

Abstract
Tuning reactivity of sulfur electrophiles is key for advancing click chemistry and chemical probe discovery. To date, activation of the sulfur electrophile for protein modification has been ascribed principally to stabilization of a fluoride leaving group (LG) in covalent reactions of sulfonyl fluorides and arylfluorosulfates. We recently introduced sulfur-triazole exchange (SuTEx) chemistry to demonstrate the triazole as an effective LG for activating nucleophilic substitution reactions on tyrosine sites of proteins. Here, we probed tunability of SuTEx for fragment-based ligand discovery by modifying the adduct group (AG) and LG with functional groups of differing electron-donating and -withdrawing properties. We discovered the sulfur electrophile is highly sensitive to the position of modification (AG versus LG), which enabled both coarse and fine adjustments in solution and proteome activity. We applied these reactivity principles to identify a large fraction of tyrosine sites (~30%) on proteins (~44%) that can be liganded across >1500 probe-modified sites quantified by chemical proteomics. Our proteomic studies identified non-catalytic tyrosine- and phosphotyrosine-sites that can be liganded by SuTEx fragments, with site specificity, in lysates and live cells to disrupt protein function. Collectively, we describe SuTEx as a versatile covalent chemistry with broad applications for chemical proteomics and protein ligand discovery.
TOC graphic
INTRODUCTION
Covalent small molecules are enabling tools for investigating protein function in biology1 and represent an important class of drug molecules2. Electrophilic or photoreactive groups embedded in fragments or high molecular weight binders have been used, in combination with proteomic technologies, to uncover ligand sites that can be exploited for pharmacological control3–5. Development of cysteine-5–8 and lysine-reactive chemistry9–12, for example, are creating new opportunities for perturbing and degrading proteins based on enzymatic and non-catalytic functions13–14. Beyond liganding sites on proteins4–5, 9, 15, covalent probes can be adapted to study post-translational modifications (PTM) including crotonolyation16, methylation17, deimination18, and phosphorylation19. New chemoselective reactions, therefore, are important for advancing chemical probes used for basic and therapeutic discovery.
We recently introduced sulfur-triazole exchange (SuTEx) chemistry as a new class of electrophiles for chemical proteomic applications19. Akin to sulfonyl-fluorides20–22 and fluorosulfates23–29 (i.e. SuFEx30), the SuTEx reaction occurs through nucleophilic attack at the sulfur center with stabilization of the leaving group (LG) as a likely driving force to facilitate protein reaction (Figure 1). In contrast with fluoride on SuFEx, the addition of a triazole LG on SuTEx molecules introduced additional capabilities for tuning reactivity of the sulfur electrophile. We demonstrated, using a collection of alkyne-modified SuTEx probes, that structural modifications to the triazole LG can dramatically enhance chemoselectivity of SuTEx reaction for tyrosine over other nucleophilic residues on proteins in both lysates and live cells19. We exploited the tyrosine reactivity of SuTEx to develop a chemical phosphoproteomics strategy for profiling activation of tyrosine phosphorylation19.
Figure 1.

Investigating reactivity of the sulfur electrophile using sulfur-triazole exchange (SuTEx) chemistry.
To date, SuTEx has been explored largely as a proteomic tool for global quantification of changes in tyrosine function and post-translational state. Our functional profiling studies revealed a subset of hyper-reactive tyrosines (~5% of all quantified tyrosines) that were localized to enzyme active sites but also prevalent in domains mediating protein-protein and -nucleotide interactions19. The availability of reactive tyrosines combined with the ability to modulate reactivity, and potentially specificity, supports SuTEx as a promising strategy for fragment-based ligand discovery31–33 (FBLD). However, the sensitivity of the sulfur electrophile to functional group modifications on the adduct- and leaving-groups and whether there is an advantage to modifying both positions for protein reaction has not been systematically evaluated. Furthermore, the functional consequences of liganding tyrosines on proteins with SuTEx electrophiles is currently unknown.
Here, we developed a library of fragment electrophiles to investigate the tunability of SuTEx in both solution and proteomes. We discovered the sulfur electrophile is highly sensitive to the position of chemical modification, which permitted both coarse and fine adjustments for activating nucleophilic substitution reactions. We applied our reactivity findings to demonstrate the versatility of SuTEx for FBLD. Through competitive studies with a SuTEx fragment library, we discovered >300 liganded tyrosine sites across hundreds of distinct protein targets quantified by chemical proteomics. Finally, we apply SuTEx to identify non-catalytic tyrosine and phosphotyrosine sites and show liganding these sites in lysates and live cells is a viable strategy for disrupting protein function.
RESULTS AND DISCUSSION
SuTEx fragment design and synthesis
We synthesized a fragment library of 1,2,4-sulfonyl triazoles to test whether SuTEx chemistry could be adapted for development of protein ligands. We selected the SuTEx probe HHS-482 as a lead scaffold for fragment development because this sulfonyl-triazole showed the highest tyrosine chemoselectivity among probes tested previously19. The common SuTEx electrophile core was structurally elaborated with diverse small molecule binding elements on both the adduct group (AG) and LG to create library members with an average molecular weight of 336 Da (Supplementary Figure 1). Fragments were created with structural elements bearing differing electron-withdrawing (EWG) or -donating (EDG) properties to test substituent effects on SuTEx reaction mechanism. Functional groups that are EWG by both resonance and polar interactions (cyano) as well as substituents (fluoro) with opposing effects from resonance (EDG) and polar (EWG) components were represented in our library34. We also included alkyl groups (cyclopropyl) for direct comparison with aryl substituents.
R-substituted phenyl amides were coupled with DMF-DMA to produce amidine intermediates that underwent cyclization in acetic acid with hydrazine hydrate to form the corresponding 1,2,4-triazole35 (Scheme 1). In general, amidine cyclization reactions proceeded with greater than 75% yields across diverse functional groups and were purified by re-crystallization to complete the entire process in ~6 hrs. AG diversity was introduced by coupling 1,2,4-triazoles with alkyl- or aryl-sulfonyl chlorides modified with respective functional groups. Interestingly, aryl sulfonyl chlorides reacted rapidly with 1,2,4-triazoles (completion in minutes at room temperature) while alkyl counterparts reacted slowly or not at all under the same conditions. See Supporting Information for details and characterization of SuTEx fragments.
Scheme 1.
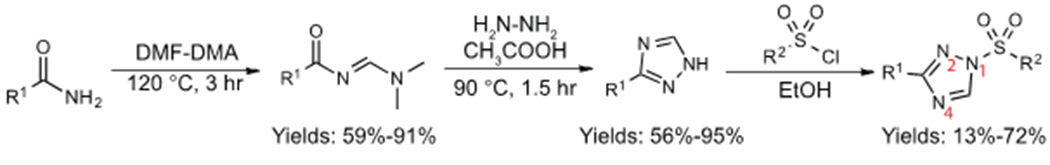
Synthetic scheme showing general strategy for synthesis of a 1,2,4-sulfonyl triazole fragment library.
