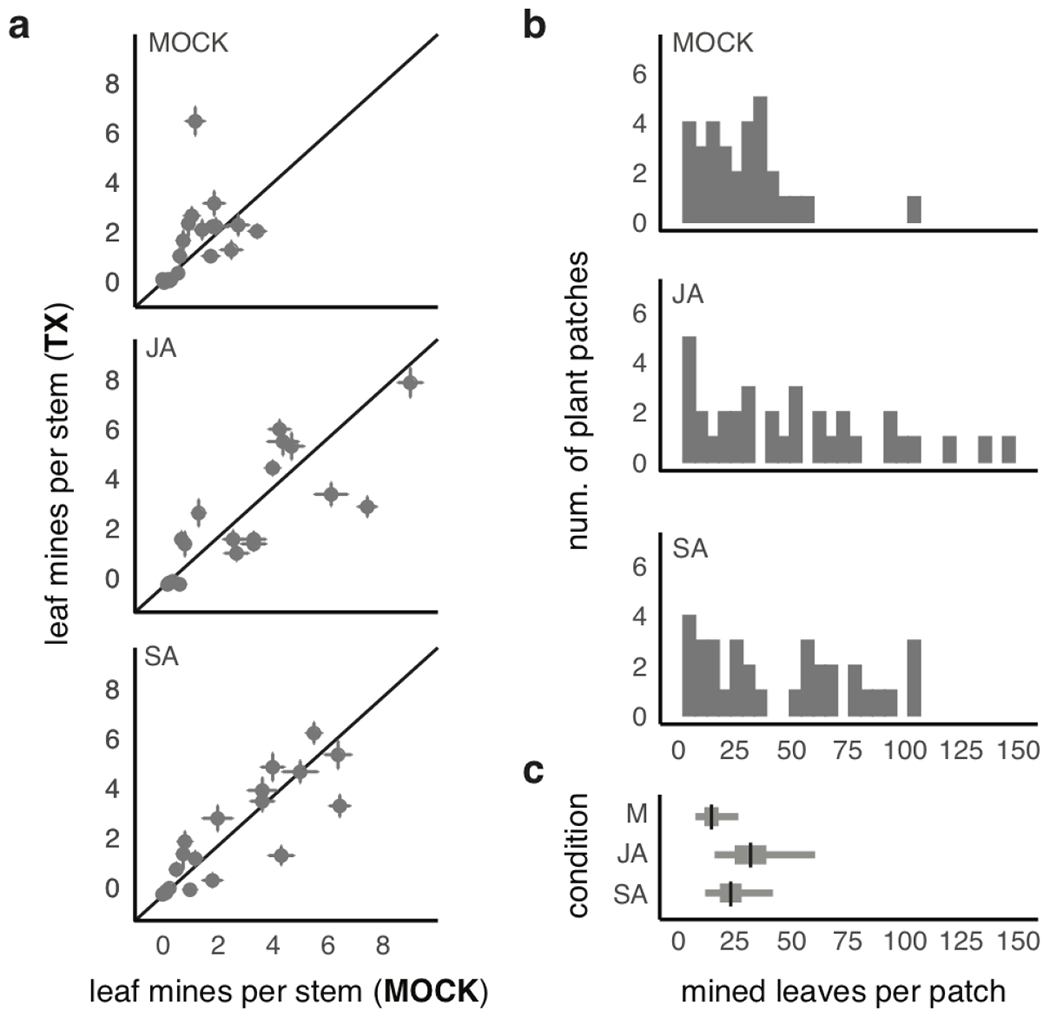
a. For each plot separately, plotted is the average (± 1 std error) leaf mines per stem calculated at the patch level (n=16 stems per patch) for mock-treated (x-axis) versus hormone-treated (y-axis) patches. The three panels represent plots assigned to each of the three conditions: mock (i.e., control), jasmonic acid (JA) or salicylic acid (SA). b. Histograms of patch-level leaf miner damage broken down by patch-level treatment. c. Marginal effects for estimates of patch-level treatment on total mined leaves per patch (see table S5 for statistical results). Black bars are posterior means, while thick and thin bars comprises middle 50%- and 95%-iles of posterior distributions of model terms marginalized over all other parameters.
Extended Data Fig. 4. Hormone pre-treatment (Mock, JA, or SA) five weeks prior to sampling does not leave a clear signature in the distribution of γ across samples for undamaged (gray) or herbivore damaged (orange) leaf samples.
