

Summary
Adult mesenchymal stem cells, including preadipocytes, possess a cellular sensory organelle called the primary cilium. Ciliated preadipocytes abundantly populate perivascular compartments in fat and are activated by high fat diet. Here, we sought to understand if preadipocytes use their cilia to sense and respond to external cues to remodel white adipose tissue. Abolishing preadipocyte cilia in mice severely impairs white adipose tissue expansion. We discover that TULP3-dependent ciliary localization of the omega-3 fatty acid receptor FFAR4/GPR120 promotes adipogenesis. FFAR4 agonists and ω−3 fatty acids, but not saturated fatty acids, trigger mitosis and adipogenesis by rapidly activating cAMP production inside cilia. Ciliary cAMP activates EPAC signaling, CTCF-dependent chromatin remodeling, and transcriptional activation of PPARγ and CEBPα to initiate adipogenesis. We propose dietary ω−3 fatty acids selectively drive expansion of adipocyte numbers to produce new fat cells and store saturated fatty acids, enabling homeostasis of healthy fat tissue.
Keywords: Primary cilia, ciliary signaling, adipogenesis, preadipocyte, FFAR4, GPR120, omega-3 fatty acid, mesenchymal stem cells, obesity, diabetes
In brief -
An omega-3 fatty acid drives adipogenesis through ciliary signaling
Graphical Abstract
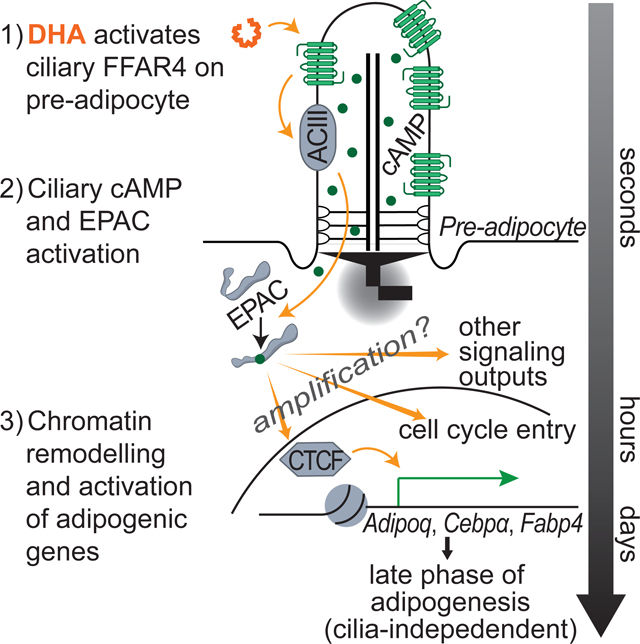
Introduction
Mesenchymal stem and progenitor cells divide to support homeostasis and expansion of four major tissues: muscle, bone, cartilage, and fat. The signals that regulate mesenchymal tissue regeneration and expansion remain unclear, including (1) how growth factors and nutritive fluxes trigger cell cycle entry and stem cell expansion; (2) what structures in tissue anchor and organize the stem cell niche; and (3) what epigenetic programs favor stem cell maintenance versus differentiation.
White adipose tissue (WAT) stores energy and controls energy homeostasis (Rosen and Spiegelman, 2014). WAT can regenerate, expand, and contract in response to tissue damage or altered nutritional flux (Sakaguchi et al., 2017). Specifically, WAT can expand by both generating more adipocytes (hyperplasia) and by storing more fat in existing adipocytes (hypertrophy) (Haczeyni et al., 2018). Excessive hypertrophy is linked to increased tissue hypoxia, fibrosis, and inflammation, leading to insulin resistance and metabolic dysfunction (Ghaben and Scherer, 2019). The adipogenic potential of preadipocytes affects the balance between hypertrophic and hyperplastic WAT expansion, and is influenced by age, sex, location of WAT depots, genetic predisposition, and nutritional fluxes (Arner et al., 2013; Jeffery et al., 2016; Karastergiou and Fried, 2017; Palmer and Kirkland, 2016).
Adipogenesis is regulated by a combination of signals including insulin and cyclic AMP. In response to adipogenic signals, preadipocytes both in vitro and in vivo exit quiescence and reenter the cell cycle to regenerate preadipocytes and generate daughter cells that differentiate into adipocytes (Jeffery et al., 2015; Wang et al., 2013). The molecular mechanism of adipogenesis has been characterized extensively in a murine cell line, 3T3-L1 cells. In response to the differentiation factors insulin, the glucocorticoid dexamethasone (Dex), and the cAMP elevating drug IBMX, 3T3-L1 cells re-enter the cell cycle and activate an adipogenic transcriptional cascade involving PPARγ and CEBPα (Rosen et al., 2000; Tang et al., 2003). Apart from insulin, the physiological factors that promote adipogenesis in vivo remain unclear. Discovering factors that shift the balance of WAT expansion to hyperplasia could provide a new therapeutic strategy for limiting the consequences of obesity.
Preadipocytes reside along the vasculature in fat tissue, where they may sense systemic changes in metabolites and couple nutritional fluxes to adipogenesis (Tang et al., 2008). Earlier studies lack definitive markers and functional identity to visualize the preadipocyte in fat tissue, although we and others have noted that preadipocytes, like other quiescent mesenchymal stem and progenitor cells, possess a single projection called a primary cilium (Kopinke et al., 2017; Lyu and Zhou, 2017; Tummala et al., 2010).
The primary cilium is a sensory organelle nucleated by the mother centriole and enriched with receptors, particularly G protein-coupled receptors (GPCRs) (Hilgendorf et al., 2016). Transport of these receptors into the cilium is regulated by at least two protein complexes, the BBSome and the TULP3-IFTA complex (Mukhopadhyay et al., 2010; Nachury et al., 2007). Recent studies have reported the potential importance of primary cilia for differentiation into adipocytes (Forcioli-Conti et al., 2015; Huang-Doran and Semple, 2010; Marion et al., 2012; Marion et al., 2009; Qiu et al., 2010; Zhu et al., 2009), osteocytes (Xiao and Quarles, 2010; Yuan and Yang, 2016), chondrocytes (Deren et al., 2016; Kelly and Jacobs, 2010), and myocytes (Fu et al., 2014; Jaafar Marican et al., 2016). The importance of primary cilia for mesenchymal stem and progenitor cell function is highlighted by human genetic disorders called ciliopathies caused by ciliary dysfunction. Some ciliopathies are characterized by tissue degeneration and metabolic dysfunctions including obesity and diabetes (Hildebrandt et al., 2011; Waters and Beales, 2011).
To discover how fat tissue organizes potential ciliated progenitor cells, we used a transgenic mouse model with fluorescently tagged primary cilia. We found that an extensive population of ciliated cells is localized along the vasculature in fat pads, positioning these cells to sense and respond to systemic fluxes of metabolites and growth factors. To determine if preadipocytes use their cilia to sense these factors, we genetically removed preadipocyte cilia and found that ciliation of preadipocytes is critically important for WAT expansion. To discover key fluxes that trigger differentiation, we screened for ciliary GPCRs as candidate adipogenic regulators, and discovered that FFAR4/GPR120 localizes to preadipocyte cilia in vitro and in vivo. The FFAR4/GPR120 ligand DHA, an ω−3 fatty acid, but not saturated fatty acids, efficiently induce adipogenesis. Addition of DHA or synthetic FFAR4 agonists to preadipocytes triggers a rapid increase in ciliary cAMP and activation of the cAMP effector EPAC critical for adipogenesis. Profiling of DHA/FFAR4-dependent transcriptional and chromatin-remodeling activities uncovered that the regulator of chromatin architecture CTCF helps activate transcriptional effectors including CEBPα to promote adipogenesis. Thus, our results reveal that preadipocyte cilia selectively sense ω−3 fatty acids through FFAR4 to induce adipogenesis.
Results
Characterizing the primary cilium of preadipocytes in vitro and in vivo
Previous studies have shown that human and murine preadipocytes are ciliated in vitro when quiescent, and that the primary cilium is important for adipogenesis, in part by displaying a highly sensitized IGF-1 receptor (Zhu et al., 2009). To confirm and extend these observations, we first assessed the ciliation status of differentiating 3T3-L1 cells. 80% of confluent 3T3-L1 cells are ciliated, and primary cilia are uniformly lost as differentiating 3T3-L1 cells accumulate lipid (Fig 1A, D). While 3T3-L1 cells in the early phase of differentiation with little or no lipid are typically ciliated, mature, lipid-laden 3T3-L1 adipocytes are not ciliated, suggesting that the primary cilium specifically functions in undifferentiated and differentiating preadipocytes. Similarly, both mouse and human preadipocytes grown to confluency are ciliated, and the primary cilium is lost during differentiation (Fig 1B, C; Fig S1A). Specifically, 80% of FACS-sorted mouse preadipocytes (Lin− CD34+ CD29+ SCA1+) are ciliated. This cell population has previously been shown capable of efficient in vitro differentiation and reconstitution of WAT depots in lipodystrophic mice (Rodeheffer et al., 2008). These findings suggest that the primary cilium may be a functional marker of preadipocytes in vivo.
Figure 1. Preadipocytes are ciliated in vitro and in vivo, and an abundant perivascular cell in fat tissue.

(A-C) Immunofluorescence staining visualizes primary cilia on (A) confluent 3T3-L1 cells, (B) primary mouse preadipocytes (Lin−, CD34+, SCA1+, CD29+), and (C) human preadipocytes. ACTUB stains axoneme, PCNT and CEP170 stain the basal body. (D) The primary cilium is lost in differentiating 3T3-L1 cells. n=number of cells counted. (E-G) Whole mount imaging of epididymal WAT from cilia glow mouse identifies ciliated perivascular cells. All cells have a CENTRIN2-GFP+ centrosome, ciliated cells are CENTRIN2-GFP+ and ARL13BmCherry+ (inset). (E) Lipid droplets are visualized by phase and (F) blood vessels are CD31+. (G) 3D surface reconstruction of blood vessel and adjacent ciliated perivascular cells. (H) Quantification of ciliation on perivascular cells in subcutaneous (dark grey) and visceral (light grey) mouse WAT of cilia glow mice. Bar graphs show average from 2 littermates ± SD. (I) HFD promotes transient deciliation in epididymal WAT of cilia glow mice after 3 days. Bar graph shows average of 4 independent experiments ± SEM, each using 1–2 littermates per diet. n=number of perivascular cells counted; p-value calculated using chi-squared test is p<0.005. (J, K) 2 weeks of HFD activates ciliated perivascular cells to re-enter the cell cycle. (J) Whole mount image and (K) quantification of BrdU+ ciliated perivascular cells in epididymal WAT. Data are percent BrdU+ of ciliated perivascular cells (n=3 mice on HFD; n=2 mice on chow) ± SEM. n=number of perivascular ciliated cells counted. Arrowheads point to perivascular ciliated cells. p-values calculated using t-test unless noted otherwise, ** p<0.01; See also Figure S1.
To test this hypothesis, we visualized ciliated cells within intact fat tissue. Of note, there has been considerable progress in identifying in vivo markers of preadipocytes, including PDGFRα for lineage tracing and sorting, but to date no unique, specific in vivo preadipocyte markers have been identified (Berry and Rodeheffer, 2013; Guimaraes-Camboa et al., 2017; Gupta et al., 2012; Hepler et al., 2017; Jiang et al., 2014; Rodeheffer et al., 2008; Vishvanath et al., 2016). Functional studies suggested that preadipocytes reside along the vasculature within WAT (Tang et al., 2008). We used a transgenic mouse model expressing fluorescently-tagged CENTRIN2 and ARL13B to visualize the base and axoneme of primary cilia, respectively (henceforth referred to as cilia glow mouse). Strikingly, ciliated cells within fat pads are organized along vascular tracts, and mature adipocytes are not ciliated (Fig 1E). The expansive populations of ciliated cells are located immediately adjacent to blood vessels (Fig 1F, G; Fig S1B). Ciliated perivascular cells account for ~30% of all perivascular cells in both visceral and subcutaneous WAT (Fig 1H). Staining and quantifying cilia in non-transgenic mice verifies the proportion of ciliated perivascular cells in WAT (Fig S1C). Macrophages, which can account for up to 40% of other cells in WAT (Weisberg et al., 2003), are not ciliated (Fig S1D). As further confirmation, we genetically marked preadipocytes using a tamoxifen-inducible Pdgfrα-CreERT allele (Kang et al., 2010) crossed to a Rosa26EYFP reporter (Srinivas et al., 2001). PDGFRα-lineage perivascular cells in murine WAT are ciliated in vivo (Fig S1E). Thus, we propose that the primary cilium marks the resident preadipocyte population along the vasculature in vivo, raising the hypothesis that ligands carried by circulation can activate these precursor cells to differentiate.
To test this hypothesis, we activated preadipocytes in vivo using high fat diet (HFD). Previously, HFD was shown to induce quiescent preadipocytes in visceral WAT to rapidly re-enter the cell cycle after 3 days, and then exit the cell cycle after 1 week (Jeffery et al., 2015). Of note, cells transiently lose their cilia during mitosis (Ford et al., 2018; Kim and Dynlacht, 2013). Consistent with the hypothesis, there is a transient decrease in the percentage of ciliated perivascular cells after 3 days on HFD (Fig 1I). After 2 weeks on HFD, we assessed ciliation and BrdU incorporation and found that 50% of all ciliated perivascular cells were BrdU+, indicating that they were activated in response to HFD to enter the adipogenic program (Fig 1J, K). Taken together, the primary cilium marks preadipocytes that are responsive to dietary fatty acids in vivo.
Preadipocyte cilia are critical for WAT expansion in vivo
To assess the in vivo function of preadipocyte cilia, we conditionally deleted Ift88, a gene required for ciliogenesis and ciliary maintenance, in preadipocytes using tamoxifen-inducible Pdgfrα-CreERT. We refer to Pdgfrα-CreERT Ift88flox/− mice treated with tamoxifen hereafter as PAno cilia mice. Control mice were tamoxifen-treated littermates lacking Pdgfrα-CreERT (Ift88flox/−) or retaining one wild-type allele of Ift88 (Pdgfrα-CreERT Ift88flox/+) (Fig 2A). Preadipocytes of PAno cilia mice treated with tamoxifen at 3 weeks of age lacked cilia (Fig S2A). Notably, loss of preadipocyte cilia dramatically reduced body weight, with both PAno cilia male and female mice weighing almost 20g less than control male and female littermates at 17 weeks post-tamoxifen administration (Fig 2A). Echo-MRI measurement revealed that this weight loss was largely due to reduced total fat mass (Fig 2B). Loss of cilia in preadipocytes resulted in a significant reduction of gonadal WAT (Fig 2B). We observed similar, but quantitatively smaller differences in body weight and fat mass when tamoxifen was administered at 6 weeks of age, arguing that preadipocyte cilia are continuously required for full WAT expansion (Fig S2B).
Figure 2. Preadipocyte cilia promote WAT expansion.

(A) Body weight measurements of control (Ift88flox/− and Pdgfrα-CreERT Ift88Δ/+) and PAno cilia mice (Pdgfrα-CreERT Ift88Δ/-) (n=5 per sex and genotype). (B) Dissected gonadal fat pads and measurements of total fat and lean mass by Echo-MRI of control and PAno cilia mice 17 weeks after tamoxifen administration. Scale bar is 1cm. (C) Serum leptin levels of control and PAno cilia mice 17 weeks after tamoxifen administration. (D) Immunofluorescence staining for lineage marker (EYFP, green) and adipocytes (LipidTox, red) of 20-week-old controllineage (Pdgfrα-CreERT Ift88Δ/+ Rosa26EYFP) and PAno cilia+lineage (Pdgfrα-CreERT Ift88Δ/- Rosa26EYFP) mice after tamoxifen administration at 3 weeks of age. PAno cilia+lineage mice contain fewer lineage-derived EYFP+ adipocytes compared to controllineage mice. Scale bar is 100 μm. All data are represented as mean ± SEM. p-values calculated using standard t-test and two-way ANOVA followed by Tukey’s multiple comparison test (**<0.01, ***<0.001 and ****<0.0001). See also Figure S2.