In summary, we developed an efficient synthetic strategy for installing chemical diversity into SuTEx molecules via both AG and LG modifications. Our findings demonstrate good functional group tolerance to build structurally diverse SuTEx fragments. Compared with SuFEx, the SuTEx scaffold offers new opportunities to simultaneously probe features of the AG and LG that affect covalent reaction of the sulfur electrophile.
Tuning SuTEx reactivity
We used high-performance liquid chromatography (HPLC) to investigate the effects of AG/LG modifications on SuTEx reactivity in solution. We selected nucleophiles that modeled tyrosine (p-cresol) and lysine (n-butylamine) side chains for our HPLC studies based on previous reports of SuTEx reaction with these residues19. We predicted that SuTEx fragments exposed to p-cresol or n-butylamine, in the presence of tetramethylguanidine (TMG) base, would undergo nucleophilic substitution reactions that could be monitored by depletion of SuTEx fragment- and appearance of the respective covalent product-signal (Supplementary Figure 2). We synthesized standards of predicted products from reaction of each SuTEx fragment to optimize chromatography and detection in our HPLC assay (see Supporting Information for details and chromatograms of HPLC assay).
A direct comparison of different AGs revealed differences in reaction of alkyl-compared with aryl-sulfonyl-triazoles. The addition of a cyclopropyl group on the AG eliminated activity of SuTEx fragments towards p-cresol (Figure 2A). Closer inspection of aryl-sulfonyl-triazoles revealed trends in reactivity that support electronic effects of substituents to facilitate covalent reaction. For example, modification with the cyano EWG group resulted in rapid reaction as determined by the calculated half-life for fragment consumption (JWB137, t1/2 = 1.1 min; Figure 2A). Substitution with another electron-deficient aromatic system such as pyridine also resulted in rapid reaction of the sulfur electrophile with p-cresol (JWB141, t1/2 = 1.6 min; Figure 2A). In contrast, substituents like the fluoro (JWB135) and biphenyl group (JWB142) characterized by mixed polar and resonance interactions34 showed attenuated reactivity (t1/2 values of ~16 min, Figure 2A). Addition of a methoxy group dramatically reduced SuTEx reactivity as evidenced by incomplete reaction in the time-frame tested (JWB136, Figure 2A).
Figure 2. Tuning reactivity of the sulfur electrophile.

SuTEx fragments were incubated with p-cresol in the presence of tetramethylguanidine (TMG, 1.1 equivalents) base and time-dependent covalent reaction monitored by the reduction of respective fragment starting material. Modifications to the adduct group (AG; A) and triazole leaving group (LG; B) could alter solution reactivity of SuTEx fragments. The calculated half-life of individual SuTEx fragments are shown in parentheses. The half-lives for all SuTEx fragments tested are listed in Table S1. Formation of the p-cresol adduct was confirmed by retention time that matched synthetic standards for respective reaction products (see Supporting Information for details of HPLC methods and data). Data shown are representative of n = 3 independent experiments.
Modifications to the LG altered SuTEx reactivity in a more graded fashion that correlated with the electron withdrawing character of the respective substituent. For example, the addition of a trifluoromethyl group to the phenyl-triazole LG accelerated solution reaction with p-cresol (compare JWB105 and JWB150; Figure 2B). In contrast with the AG, modifications to the LG resulted in more subtle alterations in SuTEx reaction as evidenced by comparing t1/2 values across the fragments tested. Comparing the range of t1/2 values across fragments demonstrated that AG modifications have a more severe impact on SuTEx reaction (t1/2 from 1 to >360 min) compared with analogous changes on the LG (t1/2 from 3 to 14 min; Figure 2 and Table S1). Finally, we found that SuTEx fragments reacted with p-cresol more rapidly compared with n-butylamine, which matched our previous findings that SuTEx chemistry is more phenol reactive19 (Table S1).
In summary, our solution studies highlight the merits of modifying the AG and LG for broad- and fine-tuning, respectively, of SuTEx reaction with nucleophiles in solution. The general enhancement of the nucleophilic substitution reaction with EWG substituents is likely due to the increased electrophilic character of the sulfur center. Importantly, the acceleration in covalent reaction did not compromise chemoselectivity of SuTEx for phenol-over amine-nucleophiles. We also identified a cyclopropyl-AG modification that largely eliminated SuTEx reactivity, which provides a means to produce inactive negative control molecules. Taken together, SuTEx chemistry offers multiple avenues for controlling electrophilicity of the sulfur center, which are key features for enabling protein ligand discovery.
Proteome-wide structure-reactivity relationships of SuTEx fragments
Next, we tailored our reported chemical proteomic method for functional tyrosine profiling19 to investigate AG/LG effects on SuTEx fragment reactivity in complex proteomes (Supplementary Figure 3). In brief, isotopically light and heavy soluble proteomes from DM93 melanoma cells cultured by stable isotopic labeling by amino acids in cell culture (SILAC36) media were used for quantitative liquid chromatography-mass spectrometry (LC-MS) studies. Light and heavy DM93 proteomes were treated with dimethyl sulfoxide (DMSO) vehicle or SuTEx fragment (50 μM, 30 min, 37 °C), respectively, followed by labeling with the tyrosine-reactive probe HHS-48219 (50 μM, 30 min, 37 °C) and copper-catalyzed azide-alkyne cycloaddition (CuAAC) conjugation of a desthiobiotin-azide enrichment tag. Proteomes were digested with trypsin protease, HHS-482-modified peptides containing a desthiobiotin tag enriched by avidin chromatography and analyzed by high-resolution LC-MS/MS and bioinformatics as previously described19.
To evaluate substituent effects on proteome activity, we compared reactivity profiles of each respective SuTEx fragment across >1500 total distinct HHS-482-modified tyrosine sites from >650 detected proteins (Supplementary Figure 4 and Table S1). Fragments were screened across independent biological replicates (n = 2-3) and high-quality tyrosine site annotations were identified by detection in at least a single biological replicate from each fragment dataset, probe-specific enrichment (HHS-482 probe/DMSO SILAC ratio (SR) >5), and quality control confidence criteria of ≥300 Byonic score37, 1% protein false discovery rate (FDR), and ≤5 ppm mass accuracy in order to minimize false positives19. SILAC ratios (SR) from competitive studies (Light – DMSO/Heavy-fragment) were used to identify fragment-competed tyrosine residues as sites showing >75% reduction in enrichment by HHS-482 compared with DMSO vehicle control (i.e. liganded tyrosines, SR >4; Figure 3A and B). In total, we identified 305 liganded tyrosines on 213 distinct proteins, which corresponded to ~30% and ~44% of total quantified tyrosines and proteins, respectively (Figure 4A); these percentages are comparable with ligandability measures reported for cysteines5. In agreement with previous SuTEx studies19, we observed a high preference for tyrosine compared with lysine sites (Y/K ratio) in our fragment ligand competition studies (average Y/K ratio of 4.5 Supplementary Figure 5).
Figure 3. Fragment-based ligand discovery using SuTEx.