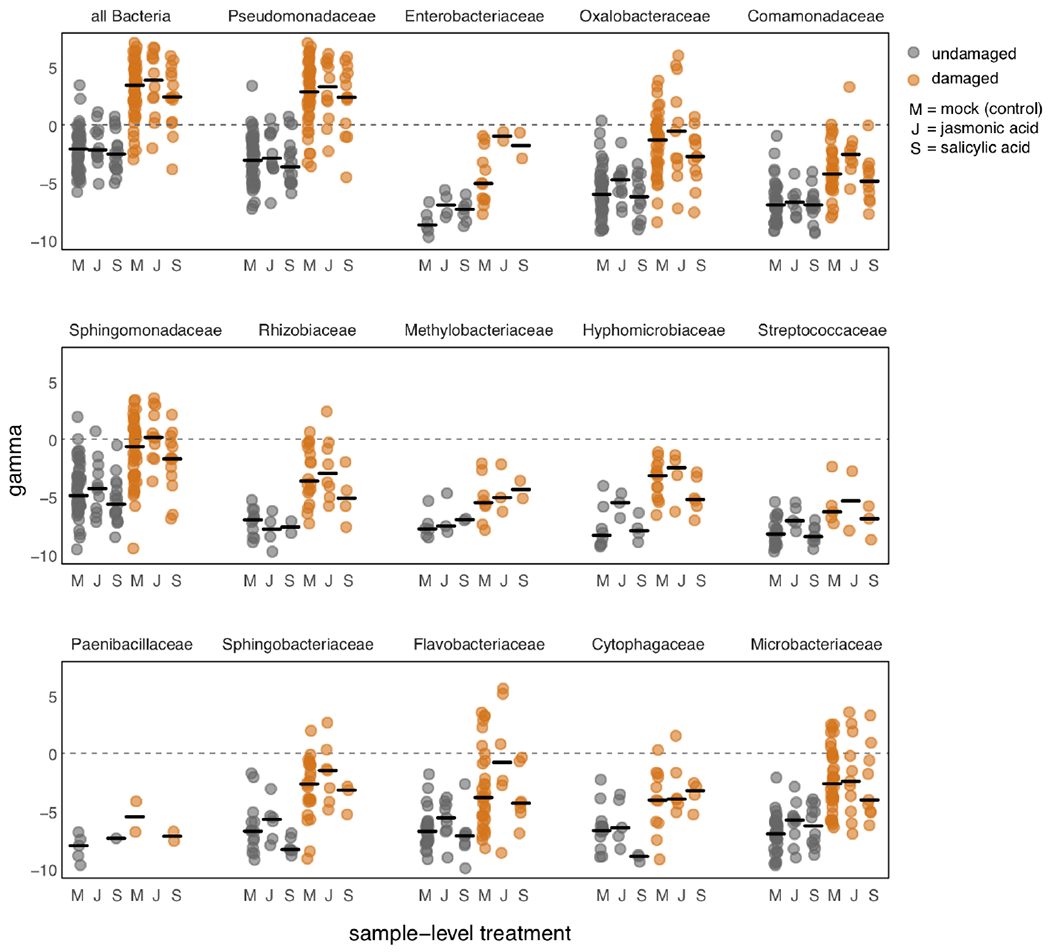
For each of the 14 most abundant bacterial families, in addition to all Bacteria, we have plotted the distributions of raw γ for damaged and undamaged samples across mock-(M), JA-, or SA-treated plant patches. Black bars represent medians for the respective distribution, and data points are slightly jittered along the x-axis. Systematic differences in γ can be easily seen between damaged versus undamaged sample classes, whereas no systematic differences can be seen between the different hormone treatment classes within each damage class. If any effects of hormone treatments are indeed present, they do not constitute a discernible feature of these data, supporting the choice to devote minimal attention to this aspect of our experiment.
Supplementary Material
Acknowledgements
PTH and NKW gratefully acknowledge funding from the National Science Foundation (DEB-1309493 to PTH, DEB-1256758 to NKW), the National Institute of General Medical Sciences of the National Institutes of Health (R35GM119816 to NKW), as well as the Rocky Mountain Biological Laboratory. We are indebted to field assistance provided by Heather Briggs, Kara Cromwell, Aaron Koning, Lucy Anderson, Kyle Niezgoda, Devon Picklum, and Nicolas Alexandre; bioinformatics advice from Tim K O’Connor; and laboratory assistance from Hoon Pyon and Amir Abidov. We thank our contacts at Argonne National Laboratory Sarah Owens and Jason Koval for their technical expertise and support.