Consistent with reduced fat mass, histological analysis showed that the average adipocyte is smaller in PAno cilia mice (Fig S2C). As expected, serum leptin levels are also reduced in PAno cilia mice (Fig 2C). Since preadipocytes, but not mature adipocytes, are ciliated (Fig 1D−E), we hypothesized that cilia are critical for the adipogenic potential of preadipocytes, accounting for the decreased WAT expansion in PAno cilia mice. To test this hypothesis, we introduced the Rosa26EYFP reporter into our control (Pdgfrα-CreERT Ift88flox/+ Rosa26EYFP, referred to hereafter as controllineage) and PAno cilia (Pdgfrα-CreERT Ift88flox/− Rosa26EYFP, referred to hereafter as PAno cilia+lineage ) mice, and followed the fate of preadipocytes with and without cilia by virtue of EYFP expression. Specifically, we quantified lineage-traced, EYFP+ adipocytes in PAno cilia+lineage and control lineage mice. Deletion of cilia in preadipocytes dramatically reduced the proportion of EYFP-expressing differentiated adipocytes (Fig 2D). Thus, preadipocyte cilia are important for adipogenesis and WAT expansion.
Despite the reduction in total fat mass in PAno cilia mice, we did not note symptoms of lipodystrophy, such as increased serum free fatty acid, insulin, or glucose levels, or hepatic fat deposition in PAno cilia mice (Fig S2D–E). We do not know if these hallmarks of lipodystrophy would become apparent with age or if PAno cilia mice were challenged with HFD.
We also interrogated other tissues to assess potential effects on WAT expansion. Pdgfrα-CreERT is active in WAT (Fig S1E), and does not induce extensive recombination in the liver, pancreas, or small or large intestine (O’Rourke et al., 2016). Within skeletal muscle, the activity of this transgenic Pdgfrα-CreERT allele is restricted to Pdgfrα-expressing fibro-adipogenic progenitors (Kopinke et al., 2017). As primary cilia in the brain have previously been shown to regulate satiety and locomotion (Davenport et al., 2007; Loktev and Jackson, 2013; Siljee et al., 2018), we assessed PAno cilia and control mice for food intake, energy expenditure, or activity using the Comprehensive Lab Animal Monitoring System (CLAMS) and observed no differences (Fig S2F). We also did not observe changes in the amount of gross brown adipose tissue in PAno cilia mice (Fig S2G), and it would be interesting to test for a role of preadipocyte cilia during β3-adrenergic induced de novo BAT formation. To assess whether differences in energy absorption might contribute to the reduction in fat mass, we evaluated levels of glucose and free fatty acids in serum or adaptive hyperphagia (Crenn et al., 2004), but found no differences between control and PAno cilia mice (Fig S2E−F). We therefore conclude that preadipocyte ciliation is important for WAT expansion and that preadipocyte cilia promote adipogenesis in vivo.
TULP3, an essential regulator of ciliary GPCR entry, is critical for adipogenesis
To assess how primary cilia promote adipogenesis, we focused on identifying GPCRs, the largest class of ciliary receptors (Hilgendorf et al., 2016), and hypothesized that preadipocyte ciliary GPCRs transduce adipogenic cues. To test this dependence, we used CRISPR-Cas9 to generate 3T3-L1 cells lacking TULP3, a protein required for transporting GPCRs into the primary cilium (Fig 3A, Fig S3A) (Loktev and Jackson, 2013; Mukhopadhyay et al., 2010). 3T3L1 cells lacking TULP3 form primary cilia at the expected frequency, but lack a ciliary protein (ARL13B), consistent with published reports (Fig S3C) (Hwang et al., 2019). We assessed adipogenesis by Oil Red O staining, quantified by absorbance of isopropanol extracted dye. Intriguingly, loss of TULP3 did not attenuate adipogenesis of 3T3-L1 cells treated with the full complement of the standard, historical differentiation factors (Fig 3B). This differentiation media (DM) includes a high concentration of Dex, a glucocorticoid receptor agonist that directly and broadly promotes transcription (Siersbaek et al., 2012), and thus may uncouple adipogenesis from the sensing of extracellular cues by GPCRs. Therefore, we titrated down the historical DM, which only slightly decreased the efficiency and kinetics of 3T3-L1 adipogenesis at 0.5 or 0.25x concentrations. Importantly, TULP3 was required for efficient adipogenesis using this more dilute DM, as shown by sgRNA knockout (Fig 3B) or siRNA knockdown of Tulp3 (Fig S3D–F). This loss of adipogenic activity following Tulp3 depletion was rescued by expression of human TULP3, confirming the specificity of the effect (Fig 3C, D; Fig S3G).
Figure 3. TULP3 knockouts support a requirement for ciliary GPCRs during adipogenesis.

(A) Immunoblot showing depletion of TULP3 protein in 3T3-L1 cells. (B) TULP3 is required for 3T3-L1 differentiation induced by reduced amounts of DM. Lipids are visualized by Oil Red O staining (top) and quantified by measuring absorbance post-isopropanol extraction of Oil Red O (bottom). (C) Loss of adipogenic potential due to TULP3 depletion is rescued by human GFP-tagged TULP3. (D) TULP3 expression levels correlate with adipogenic potential as determined by live imaging and quantification of green fluorescence intensity using BODIPY. Dotted line denotes media change. Shaded area describes 95% confidence interval. Bar graphs are normalized mean ± SD; * p<0.05; ** p<0.01; *** p<0.001. See also Figure S3.
Overexpression of TULP3 in wild type 3T3-L1 preadipocytes increased the rate and the amount of adipogenesis, as assessed by kinetic analysis of fluorescence intensity of the lipophilic green fluorescent dye BODIPY (Fig 3D). This experiment indicates that TULP3 levels may determine signaling efficiency and outcome. Thus, TULP3 expression levels, and by extension ciliary receptor trafficking, may accelerate adipogenesis in 3T3-L1 cells.
Preadipocytes display FFAR4 in the primary cilium
Next, we sought to discover which GPCRs may function at preadipocyte primary cilia. We compiled a list of candidate ciliary preadipocyte GPCRs, based on expression patterns and receptors with a known role in regulating adipogenesis (Table S1). These GPCRs were expressed as C-terminally tagged GFP fusion proteins and screened for ciliary localization in 3T3-L1 preadipocytes (Fig 4A). This analysis uncovered the free fatty acid receptor FFAR4/GPR120 as distinctively ciliary (Fig S4A).
Figure 4. FFAR4 is a ciliary GPCR displayed by preadipocytes.

(A) Schematic of screen to identify ciliary GPCR in 3T3-L1 cells. (B-D) Endogenous FFAR4 localizes to the primary cilium of undifferentiated, confluent (B) 3T3-L1 cells, (C) primary mouse preadipocytes in the stromal vascular fraction (SVF, depleted for RBCs and WBCs) from cilia glow mice, and (D) primary human preadipocytes. (E) Whole mount images of epididymal WAT from cilia glow mice shows that ciliated perivascular cells display ciliary FFAR4. (F) Loss of TULP3 prevents ciliary trafficking of FFAR4 in 3T3-L1 preadipocytes. (G) Induction of differentiation results in internalization of ciliary FFAR4. n=number of cells counted, bar graph is average ± SD; ** p<0.01. See also Figure S4.
To confirm ciliary localization of FFAR4, we generated an antibody against the protein. Notably, endogenous FFAR4 localized to the primary cilium of undifferentiated, confluent 3T3-L1 preadipocytes as well as to the primary cilium of primary preadipocytes isolated from mouse and human WAT (Fig 4B–D, Fig S4B). Moreover, FFAR4 also localized to the primary cilium of perivascular ciliated preadipocytes in vivo (Fig 4E). We confirmed the specificity of staining in 3T3-L1 cells lacking FFAR4 and in mouse primary preadipocytes isolated from Ffar4 knockout mice (Fig S4C–E). Finally, ciliary FFAR4 localization is TULP3-dependent (Fig 4F, Fig S4F).
To better understand how FFAR4 might participate in adipogenesis, we assessed the expression and localization of FFAR4 during adipogenesis. Intriguingly and consistent with previous observations, overall FFAR4 mRNA and protein levels increased dramatically during adipogenesis and as the primary cilium is lost (Fig S4G) (Gotoh et al., 2007). FFAR4 is at low levels in undifferentiated cells, except for the distinctive pool in the cilium. Moreover, FFAR4 relocalized from primary cilia to the plasma membrane as 3T3-L1 cells underwent differentiation (Fig 4G; Fig S4H). Thus, we propose that FFAR4 can localize to both the primary cilium and the plasma membrane, and that FFAR4 has a higher efficiency for ciliary targeting such that it specifically localizes to primary cilia of undifferentiated, ciliated preadipocytes when FFAR4 expression is low. As the cilium is lost and FFAR4 expression increases during adipogenesis, the protein localizes to the plasma membrane where it can regulate glucose uptake in mature adipocytes (Fig S4I) (Oh et al., 2010). Thus, FFAR4 is a newly identified ciliary GPCR that can localize to primary cilia of preadipocytes both in vitro and in vivo in a TULP3-dependent manner.
Activation of ciliary FFAR4 drives early steps in adipogenesis
Given the requirement for TULP3 in adipogenesis, we hypothesized that FFAR4 in preadipocyte cilia promotes early adipogenesis, whereas FFAR4 on the plasma membrane of mature adipocytes has a distinct role. FFAR4 is activated by long chain free fatty acids, and specifically ω−3 fatty acids (Hirasawa et al., 2005; Oh et al., 2010). We therefore assessed how supplementing DM with the ω−3 fatty acid DHA affects adipogenesis (Fig 5A). Previous studies on the effect of free fatty acids on adipogenesis have yielded conflicting results, likely due to differences in DM composition and timing of free fatty acid supplementation (Kim et al., 2006; Madsen et al., 2005; Song et al., 2016). Thus, we added DHA during the first two days of differentiation, when FFAR4 is exclusively ciliary, and titrated the individual DM components in the presence and absence of DHA to determine dosages with greatest DHA potentiation (henceforth referred to as DHA cocktail and control cocktail respectively, Fig S5A). Consistent with the requirement for TULP3 in adipogenesis, DHA potentiates adipogenesis when individual DM components are reduced (Fig 5B).
Figure 5. FFAR4 activation promotes adipogenesis.

(A) Schematic of 3T3-L1 differentiation experiment in the presence of free fatty acids. (B) 3D mesh plot of differentiation using different amounts of insulin, Dex, and IBMX in the presence or absence of 100μM DHA during the first 2 days of differentiation. Data plotted is normalized absorbance post-isopropanol extraction of Oil Red O (images in Figure S4A). (C-D) DHA cocktail enhances differentiation of 3T3-L1 cells, and this is attenuated by (C) loss of TULP3 or FFAR4 protein or (D) addition of FFAR4 antagonist (AH7614) in a dose-dependent manner. (E) FFAR4 activation promotes adipogenesis of primary mouse preadipocytes (Lin−, CD34+, SCA1+, CD29+) from wild-type, but not Ift88flox/flox post-transduction with Cre recombinase and GFP. n = number of GFP+ cells counted. Scale bar 10μm. (F) Daily FFAR4 agonist intraperitoneal injection followed by HFD increases diet-induced obesity. All data are mean ± SEM. p-values calculated using standard t-test comparing FFAR4 agonist, HFD versus vehicle, HFD. (G) Supplementing DHA, but not palmitic acid or oleic acid, enhances adipogenesis. (H) Addition of FFAR4, but not FFAR1, agonist enhances adipogenesis. Bar graphs are normalized mean ± SD. * p<0.05; ** p<0.01; *** p<0.001. See also Figure S5.
Free fatty acids have previously been proposed to be ligands of PPARγ (Krey et al., 1997). To confirm that DHA functions as an extracellular signaling molecule to promote adipogenesis, rather than as a PPARγ agonist, we stimulated 3T3-L1 cells with the DHA cocktail in the presence of the PPARγ antagonist T0070907. This antagonist is a potent inhibitor of adipogenesis when present during the entire differentiation time course (Day 0–6), consistent with PPARγ being required for adipogenesis. However, when the PPARγ antagonist was added concurrently with DHA (Day 0–2), it did not inhibit adipogenesis (Fig S5B). DHA therefore does not promote adipogenesis by directly activating PPARγ.
Instead, DHA-enhanced adipogenesis is dependent on FFAR4 and ciliary FFAR4 localization, since DHA failed to enhance adipogenesis in 3T3-L1 cells lacking TULP3 or FFAR4 (Fig 5C; Fig S3B). Similarly, addition of the FFAR4 antagonist AH-7614 (Day 0–2) inhibited DHA-enhanced adipogenesis in a dose-dependent manner (Fig 5D). 3T3-L1 differentiation in the DHA cocktail was more sensitive to the FFAR4 antagonist when compared to the standard, historical DM (Fig S5C). Thus, DHA functions as an extracellular signaling molecule to promote adipogenesis. Moreover, FFAR4 agonist promoted adipogenesis of primary mouse preadipocytes and FFAR4 antagonism inhibited adipogenesis of primary human preadipocytes, suggesting that FFAR4 is broadly required for adipogenesis (Fig S5D, E).
To further validate our in vitro findings, we isolated primary preadipocytes from Ift88flox/flox and wild-type males and induced loss of cilia by transducing with an adenovirus expressing both Cre recombinase and GFP. Cre removed cilia from primary Ift88flox/flox preadipocytes, but not from primary wild-type preadipocytes (Fig 5E). Similarly, primary Ift88flox/flox preadipocytes expressing GFP only possessed cilia (Fig S5F). Notably, loss of cilia impaired the ability of FFAR4 agonist to promote adipogenesis of primary preadipocytes (Fig 5E, Fig S5F). Thus, DHA, a physiological ligand for FFAR4, promotes adipogenesis of 3T3-L1 preadipocytes as well as primary murine preadipocytes via primary cilia.
We investigated whether FFAR4 activation also promotes adipogenesis in vivo. Mice were injected daily with FFAR4 agonist for 2 weeks, and switched to a HFD starting the second week for a total of 3 months. We sequentially administered the agonist followed by HFD, since our in vitro data showed that FFAR4 activation in the early phase of adipogenesis is sufficient to promote adipogenesis. Consistent with our ex vivo data, pretreatment with FFAR4 agonist causes a dramatic and synergistic increase in body weight when combined with HFD (Fig 5F).
We next considered whether different types of fatty acids promote adipogenesis. Intriguingly, only the FFAR4 ligand DHA, but not saturated or mono-unsaturated palmitic or oleic acid, significantly and robustly potentiated adipogenesis (Fig 5G). Thus, DHA is selectively able to drive FFAR4-mediated adipogenesis, in contrast to saturated or mono-unsaturated fatty acids which may instead drive hypertrophy of mature adipocytes. Of note, the ω−3 fatty acid DHA, can activate FFAR1 in addition to FFAR4 (Ichimura et al., 2014). Only FFAR4 localizes to cilia (data not shown). Using the specific pharmacological agonists TUG891 (FFAR4) and TUG424 (FFAR1), we found that activating FFAR4 (but not FFAR1) during the first 2 days of differentiation promoted 3T3-L1 adipogenesis (Fig 5H). Thus, we establish that DHA and FFAR4 are a physiologically-relevant ligand-receptor pair that is organized by preadipocyte cilia to promote adipogenesis.
DHA and FFAR4 promote adipogenesis by raising ciliary cAMP levels
We sought to identify the molecular mechanism by which DHA promotes adipogenesis. First, we confirmed using fluorescence microscopy that DHA cocktail promoted adipogenesis by activating more preadipocytes to differentiate. The number of lipid-containing adipocytes increased as assessed by the lipophilic green fluorescent BODIPY dye both in an endpoint assay and kinetically during the differentiation time course (Fig 6A, B). Moreover, adipogenic genes were induced and pre-adipocyte genes were downregulated in response to DHA cocktail (Fig S6A). Thus, DHA promotes more preadipocytes to differentiate.