(A) Heat map showing SILAC ratios (SR) of representative tyrosines competed by fragments and organized by hierarchical clustering. Fragment competition at tyrosine sites was quantified using the area under the curve of MS1 extracted ion chromatograms (EIC) from HHS-482-labelled peptides in DMSO (light, red) versus fragment-treated (heavy, blue) DM93 soluble proteomes. Competitive chemical proteomic studies were performed as shown in Supplementary Figure 3. Tyrosine sites shown are liganded (SR >4) by at least 2 fragments with the number of liganded sites and proteins listed for each molecule. Y-axis lists the protein name and quantified tyrosine site. (B) Representative MS1 EICs of tyrosine sites from quantitative LC-MS chemical proteomics: non-liganded (blue, SR <2), partially-liganded (orange, 2≤ SR ≤4), and liganded (yellow, SR >4). (C) Reactivity of fragments was assessed by comparing the fraction of tyrosine sites competed: non-liganded (blue, SR <2), partially-liganded (orange, 2≤ SR ≤4), and liganded (yellow, SR >4). All data shown are representative of n = 2-3 biologically independent experiments.
Figure 4. Analysis of tyrosines and proteins liganded by SuTEx fragments.
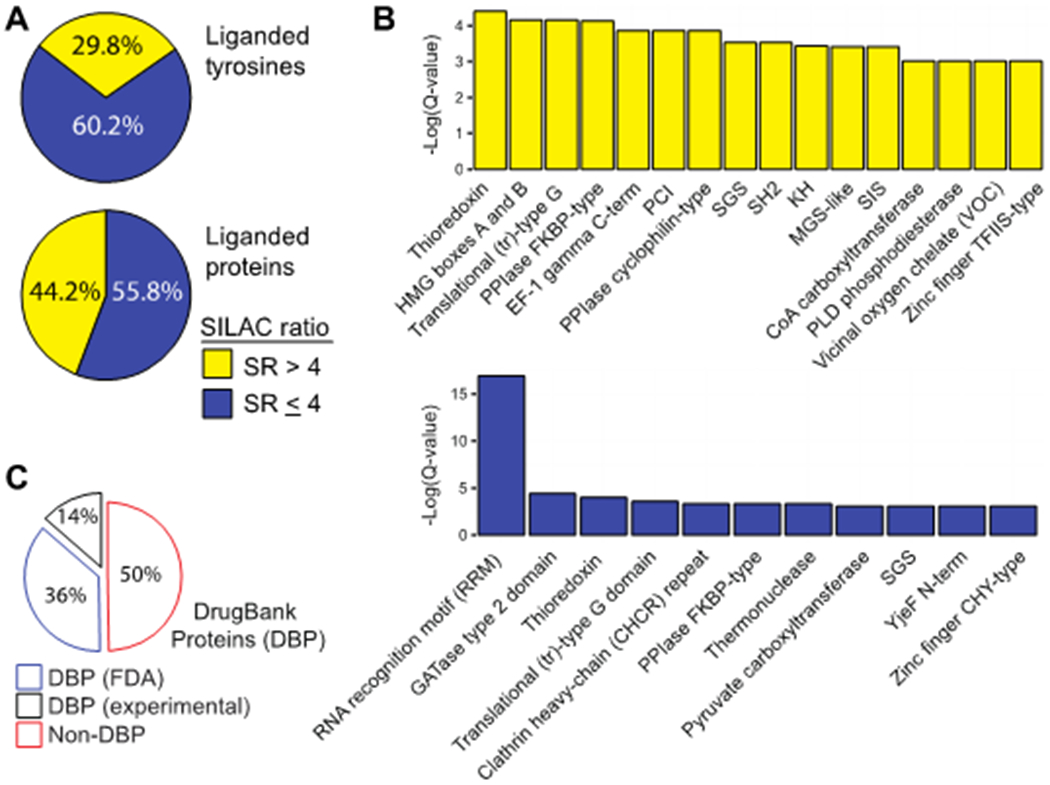
(A) Distribution of liganded and non-liganded tyrosine sites and proteins from chemical proteomic analyses of DM93 soluble proteomes. Data shown for quantified tyrosines (top) and proteins (bottom) that were liganded (SR > 4) by at least 1 fragment. (B) Enriched domain annotations as determined by Q < 0.05 after Benjamini–Hochberg correction of a two-sided binomial test. (C) Distribution of liganded proteins (SR > 4) found in DrugBank (DBP group) compared with proteins that did not match a DrugBank entry (non-DBP). All data shown are representative of n = 2-3 biologically independent experiments.
Liganded tyrosine sites were enriched for functional domains involved in nucleotide binding (PRU00267, PRU1059), protein-protein interactions (PRU00191, PRU00386), enzymatic reactions (PRU00691, PRU00277), and metal binding (PRU01163, PRU00472; Figure 4B). A large fraction of liganded tyrosines resided in proteins absent from the DrugBank database38, which supports SuTEx fragments targeting proteins that lack pharmacological probes (Figure 4C). Liganded tyrosines included enzymes such as GSTP1, for which we previously identified a hyper-reactive catalytic tyrosine in the glutathione binding site (Y8), as well as a tyrosine site (Y273) in the first catalytic cysteine half-domain (FCCH39) of the ubiquitin activating enzyme UBA140–41. Non-liganded tyrosines were enriched for domain classes that were distinct from liganded tyrosines and similar to profiles observed for SuTEx alkyne probes19 (Figure 4B). These data support the importance of molecular recognition for SuTEx fragment-tyrosine interactions at protein binding sites. Differences in reactivity were observed with individual fragment electrophiles that displayed liganded tyrosine frequencies ranging from <0.1% (JWB142) to >25% (JWB150) with a mean liganded frequency of 4.6% (Figure 3C).
Liganded tyrosines showed clear structure-activity relationships (SAR) with the SuTEx fragment library (Figure 3A). Comparison of JWB150, JWB152, and JWB146 uncovered relative trends in proteomic reactivity that suggest EWGs on the AG as a common feature of SuTEx fragments with higher liganded tyrosine frequencies (Figure 3A and C). Despite these proteomic trends, which somewhat matched our HPLC studies (Figure 2), we also observed differences that directly contrasted with general reactivity profiles of SuTEx fragments. For example, JWB152 showed a lower liganded tyrosine frequency compared with JWB150 despite exhibiting substantially higher reactivity in our HPLC assay (Supplementary Figure 6 and Table S1). These data suggest that in addition to driving reactivity, structural modifications on the AG can contribute to binding events that enhance fragment-tyrosine interactions of compounds sharing a common LG. The differences in reactivity profiles of JWB198 and JWB202, which are differentiated by AG structure on a common LG scaffold, further support recognition as a contributor of SuTEx fragment interactions on proteins (Figure 3A and Supplementary Figure 1). We also identified several fragments including JWB142 and JWB146 with a reduced liganded tyrosine frequency while retaining high activity (SR >6) against tyrosine competed sites on YWHAE42 (Y49) and PLD343 (Y437), respectively (Figure 3A and C). Finally, we discovered that the cyclopropyl-AG-modified fragment JWB131 was largely unreactive against the proteome (Figure 3A and Supplementary Figure 1).
