

Abstract
Objectives
Cytomegalovirus (CMV) is known to have a significant impact on immune recovery post‐allogeneic haemopoietic stem cell transplant (HSCT). Adoptive therapy with donor‐derived or third‐party virus‐specific T cells (VST) can restore CMV immunity leading to clinical benefit in prevention and treatment of post‐HSCT infection. We developed a mass cytometry approach to study natural immune recovery post‐HSCT and assess the mechanisms underlying the clinical benefits observed in recipients of VST.
Methods
A mass cytometry panel of 38 antibodies was utilised for global immune assessment (72 canonical innate and adaptive immune subsets) in HSCT recipients undergoing natural post‐HSCT recovery (n = 13) and HSCT recipients who received third‐party donor‐derived CMV‐VST as salvage for unresponsive CMV reactivation (n = 8).
Results
Mass cytometry identified distinct immune signatures associated with CMV characterised by a predominance of innate cells (monocytes and NK) seen early and an adaptive signature with activated CD8+ T cells seen later. All CMV‐VST recipients had failed standard antiviral pharmacotherapy as a criterion for trial involvement; 5/8 had failed to develop the adaptive immune signature by study enrolment despite significant CMV antigen exposure. Of these, VST administration resulted in development of the adaptive signature in association with CMV control in three patients. Failure to respond to CMV‐VST in one patient was associated with persistent absence of the adaptive immune signature.
Conclusion
The clinical benefit of CMV‐VST may be mediated by the recovery of an adaptive immune signature characterised by activated CD8+ T cells.
Keywords: adoptive T‐cell therapy, CyTOF, haemopoietic stem cell transplant, immune reconstitution, immunotherapy, mass cytometry
In this study, immune profiling with mass cytometry identified immune signatures associated with cytomegalovirus (CMV) reactivation after allogeneic stem cell transplant in patients undergoing natural immune recovery. Failure to control CMV was associated with failure to develop an adaptive immune signature that could be reversed with third‐party CMV virus‐specific T cells in some recipients.

Introduction
Allogeneic haemopoietic stem cell transplant (HSCT) is associated with significant toxicities despite advances in conditioning and supportive care. Even in patients with low disease risk and no comorbidities, about 15–20% of patients currently die within the first 12 months after transplant and the percentage rises steeply in patients with more advanced disease or significant comorbidities. Infection is a direct or contributing cause of death in > 80% of these patients. 1 While cytomegalovirus (CMV) is an unusual cause of death itself, infection with the virus is strongly associated with increased early mortality, despite routine use of pre‐emptive antiviral therapy. 1
We and others have explored the use of post‐HSCT adoptive T‐cell therapy with CMV virus‐specific T cells (CMV‐VST) to rapidly restore immune function in the post‐transplant period. A number of phase I and II studies have shown beneficial clinical effect of donor‐derived CMV‐VST in viral control in primary and secondary prophylaxis of viral reactivation or in the salvage setting. 2 , 3 , 4 , 5 , 6 , 7 Given logistic and cost impediments associated with donor‐derived VST, banks of off‐the‐shelf VST from third‐party donors have been established. 8 , 9 , 10 , 11 A number of trials have shown excellent clinical efficacy 10 , 12 , 13 , 14 , 15 with a majority of patients with refractory infections achieving complete virological control despite significant HLA disparity between third‐party donor and recipient. In a recent study from our institution, 74% of patients with treatment‐resistant viral infections experienced complete resolution of viraemia post‐third‐party VST infusion, 14 a finding similar to outcomes observed in other trials. 10 , 16 , 17 Peripheral blood from a subset of participants from this clinical trial was used for the current study. 11 , 14
The biological mechanisms of this clinical benefit are incompletely understood. Immunological changes post‐VST have been studied using standard flow cytometry and antigen‐specific immunoassays (tetramer and intracellular flow cytometry, enzyme‐linked immunospot (ELISPOT) and cytokine secretion assays). We have previously observed a rise in the CD8+ T‐cell count at the time of viral clearance with third‐party VST, 14 and that CMV antigenaemia is required for the induction of CMV‐specific cellular immune responses in recipients of prophylactic donor‐derived CMV‐VST. 5 More recently, the development of high‐dimensional flow cytometry and mass cytometry (cytometry by time of flight or CyTOF) 18 , 19 , 20 offers the opportunity to assess both the breadth and depth of immunological alterations that occur in patients receiving immunotherapies.
Here, we developed a mass cytometry platform and analysis strategy using existing bioinformatics tools for global immunological assessment and have used it to explore immune reconstitution in HSCT recipients with and without VST treatment.
Results
Patients and sample characteristics
Mass cytometry was performed on 79 PBMC samples from 34 individuals, of whom 13 were healthy individuals and 21 were allogeneic HSCT recipients (Tables 1 and 2 for participant characteristics; Figure 1 for study schema). Of the transplant recipients, 13 were recipients of HSCT alone (without VST) and eight were treated with HSCT plus third‐party CMV‐VST. While this was a non‐randomised study, the HSCT‐alone and VST recipients were similar, with no significant differences in baseline characteristics. Of all HSCT recipients, CMV reactivation was detected in 17/21 (81%) patients at a median of 31 days post‐transplant (range 17–45). The VST patients had CMV reactivation with inadequate response to first‐line antiviral treatment as an eligibility for trial recruitment.