Figure 6. FFAR4 regulates initiation of adipogenesis via cAMP.

(A, B) DHA cocktail initiates adipogenesis in 3T3-L1 cells (A) after 6 days of differentiation, and (B) as assessed by the increase in lipid droplets (BODIPY) over time. Data is average of 3 wells ± SD, shaded area is 95% confidence interval. Dotted line denotes media change. n=number of cells counted. Scale bar is 50μm. (C) DHA cocktail results in cell cycle re-entry and this requires TULP3 protein. Bar graphs show normalized average from 3 experiments ± SD; (D) 3T3-L1 cells were exposed to individual components of the modified cocktail ± FFAR4 agonist for 48h. FFAR4 activation partially replaces IBMX. (E, F) FFAR4 activation elevates ciliary cAMP levels. 3T3-L1 cells were transduced with a ciliary cAMP sensor (green) and reference marker (red). Addition of FFAR4 agonist (denoted by arrow) results in decreased ciliary green fluorescence intensity, indicating that ciliary cAMP levels increase. (E) Representative images showing cAMP sensor (green) and cilia (red) offset. Scale bar 5μm. (F) Background subtracted ratio of fluorescence intensities are normalized to DMSO control and 0 second time point. n=6 for FFAR4 agonist and n=4 for DMSO control ± SD, where n is the average of all cilia measured per well; (G) 3T3-L1 cells were differentiated with DHA cocktail in the presence of an inhibitor against EPAC (ESI-09) or PKA (Rp-cAMPS) for the first 2 days. Lipid accumulation was assessed on Day 4. Inhibition of EPAC, but not PKA, attenuates DHA enhanced adipogenesis in a dose-dependent manner. * p<0.05; ** p<0.01; *** p<0.001. See also Figure S6.
We investigated at which point in the adipogenic program DHA acts. Activation of quiescent preadipocytes in vitro and in vivo causes rapid cell cycle re-entry prior to induction of PPARγ. In 3T3-L1 cells, mitoses occur during the first 2 days of differentiation. To determine if DHA promotes cell cycle re-entry in 3T3-L1 cells, we assessed EdU incorporation 40 hours after addition of DHA or control cocktail and found that DHA promoted re-entry into the cell cycle (Fig 6C, Fig S6B). Thus, DHA is mitogenic in this context and initiates the adipogenic program. Importantly, the mitogenic function of DHA is dependent on ciliary FFAR4 localization, since 3T3-L1 cells lacking TULP3 fail to undergo mitosis in response to DHA cocktail. In contrast, cell cycle re-entry in response to the historical DM is independent of TULP3 (Fig S6B, C). These data are consistent with our previous results showing that high levels of glucocorticoids uncouple initiation of adipogenesis from sensing of extracellular cues (Fig 3B), and strongly argue that the DHA cocktail described here is more physiologically relevant to adipogenesis.
In particular, we considered that free fatty acids, unlike pharmacological components in DM such as Dex and IBMX, may be in vivo regulators of adipogenesis. We therefore hypothesized that one or more components in DM (insulin, Dex, and IBMX) mimic the effect of DHA to initiate adipogenesis. To test this hypothesis, we serially omitted each component in the presence or absence of FFAR4 agonist. We note that some insulin and IGF1 are present in the serum, although at amounts insufficient to trigger robust adipogenesis. FFAR4 activation can replace IBMX, a phosphodiesterase inhibitor that raises cellular levels of the second messenger cAMP, and can also partially substitute for insulin (Fig 6D, Fig S6D). We therefore hypothesized that FFAR4 activation promotes adipogenesis by signaling via cAMP specifically in the primary cilium.
To test our hypothesis, we transduced 3T3-L1 preadipocytes with a ciliary-targeted cAMP sensor (cilia cADDIS) optimized for live cell imaging (Moore et al., 2016). Increased ciliary cAMP caused decreased ciliary green fluorescence signal of the cADDIS sensor. We quantified dynamic changes in cAMP levels as the ratio of green fluorescence to red fluorescence reference (generated by a constitutively ciliary-localized mCherry fusion protein). Strikingly, addition of FFAR4 agonist TUG-891 rapidly increased ciliary cAMP levels in confluent 3T3-L1 preadipocytes (Fig 6E, F). This increase occurred within seconds of treatment and was dependent on ciliary localization of FFAR4, as loss of TULP3 prevented ciliary cAMP signaling in 3T3-L1 cells (Fig S6E).
We investigated which cAMP second messenger system mediates the cAMP signal. cAMP can activate at least two downstream pathways, protein kinase A (PKA) and exchange factor directly activated by cAMP (EPAC). To determine which of these pathways is activated by ciliary FFAR4, we titrated increasing amounts of established PKA or EPAC inhibitors onto 3T3-L1 cells differentiating in DHA cocktail. Inhibition of EPAC, but not PKA, prevented DHA-enhanced adipogenesis (Fig 6G). Specifically, EPAC inhibitor ESI-09 attenuated adipogenesis induced by DHA cocktail at levels consistent with its reported IC50 (~5μM), whereas high level (125μM) PKA inhibitor Rp-cAMPs (IC50 ~5μM) did not. Thus, DHA initiates the adipogenic program through activation of localized ciliary cAMP and EPAC, resulting in cell cycle re-entry. Moreover, DHA can replace IBMX as a differentiation factor to induce adipogenesis in vitro.
DHA and FFAR4-induced adipogenesis requires CTCF, a regulator of chromatin architecture
Having identified ciliary cAMP and EPAC as mediators of DHA cocktail-induced adipogenesis, we examined how FFAR4 affects the adipogenic transcriptional program. Adipogenic transcriptional regulators collectively activate two adipogenic master transcription factors, PPARγ and CEBPα. DHA cocktail induces PPARγ and CEBPα (Fig 7A, B; Fig S7A). To understand the kinetics of PPARγ activation, we generated a reporter 3T3-L1 cell line, appending an open reading frame encoding a self-cleaving peptide T2A and green fluorescent protein to the 3’ end of the endogenous PPARγ locus, such that induction of PPARγ can be monitored by assessing green fluorescence intensity over time. PPARγ was induced after 48h of DHA cocktail treatment, and this was FFAR4 dependent (Fig S7B).
Figure 7. FFAR4 activates PPARγ and CEBPα via CTCF-dependent chromatin remodeling.

(A-B) Addition of DHA cocktail to 3T3-L1 cells increased (A) PPARγ and (B) CEBPα protein levels. 3 independent samples shown per time points. (C-D) Schematic of next generation RNA sequencing experiment. 3T3-L1 cells were treated for 24h with ctrl cocktail, DHA cocktail, or historical DM. (C) Venn-diagram of all significantly altered genes (q<0.05) with greater than 2 fold up- or downregulation compared to 0h. Data describes 3 independent experiments. (D) Gene ontology enrichment analysis of all genes significantly upregulated more than 2 fold in response to both DHA cocktail and historical DM. (E) Chromatin accessibility as assessed by ATAC-seq shows enrichment of CTCF binding motifs in open chromatin after 4h of DHA only treatment. (F) Known enhancer-adipogenic gene promoter loops that contain a CTCF binding motif and are opened by DHA. Fold change describes average increase in accessibility from 2 independent experiments. (G) Loss of CTCF attenuates expression of adipogenic genes in response to FFAR4 activation. Bar graph is average of three independent experiments ± SD. (H) Immunoblot showing depletion of CTCF protein in 3T3-L1 cells. (I) CTCF is required for 3T3L1 adipogenesis induced by FFAR4 agonist cocktail. Bar graphs are normalized mean ± SD. * p<0.05; *** p<0.001. See also Figure S7.
We assessed the expression and activation of known early adipogenic transcription factors and regulators. DHA cocktail induced many known adipogenic factors, including the CEBPβ transcription factor and phosphorylation of AKT, ERK, and CREB (Fig S7C, D). However, while these adipogenic mediators were required for DHA cocktail-induced adipogenesis (Fig S7E), control cocktail (-DHA) induced equivalent phosphorylation of AKT, ERK, and CREB (Fig S7C, D), arguing that these previously described adipogenic regulators are not specifically induced by DHA.
We therefore performed unbiased next generation sequencing to identify gene expression changes specifically induced by DHA. Specifically, we compared gene expression of confluent, undifferentiated 3T3-L1 cells to 3T3-L1 cells treated with historical DM, DHA cocktail, or control cocktail for 24 hours (Table S2). There was a dramatic shift in gene expression for all three induction conditions even at this early time point (Fig 7C). Since both DM and DHA cocktail, but not control cocktail promote robust adipogenesis, these data argue that most of the gene expression changes observed are not important for adipogenesis. Specifically, the ~1300 gene changes induced by DM only and the ~100 genes controlled by the DHA cocktail only may not be relevant to adipogenesis, focusing us on the 136 genes that were induced by both. The smaller number of genes affected by the DHA cocktail provides additional evidence that this formulation may be more physiologically relevant to the initiation of adipogenesis.
To further explore the genes upregulated by both DM and DHA cocktail, we analyzed Gene Ontology, which suggested that chromatin binding may be regulated (Fig 7D). We assessed how DHA treatment affects chromatin accessibility by performing an assay for transposase-accessible chromatin sequencing (ATACseq) at different differentiation time points followed by transcription factor motif analysis (Table S3). We specifically compared chromatin accessibility of confluent, undifferentiated 3T3-L1 cells with 3T3-L1 cells treated with DHA only for 4 hours, since this is the earliest time point when we observe consistent changes in chromatin accessibility (data not shown), and we wanted to interrogate the direct effects of DHA and not secondary effects due to initiation of the adipogenic transcriptional program. As a general control for the ATACseq method, we also assessed chromatin accessibility of 3T3-L1 cells treated with Dex alone for 4 hours, a well-established glucocorticoid receptor agonist that mediates DNA-binding of its receptor. Transcription factor motif analysis confirmed that Dex treatment caused a dramatic increase in chromatin opening at glucocorticoid receptor (Nr3c1) binding sites, as well at sites for adipogenic transcription factors such as CEBP proteins and KLF proteins (Fig 7E). Intriguingly, DHA treatment increased chromatin access at CTCF binding motifs (Fig 7E). CTCF is a regulator of transcription and chromatin architecture. Thus, these data suggest that DHA promotes adipogenesis by inducing CTCF-mediated chromatin remodeling to activate the adipogenic transcriptional program.
Previous studies have shown dynamic chromatin remodeling of promoter-enhancer interactions during 3T3-L1 differentiation including at CTCF binding sites, and that these interactions correlate with adipogenic gene expression (Dubois-Chevalier et al., 2014; Siersbaek et al., 2017). We hypothesized that DHA induces adipogenesis by promoting formation of chromatin loops between enhancers and promoters of adipogenic genes in a CTCF-dependent manner. To test this hypothesis, we identified which CTCF motif-containing regions were opened in response to DHA, as well as the proximal promoters (Table S4). CTCF was recruited to regulatory regions for Adipoq, Cebpα, and Fabp4 (Fig 7F).
To further test our hypothesis that CTCF mediates DHA-enhanced adipogenesis, we generated 3T3-L1 cells lacking CTCF (Fig 7H; Fig S7I). Loss of CTCF did not affect cell proliferation (Fig S7F). We induced adipogenesis of control and Ctcf knockout cells with DHA cocktail. Expression of Adipoq, Cebpα, and Fabp4 were induced, and this was partially dependent on CTCF (Fig 7G). Moreover, loss of CTCF significantly attenuated DHA-mediated adipogenesis (Fig 7I), but had no effect on adipogenesis induced by the historical DM (Fig S7G). We confirmed that CTCF is required for DHA-enhanced adipogenesis in multiple independent 3T3L1 cell lines lacking CTCF (Fig S7H–I). Thus, DHA may promote adipogenesis by inducing formation of chromatin loops between enhancers and the promoters of adipogenic genes via CTCF.
Discussion
By focusing on the primary cilium of preadipocytes, we have identified a strong candidate for a physiologically relevant ligand-receptor pair that activates early events in adipogenesis. We found that primary cilia identify preadipocytes in vivo. Genetic ablation of cilia in preadipocytes blocks adipogenesis in vivo and strongly reduces fat mass. The underlying molecular mechanism requires signaling by the ω−3 fatty acid receptor FFAR4/GPR120 at preadipocyte cilia. DHA activates ciliary FFAR4, resulting in a rapid increase in ciliary cAMP levels. Ciliary cAMP in turn promotes adipogenesis by activating the EPAC effector protein, a guanine-nucleotide exchange factor. DHA induces chromatin remodeling through CTCF, inducing adipogenic genes and adipogenesis. Several key conclusions emerge.
ω−3 fatty acids promote adipogenesis through FFAR4
This current study establishes a cell autonomous signaling function for FFAR4 in initiating adipogenesis in preadipocytes. Specifically, in vivo, ex vivo, and in vitro loss of preadipocyte ciliation impairs adipogenesis, while activation of ciliary FFAR4 signaling dramatically promotes adipogenesis. We do not understand why we did not observe systemic hallmarks of lipodystrophy despite the dramatic failure to expand WAT tissue in PAno cilia mice, and we propose that the residual amount of WAT in PAno cilia mice (~5g) was sufficient to buffer the extra expended energy within the experimental time frame. We further postulate that removing preadipocyte cilia prior to 3 weeks of age, extension of the physiological analysis beyond 20 weeks of age, or feeding mice a HFD could reveal systemic metabolic consequences of preadipocyte ciliation loss.
Previous studies have described the metabolic role of FFAR4 in promoting glucose uptake in adipocyte and in mediating the systemic anti-diabetic effects of ω−3 fatty acids, including DHA (Ichimura et al., 2012; Oh et al., 2010; Oh et al., 2014; Suckow et al., 2014). Taken together, our data suggest that the anti-diabetic effects of DHA and FFAR4 are mediated in part by shifting WAT expansion towards hyperplasia rather than adipocyte hypertrophy or deposition of fat in other tissues including liver. These data provide a possible understanding of the anti-diabetic effects of dietary DHA supplementation: DHA increases the generation of more adipocytes with lower fat content per adipocyte.
A mitogenic role for ω−3 fatty acids: nutritive fluxes trigger cell cycle entry and stem cell expansion
In preadipocytes, cAMP is important for cell cycle re-entry and differentiation, reflected by the inclusion of IBMX in the historical DM. By replacing IBMX with DHA and establishing a direct and rapid link to cAMP elevation in the primary cilium, we link a physiological ligand to cAMP production in preadipocytes. This finding raises critical questions. What is the threshold for DHA signaling in vivo? Are there differential responses in different WAT depots? How do other adipogenic factors such as insulin affect DHA signaling?
Epigenetic programs for adipogenesis
The importance of transcription factors in controlling adipogenesis is well-characterized (Rosen et al., 2000). The chromatin regulatory factors that cooperate with adipogenic transcription factors are less well identified. A recent study highlighted a dynamic rewiring of promoter-enhancer loops by 3T3-L1 preadipocytes within four hours of initiation of adipogenesis (Siersbaek et al., 2017). We found that four hours of Dex only treatment opens chromatin and affects transcription factor binding, consistent with previous reports (Siersbaek et al., 2012). CTCF, a factor important for organizing chromatin loops, mediates DHA-dependent adipogenesis by promoting expression of the adipogenic genes such as Adipoq, Cebpα, and Fabp4. Further investigation may determine how adipogenic signals including DHA control CTCF function and chromatin looping.
The primary cilium as a sensor of multiple growth factors and nutrients
There is growing appreciation for the primary cilium as a sensory organelle. We show that preadipocytes in vitro and in vivo are ciliated, and that ciliated perivascular cells account for almost 30% of all perivascular cells. It remains to be elucidated how uniform or heterogeneous this population may be with regard to stemness and adipogenic potential, and further studies to define stem cell and signaling populations will be important. It would be particularly interesting to examine ciliation in preadipocyte subpopulations within fat tissue, as recently revealed through the use of single cell sequencing (Burl et al., 2018; Hepler et al., 2018; Merrick et al., 2019; Schwalie et al., 2018).