In summary, our chemical proteomic studies highlight the advantage of modifying the AG and LG on SuTEx fragments for tuning reactivity and specificity at tyrosine sites on proteins (Figure 3 and 4). In contrast with previous efforts to develop globally reactive probes19, our current efforts identified SuTEx fragments with reduced proteome reactivity while retaining high efficiency for competing at tyrosine sites on select proteins (JWB202, and JWB198; Figure 3 and Supplementary Figure 4). The latter finding supports AG and/or LG modification as a strategy for not only controlling electrophilicity (Figure 2) but also to alter molecular recognition at protein binding sites as evidenced by the distinct profile of enriched domains in liganded (fragment activity) compared with non-liganded sites (general probe enrichment; Figure 4B). Importantly, the chemoselectivity for tyrosine over lysine in proteomes is retained in structurally diverse fragments that, combined with the ability to prioritize tyrosine sites based on hyper-reactivity19, positions SuTEx as a promising strategy for FBLD31–33.
Liganding a non-catalytic tyrosine to disrupt protein function
To determine the functional impact of tyrosine-ligand interactions identified by SuTEx, we selected human DPP3 because it contains a single probe-modified tyrosine site (Y417) that is not catalytic but near the zinc binding region of this metallopeptidase19, 44 (Figure 5A). Our goal was to test whether liganding a non-catalytic tyrosine is a viable strategy for developing inhibitors of enzymes like DPP3. We screened our SuTEx fragment library for DPP3 ligands by competitive gel-based chemical proteomic profiling with HHS-482 (100 μM fragment, 37 °C, 30 min; Supplementary Figure 7A and B). We quantified results from our gel-based competition screens to identify fragment hits that showed activity against DPP3 while maintaining reasonable selectivity (i.e. not broadly reactive) across the proteome (Supplementary Figure 7C). DPP3 fragment hits were verified as inhibitors using an established peptidase assay19, which led to identification of JWB142 as our lead DPP3 inhibitor based on good inhibitory activity and increased selectivity compared with other candidate molecules (Supplementary Figure 7D).
Figure 5. Liganding non-catalytic tyrosines for blockade of protein activity.
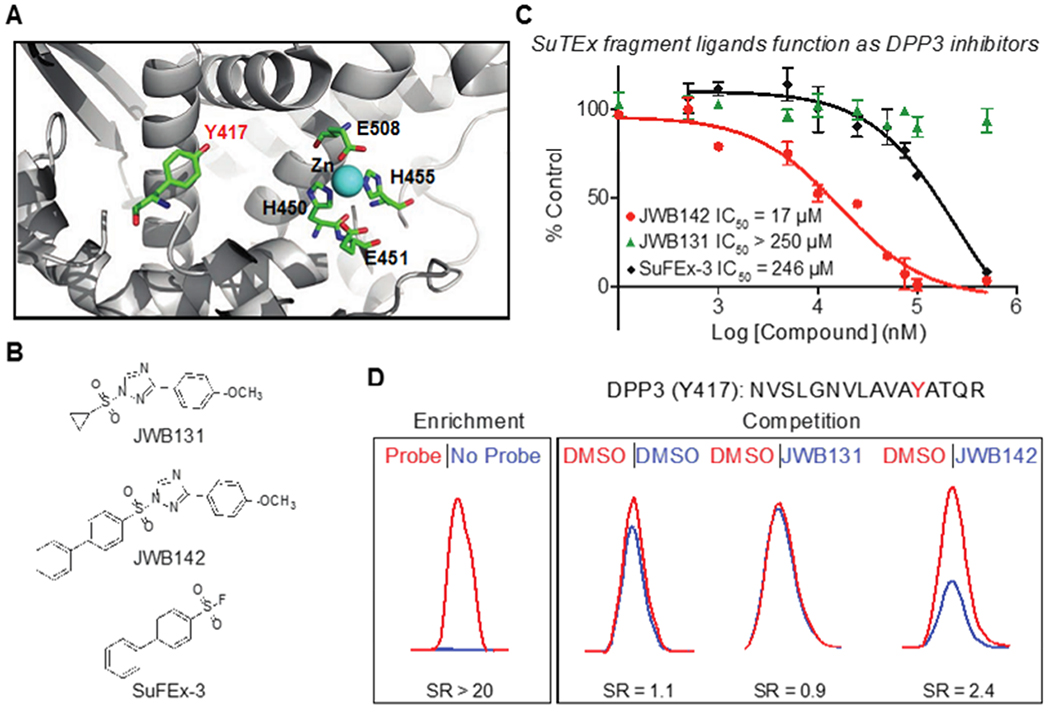
(A) Crystal structure of human DPP3 active site (PDB accession code 3FVY). The location of residues involved in zinc metal binding (H450, H455, E508), the catalytic glutamate (E451), and a non-catalytic tyrosine 417 (Y417) identified by SuTEx are highlighted. (B) Lead SuTEx fragments (JWB142) and negative control probe (JWB131) identified from a gel-based chemical proteomic screen against recombinant DPP3 proteomes (Supplementary Figure 7). (C) JWB142 but not JWB131 blocked catalytic activity of purified DPP3 in a concentration-dependent manner as measured by substrate assay: JWB142, IC50 = 17 μM, 95% confidence intervals: 11-27 μM. JWB142 showed >10-fold increase in inhibitory activity compared with the SuFEx counterpart: SuFEx-3, IC50 = 246 μM, 95% confidence intervals: 117-519 μM. Data are shown as mean ± s.e.m.; n = 3 biologically independent experiments. (D) DPP3 Y417 site is liganded (~50% blockade) by JWB142 but not JWB131 fragment as judged by quantitative chemical proteomic analysis of recombinant human DPP3-HEK293T soluble cell proteome. All data shown are representative of n = 2 biologically independent experiments.
Given the proximity of Y417 to the catalytic zinc in the active site (Figure 5A), we predicted that JWB142 disrupts DPP3 peptidase function by liganding the Y417 site. First, we demonstrated that pretreatment with JWB142 resulted in concentration-dependent blockade of recombinant DPP3 peptidase activity (IC50 = 17 μM, Figure 5B and C). We included a structurally analogous negative control molecule that contained a cyclopropyl-modified AG that rendered JWB131 inactive against DPP3 to determine site specificity of inhibitory activity for Y417 (Figure 5B and C). In support of our hypothesis, we demonstrated the ability of our lead fragment to ligand the Y417 site by LC-MS chemical proteomic analysis of recombinant DPP3-HEK293T proteomes (50 μM fragment, 37 °C, 30 min). We observed ~50% blockade of HHS-482 labeling of DPP3 Y417 with JWB142 but not JWB131 competition (SR = 2.4, Figure 5D).
We also evaluated a biphenyl sulfonyl-fluoride analog of JWB142 to compare potency of SuTEx and SuFEx for development of protein ligands (SuFEx-3, Figure 5B). In agreement with reduced activity of sulfonyl-fluoride compared -triazole compounds19, SuFEx-3 showed >10-fold reduced potency against DPP3 compared with JWB142 (IC50 = 246 μM, Figure 5C). The difference in biochemical activity was also reflected by HPLC assays, which showed completion of JWB142 reaction within ~6 hours while SuFEx-3 was largely unreactive for the same time period (Supplementary Figure 8).