Footnotes
Competing Interests Statement: The authors declare that they have no conflicts of interest.
Code Availability
Code to reproduce all analyses, figures, and tables is freely available at https://github.com/phumph/coinfection.
Data Availability
All sequence data has been deposited to the NIH Sequence Read Archive (SRA) under accession SUB5541275 and can also be accessed via BioProject ID PRJNA587302 (http://www.ncbi.nlm.nih.gov/bioproject/587302). Saved model objects are freely available for download from Dryad digital repository44.
References
- [1].Lloyd-Smith JO, Poss M, and Grenfell BT Hiv-1/parasite co-infection and the emergence of new parasite strains. Parasitology, 135(7):795–806, June 2008. doi: 10.1017/S0031182008000292. [DOI] [PubMed] [Google Scholar]
- [2].Laine A-L Context-dependent effects of induced resistance under co-infection in a plant-pathogen interaction. Evol Appl, 4(5):696–707, September 2011. doi: 10.1111/j.1752-4571.2011.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tollenaere C, Susi H, and Laine A-L Evolutionary and epidemiological implications of multiple infection in plants. Trends Plant Sci, 21(1):80–90, January 2016. [DOI] [PubMed] [Google Scholar]
- [4].Karvonen A, Jokela J, and Laine A-L Importance of sequence and timing in parasite coinfections. Trends Parasitol, December 2018. doi: 10.1016/j.pt.2018.11.007. [DOI] [PubMed] [Google Scholar]
- [5].Halliday FW, Umbanhowar J, and Mitchell CE Interactions among symbionts operate across scales to influence parasite epidemics. Ecol Lett, 20:1285–1294, September 2017. [DOI] [PubMed] [Google Scholar]
- [6].Susi H, Barres B, Vale PF, and Laine A-L Co-infection alters population dynamics of infectious disease. Nat Commun, 6:5975, January 2015. doi: 10.1038/ncomms6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Horton MW, Bodenhausen N, Beilsmith K, Meng D, Muegge BD, Subramanian S, Vetter MM, Vilhjalmsson BJ, Nordborg M, Gordon JI, and Bergelson J Genome-wide association study of arabidopsis thaliana leaf microbial community. Nat Commun, 5:5320, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bulgarelli D, Rott M, Schlaeppi K, Ver Loren van Themaat E. , Ahmadinejad N., Assenza F, Rauf P, Huettel B, Reinhardt R, Schmelzer E, Peplies J, Gloeckner FO, Amann R, Eickhorst T, and Schulze-Lefert P. Revealing structure and assembly cues for arabidopsis root-inhabiting bacterial microbiota. Nature, 488(7409):91–5, Aug 2012. [DOI] [PubMed] [Google Scholar]
- [9].Bodenhausen N, Bortfeld-Miller M, Ackermann M, and Vorholt JA A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet, 10(4):e1004283, Apr 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Edwards J, Johnson C, Santos-Medellin C, Lurie E, Podishetty NK, Bhatnagar S, Eisen JA, and Sundaresan V Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci USA, 112(8):E911–20, Feb 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wagner MR, Lundberg DS, Del Rio TG, Tringe SG, Dangl JL, and Mitchell-Olds T Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat Commun, 7: 12151, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Finkel OM, Castrillo G, Herrera Paredes S, Salas Gonzalez I, and Dangl JL Understanding and exploiting plant beneficial microbes. Curr Opin Plant Biol, 38:155–163, 08 2017. doi: 10.1016/j.pbi.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Orozco-Mosqueda MDC, Rocha-Granados MDC, Glick BR, and Santoyo G Microbiome engineering to improve biocontrol and plant growth-promoting mechanisms. Microbiol Res, 208:25–31, Mar 2018. doi: 10.1016/j.micres.2018.01.005. [DOI] [PubMed] [Google Scholar]
- [14].Maron JL and Crone E Herbivory: effects on plant abundance, distribution and population growth. Proc Biol Sci, 273(1601):2575–84, October 2006. doi: 10.1098/rspb.2006.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Agrawal AA Induced responses to herbivory and increased plant performance. Science, 279(5354): 1201–2, February 1998. [DOI] [PubMed] [Google Scholar]