Mice lacking ciliated preadipocytes fail to expand WAT in vivo, highlighting the importance of cilia to a mesenchymal progenitor cell function and raising the possibility that defects in mesenchymal progenitor cells may contribute to human ciliopathies. The ciliary DHA/FFAR4 signaling pathway provides a molecular hypothesis about how ciliary dysfunction in ciliopathies may contribute to metabolic dysfunction. A recent study also described that adipose-derived mesenchymal stem cells from WAT of obese individuals display shorter, signaling defective cilia compared to ASCs isolated from lean WAT, suggesting that obesity may also impact ciliary signaling (Ritter et al., 2018). Future investigations are required to determine whether defective ciliary FFAR4 signaling leads to metabolic dysfunction.
The primary cilium likely senses multiple pro- and anti-adipogenic signals, but the interplay of different ciliary signaling pathways within a single cilium is unclear. Conceivably, the primary cilium integrates multiple signals, organizing outputs to secondary messengers, including cAMP and calcium. 3T3-L1 primary cilia, in addition to FFAR4, display IGF-1 receptor (Zhu et al., 2009). Our data show that DHA can partially substitute for insulin, raising the possibility of crosstalk between DHA and insulin signaling at the cilium.
FFAR4 signaling in ciliated preadipocytes versus mature, unciliated adipocytes
FFAR4 expression increases dramatically during adipogenesis, and the receptor is highly expressed in mature unciliated adipocytes. As the primary cilium is a cylinder with an approximate diameter of 200–300nm and a median length of 3–5μm, an average adipocyte with a diameter of 100μm would need to express almost 10,000 times more FFAR4 receptors to achieve the same receptor density as is achieved by ciliary FFAR4 in preadipocytes. We propose that concentrating FFAR4 in the cilium sensitizes its responses to enable the initiation of adipogenesis in perivascular preadipocytes, and that DHA and FFAR4-mediated activation of CTCF may provide a robust mechanism for sustaining expression of high levels of FFAR4 in mature adipocytes.
Differentiated unciliated cells may therefore repurpose formerly ciliary receptors, and in the same tissue, ciliated and unciliated cells may use the same receptor to activate distinct effector pathways at different subcellular locales to coordinate the overall tissue response to a ligand. DHA can promote tissue expansion by initiating adipogenesis of preadipocytes and promoting hypertrophy of adipocytes (Oh et al., 2010). Confining FFAR4 to the primary cilium in preadipocytes, versus the plasma membrane in mature adipocyte, may selectively activate distinct effector pathways; our data suggests that ciliary FFAR4 in preadipocytes couples to Gαs, while FFAR4 in the plasma membrane of mature adipocytes has previously been shown to couple to Gαq (Oh et al., 2010). This differential coupling may allow for distinct responses to DHA for cells of different states within the same tissue.
Saturated versus unsaturated fatty acids: how to make healthy fat
The ability to both deposit and mobilize fatty acids in response to nutritive cues is critical for fat to remain metabolically healthy (Ghaben and Scherer, 2019). Defective fatty acid transport can lead to adipose tissue inflammation, fibrosis, and diabetes. We have found that a ω−3 fatty acid, DHA, stimulates expansion of preadipocytes, but that saturated and mono-unsaturated fatty acids do not. Accordingly, the proportion of ω−3 and saturated fatty acids, and the ability to sense this ratio, may determine the balance between hyperplastic and hypertrophic adipose tissue expansion. Consistently, ω−3 fatty acid supplementation results in improved insulin sensitivity and decreased adipose tissue inflammation in humans and mice (Gao et al., 2017; Oh et al., 2010; Spencer et al., 2013).
Star Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Jackson (pjackson@stanford.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In vivo animal studies
Gt(ROSA)26Sortm1(EYFP)Cos and Pdgfrα-CreERT mice (Tg(Pdgfrα-cre/ERT)467Dbe) have been described previously (Kang et al., 2010; Srinivas et al., 2001). Ffar4 knockout mice in a C56BL/6 background were generated by in vitro fertilization using sperm from Ffar4tm1(KOMP)Vlcg obtained from KOMP. Cilia-Glow mice (Tg(CAG-Arl13b/mCherry)1Kv) and Tg(CAG-EGFP/CETN2)3−4Jgg/KvandJ)) were purchased from Jackson Laboratory (027967). Ift88flox/flox mice (B6.129P2-Ift88tm1Bky/J)) were purchased from Jackson Laboratory (022409). C57Bl/6J mice were purchased from Jackson Laboratory (000664).
All mice were maintained under specific pathogen-free conditions at the Stanford and UCSF animal care facility. Mice were cared for and all experiments were approved by the Administrative Panel on Laboratory Care and the Institutional Animal Care and Use Committee (IACUC) of at Stanford University and UCSF. Animals were housed in groups of 5 adults per cage. Male mice between 6–8 weeks of age were used for isolation of murine primary preadipocytes and whole mount WAT imaging. Unless otherwise stated, mice were kept on normal chow diet. For the high fat diet experiments, male littermates between the ages of 6–8 were split and put on 60% HFD or the matched control diet (ResearchDiets D12492, D12450J). To measure in vivo activation of preadipocytes in response to HFD, mice were given BrdU (Abcam) in the drinking water at 0.8mg/ml for the entire duration of the HFD experiment (2 weeks), with fresh BrdU in the water provided every 2–3 days. To assess effects of TUG‐891 on body weight, 9‐week‐old male C57Bl/6J mice (JAX) were randomized to receive an intraperitoneal injection with either TUG‐891 (35 mg/kg) or 10% dimethyl sulfoxide (DMSO) vehicle dissolved in corn oil once daily for 2 weeks. Starting the second week, mice were split and put on 60% HFD or the matched control diet (ResearchDiets D12492, D12450J) for 3 months. To assess the importance of preadipocyte ciliation, Tamoxifen (Sigma T-5648), dissolved in corn oil, was administered to Pdgfrα-CreERT Ift88 conditional mice by oral gavage (200–250 mg/kg) on two consecutive days at 3 or 6 weeks of age and sex as indicated. In addition, all control mice regardless of gender or genotype also received tamoxifen. At day of tamoxifen treatment, mice were put on breeder chow (Lab Diet #5058). Pdgfrα-CreERT Ift88 conditional mice were maintained by crossing to CD1 mice (Charles River).
Cell line models
3T3-L1 cells were cultured in DMEM medium containing 10% Bovine Calf Serum, 1% Pen/Strep, and 1% GlutaMAX and switched to DMEM containing 10% FBS, 1% Pen/Strep, and 1% GlutaMAX during adipogenesis.
Human white preadipocytes were cultured in Preadipocyte Growth Medium, differentiated in Preadipocyte Differentiation Medium, and adipocytes were maintained in Adipocyte Nutrition Medium (PromoCell).
Murine primary preadipocytes were isolated from inguinal or epididymal white adipose tissue from wild-type, cilia-glow, Ffar4 knockout, and Ift88flox/flox male mice using mouse protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Stanford University. Primary preadipocytes were maintained and differentiated in DMEM medium containing 10% FBS, 1% Pen/Strep, and 1% GlutaMAX.
METHOD DETAILS
Plasmids
pMCB306 (lentiviral vector, loxP-mU6-sgRNAs-puro resistance-EGFP-loxP) and p293 Cas9BFP were gifts from Prof. Michael Bassik (Stanford University). pCMV-VSV-G and pCMV-dR8.2 dvpr were gifts from Prof. Bob Weinberg (Addgene plasmid #8454 and #8455) (Stewart et al., 2003). Lentiviral vectors containing sgRNA were generated by ligating annealed sgRNA oligonucleotides into pMCB306 vector digested with BstXI and BlpI restriction enzymes.
pBabe LAPN and LAPC plasmids are retroviral gateway destination vectors containing N- and C-terminal LAP (eGFP-TEV cleavage site-S tag-Precision cleavage) tags as previously described (Mukhopadhyay et al., 2010). Expression constructs were generated by Gateway LR reactions between entry clones and destination vectors.
Cell Line generation
Retroviral vectors carrying the gene of interest were transfected into Phoenix Eco-Env cells using Fugene6 (Promega), and lentiviral vectors carrying the gene of interest were co-transfected with pCMV-VSV-G and pCMV-dR8.2 dvpr into 293T cells. Media was replaced after 24h and virus was harvested 48 and 72h post-transfection. Virus was filtered with a 0.45μm OVDF filter (Millipore) and used to infect cell lines with 10μg/ml polybrene (Millipore). Media was replaced after 24h and cells were sorted for GFP positivity after 48–72h post-infection.
To generate Crispr/Cas9 knockout cells, 3T3-L1 Cas9-BFP cells were infected with lentivirus containing the sgRNA of interest. Knockout efficiency was determined 10 days post-infection by TIDE analysis (Brinkman et al., 2014). Primers listed in Table S5.
3T3-L1 cells expressing Cas9-BFP were generated by infection of virus harvested from 293T cells transfected with p293 Cas9-BFP, pCMV-VSV-G and pCMV-dR8.2 dvpr. 3T3-L1 Cas9-BFP cells were sorted for BFP positivity.
For rescue experiments of 3T3-L1 sgRNA cells, the sgRNA was removed by adenoviral infection with recombinant Cre, followed by sorting for GFP negativity.
For knockdown experiments using siRNA, 3T3-L1 cells were plated and transfected with 25nM siRNA after 24h using DharmaFECT Reagent 1. Media was changed after 24h, and differentiation was initiated 48h post-transfection.
Immunofluorescence staining
Cells were grown on 12mm round coverslips and fixed with 4% paraformaldehyde (433689M, AlfaAesar) in PBS at room temperature for 10min. Samples were blocked with 5% normal donkey serum (017–000-121, Jackson ImmunoResearch) in IF buffer (for FFAR4 staining: 3% BSA and 0.4% saponin in PBS; for all else: 3% BSA and 0.1% NP-40 in PBS) at room temperature for 30min. Samples were incubated with primary antibody in IF buffer at room temperature for 1h, followed by 5 washes with IF buffer. Samples were incubated with fluorescent-labeled secondary antibody at room temperature for 30min, followed by a 5 min incubation with 4’,6-diamidino-2-phenylindole (DAPI) in PBS at room temperature for 5min and 5 washes with IF buffer. Coverslips were mounted with Fluoromount-G (0100–01, SouthernBiotech) onto glass slides followed by image acquisition. Antibodies were used as follows: Pericentrin (Covance, PRB-432C, 1:500), Acetylated tubulin (Sigma, T7451, 1:2000), CEP170 (Thermo, 41–3200, 1:500), ARL13B (UC Davis/NIH NeuroMab Facility, 73–287, 1:1000), FFAR4 (Yenzym, 1:500), FFAR4 (Santa Cruz, sc-390752, 1:100), FGFR1OP (Novus, H00011116-M01, 1:1000), GFP (Invitrogen, 1:2000, A10262).
Immunohistochemistry
WAT tissue was fixed in Zink-buffered formalin overnight at RT, paraffin embedded and sectioned. Tissue sections were de-waxed and rehydrated before undergoing standard Haemotoxylin and Eosin (H&E) staining. To assess the area of individual adipocytes, H&E stained WAT sections were thresholded and adipocyte area quantified using Analyze Particle function in ImageJ.
Antibody generation
The rabbit polyclonal antibody against FFAR4 was generated by rabbit injections (Yenzym, Cys-PILYNMSLFRNEWRK) followed by affinity purification using standard protocols.
Whole mount white adipose staining
Mice were perfused with PBS followed by 4% PFA in PBS. White adipose tissue was removed and cut into narrow strips and further fixed in 4% PFA and 0.3% Triton-X in PBS for 15min at room temperature. WAT strips were washed 3 times for a total of 1h in 0.3% Triton-X/PBS (PBST) at room temperature and blocked overnight at 4C in 3% normal donkey serum/PBST. WAT strips were incubated with primary antibody in PBST for at least 6h at room temperature, followed by 3 30min washes in PBST at room temperature. WAT strips were incubated with fluorescent-labeled secondary antibody (1:500) in PBST overnight at 4C, followed by incubation with 2μg/ml DAPI/PBST for 30min at room temperature and 2 more 30min washes in PBST at room temperature. WAT strips were cut into small pieces (~2mm3) and mounted with Glycergel Mounting media (Dako, C056330–2) containing 0.02g/ml 1,4-Diazabicyclo[2.2.2]octane onto glass slides into a chamber created by stacking 2 coverslips.
For BrdU staining, the protocol was amended as follows: Strips of WAT post-fixation with PFA and 3 washes in PBST were incubated in 1N HCl at 37 °C for 30min. WAT strips were then washed 3 times in PBS and incubated in 0.1M borate buffer (pH 8.5) for 10min. WAT strips were washed 3 times for 10min each in PBST and then blocked overnight followed by staining as described above.
Antibodies were used as follows: CD31 (BD, 553370, 1:200), Pericentrin (Covance, PRB-432C, 1:500), Acetylated tubulin (Sigma, T7451, 1:1000), BrdU (Cell Signaling, 5292, 1:500), GFP (1:1000, Avis lab #1020), PDGFRα (1:2500, R&D Systems #AF1062), ARL13B (1:1000, Proteintech #17711–1-AP), LipidTox (1:250, Invitrogen, H34475).
Epi-fluorescence and confocal imaging
Images were acquired on an Everest deconvolution workstation (Intelligent Imaging Innovations) equipped with a Zeiss AxioImager Z1 microscope and a CoolSnapHQ cooled CCD camera (Roper Scientific) and a 40x NA1.3 Plan-Apochromat objective lens (420762–9800, Zeiss) was used.
Confocal images were acquired on a Marianas spinning disk confocal (SDC) microscopy (Intelligent Imaging Innovations).
For Figs 2 and S2, images were acquired using a Leica DMi8 microscope equipped with a DFC7000T color camera (bright field images) as well as the SPE confocal system (immunofluorescence).
In vitro Adipogenesis
3T3-L1 cells were grown to confluency in DMEM containing 10% Bovine Calf Serum, followed by another 2 days at confluency in DMEM containing 10% Bovine Calf Serum. Adipogenesis was then induced using DMEM containing 10% FBS and differentiation cocktail consisting of insulin, dexamethasone, IBMX and/or DHA. 1x DM is 1μg/ml insulin, 1μM Dex, 0.5mM IBMX. DHA cocktail is 0.4μg/ml insulin, 0.02mM IBMX, 0.1μM Dex, 100 μM DHA. FFAR4 agonist cocktail is 0.4μg/ml insulin, 0.02mM IBMX, 0.1μM Dex, 100 μM TUG891. Ctrl cocktail is 0.4μg/ml insulin, 0.02mM IBMX, 0.1μM Dex, EtOH/DMSO. Where noted, palmitic acid was added at 100μM, oleic acid was added at 100μM, FFAR4 agonist TUG891 was added at 100μM, and FFAR1 agonist TUG424 was added at 100μM for the first 2 days of differentiation. After 2 days of differentiation cocktail, media was changed to DMEM containing 10% FBS and 1μg/ml insulin. Maintenance media was changed every 2–3 days for a total differentiation time of 4–8 days.
Mouse primary preadipocytes were grown to confluency in DMEM containing 10% FBS. Cells were not grown for another 2 days at confluency. Adipogenesis was induced using DMEM containing 10% FBS and differentiation cocktail consisting of insulin, dexamethasone, IBMX and/or DHA or TUG891. After 3 days of differentiation cocktail, media was changed to DMEM containing 10% FBS and 1μg/ml insulin. Maintenance media was changed every 2–3 days for a total differentiation time of 4–8 days.
Human primary preadipocytes were grown to confluency in Growth Media (PromoCell). Adipogenesis was induced using Differentiation Media (PromoCell). After 3 days, media was changed to Nutrition Media (PromoCell) for another 12–14 days with media changes every 2–3 days.