Our findings identified JWB142 as a DPP3 ligand that blocks biochemical function via covalent modification of Y417 located adjacent to the catalytic zinc-binding site. Akin to targeting non-catalytic cysteines for inhibitor development45, we demonstrated that liganding a non-catalytic tyrosine is a viable strategy for blocking protein activity (Figure 5). Specifically, we included a matching inactive control molecule JWB131 to demonstrate site specificity for JWB142 blockade of DPP3 biochemical activity (Figure 5C and D). We also demonstrated that SuTEx can dramatically enhance potency of sulfur electrophiles (compare JWB142 and SuFEx-3, Figure 5B and C) while maintaining reasonable specificity across the proteome (JWB142, Figure 3A). Future efforts will focus on further optimization of JWB142 to improve affinity and specificity for inactivation of DPP3, which has been implicated in nociception (via N-terminal cleavage of opioid peptides) and human cancers including ovarian46 and squamous cell lung carcinomas47 through increased enzymatic or protein-protein interaction function, respectively.
Liganding a phosphotyrosine site in live cells
We next tested whether SuTEx fragments could serve as protein ligands in live cells. We chose glutathione S-transferase Pi (GSTP1) for proof-of-concept studies because it possesses a single hyper-reactive tyrosine that is catalytic and a reported phosphorylation site (Y819, 48). Consistent with its hyper-reactive character, we showed robust HHS-482-labeling of recombinant WT GSTP1 that was lost in Y8F mutant and validates use of this probe for a gel-based competitive assay screen of potential GSTP1 inhibitors (Supplementary Figure 9). We screened recombinant human GSTP1-HEK293T proteomes against our SuTEx library (50 μM, 37 °C, 30 min) and identified several hit fragments that showed >80% blockade of HHS-482-labeling (Supplementary Figure 9). We chose to focus on JWB152 and JWB198 for further studies because of the availability of structurally analogous negative control compounds to evaluate specificity in our pharmacological experiments (JWB146 and JWB191, respectively; Figure 6A).
Figure 6. Liganding a hyper-reactive phosphotyrosine site of GSTP1 in live cells.

(A) Gel-based chemical proteomic analysis of GSTP1-HEK293T soluble proteomes pretreated with vehicle or fragment electrophiles (50 μM, 30 min, 37 °C) followed by labeling with HHS-482 under the same treatment conditions. GSTP1 Y8F mutant shows >90% reductions in probe labeling compared with wild-type protein. JWB152 and JWB198 but not JWB146 or JWB191 block HHS-482 labeling to levels comparable with Y8F mutant. Western blot analyses (α-FLAG) confirm equivalent FLAG-tagged GSTP1 expression across all conditions tested. (B) In vitro potency of JWB152 and JWB198 against recombinant GSTP1 lysates as evaluated by GSH substrate assay (JWB152, IC50 = 23 μM, 95% confidence intervals: 14-39 μM; JWB198, IC50 = 16 μM, 95% confidence intervals: 11-22 μM . The negative control probes JWB146 and JWB191 did not show inhibitory activity even at the highest concentration tested (250 μM). The SuFEx analog (SuFEx-2) showed moderate inhibition of GSTP1 activity at the highest concentration tested (250 μM). Data are shown as mean ± s.e.m,; n=3 biologically independent experiments. (C) GSTP1 Y8 site is liganded (~70% blockade) by JWB198 but not JWB146 in live DM93 cells treated with SuTEx fragments followed by quantitative chemical proteomic analysis. (D) Heat map showing quantified tyrosine sites on GSTP1 and the ability of JWB198 to ligand Y8 with site specificity in live cells. JWB146 was inactive against all GSTP1 tyrosine sites quantified. See Supplementary Figure 11 for details on location of quantified tyrosine sites in the GSTP1 crystal structure. All data shown are representative of n=2 biologically independent experiments.
We used a biochemical substrate assay19 to test whether our fragment lead molecules blocked GSTP1 catalytic activity. Pretreatment with JWB152 or JWB198 inactivated GSTP1 in a concentration dependent manner (IC50 = 23 and 16 μM, respectively; Figure 6B). Specificity of inhibition against recombinant GSTP1 was confirmed by lack of activity of the negative control fragments JWB146 and JWB191 (Figure 6B). We also used a sulfonyl-fluoride analog SuFEx-2 to directly compare SuFEx and SuTEx activity against recombinant GSTP1. Consistent with our DPP3 findings, the SuTEx fragment showed a >10-fold increase in potency compared with the SuFEx analog in the GSTP1 activity assay (Figure 6B).
Next, we treated SILAC DM93 cells with JWB152 or JWB198 to determine whether these SuTEx fragments could ligand Y8 of endogenous GSTP1 in living systems (50 μM compound, 1.5 hr, 37 °C). Cells were pretreated with DMSO vehicle or SuTEx fragments followed by cells lysis, HHS-482 labeling of proteomes, and quantitative chemical proteomics (Supplementary Figure 3). Proteomes from JWB198-treated cells showed ~70% blockade of HHS-482 labeling of native GSTP1 Y8 (Figure 6C). Inhibitory activity of JWB198 was site specific as determined by lack of activity against other GSTP1 probe-modified sites (Y50, Y64, Y80, Y119, and Y199, SR ~1; Figure 6D and Supplementary Figure 10A). Several of the probe-modified tyrosines sites (Y50 and Y64) were in equivalent proximity from the GSH substrate compared with Y8 as determined by co-crystal structures of GSTP1 (5GSS, Supplemental Figure 11). In contrast, we observed mild in situ activity for JWB152 against GSTP1 Y8 (~20% inhibition) despite comparable in vitro potency compared with JWB198 (Figure 6B and Supplementary Figure 10B).
A potential explanation for differences in cellular activity of JWB152 compared with JWB198 is cell permeability. We tested this hypothesis by performing a subcellular location analysis of liganded proteins from our DM93 live cell studies (see Supporting Information for details of subcellular analysis). Our findings revealed that JWB152 and JWB198 showed comparable ability to modify proteins found in intracellular compartments including the cytosol and nuclear lumen (Supplementary Figure 12). An alternative interpretation is the higher reactivity of JWB152 compared with JWB198 reduces the intracellular fraction of the former inhibitor to effectively engage GSTP1 Y8 because of occupancy at additional cellular proteins. In support of this hypothesis, we compared proteome-wide activity of JWB198 and JWB152 and showed the latter compound reacted more broadly against tyrosine sites (>3-fold) in our live DM93 studies (Supplementary Figure 13 and 14, Table S1). Future studies aimed at understanding structural modifications that influence intracellular bioavailability49 of SuTEx molecules will further facilitate development of cell-active ligands.
Collectively, we identified JWB198 as a SuTEx fragment that is capable of liganding Y8 of GSTP1 in lysates and live cells. We demonstrate that development of tyrosine-reactive SuTEx fragments presents a unique opportunity to site-specifically perturb tyrosines that are known to be regulated by phosphorylation on protein targets involved in drug resistance in cancer50.