- [16].Bressan M, Roncato M-A, Bellvert F, Comte G, Haichar FZ, Achouak W, and Berge O Exogenous glucosinolate produced by arabidopsis thaliana has an impact on microbes in the rhizosphere and plant roots. ISME J, 3(11):1243–57, November 2009. [DOI] [PubMed] [Google Scholar]
- [17].Thaler JS, Humphrey PT, and Whiteman NK Evolution of jasmonate and salicylate signal crosstalk. Trends Plant Sci, 17(5):260–70, May 2012. doi: 10.1016/j.tplants.2012.02.010. [DOI] [PubMed] [Google Scholar]
- [18].Wagner MR, Lundberg DS, Coleman-Derr D, Tringe SG, Dangl JL, and Mitchell-Olds T Natural soil microbes alter flowering phenology and the intensity of selection on flowering time in a wild arabidopsis relative. Ecol Lett, 17(6):717–26, June 2014. doi: 10.1111/ele.12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Humphrey PT, Gloss AD, Alexandre NM, Villalobos MM, Fremgen MR, Groen SC, Meihls LN, Jander G, and Whiteman NK Aversion and attraction to harmful plant secondary compounds jointly shape the foraging ecology of a specialist herbivore. Ecol Evol, 6(10):3256–68, 05 2016. doi: 10.1002/ece3.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Humphrey PT, Nguyen TT, Villalobos MM, and Whiteman NK Diversity and abundance of phyllosphere bacteria are linked to insect herbivory. Mol Ecol, 23(6):1497–515, March 2014. doi: 10.1111/mec.12657. [DOI] [PubMed] [Google Scholar]
- [21].Vandeputte D, Kathagen G, D’hoe K, Vieira-Silva S, Valles-Colomer M, Sabino J, Wang J, Tito RY, De Commer L, Darzi Y, Vermeire S, Falony G, and Raes J Quantitative microbiome profiling links gut community variation to microbial load. Nature, 551(7681):507–511, 11 2017. doi: 10.1038/nature24460. [DOI] [PubMed] [Google Scholar]
- [22].Turcotte MM, Davies TJ, Thomsen CJM, and Johnson MTJ Macroecological and macroevolutionary patterns of leaf herbivory across vascular plants. Proc Biol Sci, 281(1787), July 2014. doi: 10.1098/rspb.2014.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hirano SS and Upper CD Bacteria in the leaf ecosystem with emphasis on pseudomonas syringae-a pathogen, ice nucleus, and epiphyte. Microbiol Mol Biol Rev, 64(3):624–53, September 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lindow SE and Brandl MT Microbiology of the phyllosphere. Appl Environ Microbiol, 69(4): 1875–83, April 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cui J, Bahrami AK, Pringle EG, Hernandez-Guzman G, Bender CL, Pierce NE, and Ausubel FM Pseudomonas syringae manipulates systemic plant defenses against pathogens and herbivores. Proc Natl Acad Sci USA, 102(5):1791–6, February 2005. doi: 10.1073/pnas.0409450102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ziist T, Heichinger C, Grossniklaus U, Harrington R, Kliebenstein DJ, and Turnbull LA Natural enemies drive geographic variation in plant defenses. Science, 338(6103):116–9, October 2012. doi: 10.1126/science.1226397. [DOI] [PubMed] [Google Scholar]
- [27].Underwood N, Anderson K, and Inouye BD Induced vs. constitutive resistance and the spatial distribution of insect herbivores among plants. Ecology, 86(3):594–602, 2005. [Google Scholar]
- [28].Shaw DJ and Dobson AP Patterns of macroparasite abundance and aggregation in wildlife populations: a quantitative review. Parasitology, 111 Suppl:S111–27, 1995. [DOI] [PubMed] [Google Scholar]
- [29].Karban R and Baldwin IT Induced Responses to Herbivory . University of Chicago Press, 1997. [Google Scholar]