Isolation of primary preadipocytes
Inguinal or epididymal white adipose tissue was removed and minced. Minced tissue was incubated in Collagenase Buffer (3,000 U/ml type II collagenase powder (Sigma, C6885), 100 U/ml DNase (Worthington, LS006344), 1 mg/ml poloxamer 188 (P-188) (Sigma P5556), 1 mg/ml BSA, 20 mM HEPES buffer, and 1 mM CaCl2 in Medium 199 with Earle’s salts (Sigma, M4530)) for 10min at 37C, followed by 20min shaking (250rpm) at 37C. Digested samples were strained through a 100μm filter and diluted 1:1 in cell suspension buffer (2% FBS, 1 mg/ml P-188, and 1% pen/strep in PBS), followed by centrifugation (1300rpm, 5min).
For isolation of SVF cells depleted for RBCs and WBCs, the cell pellet was resuspended in cell suspension buffer and layered onto Histopaq-1077 (Sigma, 10771), followed by 20min centrifugation at 1300rpm. PBMC layer was removed and centrifuged (5min, 1300rpm), and cell pellet was resuspended in cell suspension buffer containing anti-CD45 microbeads (Miltenyi Biotec, 130–052-301) and anti-TER119 microbeads (Miltenyi Biotec, 130–049-901) for 30min at 4C, followed by MACS depletion using an LD column. Flow-through was collected and centrifuged (1300rpm, 5min). Cell pellet containing primary preadipocytes were resuspended in DMEM containing 10% FBS, 1% Pen/Strep, and 1% GlutaMAX.
For isolation of Lin− (CD45− CD31− TER119−) CD34+ CD29+ SCA1+, the cell pellet post-digestion was resuspended in cell suspension buffer and stained with CD45 PE-Cy7 (eBioscience, 250451–81, 30-F11 clone, 1:800), CD31 PE-Cy7 (eBioscience, 25–0311-81, 1:800), TER119 PE-Cy7 (eBioscience, 25–5921-81, 1:800), CD34 Alexa Fluor 700 (eBioscience, 56–0341-82, 1:200), CD29 APC (eBioscience, 17–0291-80, 1:400), SCA1 Pacific Blue (Biolegend, 108120, 1:1000), propidium iodide for 20min, washed twice in cell suspension buffer, filtered and sorted on a FACSAria Fusion sorter.
To knock-out Ift88 in isolated primary preadipocytes, cells were sorted for Lin− (CD45− CD31− TER119−) CD34+ CD29+ SCA1+, and then plated on collagen-coated plates. Cells were grown to confluency and then transduced with an adenovirus expressing GFP only or GFP and Cre Recombinase (Vector BioLabs, 1060, 1700) at 3e7 PFU/cm2 of plate surface area. After 48h, cells were either differentiated, or trypsinized and plated onto a coverslip for differentiation or ciliation assays. GFP signal was amplified using an antibody against GFP and lipid droplets were visualized using LipidTox (Thermo, H34477, 1:200). Percent ciliation was determined by FGFR1OP (basal body) and ARL13B (axoneme) staining of GFP+ cells.
Sample preparation and immunoblot
Cells were lysed in 1x LDS buffer containing DTT and incubated at 95C for 5min. Proteins were separated using NuPage Novex 4–12% Bis-Tris protein gels (Thermo Fisher Scientific, WG1402BOX) in NuPage MOPS SDS running buffer (50 mM MOPS, 50 mM Tris Base, 0.1% SDS, 1 mM EDTA, pH 7.7), followed by transfer onto nitrocellulose membranes (Life Technologies, LC2001) in Towbin Buffer (25 mM Tris, 192 mM glycine, pH 8.3) containing 10% methanol. Membranes were blocked in LI-COR Odyssey Blocking Buffer (LI-COR, NC9232238) for 30 min at room temperature, followed by incubation with primary antibody in blocking buffer for at least 1h at room temperature. The membrane was washed 3 times for 10min in TBST buffer (20 mM Tris, 150 mM NaCl, 0.1% Tween 20, pH 7.5) at room temperature, incubated with secondary IRDye antibodies (LI-COR) in blocking buffer for 30 min at room temperature, and then washed 3 times for 10min in TBST buffer. Membranes were scanned on an Odyssey CLx Imaging System (LI-COR), with protein detection at 680 and 800 nm. Antibodies were used as follows: TULP3 (Yenzym, 1:500), PPARγ (Cell signaling, 2435, 1:1000), Tubulin (Sigma, 9026, 1:5000), CEBPα (Cell signaling, 8178, 1:1000), CTCF (Cell signaling, 2899, 1:1000), ERK (Cell signaling, 9107, 1:1000), pERK (Cell signaling, 9101, 1:1000), AKT (Cell signaling, 9272, 1:1000), pAKT (Cell signaling, 9271, 1:1000), CREB (Cell signaling, 9104, 1:1000), pCREB (Cell signaling, 9198, 1:1000)
Glucose and serum analysis
To measure glucose levels, a small drop of blood from the tail was placed on a human glucometer (Bayer). Food was removed in the late afternoon and fasting glucose levels measured the next day. To minimize stress, mice were handled for one week to acclimate. For serum analysis, blood was collected via cardiac puncture and serum harvested. ELISAs for insulin (ALPCO, 80-INSMSU-E01), leptin (ALPCO, 22-LEPMS-E01) and free fatty acids (Wako, NEFA-HR2) was done according to manufacturer’s instructions. Data were plotted in GraphPad.
Oil red O staining for lipid visualization and quantification
Cells are fixed in 4% PFA/PBS for 10min at room temperature, followed by 3 rinses in PBS. Samples were incubated in 60% isopropanol for 5min at room temperature and then allowed to dry completely. Samples are then incubated in freshly diluted 60% Oil Red O staining solution in water (stock is 0.5% Oil Red O (Sigma, 00625) in isopropanol) for 20min at room temperature, followed by 3 rinses in water. Samples were allowed to dry completely and imaged.
For quantification, Oil Red O was extracted by incubating dried samples stained on the same day in 100% isopropanol for 5min at room temperature and absorbance was measured at 510nm.
To visualize lipids in liver tissue section, liver samples were fixed in 4% PFA for 2 hrs at 4C, cryprotected in 30% sucrose overnight before cyrosectioning. Tissue sections were air dried, before staining was performed. Slides were mounted with FluoroMount G before visualized. To quantify the area occupied by Oil Red O, images were converted to binary images using the Threshold function and quantified using the Analyze Particle function in ImageJ.
Live Cell Imaging Analysis for adipogenesis and proliferation
Live imaging for kinetics quantification of adipogenesis and for proliferation assay was performed using the IncuCyte Live Cell Analysis Imaging System (Essen Bioscience) with images acquired every 2h using the 10x objective. Proliferation was assessed using a confluency mask generated by the IncuCyte Zoom Analysis Software using phase images. For the kinetic quantification of adipogenesis, 3T3-L1 cells were differentiated as described above and supplemented with 200nM BODIPY 493/503, and green fluorescence images were acquired every 2h using default IncuCyte setting. Total green fluorescence intensity was determined from a green fluorescent mask generated by the IncuCyte Zoom Analysis Software.
Echo-MRI and CLAMS
To assess fat vs. lean mass, whole composition analysis was performed using the EchoMRI3in1™ machine from Echo Medical System according to manufacturer’s instructions. To assess any changes in metabolism, we used the Comprehensive Lab Animal Monitoring System (CLAMS) made by Columbus Instruments, Inc. In brief, mice were single-housed in a large enclosure with precise control over the temperature and light / dark cycle. All values are normalized to body weight. Data were analyzed using the CLAX software from Columbus Instruments, exported into Excel and plotted in GraphPad.
Quantitative Real time PCR
RNA was extracted using the RNeasy Lipid Tissue Kit (QIAGEN) and cDNA was synthesized using M-MLV Reverse Transcriptase (Invitrogen, 28025–013). Quantitative real time PCR was performed using TaqMan Probes (Invitrogen) and the TaqMan Gene Expression Master Mix (Applied Biosystems, 4369016) in 96-well MicroAmp Optical reaction plates (Applied Biosystems, N8010560).
For Fig S2A & S2H, whole white or brown adipose tissue was placed in in TRIzol (Thermo Fisher) and disrupted using the TissueLyser LT (Qiagen) and RNA was isolated using the Qiagen RNeasy kit. cDNA was synthesized using the qScript cDNA synthesis kit (Quantabio). RT-qPCR was performed in technical triplicates on a Quant Studio 6 real-time PCR machine (Applied Biosystems) using the PowerUp master mix (Invitrogen). Fold changes were calculated using the 2−ΔΔCT method (Schmittgen & Link 2008) and expression levels normalized to the average of the housekeeping genes Hprt and Pde12.
EdU incorporation for cell cycle analysis
EdU incorporation was assessed using the Click-iT EdU Imaging Kit (Thermo, C10340) according to manufacturer’s recommendation. Briefly, 3T3-L1 were grown to confluency in a 96well cell imaging plate (Eppendorf, 0030741013) and kept at confluency for 2 days.
Differentiation was initiated using the differentiation cocktail indicated in the figure legend, and EdU was added for a final concentration of 1μM after 16h of differentiation. After a total of 40h of differentiation, cells were fixed in 4% PFA/PBS for 15min at room temperature, followed by 2 rinses in 3%BSA/PBS. Cell were permeabilized in 0.5% Triton-X/PBS for 20min at room temperature, followed by 2 rinses in 3%BSA/PBS. Samples were then incubated in freshly made Click-iT reaction cocktail for 30min at room temperature, followed by 1 rinse in 3%BSA/PBS and 1 rinse in PBS. Samples were then incubated in 2μg/ml Dapi in PBS for 30min at room temperature followed by 1 rinse in PBS. Samples were stored in PBS and images were acquired using a Keyence fluorescent microscope (10x objective). EdU positivity was assessed using the CellProfiler pipeline.
Live Cell Ciliary cAMP assay
3T3-L1 cells were seeded at 1e4 cells/well in a 96-well cell imaging plate (Eppendorf, 0030741013) and transduced the following day with the ratiometric cilia-targeted cADDis BacMam (Molecular Montana, D0211G) according to manufacturer’s recommendation. Briefly, cell were infected with 25ul of BacMam sensor stock in a total of 150ul of media containing 2mM Sodium Butyrate (Molecular Montana) for 30min at room temperature followed by 6h in the 37C tissue culture incubator. BacMam was removed and replaced with DM containing 1mM Sodium Butyrate for 16–24h. Prior to imaging, cells were incubated in PBS for 20min at room temperature. Images were acquired on a Marianas spinning disk confocal (SDC) microscopy (Intelligent Imaging Innovations) (40x, epi-fluorescence) every 10 seconds for 5min with agonist added after 30sec. Red fluorescence was used to determine a mask and background subtracted green and red fluorescent intensity over time was determined using Slidebook (Intelligent Imaging Innovations).
PPARγ reporter cell line generation
The donor plasmid containing a 5’ homology arm (with stop codon mutated), T2A, EGFP, and 3’ homology arm was generated by Gibson Assembly of pUC19 plasmid linearized by BamHI and EcoRI digestion, Gibson fragment 1 (GCTTGCATGCCTGCAGGTCGACTCTAGAGGACCCCTCCAAAGTGAAGCAGTCTTTTATCT TGCTGGTAATGGGATGTGTTTGTGCTAAGCAAAAAAACAGCAAGGCTTACACTTGAAAAAT CTTATTTGAAGCTGTTATGAGAATGTAGGTAGAAGTTTAAAAAGGAAAACAAAATAAAACAA AACAAAAACAAACAAACAAACAAAACACTCCTCCAGGAGCAAAGGTTGGTAATGTGATTTCT GTGAGGAGAAGCATGTTGCCAGAAGAGGCCTTGGGTTGAAGACAGGATGCTCCTGATGGC ATTGCATCAGTTAGTCCTGGTTGGGAAGGGCTTGAGCCTTTGATTCCATCCTTGGGTCTTG CCCATTAGCTGCAGAGCCTCGCTCCACGAACCTGCTTAAGAAACAGGGCGGTGATGGGGT CTTGGCTCTTCGGTAAAGCATGTGCCTAACAGCTCGAGAACTGGGTTTCTTCTCCAGCCTG GGGAGCAGGGAATCTGAAGCTGCACTCCTTAGAGCCCCAGAGGAGGTCTGACAAAGCCTC TTTTTGTCTTCTCATTCTCCCAGACCGCCCAGGCTTGCTGAACGTGAAGCCCATCGAGGAC ATCCAAGACAACCTGCTGCAGGCCCTGGAACTGCAGCTCAAGCTGAATCACCCAGAGTCC TCTCAGCTGTTCGCCAAGGTGCTCCAGAAGATGACAGACCTCAGGCAGATCGTCACAGAG CACGTGCAGCTACTGCATGTGATCAAGAAGACAGAGACAGACATGAGCCTTCACCCCCTG CTCCAGGAGATCTACAAGGACTTGTATTTGCAGGAAAGTCCCGGCTCCGGAGAGGGCAGA GGAAGTCTGCTAACATGCGGTGACGTCGAGGAGAATCCTGGCCCAATGGTGAGCAAGGG CGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACG GCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACC CTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACC CTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTC TTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGAC GGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCAT CGAGCTGAAGGG) and Gibson fragment 2 (CGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACG GCAACATCCTGGGGCACAAGCTGGAGTACAACTACAACAGCCACAACGTCTATATCATGG CCGACAAGCAGAAGAACGGCATCAAGGTGAACTTCAAGATCCGCCACAACATCGAGGACG GCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTG CTGCTGCCCGACAACCACTACCTGAGCACCCAGTCCGCCCTGAGCAAAGACCCCAACGAG AAGCGCGATCACATGGTCCTGCTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATG GACGAGCTGTACAAGTAATTCTATTGATTGCACTATTATTTTGAGGGAAAAAAATCTGACAC CTAAGAAATTTACTGTGAAAAAGCATTTAAAAACAAAAAGTTTTAGAACATGATCTATTTTAT GCATATTGTTTATAAAGATACATTTACAATTTACTTTTAATATTAAAAATTACCACATTATAAA ATTGTTTATAGTATTTAAAGAATGAATGCATGTCTCCCATTCTGCCATCACAGATGGGGGAA AGGATTTCCTCATTTTATTTCTTTTCTTCTTTCTTTTCTTCATCCTTTGACATGCATATGTGCT CATATATATGTTTATGTGTGAGTAGATGCTGTGTATGTGTATGGAGGCCCAGAGACAGCTC TAAAGCTGGCTCATTCTCAGGACTGTCATCTACCCACTTTTAGACAGGGTCTTTCATTTGGC CTAAATTTAACCAATTAGGTTAGACTAGAGGGTCAGTGAGCCCCGGGGAACGGCCTGGCC CCACCCGCCCATCACTACCCTGTCTGGCATGTTTAGGTGGATTCGAGAGGAGGGGAGGAG CTGGACTCAGGTCCTCATGCTCTCAAGGCTTCACAAGAACCCAAACTTCTCCCACGACTCT GAGTTTTCACGAATCACCAGCAACATGTTTAACATATTGTCATCCCTCTATACTCATGGGGT CTGGCTCCAGGCCTCCTGAAGGAGCCATGGGTGTTCAAGTTTTCATAACAGGAGTTTCAAG TAAACTCTGCATACTGTCCCCTATCCTTTGAACTGTCTCTAGATACCTACAATACCTAACAC AATGGAAATGTTGCACTGGCCGTCGTTTTACAACGTCGTGAC).
A p306 plasmid containing a sgRNA (GGAACACGTTGTCAGCGGGT) targeting the 3’UTR of Pparγ was generated by annealing the top and bottom oligos
(TTGGGAACACGTTGTCAGCGGGTGTTTAAGAGC and TTAGCTCTTAAACACCCGCTGACAACGTGTTCCCAACAAG) and ligating into BlpI/BstXI digested MCB306.