Conclusions
Here, we systematically evaluated functional group modifications for tuning the sulfur electrophile in nucleophilic substitution reactions. We applied our reactivity findings to demonstrate the versatility of SuTEx chemistry for developing ligands to disrupt functional tyrosine sites on proteins. Although our previous report described SuTEx as a global tyrosine profiling platform19, the current study highlights the broad potential for developing protein-targeted ligands using this chemistry. The capability for simultaneous modification on the AG and LG of SuTEx fragments uncovered key insights to functional changes required for tuning sulfur electrophiles in solution and proteomes (Figure 2 and 3). We discovered the EWG and EDG character of functional groups can affect reactivity of SuTEx fragments with nucleophiles albeit to differing extents depending on the location of modification. Specifically, we showed that the sulfur electrophile was generally more sensitive to AG compared with LG modifications (Figure 2). A prominent example was addition of a cyclopropyl functional group, which eliminated reactivity of the resulting SuTEx fragments both in solution and proteomes (JWB131, Figure 2A and 3A). These findings support the concept of “coarse” and “fine” tuning of SuTEx reactivity through AG and LG modifications, respectively.
Our findings also revealed the importance of binding recognition in development of SuTEx protein ligands. Evaluation of probe-enriched domains from the liganded and non-liganded protein groups revealed distinct profiles. These data support SuTEx fragments targeting a different subset of the proteome (liganded group) compared with protein sites generally labeled by HHS-482 probe (non-liganded group, Figure 4A and B). Our hypothesis is supported by the high overlap of enriched domains identified by HHS-482 in this study compared with a similar domain profile observed for SuTEx alkyne probes (HHS-465 and -475) from our previous report19. Further support for molecular recognition in SuTEx activity in proteomes was provided by the disparity in activity of JWB152 and JWB150 in solution compared with proteomes. Although JWB152 was more reactive in solution, we observed dramatically reduced as well as orthogonal tyrosine binding sites compared with JWB150 in our LC-MS chemical proteomic studies (Figure 3A and C, Supplementary Figure 6). Additional examples include the differences in HPLC and proteome reactivity of JWB198, JWB202, and JWB152. In solution, these fragments showed comparable reactivity with cresol based on half-life values of ~ 1 min for all three molecules (Table S1). In contrast, our proteomic findings revealed clear differences in activity of these SuTEx fragments with protein sites. Specifically, JWB152 showed a >4-fold increase in the number of liganded tyrosines compared with JWB198 and JWB202 (Figure 3A).
We presented two examples for developing ligands to perturb functional tyrosine sites on proteins. First, we discovered fragment ligands for a tyrosine site located near the zinc-binding region of DPP3 (Y417). We leveraged the Y417 binding site of DPP3 to develop JWB142 as a first in class covalent DPP3 inhibitor that blocks biochemical activity by liganding a non-catalytic tyrosine site19 (Figure 5). Given the lack of ligands and inhibitors for DPP3, our findings support application of SuTEx for covalent FBLD31–33 of challenging protein targets (Non-DBP group, Figure 4C). Considering the success of covalent ligands targeting non-catalytic cysteine residues of kinases45 and other protein classes45, 51, future studies will focus on expanding our SuTEx fragment library to determine the full inventory of tyrosines that can liganded for development of protein modulators (inhibitors or activators) for biological investigations.
We demonstrated that SuTEx fragments can ligand tyrosines sites in live cells. The discovery of JWB152 and JWB198 as ligands of GSTP1 Y8 presented an opportunity to target a hyper-reactive tyrosine that is also a known site for phosphorylation48. Despite equivalent inhibitory activity in vitro, we discovered that only JWB198 could ligand the Y8 site of GSTP1 in live cells (Figure 6). Evaluation of proteome-wide reactivity showed that JWB198 was substantially less reactive (Supplementary Figure 13 and 14) while maintaining ~70% blockade of GSTP1 Y8 in live DM93 cells (Figure 6). Notably, JWB198 showed negligible activity against other quantified tyrosine sites and supports the ability of SuTEx fragments to achieve site specificity on a target protein (Figure 6, Supplementary Figure 10 and 11). Taken together, these studies highlight the advantage of tunability afforded by SuTEx when optimizing protein ligands for cellular activity.
While key for demonstrating the utility of SuTEx for FBLD, we recognize that the SAR of our current sulfonyl-triazole library could be further improved. We utilized HHS-482 as a lead scaffold for developing tyrosine-reactive ligands because this SuTEx probe showed high tyrosine chemoselectivity19. As a result of this focused SAR approach, the SuTEx fragments evaluated in our current studies bear some overlapping structural features. For example, AG and LG modifications with aryl substituents was used to evaluate SuTEx reactivity. While needed for understanding EWG and EDG effects, the outcome was inclusion of aromatic rings as a common element of our fragment structures. Future studies aimed at incorporating a larger content of sp3-hybridized and stereogenic atoms will increase the three-dimensional character of our current fragment library and facilitate a broader exploration of chemical space52. As a complementary strategy to FBLD, we can pursue late stage functionalization of bioactive molecules28 to develop SuTEx ligands with elaborated binding structures and drug-like features for systematic exploration of ‘ligandability’ of tyrosine-containing binding pockets.
In summary, we describe SuTEx as an enabling chemistry for profiling and targeting catalytic and non-catalytic tyrosine sites across the proteome. The ability to simultaneously alter reactivity of the sulfur electrophile and incorporate binding recognition through AG and LG modifications will facilitate development of protein ligands with carefully tuned reactivity and binding affinity. Future studies will focus on incorporating more structurally diverse scaffolds (e.g. by increasing sp3 content and drug-like features) to further advance SuTEx electrophiles for perturbing protein function in living systems.
METHODS
Detailed Methods are provided in the Supporting Information
Supplementary Material
ACKNOWLEDGMENTS
We thank H.S. Hahm and E.K. Toroitich for helpful discussions on the SuTEx project. We thank M. Ross for his assistance with mass spectrometry experiments and data analysis. We thank all members of the Hsu Lab and colleagues at the University of Virginia for helpful discussions. We thank T.B. Ware and R.L. McCloud for help with site-directed mutagenesis and cloning. This work was supported by the University of Virginia Cancer Center (NCI Cancer Center Support Grant grant no. 5P30CA044579-27 to A.H.L. and K.-L.H), National Institutes of Health Grants DA043571 (K.-L.H), GM007055 (J.W.B.), and CA009109 (A.L.B.), and U.S. Department of Defense (W81XWH-17-1-0487 to K.-L.H.).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental methods, supplementary figures, table S1, chemical synthesis, NMR spectra, and HPLC analysis of compound purity and reactivity are supplied as Supporting Information.
The authors declare no competing financial interest.