- [30].Wittstock U, Agerbirk N, Stauber EJ, Olsen CE, Hippler M, Mitchell-Olds T, Gershenzon J, and Vogel H Successful herbivore attack due to metabolic diversion of a plant chemical defense. Proc Natl Acad Sci USA, 101(14):4859–64, April 2004. doi: 10.1073/pnas.0308007101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Alexandre NM, Humphrey PT, Gloss AD, Lee J, Frazier J, Affeldt HA 3rd, and Whiteman NK Habitat preference of an herbivore shapes the habitat distribution of its host plant. Ecosphere, 9 (9), September 2018. doi: 10.1002/ecs2.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gloor GB, Macklaim JM, Pawlowsky-Glahn V, and Egozcue JJ Microbiome datasets are compositional: And this is not optional. Frontiers in Microbiology, 8:2224, 2017. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, Kurilshikov A, Bonder MJ, Valles-Colomer M, Vandeputte D, Tito RY, Chaffron S, Rymenans L, Verspecht C, De Sutter L, Lima-Mendez G, D’hoe K, Jonckheere K, Homola D, Garcia R, Tigchelaar EF, Eeckhaudt L, Fu J, Henckaerts L, Zhernakova A, Wijmenga C, and Raes J Population-level analysis of gut microbiome variation. Science, 352(6285):560–4, April 2016. doi: 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- [34].Raes J Editorial overview: It’s the ecology, stupid: microbiome research in the post-stamp collecting age. Curr Opin Microbiol, 44:iv–v, August 2018. doi: 10.1016/j.mib.2018.07.008. [DOI] [PubMed] [Google Scholar]
- [35].Stammler F, Glasner J, Hiergeist A, Holler E, Weber D, Oefner PJ, Gessner A, and Spang R Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome, 4(1): 28, June 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lovell D, Pawlowsky-Glahn V, Egozcue JJ, Marguerat S, and Bahler J Proportionality: a valid alternative to correlation for relative data. PLoS Comput Biol, 11(3):e1004075, March 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Foster KR and Bell T Competition, not cooperation, dominates interactions among culturable microbial species. Curr Biol, 22(19):1845–50, October 2012. doi: 10.1016/j.cub.2012.08.005. [DOI] [PubMed] [Google Scholar]
- [38].Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, Navas-Molina JA, Janssen S, Kopylova E, Vazquez-Baeza Y, Gonzalez A, Morton JT, Mirarab S, Zech Xu Z, Jiang L, Haroon MF, Kanbar J, Zhu Q, Jin Song S, Kosciolek T, Bokulich NA, Lefler J, Brislawn CJ, Humphrey G, Owens SM, Hampton-Marcell J, Berg-Lyons D, McKenzie V, Fierer N, Fuhrman JA, Clauset A, Stevens RL, Shade A, Pollard KS, Goodwin KD, Jansson JK, Gilbert JA, Knight R, and Earth Microbiome Project Consortium. A communal catalogue reveals earth’s multiscale microbial diversity. Nature, 551(7681):457–463, 11 2017. doi: 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lundberg DS, Yourstone S, Mieczkowski P, Jones CD, and Dangl JL Practical innovations for high-throughput amplicon sequencing. Nat Methods, 10(10):999–1002, October 2013. doi: 10.1038/nmeth.2634. [DOI] [PubMed] [Google Scholar]
- [40].Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP Dada2: High-resolution sample inference from illumina amplicon data. Nat Methods, 13(7):581–3, 07 2016. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vehtari A, Gelman A, and Gabry J Practical bayesian model evaluation using leave-one-out cross-validation and waic. Statistics and Computing, 27(5):1413–1432, September 2017. ISSN 1573–1375. doi: 10.1007/s11222-016-9696-4. [DOI] [Google Scholar]
- [42].Humphrey PT, Gloss AD, Frazier J, Nelson-Dittrich AC, Faries S, and Whiteman NK Heritable plant phenotypes track light and herbivory levels at fine spatial scales. Oecologia, 187(2): 427–445, June 2018. doi: 10.1007/s00442-018-4116-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lebeis SL, Paredes SH, Lundberg DS, Breakfield N, Gehring J, McDonald M, Malfatti S, Rio T Glavina del , Jones CD., Tringe SG, and Dangl JL. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science, 349(6250):860–4, August 2015. [DOI] [PubMed] [Google Scholar]
- [44].Humphrey PT and Whiteman NK Data from: Insect herbivory reshapes a native leaf microbiome. Dryad,, Dataset, 2019. doi: 10.5061/dryad.qz612jm95. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.