3T3-L1 Cas9-BFP cells were co-transfected with p306 sgPparγ and donor plasmid, sorted for GFP positivity (from p306 plasmid) 48h post-infection and allowed to grow for 10 days. 100 GFP negative cells (since expression was transient) were sorted into each well of a 96 well plate and allowed to grow. Cells were duplicate plated and one set was induced to differentiate using the traditional DM(1μg/ml insulin, 1μM dexamethasone, 0.5mM IBMX) in the IncuCyte live-imaging system to identify wells containing cells that became GFP positive during adipogenesis. Cells in positive wells were single cell sorted, allowed to grow, duplicate plated, and differentiated in the IncuCyte live-imaging system. In total, we obtained 5 3T3-L1 PPARγ T2A reporter cell lines.
Next generation RNA sequencing and analysis
3T3-L1 cells were grown to confluency, kept confluency-arrested for 2 days, and differentiated in triplicates using historical DM, ctrl (-DHA) differentiation cocktail, or DHA cocktail. After 24h, RNA was extracted using the Qiagen RNeasy kit, followed by library construction. Sequencing data was generated on an Illumina HiSeq4000 (purchased with funds from NIH under award number S10OD018220).
Sequencing data QC was performed using fastqc and trimmomatic. Differential gene expression compared to undifferentiated (0h) was assessed using three pipelines: Kallisto (reference refseq), followed by sleuth; Star (reference gencode) followed by cutdiff; and star (reference gencode) followed by htseq and deseq2. Differential gene expression was called significant if qvalue <0.05 for all three pipelines.
ATACseq and TF motif enrichment analysis
3T3-L1 cells were grown to confluency, kept confluency-arrested for 2 days, and in duplicates left untreated, treated with 100μM DHA, or treated with 1μM Dex for 4h. Aliquots of 70,000 to 80,000 cells were taken and processed as previously described (Corces et al., 2017). Briefly, 200 U/ml DNase (Worthington Biochemical, NJ, USA) were added to the cell aliquot and incubated at 37C for 30 min, centrifuged at 500 g for 5 min at room temperature and resuspended in 1 ml cold PBS. Resuspended samples were centrifuged at 500 g for 5 min at 4C and washed in 500 μl cold RSB buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, and 3 mM MgCl2) followed by centrifugation at 500 g for 5 min at 4C. Samples were resuspended and lysed for 3 min on ice in 50 μl RSB buffer containing 0.01% Digitonin (Promega, WI, USA), 0.1% NP40 and 0.1% Tween20. After lysis, 950 μl RSB with 0.1% Tween20 were added to the sample and mixed by inverting. Samples were centrifuged at 500 g for 5 min at 4C, resuspended in 50 μl of transposition mix (25 μl TD buffer [Illumina], 2.5 μl Tn5 [Illumina], 16.5 μl PBS, 0.5 μl 1% Digitonin, 0.5 μl 10% Tween20 and 5 μl water) and incubated at 300 rpm at 37C. All samples were purified using MinElute PCR Purification columns (Qiagen, Germany) and amplified and barcoded with custom Nextera primers (Table S5) as previously described (Buenrostro et al., 2015). ATAC libraries were paired-end sequenced (2× 76 bp) on a NextSeq500 (Illumina) using a high output v2 150 cycle kit (Illumina).
BCL files were demultiplexed using bcl2fastq (Illumina), adapters were trimmed using cutadapt and reads were aligned to the mouse genome (mm9) using bowtie2. Duplicates were removed using Picard MarkDuplicates. Mitochondrial reads and reads mapping to a modified version of the mm9 ENCODE blacklist (Table S6) were removed using samtools. Peaks were called using MACS2 and filtered by q value for the top 100K peaks. The peak set was then reduced to nonoverlapping peaks and peak width was adjusted to 500 bp. Transcription factor motif analysis was performed using chromVAR. Bias-corrected deviations were plotted for the 50 most variable transcription factor motifs using the R pheatmap package.
Filtered and normalized enhancer regions from (Siersbaek et al., 2017) were intersected with CTCF-binding sites from atac-seq. The resulting data was further filtered to keep only enhancer sites that were also detected by CTCF CHIP-seq from (Siersbaek et al., 2017). Corresponding promoter sites were used to infer genes regulated by these promoters.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the statistical test used, exact value of n, what n represents, and the distribution and deviation are reported in the figures and corresponding figure legends. Most data are represented as the mean ± standard deviation and the p-value was determined using two-tailed Student’s t tests.
Unless otherwise stated, statistical analyses were performed in Microsoft Excel and GraphPad Prism.
DATA AND SOFTWARE AVAILABILITY
GEO accession numbers are GSE118470 (ATACseq of 3T3L1 cells undergoing differentiation) and GSE118471 (RNAseq of 3T3L1 cells undergoing differentiation)
Supplementary Material
Table S2. Related to Figure 7; RNAseq data
Table S3. Related to Figure 7; ATACseq data
Table S4. Related to Figure 7; Enhancer Promoter data
Table S5. Related to STAR Methods; Primers used in this study
Table S6. Related to STAR Methods; Blacklist
(A) Primary preadipocytes in the SVF (depleted for RBCs and WBCs) from the inguinal WAT of CENTRIN2-GFP, ARL13BmCherry (cilia glow) mice were differentiated using the historical DM (1μg/ml insulin, 1μM dexamethasone, 0.5mM IBMX). Ciliation status was quantified for different differentiation time points (D-1 refers to Day −1 when cells are not confluent (1 day post-isolation), D0 refers to Day 0 when cells are confluent and differentiation is initiated, D3 refers to Day 3 of differentiation when cells are switched into maintenance media (1μg/ml insulin), and D5 refers to Day 5 of differentiation when lipid starts being accumulated). n=number of cells quantified per time point; cilia are categorized by length of axoneme since there was a wide range of ciliary lengths; bar graphs are average number of ciliated cells ± SD. (B) Cross-section of a blood vessel in epididymal WAT from a wild type mouse shows that ciliated cells are perivascular. Yellow dotted line outlines blood vessel as determined by Cd31 staining. (C) Whole mount image of epididymal WAT from wild type mouse. BODIPY visualizes lipid droplets. Arrow heads point to ciliated perivascular cells. (D) Whole mount image of epididymal WAT from cilia glow mouse showing that perivascular macrophages as visualized by MAC2 staining are not ciliated. (E) Immunofluorescence for preadipocytes (PDGFRα, red), cilia (ARL13B, marked by arrowheads) and lineage tracer (EYFP, green) 5 weeks post induction of lineage label in Pdgfrα-CreERT Rosa26EYFP mice. Scale bar is 25 μm. CD31 visualizes blood vessels, pericentrin visualizes the ciliary base, and acetylated tubulin or ARL13B visualize the ciliary axoneme.
{kind=link}
(A) TIDE analysis showing genomic alterations in the engineered 3T3-L1 sgTulp3 cell line. (B) TIDE analysis showing genomic alterations in the engineered 3T3-L1 sgFfar4 cell line. (C) Loss of TULP3 has no effect on ciliary structure, as determined by acetylated tubulin staining, but prevents trafficking of ciliary proteins, including ARL13B. (D) Loss of TULP3 using siRNA-mediated knockdown results in attenuated adipogenesis. 3 different siRNAs targeting TULP3 were used, siRNA targeting PPARγ is positive control. Lipid accumulation visualized by Oil Red O staining (E) Quantification of adipogenesis by isopropanol extraction of Oil Red O. (F) Quantification of TULP3 knockdown efficiency using siRNA. (G) Immunoblot showing depletion of TULP3 in 3T3-L1 sgTulp3 cell line, and overexpression of GFP-TULP3 fusion protein. P-values calculated using t test, * p<0.05;
{kind=link}
(A) Left: Immunofluorescence for preadipocytes (PDGFRα, red) and cilia (ARL13B, green) 5 weeks post tamoxifen administration in control and PAno cilia mice. Ciliated PAs are marked by arrowheads. Scale bar is 25 μm. Right: Quantifications of the percentage of ciliated PAs present 5 weeks after conditional removal of PA cilia. Far right: Comparison of Ift88 expression between control and PAno cilia mice via RT-qPCR from whole gonadal WAT (n=4 per genotype). (B) Left: Body weight measurements in control and PAno cilia mice (n=4 for control and n=3 for PAno cilia mice). Right: Picture of gonadal fat pad and Echo-MRI measurements of total fat and lean mass in control and PAno cilia mice 11 weeks after tamoxifen administration. Scale bar is 1cm. (C) H&E staining of gonadal WAT tissue sections and the respective quantifications of the mean area of adipocytes in control and PAno cilia mice. Scale bar is 100 μm. (D) Left: Oil Red O-stained liver sections from control and PAno cilia mice. Right: Quantification of area occupied by Oil Red O between genotypes. Scale bar is 100 μm. (E) Serum levels for insulin and free fatty acids (FFA) were measured using ELISA and glucose levels using a glucometer between control and PAno cilia mice at either fed state or after an overnight fasting period as indicated. (F) Plotting of hourly oxygen consumption (VO2), food intake and total movement over 4 days of control (n=♂14 & ♀12) and PAno cilia mice (n=♂7 & ♀9). (G) Dissected interscapular brown adipose tissue (BAT) of two control and two PAno cilia mice. Scale bar is 1 cm. Comparison of Ift88, Ucp1, Pparγ and Perilipin expression between control (n=6) and PAno cilia mice (n=5) via RT-qPCR from whole interscapular BAT. All data are represented as mean ± SEM. p-values were calculated using standard t-test and two-way ANOVA followed by Tukey’s multiple comparison test (*<0.05, **<0.01, ***<0.001 and ****<0.0001). Of note, all mice depicted are littermates and maintained on breeder chow starting the day of tamoxifen administration.
{kind=link}
(A) 3T3-L1 FFAR4-GFP fusion protein localizes to primary cilium. SMO-GFP is positive control. (B) Validation of ciliary localization of FFAR4 using second independent antibody (Santa Cruz). 3T3-L1 cells on Day 0 and Day 2 of differentiation. (C-E) Validation of FFAR4 antibody in (C) 3T3-L1 cells using Crispr/Cas9, (D) SVF isolated from inguinal and (E) epididymal white adipose tissue of wild-type and Ffar4 knockout littermates. We note that there is some non-specific ciliary FFAR4 background staining with this antibody. (F) Representative immunofluorescence images showing depletion of ciliary FFAR4 with loss of TULP3 protein. (G) FFAR4 expression by mRNA and protein increases during differentiation. 20x objective was used to visualize the whole cell. This makes visualization of individual cilia difficult. (H) Endogenous FFAR4 is ciliary during 3T3-L1 differentiation. D0, 2, 4, 6 are Day 0, 2, 4 and 6 of differentiation. (I) Endogenous FFAR4 localizes to the primary cilium of preadipocytes and to the plasma membrane of mature adipocytes in vivo in epididymal WAT whole mounts from cilia glow male mice. The fluorescence intensity of FFAR4 localized to the primary cilia is higher than the fluorescence intensity of FFAR4 localized to the adipocyte plasma membrane, consistent with FFAR4 receptor density being higher in the primary cilium. White arrows and boxes highlight cilia.
{kind=link}
(A) DHA cocktail promotes activation of adipogenic genes Adiponectin, Fabp4, and Plin1, and represses expression of preadipocyte genes Col1a and Dlk1. These data show that DHA cocktail initiates adipogenesis of 3T3-L1 preadipocytes. Bar graph is average of three independent experiments ± SD. (B) DHA cocktail results in cell cycle re-entry and this is dependent on Tulp3 protein. Representative images showing EdU incorporation of confluency-arrested 3T3-L1 cells maintained in growth media, or differentiated using the historical DM, the ctrl cocktail (-DHA), or the DHA cocktail. EdU was added at 16h, and cells were fixed and stained at 40h. (C) Addition of historical DM (1μg/ml insulin, 1μM dexamethasone, 0.5mM IBMX) to 3T3-L1 cells lacking Tulp3 promotes cell cycle re-entry. Tulp3 protein is only required for DHA-mediated mitotic differentiative expansion and adipogenesis. (D) Quantification of adipogenesis in the presence or absence of FFAR4 agonist with one component of the modified cocktail omitted per condition. Images shown in Figure 5B. FFAR4 agonist can partially substitute for Dex, insulin, and IBMX, but has quantitatively the largest effect replacing IBMX. This suggests that FFAR4 activation results in elevation of cAMP second messenger. This can enhance adipogenesis in the absence of any single cocktail component, but has the greatest effect replacing the phosphodiesterase inhibitor IBMX. (E) Loss of Tulp3 prevents increase in ciliary cAMP by addition of FFAR4 agonist. cAMP sensor with red fluorescent constitutive ciliary marker was expressed in 3T3-L1 sgGFP (wild type control) and sgTulp3 cells. Addition of FFAR4 agonist, but not DMSO control, in wildtype but not Tulp3 knockout cells results in a significant decrease in the ratio of green fluorescence (cAMP sensor) to red fluorescence (constitutive ciliary marker) intensities, showing that FFAR4 activation elevates ciliary cAMP and that this is dependent on Tulp3 and thus ciliary localization of FFAR4. Bar graphs represent mean ± SD. *<0.05; ** p<0.01; *** p<0.001.
{kind=link}
(A) DM component titration in the presence or absence of 100μM DHA to determine the optimal concentrations of insulin, dexamethasone, and IBMX to observe enhancement of differentiation by DHA. DHA and DM components were added for 48h to 2 day post-confluent 3T3-L1 cells. At 0.4μg/ml insulin, 0.02mM IBMX, 0.04μM dexamethasone (red circle), there is little differentiation with EtOH control and robust differentiation in the presence of DHA, as visualized by Oil Red O. (B) DHA does not promote adipogenesis by directly activating PPARγ. 3T3-L1 preadipocytes were differentiated using the DHA cocktail for 2 days followed by maintenance media (1μg/ml insulin) for 4 days. Lipid accumulation was assessed by green fluorescent BODIPY dye. The PPARγ antagonist T0070907 was added either concurrently with DHA (D0–2) or throughout the differentiation time course (D0–6). Presence of the PPARγ antagonist throughout the differentiation time course (D0–6) inhibits adipogenesis in a dose-dependent manner, consistent with PPARγ being required during the second phase of adipogenesis. The presence of the PPARγ concurrently with DHA (D0–2) does not inhibit adipogenesis, arguing that DHA does not promote adipogenesis by directly activating PPARγ. (C) FFAR4 antagonist (AH7614) has a lower IC50 when 3T3-L1 adipogenesis is induced by DHA cocktail versus historical DM. (D) Addition of FFAR4 agonist TUG891 (100μM) during the first 72h of differentiation enhances differentiation of primary murine preadipocytes in the SVF from inguinal WAT. (E) Addition of FFAR4 antagonist AH7614 (10μM) to differentiating human preadipocytes during the first 72h attenuates differentiation. (F) FFAR4 activation promotes adipogenesis of primary mouse preadipocytes via the primary cilium. Preadipocytes (Lin−, CD34+, SCA1+, CD29+) isolated from Ift88flox/flox males were transduced with an adenovirus expressing GFP only or GFP and Cre recombinase (+Cre). IFT88 is required for ciliation and percent ciliation was determined by FGFR1OP (basal body) and ARL13B (axoneme) staining of GFP+ confluent preadipocytes. Percent differentiation was determined by LipidTox Neutral Red staining of GFP+ cells differentiated with modified cocktail in the presence or absence of 100μM TUG891. Representative images shown. n = number of GFP+ cells counted. Differentiation data is normalized to ctrl ± SD. ** p<0.01; *** p<0.001.