References
- 1.Schreiber SL; Kotz JD; Li M; Aube J; Austin CP; Reed JC; Rosen H; White EL; Sklar LA; Lindsley CW; Alexander BR; Bittker JA; Clemons PA; de Souza A; Foley MA; Palmer M; Shamji AF; Wawer MJ; McManus O; Wu M; Zou B; Yu H; Golden JE; Schoenen FJ; Simeonov A; Jadhav A; Jackson MR; Pinkerton AB; Chung TD; Griffin PR; Cravatt BF; Hodder PS; Roush WR; Roberts E; Chung DH; Jonsson CB; Noah JW; Severson WE; Ananthan S; Edwards B; Oprea TI; Conn PJ; Hopkins CR; Wood MR; Stauffer SR; Emmitte KA; Team, N. I. H. M. L. P., Advancing Biological Understanding and Therapeutics Discovery with Small-Molecule Probes. Cell 2015, 161 (6), 1252–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh J; Petter RC; Baillie TA; Whitty A, The resurgence of covalent drugs. Nat Rev Drug Discov 2011, 10 (4), 307–17. [DOI] [PubMed] [Google Scholar]
- 3.Parker CG; Galmozzi A; Wang Y; Correia BE; Sasaki K; Joslyn CM; Kim AS; Cavallaro CL; Lawrence RM; Johnson SR; Narvaiza I; Saez E; Cravatt BF, Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 2017, 168 (3), 527–541 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y; Dix MM; Bianco G; Remsberg JR; Lee HY; Kalocsay M; Gygi SP; Forli S; Vite G; Lawrence RM; Parker CG; Cravatt BF, Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat Chem 2019, 11 (12), 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Backus KM; Correia BE; Lum KM; Forli S; Horning BD; Gonzalez-Paez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF, Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534 (7608), 570–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weerapana E; Wang C; Simon GM; Richter F; Khare S; Dillon MB; Bachovchin DA; Mowen K; Baker D; Cravatt BF, Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468 (7325), 790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradshaw JM; McFarland JM; Paavilainen VO; Bisconte A; Tam D; Phan VT; Romanov S; Finkle D; Shu J; Patel V; Ton T; Li X; Loughhead DG; Nunn PA; Karr DE; Gerritsen ME; Funk JO; Owens TD; Verner E; Brameld KA; Hill RJ; Goldstein DM; Taunton J, Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat Chem Biol 2015, 11 (7), 525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoo E; Stokes BH; de Jong H; Vanaerschot M; Kumar T; Lawrence N; Njoroge M; Garcia A; Van der Westhuyzen R; Momper JD; Ng CL; Fidock DA; Bogyo M, Defining the Determinants of Specificity of Plasmodium Proteasome Inhibitors. J Am Chem Soc 2018, 140 (36), 11424–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hacker SM; Backus KM; Lazear MR; Forli S; Correia BE; Cravatt BF, Global profiling of lysine reactivity and ligandability in the human proteome. Nat Chem 2017, 9 (12), 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Q; Ouyang X; Wan X; Gajiwala KS; Kath JC; Jones LH; Burlingame AL; Taunton J, Broad-Spectrum Kinase Profiling in Live Cells with Lysine-Targeted Sulfonyl Fluoride Probes. J Am Chem Soc 2017, 139 (2), 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patricelli MP; Nomanbhoy TK; Wu J; Brown H; Zhou D; Zhang J; Jagannathan S; Aban A; Okerberg E; Herring C; Nordin B; Weissig H; Yang Q; Lee JD; Gray NS; Kozarich JW, In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol 2011, 18 (6), 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shannon DA; Banerjee R; Webster ER; Bak DW; Wang C; Weerapana E, Investigating the proteome reactivity and selectivity of aryl halides. J Am Chem Soc 2014, 136 (9), 3330–3. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X; Crowley VM; Wucherpfennig TG; Dix MM; Cravatt BF, Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat Chem Biol 2019, 15 (7), 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spradlin JN; Hu X; Ward CC; Brittain SM; Jones MD; Ou L; To M; Proudfoot A; Ornelas E; Woldegiorgis M; Olzmann JA; Bussiere DE; Thomas JR; Tallarico JA; McKenna JM; Schirle M; Maimone TJ; Nomura DK, Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat Chem Biol 2019, 15 (7), 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Resnick E; Bradley A; Gan J; Douangamath A; Krojer T; Sethi R; Geurink PP; Aimon A; Amitai G; Bellini D; Bennett J; Fairhead M; Fedorov O; Gabizon R; Gan J; Guo J; Plotnikov A; Reznik N; Ruda GF; Diaz-Saez L; Straub VM; Szommer T; Velupillai S; Zaidman D; Zhang Y; Coker AR; Dowson CG; Barr HM; Wang C; Huber KVM; Brennan PE; Ovaa H; von Delft F; London N, Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J Am Chem Soc 2019, 141 (22), 8951–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bos J; Muir TW, A Chemical Probe for Protein Crotonylation. J Am Chem Soc 2018, 140 (14), 4757–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang R; Islam K; Liu Y; Zheng W; Tang H; Lailler N; Blum G; Deng H; Luo M, Profiling genome-wide chromatin methylation with engineered posttranslation apparatus within living cells. J Am Chem Soc 2013, 135 (3), 1048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bicker KL; Subramanian V; Chumanevich AA; Hofseth LJ; Thompson PR, Seeing citrulline: development of a phenylglyoxal-based probe to visualize protein citrullination. J Am Chem Soc 2012, 134 (41), 17015–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahm HS; Toroitich EK; Borne AL; Brulet JW; Libby AH; Yuan K; Ware TB; McCloud RL; Ciancone AM; Hsu KL, Global targeting of functional tyrosines using sulfur-triazole exchange chemistry. Nat Chem Biol 2020, 16 (2), 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narayanan A; Jones LH, Sulfonyl fluorides as privileged warheads in chemical biology. Chem Sci 2015, 6 (5), 2650–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grimster NP; Connelly S; Baranczak A; Dong J; Krasnova LB; Sharpless KB; Powers ET; Wilson IA; Kelly JW, Aromatic sulfonyl fluorides covalently kinetically stabilize transthyretin to prevent amyloidogenesis while affording a fluorescent conjugate. J Am Chem Soc 2013, 135 (15), 5656–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fahrney DE; Gold AM, Sulfonyl Fluorides as Inhibitors of Esterases. I. Rates of Reaction with Acetylcholinesterase, α-Chymotrypsin, and Trypsin. Journal of the American Chemical Society 1963, 85 (7), 997–1000. [Google Scholar]