{kind=link}
(A) Quantification of mRNA levels of Cebpβ, Pparγ, and Cebpα at different differentiation time points using control cocktail (EtOH) or DHA cocktail (DHA). We observe significant DHA-dependent transcriptional activation of Pparγ and Cebpα within 24h of differentiation, but not of CEBPβ. (B) A PPARγ-T2A-GFP knockin reporter generated using Crispr/Cas9 shows increased expression of PPARγ in response to DHA cocktail (DHA) compared to control cocktail (EtOH) within 3 days of differentiation. Addition of the FFAR4 antagonist AH7614 inhibits PPARγ expression in a dose-dependent manner. Dotted line denotes media changes, images are taken every 2h. (C) Immunoblot showing CEBPβ and CEBPα protein levels at different time points of differentiation using control cocktail or DHA cocktail. We consistently observe a DHA-dependent increase in expression of CEBPα, but not of CEBPβ. (D) Immunoblot showing phosphorylated and overall levels of ERK, AKT, and CREB levels at different differentiation time points using control cocktail, DHA cocktail, or FFAR4 agonist (TUG891) cocktail. While there is activation of these adipogenic regulators during differentiation, this is not DHA-dependent, suggesting that cocktail components other than DHA, notably insulin, activate these adipogenic regulators. (E) While ERK and AKT activation is not mediated by DHA, activation of these pathways is required for DHA-enhanced adipogenesis, consistent with the notion that cocktail components other than DHA activate these adipogenic regulators and this is required for adipogenesis. (F) 3T3L1 cells lacking CTCF show only a minor, not significant growth defect. Cell growth was assessed using Essen IncuCyte Live Cell Imaging Analysis in normal growth media; images taken every 2h. (G) Loss of CTCF does not affect adipogenesis induced by the historical DM (1μg/ml insulin, 1μM dexamethasone, 0.5mM IBMX). (H-I) Three different 3T3-L1 Ctcf knockout cells lines were generated by Crispr/Cas9 using 3 different sgRNAs. The cell line sgCtcf-1 was used in Fig 6F–G. (H) All three lines show an adipogenesis defect in response to DHA cocktail. (I) TIDE analysis describing genomic rearrangement of all three engineered 3T3-L1 sgCtcf cell lines.
{kind=link}
Preadipocytes, located along blood vessels, are ciliated in vitro and in vivo
Loss of preadipocyte ciliation strongly impairs white adipose tissue expansion
Ciliary GPCRs are critical for adipogenesis and FFAR4/GPR120 localizes to cilia
ω−3 fatty acids activate ciliary FFAR4 and trigger adipogenesis via ciliary cAMP
Acknowledgements
PKJ was supported by NIH grants 5R01GM114276, 5U01CA199216, 5UL1TR00108502 and the Stanford Department of Research and Baxter Laboratory. JFR was supported by NIH grants AR054396, DK106404 and GM095941. DK was supported by departmental startup funds. KIH is a Layton Family Fellow of the Damon Runyon Cancer Research Foundation (DRG-2210-14). CTJ is supported by an NIH training grant (TG2-01159). AM is supported by the Swedish Research Council (grant 2015-06403). JFR and WJG are Chan Zuckerberg Biohub investigators. We thank N. Mooney for animal husbandry support. We thank the UCSF Diabetes Research Center, especially Chris Paillart, for help with the CLAMS and EchoMRI studies. We thank Kevin Corbit and Jennifer Tran for assistance with serum analysis.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anders S, Pyl PT, and Huber W. (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner P, Andersson DP, Thorne A, Wiren M, Hoffstedt J, Naslund E, Thorell A, and Ryden M. (2013). Variations in the size of the major omentum are primarily determined by fat cell number. J Clin Endocrinol Metab 98, E897–901. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs FK, Schrode N, Hadjantonakis AK, and Anderson KV (2015). Lineage specificity of primary cilia in the mouse embryo. Nat Cell Biol 17, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry R, and Rodeheffer MS (2013). Characterization of the adipocyte cellular lineage in vivo. Nat Cell Biol 15, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B. (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burl RB, Ramseyer VD, Rondini EA, Pique-Regi R, Lee YH, and Granneman JG (2018). Deconstructing Adipogenesis Induced by beta3-Adrenergic Receptor Activation with Single-Cell Expression Profiling. Cell Metab 28, 300–309 e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crenn P, Morin MC, Joly F, Penven S, Thuillier F, and Messing B. (2004). Net digestive absorption and adaptive hyperphagia in adult short bowel patients. Gut 53, 1279–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, and Yoder BK (2007). Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol 17, 1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deren ME, Yang X, Guan Y, and Chen Q. (2016). Biological and Chemical Removal of Primary Cilia Affects Mechanical Activation of Chondrogenesis Markers in Chondroprogenitors and Hypertrophic Chondrocytes. Int J Mol Sci 17, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois-Chevalier J, Oger F, Dehondt H, Firmin FF, Gheeraert C, Staels B, Lefebvre P, and Eeckhoute J. (2014). A dynamic CTCF chromatin binding landscape promotes DNA hydroxymethylation and transcriptional induction of adipocyte differentiation. Nucleic Acids Res 42, 10943–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcioli-Conti N, Lacas-Gervais S, Dani C, and Peraldi P. (2015). The primary cilium undergoes dynamic size modifications during adipocyte differentiation of human adipose stem cells. Biochem Biophys Res Commun 458, 117–122. [DOI] [PubMed] [Google Scholar]
- Ford MJ, Yeyati PL, Mali GR, Keighren MA, Waddell SH, Mjoseng HK, Douglas AT, Hall EA, Sakaue-Sawano A, Miyawaki A, et al. (2018). A Cell/Cilia Cycle Biosensor for Single-Cell Kinetics Reveals Persistence of Cilia after G1/S Transition Is a General Property in Cells and Mice. Dev Cell 47, 509–523 e505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Asp P, Canter B, and Dynlacht BD (2014). Primary cilia control hedgehog signaling during muscle differentiation and are deregulated in rhabdomyosarcoma. Proc Natl Acad Sci U S A 111, 9151–9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Geng T, Huang T, and Zhao Q. (2017). Fish oil supplementation and insulin sensitivity: a systematic review and meta-analysis. Lipids Health Dis 16, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaben AL, and Scherer PE (2019). Adipogenesis and metabolic health. Nature reviews Molecular cell biology 20, 242–258. [DOI] [PubMed] [Google Scholar]
- Gotoh C, Hong YH, Iga T, Hishikawa D, Suzuki Y, Song SH, Choi KC, Adachi T, Hirasawa A, Tsujimoto G, et al. (2007). The regulation of adipogenesis through GPR120. Biochem Biophys Res Commun 354, 591–597. [DOI] [PubMed] [Google Scholar]
- Guimaraes-Camboa N, Cattaneo P, Sun Y, Moore-Morris T, Gu Y, Dalton ND, Rockenstein E, Masliah E, Peterson KL, Stallcup WB, et al. (2017). Pericytes of Multiple Organs Do Not Behave as Mesenchymal Stem Cells In Vivo. Cell Stem Cell 20, 345–359 e345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Mepani RJ, Kleiner S, Lo JC, Khandekar MJ, Cohen P, Frontini A, Bhowmick DC, Ye L, Cinti S, et al. (2012). Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab 15, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haczeyni F, Bell-Anderson KS, and Farrell GC (2018). Causes and mechanisms of adipocyte enlargement and adipose expansion. Obes Rev 19, 406–420. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, and Yoder BK (2007). Intraflagellar transport is essential for endochondral bone formation. Development 134, 307–316. [DOI] [PubMed] [Google Scholar]
- Hepler C, Shan B, Zhang Q, Henry GH, Shao M, Vishvanath L, Ghaben AL, Mobley AB, Strand D, Hon GC, et al. (2018). Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler C, Vishvanath L, and Gupta RK (2017). Sorting out adipocyte precursors and their role in physiology and disease. Genes Dev 31, 127–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, and Katsanis N. (2011). Ciliopathies. N Engl J Med 364, 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorf KI, Johnson CT, and Jackson PK (2016). The primary cilium as a cellular receiver: organizing ciliary GPCR signaling. Curr Opin Cell Biol 39, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, and Tsujimoto G. (2005). Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med 11, 90–94. [DOI] [PubMed] [Google Scholar]
- Huang-Doran I, and Semple RK (2010). Knockdown of the Alstrom syndrome-associated gene Alms1 in 3T3-L1 preadipocytes impairs adipogenesis but has no effect on cell-autonomous insulin action. Int J Obes (Lond) 34, 1554–1558. [DOI] [PubMed] [Google Scholar]
- Hwang SH, Somatilaka BN, Badgandi H, Palicharla VR, Walker R, Shelton JM, Qian F, and Mukhopadhyay S. (2019). Tulp3 Regulates Renal Cystogenesis by Trafficking of Cystoproteins to Cilia. Curr Biol 29, 790–802 e795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura A, Hasegawa S, Kasubuchi M, and Kimura I. (2014). Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front Pharmacol 5, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura A, Hirasawa A, Poulain-Godefroy O, Bonnefond A, Hara T, Yengo L, Kimura I, Leloire A, Liu N, Iida K, et al. (2012). Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 483, 350–354. [DOI] [PubMed] [Google Scholar]
- Jaafar Marican NH, Cruz-Migoni SB, and Borycki AG (2016). Asymmetric Distribution of Primary Cilia Allocates Satellite Cells for Self-Renewal. Stem Cell Reports 6, 798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery E, Church CD, Holtrup B, Colman L, and Rodeheffer MS (2015). Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol 17, 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery E, Wing A, Holtrup B, Sebo Z, Kaplan JL, Saavedra-Pena R, Church CD, Colman L, Berry R, and Rodeheffer MS (2016). The Adipose Tissue Microenvironment Regulates Depot-Specific Adipogenesis in Obesity. Cell Metab 24, 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Berry DC, Tang W, and Graff JM (2014). Independent stem cell lineages regulate adipose organogenesis and adipose homeostasis. Cell reports 9, 1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TR KI, Wheeler DB, Lindquist RA, Papallo A, Sabatini DM, Golland P, Carpenter AE (2008). CellProfiler Analyst: data exploration and analysis software for complex image-based screens. BMC Bioinformatics 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, and Bergles DE (2010). NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karastergiou K, and Fried SK (2017). Cellular Mechanisms Driving Sex Differences in Adipose Tissue Biology and Body Shape in Humans and Mouse Models. Adv Exp Med Biol 1043, 29–51. [DOI] [PubMed] [Google Scholar]
- Kelly DJ, and Jacobs CR (2010). The role of mechanical signals in regulating chondrogenesis and osteogenesis of mesenchymal stem cells. Birth Defects Res C Embryo Today 90, 75–85. [DOI] [PubMed] [Google Scholar]
- Kim HK, Della-Fera M, Lin J, and Baile CA (2006). Docosahexaenoic acid inhibits adipocyte differentiation and induces apoptosis in 3T3-L1 preadipocytes. J Nutr 136, 2965–2969. [DOI] [PubMed] [Google Scholar]
- Kim S, and Dynlacht BD (2013). Assembling a primary cilium. Curr Opin Cell Biol 25, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopinke D, Roberson EC, and Reiter JF (2017). Ciliary Hedgehog Signaling Restricts Injury-Induced Adipogenesis. Cell 170, 340–351 e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey G, Braissant O, L’Horset F, Kalkhoven E, Perroud M, Parker MG, and Wahli W. (1997). Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Molecular endocrinology 11, 779–791. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loktev AV, and Jackson PK (2013). Neuropeptide Y family receptors traffic via the Bardet-Biedl syndrome pathway to signal in neuronal primary cilia. Cell reports 5, 1316–1329. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu R, and Zhou J. (2017). The Multifaceted Roles of Primary Cilia in the Regulation of Stem Cell Properties and Functions. Journal of cellular physiology 232, 935–938. [DOI] [PubMed] [Google Scholar]
- M. M. (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnetjournal 17, 10–12. [Google Scholar]
- Madsen L, Petersen RK, and Kristiansen K. (2005). Regulation of adipocyte differentiation and function by polyunsaturated fatty acids. Biochimica et biophysica acta 1740, 266–286. [DOI] [PubMed] [Google Scholar]
- Marion V, Mockel A, De Melo C, Obringer C, Claussmann A, Simon A, Messaddeq N, Durand M, Dupuis L, Loeffler JP, et al. (2012). BBS-induced ciliary defect enhances adipogenesis, causing paradoxical higher-insulin sensitivity, glucose usage, and decreased inflammatory response. Cell Metab 16, 363–377. [DOI] [PubMed] [Google Scholar]
- Marion V, Stoetzel C, Schlicht D, Messaddeq N, Koch M, Flori E, Danse JM, Mandel JL, and Dollfus H. (2009). Transient ciliogenesis involving Bardet-Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc Natl Acad Sci U S A 106, 1820–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick D, Sakers A, Irgebay Z, Okada C, Calvert C, Morley MP, Percec I, and Seale P. (2019). Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BS, Stepanchick AN, Tewson PH, Hartle CM, Zhang J, Quinn AM, Hughes TE, and Mirshahi T. (2016). Cilia have high cAMP levels that are inhibited by Sonic Hedgehog-regulated calcium dynamics. Proc Natl Acad Sci U S A 113, 13069–13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, and Jackson PK (2010). TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G proteincoupled receptors into primary cilia. Genes Dev 24, 2180–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, et al. (2007). A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213. [DOI] [PubMed] [Google Scholar]
- O’Rourke M, Cullen CL, Auderset L, Pitman KA, Achatz D, Gasperini R, and Young KM (2016). Evaluating Tissue-Specific Recombination in a Pdgfralpha-CreERT2 Transgenic Mouse Line. PLoS One 11, e0162858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, and Olefsky JM (2010). GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Walenta E, Akiyama TE, Lagakos WS, Lackey D, Pessentheiner AR, Sasik R, Hah N, Chi TJ, Cox JM, et al. (2014). A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat Med 20, 942–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AK, and Kirkland JL (2016). Aging and adipose tissue: potential interventions for diabetes and regenerative medicine. Exp Gerontol 86, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu N, Cao L, David V, Quarles LD, and Xiao Z. (2010). Kif3a deficiency reverses the skeletal abnormalities in Pkd1 deficient mice by restoring the balance between osteogenesis and adipogenesis. PLoS One 5, e15240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R. K. (2015). Pheatmap: Pretty Heatmaps. R package, version 108. [Google Scholar]
- Ritter A, Friemel A, Kreis NN, Hoock SC, Roth S, Kielland-Kaisen U, Bruggmann D, Solbach C, Louwen F, and Yuan J. (2018). Primary Cilia Are Dysfunctional in Obese Adipose-Derived Mesenchymal Stem Cells. Stem Cell Reports 10, 583–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodeheffer MS, Birsoy K, and Friedman JM (2008). Identification of white adipocyte progenitor cells in vivo. Cell 135, 240–249. [DOI] [PubMed] [Google Scholar]
- Rosen ED, and Spiegelman BM (2014). What we talk about when we talk about fat. Cell 156, 20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Walkey CJ, Puigserver P, and Spiegelman BM (2000). Transcriptional regulation of adipogenesis. Genes Dev 14, 1293–1307. [PubMed] [Google Scholar]
- Sakaguchi M, Fujisaka S, Cai W, Winnay JN, Konishi M, O’Neill BT, Li M, Garcia-Martin R, Takahashi H, Hu J, et al. (2017). Adipocyte Dynamics and Reversible Metabolic Syndrome in Mice with an Inducible Adipocyte-Specific Deletion of the Insulin Receptor. Cell Metab 25, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcriptionfactor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalie PC, Dong H, Zachara M, Russeil J, Alpern D, Akchiche N, Caprara C, Sun W, Schlaudraff KU, Soldati G, et al. (2018). A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature 559, 103–108. [DOI] [PubMed] [Google Scholar]
- Siersbaek R, Madsen JGS, Javierre BM, Nielsen R, Bagge EK, Cairns J, Wingett SW, Traynor S, Spivakov M, Fraser P, et al. (2017). Dynamic Rewiring of Promoter-Anchored Chromatin Loops during Adipocyte Differentiation. Mol Cell 66, 420–435 e425. [DOI] [PubMed] [Google Scholar]
- Siersbaek R, Nielsen R, and Mandrup S. (2012). Transcriptional networks and chromatin remodeling controlling adipogenesis. Trends Endocrinol Metab 23, 56–64. [DOI] [PubMed] [Google Scholar]
- Siljee JE, Wang Y, Bernard AA, Ersoy BA, Zhang S, Marley A, Von Zastrow M, Reiter JF, and Vaisse C. (2018). Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity. Nat Genet 50, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song T, Zhou Y, Peng J, Tao YX, Yang Y, Xu T, Peng J, Ren J, Xiang Q, and Wei H. (2016). GPR120 promotes adipogenesis through intracellular calcium and extracellular signal-regulated kinase ½ signal pathway. Mol Cell Endocrinol 434, 1–13. [DOI] [PubMed] [Google Scholar]
- Spencer M, Finlin BS, Unal R, Zhu B, Morris AJ, Shipp LR, Lee J, Walton RG, Adu A, Erfani R, et al. (2013). Omega-3 fatty acids reduce adipose tissue macrophages in human subjects with insulin resistance. Diabetes 62, 1709–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, and Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckow AT, Polidori D, Yan W, Chon S, Ma JY, Leonard J, and Briscoe CP (2014). Alteration of the glucagon axis in GPR120 (FFAR4) knockout mice: a role for GPR120 in glucagon secretion. The Journal of biological chemistry 289, 15751–15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Otto TC, and Lane MD (2003). Mitotic clonal expansion: a synchronous process required for adipogenesis. Proc Natl Acad Sci U S A 100, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Zeve D, Suh JM, Bosnakovski D, Kyba M, Hammer RE, Tallquist MD, and Graff JM (2008). White fat progenitor cells reside in the adipose vasculature. Science 322, 583–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummala P, Arnsdorf EJ, and Jacobs CR (2010). The Role of Primary Cilia in Mesenchymal Stem Cell Differentiation: A Pivotal Switch in Guiding Lineage Commitment. Cell Mol Bioeng 3, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishvanath L, MacPherson KA, Hepler C, Wang QA, Shao M, Spurgin SB, Wang MY, Kusminski CM, Morley TS, and Gupta RK (2016). Pdgfrbeta+ Mural Preadipocytes Contribute to Adipocyte Hyperplasia Induced by High-Fat-Diet Feeding and Prolonged Cold Exposure in Adult Mice. Cell Metab 23, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QA, Tao C, Gupta RK, and Scherer PE (2013). Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med 19, 1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM, and Beales PL (2011). Ciliopathies: an expanding disease spectrum. Pediatric nephrology 26, 1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, and Ferrante AW Jr. (2003). Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112, 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao ZS, and Quarles LD (2010). Role of the polycytin-primary cilia complex in bone development and mechanosensing. Ann N Y Acad Sci 1192, 410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, and Yang S. (2016). Primary Cilia and Intraflagellar Transport Proteins in Bone and Cartilage. J Dent Res 95, 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Shi S, Wang H, and Liao K. (2009). Growth arrest induces primary-cilium formation and sensitizes IGF-1-receptor signaling during differentiation induction of 3T3-L1 preadipocytes. Journal of cell science 122, 2760–2768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Related to Figure 7; RNAseq data
Table S3. Related to Figure 7; ATACseq data
Table S4. Related to Figure 7; Enhancer Promoter data
Table S5. Related to STAR Methods; Primers used in this study
Table S6. Related to STAR Methods; Blacklist
(A) Primary preadipocytes in the SVF (depleted for RBCs and WBCs) from the inguinal WAT of CENTRIN2-GFP, ARL13BmCherry (cilia glow) mice were differentiated using the historical DM (1μg/ml insulin, 1μM dexamethasone, 0.5mM IBMX). Ciliation status was quantified for different differentiation time points (D-1 refers to Day −1 when cells are not confluent (1 day post-isolation), D0 refers to Day 0 when cells are confluent and differentiation is initiated, D3 refers to Day 3 of differentiation when cells are switched into maintenance media (1μg/ml insulin), and D5 refers to Day 5 of differentiation when lipid starts being accumulated). n=number of cells quantified per time point; cilia are categorized by length of axoneme since there was a wide range of ciliary lengths; bar graphs are average number of ciliated cells ± SD. (B) Cross-section of a blood vessel in epididymal WAT from a wild type mouse shows that ciliated cells are perivascular. Yellow dotted line outlines blood vessel as determined by Cd31 staining. (C) Whole mount image of epididymal WAT from wild type mouse. BODIPY visualizes lipid droplets. Arrow heads point to ciliated perivascular cells. (D) Whole mount image of epididymal WAT from cilia glow mouse showing that perivascular macrophages as visualized by MAC2 staining are not ciliated. (E) Immunofluorescence for preadipocytes (PDGFRα, red), cilia (ARL13B, marked by arrowheads) and lineage tracer (EYFP, green) 5 weeks post induction of lineage label in Pdgfrα-CreERT Rosa26EYFP mice. Scale bar is 25 μm. CD31 visualizes blood vessels, pericentrin visualizes the ciliary base, and acetylated tubulin or ARL13B visualize the ciliary axoneme.