- 23.Gao B; Zhang L; Zheng Q; Zhou F; Klivansky LM; Lu J; Liu Y; Dong J; Wu P; Sharpless KB, Bifluoride-catalysed sulfur(VI) fluoride exchange reaction for the synthesis of polysulfates and polysulfonates. Nat Chem 2017, 9 (11), 1083–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong J; Sharpless KB; Kwisnek L; Oakdale JS; Fokin VV, SuFEx-based synthesis of polysulfates. Angew Chem Int Ed Engl 2014, 53 (36), 9466–9470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Q; Woehl JL; Kitamura S; Santos-Martins D; Smedley CJ; Li G; Forli S; Moses JE; Wolan DW; Sharpless KB, SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc Natl Acad Sci U S A 2019, 116 (38), 18808–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen W; Dong J; Plate L; Mortenson DE; Brighty GJ; Li S; Liu Y; Galmozzi A; Lee PS; Hulce JJ; Cravatt BF; Saez E; Powers ET; Wilson IA; Sharpless KB; Kelly JW, Arylfluorosulfates Inactivate Intracellular Lipid Binding Protein(s) through Chemoselective SuFEx Reaction with a Binding Site Tyr Residue. J Am Chem Soc 2016, 138 (23), 7353–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang B; Wang N; Schnier PD; Zheng F; Zhu H; Polizzi NF; Ittuveetil A; Saikam V; DeGrado WF; Wang Q; Wang PG; Wang L, Genetically Introducing Biochemically Reactive Amino Acids Dehydroalanine and Dehydrobutyrine in Proteins. J Am Chem Soc 2019, 141 (19), 7698–7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z; Li J; Li S; Li G; Sharpless KB; Wu P, SuFEx Click Chemistry Enabled Late-Stage Drug Functionalization. J Am Chem Soc 2018, 140 (8), 2919–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mortenson DE; Brighty GJ; Plate L; Bare G; Chen W; Li S; Wang H; Cravatt BF; Forli S; Powers ET; Sharpless KB; Wilson IA; Kelly JW, “Inverse Drug Discovery” Strategy To Identify Proteins That Are Targeted by Latent Electrophiles As Exemplified by Aryl Fluorosulfates. J Am Chem Soc 2018, 140 (1), 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong J; Krasnova L; Finn MG; Sharpless KB, Sulfur(VI) fluoride exchange (SuFEx): another good reaction for click chemistry. Angew Chem Int Ed Engl 2014, 53 (36), 9430–48. [DOI] [PubMed] [Google Scholar]
- 31.Erlanson DA; Fesik SW; Hubbard RE; Jahnke W; Jhoti H, Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov 2016, 15 (9), 605–19. [DOI] [PubMed] [Google Scholar]
- 32.Hopkins AL; Keseru GM; Leeson PD; Rees DC; Reynolds CH, The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov 2014, 13 (2), 105–21. [DOI] [PubMed] [Google Scholar]
- 33.Scott DE; Coyne AG; Hudson SA; Abell C, Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51 (25), 4990–5003. [DOI] [PubMed] [Google Scholar]
- 34.Electrical Effect Substituent Constants for Correlation Analysis In Progress in Physical Organic Chemistry, pp 119–251. [Google Scholar]
- 35.Lin Y-I; Lang SA; Lovell MF; Perkinson NA, New synthesis of 1,2,4-triazoles and 1,2,4-oxadiazoles. The Journal of Organic Chemistry 1979, 44 (23), 4160–4164. [Google Scholar]
- 36.Mann M, Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol 2006, 7 (12), 952–8. [DOI] [PubMed] [Google Scholar]
- 37.Bern M; Kil YJ; Becker C, Byonic: advanced peptide and protein identification software. Curr Protoc Bioinformatics 2012, Chapter 13, Unit13 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wishart DS; Feunang YD; Guo AC; Lo EJ; Marcu A; Grant JR; Sajed T; Johnson D; Li C; Sayeeda Z; Assempour N; Iynkkaran I; Liu Y; Maciejewski A; Gale N; Wilson A; Chin L; Cummings R; Le D; Pon A; Knox C; Wilson M, DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018, 46 (D1), D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee I; Schindelin H, Structural insights into E1-catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell 2008, 134 (2), 268–78. [DOI] [PubMed] [Google Scholar]
- 40.Chang TK; Shravage BV; Hayes SD; Powers CM; Simin RT; Wade Harper J; Baehrecke EH, Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol 2013, 15 (9), 1067–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X; Zhao B; Sun L; Bhuripanyo K; Wang Y; Bi Y; Davuluri RV; Duong DM; Nanavati D; Yin J; Kiyokawa H, Orthogonal ubiquitin transfer identifies ubiquitination substrates under differential control by the two ubiquitin activating enzymes. Nat Commun 2017, 8, 14286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park E; Rawson S; Li K; Kim BW; Ficarro SB; Pino GG; Sharif H; Marto JA; Jeon H; Eck MJ, Architecture of autoinhibited and active BRAF-MEK1–14-3–3 complexes. Nature 2019, 575 (7783), 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gavin AL; Huang D; Huber C; Martensson A; Tardif V; Skog PD; Blane TR; Thinnes TC; Osborn K; Chong HS; Kargaran F; Kimm P; Zeitjian A; Sielski RL; Briggs M; Schulz SR; Zarpellon A; Cravatt B; Pang ES; Teijaro J; de la Torre JC; O’Keeffe M; Hochrein H; Damme M; Teyton L; Lawson BR; Nemazee D, PLD3 and PLD4 are single-stranded acid exonucleases that regulate endosomal nucleic-acid sensing. Nat Immunol 2018, 19 (9), 942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bezerra GA; Dobrovetsky E; Viertlmayr R; Dong A; Binter A; Abramic M; Macheroux P; Dhe-Paganon S; Gruber K, Entropy-driven binding of opioid peptides induces a large domain motion in human dipeptidyl peptidase III. Proc Natl Acad Sci U S A 2012, 109 (17), 6525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Q; Sabnis Y; Zhao Z; Zhang T; Buhrlage SJ; Jones LH; Gray NS, Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol 2013, 20 (2), 146–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simaga S; Babic D; Osmak M; Ilic-Forko J; Vitale L; Milicic D; Abramic M, Dipeptidyl peptidase III in malignant and non-malignant gynaecological tissue. Eur J Cancer 1998, 34 (3), 399–405. [DOI] [PubMed] [Google Scholar]
- 47.Hast BE; Goldfarb D; Mulvaney KM; Hast MA; Siesser PF; Yan F; Hayes DN; Major MB, Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res 2013, 73 (7), 2199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okamura T; Singh S; Buolamwini J; Haystead T; Friedman H; Bigner D; Ali-Osman F, Tyrosine phosphorylation of the human glutathione S-transferase P1 by epidermal growth factor receptor. J Biol Chem 2009, 284 (25), 16979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mateus A; Gordon LJ; Wayne GJ; Almqvist H; Axelsson H; Seashore-Ludlow B; Treyer A; Matsson P; Lundback T; West A; Hann MM; Artursson P, Prediction of intracellular exposure bridges the gap between target- and cell-based drug discovery. Proc Natl Acad Sci U S A 2017, 114 (30), E6231–E6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Allocati N; Masulli M; Di Ilio C; Federici L, Glutathione transferases: substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7 (1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horning BD; Suciu RM; Ghadiri DA; Ulanovskaya OA; Matthews ML; Lum KM; Backus KM; Brown SJ; Rosen H; Cravatt BF, Chemical Proteomic Profiling of Human Methyltransferases. J Am Chem Soc 2016, 138 (40), 13335–13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Over B; Wetzel S; Grutter C; Nakai Y; Renner S; Rauh D; Waldmann H, Natural-product-derived fragments for fragment-based ligand discovery. Nat Chem 2013, 5 (1), 21–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.