(A) TIDE analysis showing genomic alterations in the engineered 3T3-L1 sgTulp3 cell line. (B) TIDE analysis showing genomic alterations in the engineered 3T3-L1 sgFfar4 cell line. (C) Loss of TULP3 has no effect on ciliary structure, as determined by acetylated tubulin staining, but prevents trafficking of ciliary proteins, including ARL13B. (D) Loss of TULP3 using siRNA-mediated knockdown results in attenuated adipogenesis. 3 different siRNAs targeting TULP3 were used, siRNA targeting PPARγ is positive control. Lipid accumulation visualized by Oil Red O staining (E) Quantification of adipogenesis by isopropanol extraction of Oil Red O. (F) Quantification of TULP3 knockdown efficiency using siRNA. (G) Immunoblot showing depletion of TULP3 in 3T3-L1 sgTulp3 cell line, and overexpression of GFP-TULP3 fusion protein. P-values calculated using t test, * p<0.05;
(A) Left: Immunofluorescence for preadipocytes (PDGFRα, red) and cilia (ARL13B, green) 5 weeks post tamoxifen administration in control and PAno cilia mice. Ciliated PAs are marked by arrowheads. Scale bar is 25 μm. Right: Quantifications of the percentage of ciliated PAs present 5 weeks after conditional removal of PA cilia. Far right: Comparison of Ift88 expression between control and PAno cilia mice via RT-qPCR from whole gonadal WAT (n=4 per genotype). (B) Left: Body weight measurements in control and PAno cilia mice (n=4 for control and n=3 for PAno cilia mice). Right: Picture of gonadal fat pad and Echo-MRI measurements of total fat and lean mass in control and PAno cilia mice 11 weeks after tamoxifen administration. Scale bar is 1cm. (C) H&E staining of gonadal WAT tissue sections and the respective quantifications of the mean area of adipocytes in control and PAno cilia mice. Scale bar is 100 μm. (D) Left: Oil Red O-stained liver sections from control and PAno cilia mice. Right: Quantification of area occupied by Oil Red O between genotypes. Scale bar is 100 μm. (E) Serum levels for insulin and free fatty acids (FFA) were measured using ELISA and glucose levels using a glucometer between control and PAno cilia mice at either fed state or after an overnight fasting period as indicated. (F) Plotting of hourly oxygen consumption (VO2), food intake and total movement over 4 days of control (n=♂14 & ♀12) and PAno cilia mice (n=♂7 & ♀9). (G) Dissected interscapular brown adipose tissue (BAT) of two control and two PAno cilia mice. Scale bar is 1 cm. Comparison of Ift88, Ucp1, Pparγ and Perilipin expression between control (n=6) and PAno cilia mice (n=5) via RT-qPCR from whole interscapular BAT. All data are represented as mean ± SEM. p-values were calculated using standard t-test and two-way ANOVA followed by Tukey’s multiple comparison test (*<0.05, **<0.01, ***<0.001 and ****<0.0001). Of note, all mice depicted are littermates and maintained on breeder chow starting the day of tamoxifen administration.
(A) 3T3-L1 FFAR4-GFP fusion protein localizes to primary cilium. SMO-GFP is positive control. (B) Validation of ciliary localization of FFAR4 using second independent antibody (Santa Cruz). 3T3-L1 cells on Day 0 and Day 2 of differentiation. (C-E) Validation of FFAR4 antibody in (C) 3T3-L1 cells using Crispr/Cas9, (D) SVF isolated from inguinal and (E) epididymal white adipose tissue of wild-type and Ffar4 knockout littermates. We note that there is some non-specific ciliary FFAR4 background staining with this antibody. (F) Representative immunofluorescence images showing depletion of ciliary FFAR4 with loss of TULP3 protein. (G) FFAR4 expression by mRNA and protein increases during differentiation. 20x objective was used to visualize the whole cell. This makes visualization of individual cilia difficult. (H) Endogenous FFAR4 is ciliary during 3T3-L1 differentiation. D0, 2, 4, 6 are Day 0, 2, 4 and 6 of differentiation. (I) Endogenous FFAR4 localizes to the primary cilium of preadipocytes and to the plasma membrane of mature adipocytes in vivo in epididymal WAT whole mounts from cilia glow male mice. The fluorescence intensity of FFAR4 localized to the primary cilia is higher than the fluorescence intensity of FFAR4 localized to the adipocyte plasma membrane, consistent with FFAR4 receptor density being higher in the primary cilium. White arrows and boxes highlight cilia.
(A) DHA cocktail promotes activation of adipogenic genes Adiponectin, Fabp4, and Plin1, and represses expression of preadipocyte genes Col1a and Dlk1. These data show that DHA cocktail initiates adipogenesis of 3T3-L1 preadipocytes. Bar graph is average of three independent experiments ± SD. (B) DHA cocktail results in cell cycle re-entry and this is dependent on Tulp3 protein. Representative images showing EdU incorporation of confluency-arrested 3T3-L1 cells maintained in growth media, or differentiated using the historical DM, the ctrl cocktail (-DHA), or the DHA cocktail. EdU was added at 16h, and cells were fixed and stained at 40h. (C) Addition of historical DM (1μg/ml insulin, 1μM dexamethasone, 0.5mM IBMX) to 3T3-L1 cells lacking Tulp3 promotes cell cycle re-entry. Tulp3 protein is only required for DHA-mediated mitotic differentiative expansion and adipogenesis. (D) Quantification of adipogenesis in the presence or absence of FFAR4 agonist with one component of the modified cocktail omitted per condition. Images shown in Figure 5B. FFAR4 agonist can partially substitute for Dex, insulin, and IBMX, but has quantitatively the largest effect replacing IBMX. This suggests that FFAR4 activation results in elevation of cAMP second messenger. This can enhance adipogenesis in the absence of any single cocktail component, but has the greatest effect replacing the phosphodiesterase inhibitor IBMX. (E) Loss of Tulp3 prevents increase in ciliary cAMP by addition of FFAR4 agonist. cAMP sensor with red fluorescent constitutive ciliary marker was expressed in 3T3-L1 sgGFP (wild type control) and sgTulp3 cells. Addition of FFAR4 agonist, but not DMSO control, in wildtype but not Tulp3 knockout cells results in a significant decrease in the ratio of green fluorescence (cAMP sensor) to red fluorescence (constitutive ciliary marker) intensities, showing that FFAR4 activation elevates ciliary cAMP and that this is dependent on Tulp3 and thus ciliary localization of FFAR4. Bar graphs represent mean ± SD. *<0.05; ** p<0.01; *** p<0.001.
(A) DM component titration in the presence or absence of 100μM DHA to determine the optimal concentrations of insulin, dexamethasone, and IBMX to observe enhancement of differentiation by DHA. DHA and DM components were added for 48h to 2 day post-confluent 3T3-L1 cells. At 0.4μg/ml insulin, 0.02mM IBMX, 0.04μM dexamethasone (red circle), there is little differentiation with EtOH control and robust differentiation in the presence of DHA, as visualized by Oil Red O. (B) DHA does not promote adipogenesis by directly activating PPARγ. 3T3-L1 preadipocytes were differentiated using the DHA cocktail for 2 days followed by maintenance media (1μg/ml insulin) for 4 days. Lipid accumulation was assessed by green fluorescent BODIPY dye. The PPARγ antagonist T0070907 was added either concurrently with DHA (D0–2) or throughout the differentiation time course (D0–6). Presence of the PPARγ antagonist throughout the differentiation time course (D0–6) inhibits adipogenesis in a dose-dependent manner, consistent with PPARγ being required during the second phase of adipogenesis. The presence of the PPARγ concurrently with DHA (D0–2) does not inhibit adipogenesis, arguing that DHA does not promote adipogenesis by directly activating PPARγ. (C) FFAR4 antagonist (AH7614) has a lower IC50 when 3T3-L1 adipogenesis is induced by DHA cocktail versus historical DM. (D) Addition of FFAR4 agonist TUG891 (100μM) during the first 72h of differentiation enhances differentiation of primary murine preadipocytes in the SVF from inguinal WAT. (E) Addition of FFAR4 antagonist AH7614 (10μM) to differentiating human preadipocytes during the first 72h attenuates differentiation. (F) FFAR4 activation promotes adipogenesis of primary mouse preadipocytes via the primary cilium. Preadipocytes (Lin−, CD34+, SCA1+, CD29+) isolated from Ift88flox/flox males were transduced with an adenovirus expressing GFP only or GFP and Cre recombinase (+Cre). IFT88 is required for ciliation and percent ciliation was determined by FGFR1OP (basal body) and ARL13B (axoneme) staining of GFP+ confluent preadipocytes. Percent differentiation was determined by LipidTox Neutral Red staining of GFP+ cells differentiated with modified cocktail in the presence or absence of 100μM TUG891. Representative images shown. n = number of GFP+ cells counted. Differentiation data is normalized to ctrl ± SD. ** p<0.01; *** p<0.001.
(A) Quantification of mRNA levels of Cebpβ, Pparγ, and Cebpα at different differentiation time points using control cocktail (EtOH) or DHA cocktail (DHA). We observe significant DHA-dependent transcriptional activation of Pparγ and Cebpα within 24h of differentiation, but not of CEBPβ. (B) A PPARγ-T2A-GFP knockin reporter generated using Crispr/Cas9 shows increased expression of PPARγ in response to DHA cocktail (DHA) compared to control cocktail (EtOH) within 3 days of differentiation. Addition of the FFAR4 antagonist AH7614 inhibits PPARγ expression in a dose-dependent manner. Dotted line denotes media changes, images are taken every 2h. (C) Immunoblot showing CEBPβ and CEBPα protein levels at different time points of differentiation using control cocktail or DHA cocktail. We consistently observe a DHA-dependent increase in expression of CEBPα, but not of CEBPβ. (D) Immunoblot showing phosphorylated and overall levels of ERK, AKT, and CREB levels at different differentiation time points using control cocktail, DHA cocktail, or FFAR4 agonist (TUG891) cocktail. While there is activation of these adipogenic regulators during differentiation, this is not DHA-dependent, suggesting that cocktail components other than DHA, notably insulin, activate these adipogenic regulators. (E) While ERK and AKT activation is not mediated by DHA, activation of these pathways is required for DHA-enhanced adipogenesis, consistent with the notion that cocktail components other than DHA activate these adipogenic regulators and this is required for adipogenesis. (F) 3T3L1 cells lacking CTCF show only a minor, not significant growth defect. Cell growth was assessed using Essen IncuCyte Live Cell Imaging Analysis in normal growth media; images taken every 2h. (G) Loss of CTCF does not affect adipogenesis induced by the historical DM (1μg/ml insulin, 1μM dexamethasone, 0.5mM IBMX). (H-I) Three different 3T3-L1 Ctcf knockout cells lines were generated by Crispr/Cas9 using 3 different sgRNAs. The cell line sgCtcf-1 was used in Fig 6F–G. (H) All three lines show an adipogenesis defect in response to DHA cocktail. (I) TIDE analysis describing genomic rearrangement of all three engineered 3T3-L1 sgCtcf cell lines.
Data Availability Statement
GEO accession numbers are GSE118470 (ATACseq of 3T3L1 cells undergoing differentiation) and GSE118471 (RNAseq of 3T3L1 cells undergoing differentiation)