Table 1.
Patient characteristics
| Healthy individuals | HSCT‐alone | HSCT with third‐party VST | P‐value | |
|---|---|---|---|---|
| Number of individuals | 13 | 13 | 8 | |
| Age (median, range) | 55 (28–83) | 55 (32–70) | 58.5 (12–52) | 0.55 |
| Sex (M:F) | 9:4 | 5:8 | 4:4 | 0.67 |
| Diagnosis | ||||
| AML | 10 | 5 | 0.78 | |
| ALL | 1 | 1 | ||
| Other malignant | 2 | 2 | ||
| Conditioning | ||||
| MAC | 3 | 2 | 0.92 | |
| RIC | 10 | 6 | ||
| T‐cell depletion | 7 | 7 | 0.17 | |
| Donor | ||||
| Cord | 0 | 1 | 0.35 | |
| Haplo | 1 | 0 | ||
| MUD | 10 | 5 | ||
| Sib | 2 | 2 | ||
| CMV serostatus (R/D) | ‐ | |||
| Neg/Neg | 3 | 0 | 0.16 | |
| Neg/Pos | 0 | 0 | ||
| Pos/Neg | 6 | 6 | ||
| Pos/Pos | 4 | 2 | ||
| CMV reactivation | 9 (69%) | 8 (100%) | 0.08 | |
| Median day of reactivation (range) | 31 (25–45) | 29 (17–43) | 0.33 | |
| CMV DNA log10AUC a (range) | 2.4 (0–4.34) | 5.7 (5.06–6.42) | 0.001 | |
| CMV tissue disease | 1 (8%) | 1 (13%) | 1.0 | |
| Acute GVHD | ||||
| Overall (grade 2–4) | 6 (46%) | 2 (25%) | 0.40 | |
| Severe (grade 3–4) | 3 (23%) | 1 (13%) | 1.0 | |
| Chronic GVHD | 6 (46%) | 3 (37%) | 1.0 | |
| Relapse | 2 (15%) | 1 (13%) | 1.0 | |
| Death | 0 (0%) | 2 (25%) | 0.13 | |
| Number of samples | 13 | 42 | 24 | |
| Collection day of sample post‐transplant, median (range) | 74.5 (26–132) | 128 (63–255) | < 0.0001 | |
ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; CMV, cytomegalovirus; GVHD, graft‐versus‐host disease; Haplo, haploidentical; MAC, myeloablative conditioning; MUD, matched unrelated donor; R/D, recipient/donor; RIC, reduced intensity conditioning; Sib, sibling.
Mean log10 of CMV viral load area under the curve (AUC) in copies/mL prior to the first study timepoint. AUC is used as a measure of total viral antigen exposure.
Table 2.
Clinical detail of VST recipients
| Patient | Age (years) | Sex | Transplant indication | Type of transplant, HLA mismatch, (cell source other than PBSC) | Conditioning | T‐cell depletion | R/D CMV serostatus | CMV tissue disease | Days of prior antiviral therapy (days) | CMV AUC (log10) prior to first sample for this study | Prior lines of antiviral pharmacotherapy | Degree of HLA match in VSTs | Best virologic response |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 63 | M | AML/MDS | MUD | RIC | ATG | Pos/Neg | Yes | 61 | 6.42 | 2 | 4/6 and 3/6 | PR then death from CMV |
| 2 | 41 | M | T‐NHL | MUD | MAC | Alem | Pos/Neg | No | 27 | 5.06 | 1 | 2/6 and 3/6 | CR |
| 3 | 64 | M | AML | MUD | RIC | ATG | Pos/Neg | No | 23 | 5.83 | 1 | 4/6 and 2/6 | CR |
| 6 | 36 | F | MDS | MSD | RIC | None | Pos/Pos | No | 22 | 5.37 | 2 | 3/6 | CR |
| 7 | 58 | F | ALL | MSD | RIC | ATG | Pos/Neg | No | 44 | 5.74 | 1 | 4/6 | CR |
| 8 | 59 | F | AML | MUD | RIC | ATG | Pos/Neg | No | 31 | 5.69 | 1 | 3/6 | CR |
| 21 | 59 | F | AML | MUD | RIC | ATG | Pos/Neg | No | 50 | NA | 1 | 3/6 and 3/6 | CR |
| 25 | 12 | M | AML | MUD (Cord) | MAC | ATG | Pos/Pos | No | 60 | NA | 1 | 2/6 | CR |
Viral response was defined to be complete response (CR) – complete disappearance of viraemia and partial response (PR) – 50% reduction in viral copy number in the blood.
alem, alemtuzumab; ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; ATG, anti‐thymocyte globulin; MAC, myeloablative conditioning; MDS, myelodysplastic syndrome; MSD, matched sibling donor; MUD, matched unrelated donor, NA, not available because of the format of reporting of the CMV PCR at the local site; the level is expected to be high given the prolonged use of CMV antiviral therapy prior to study recruitment.
Figure 1.
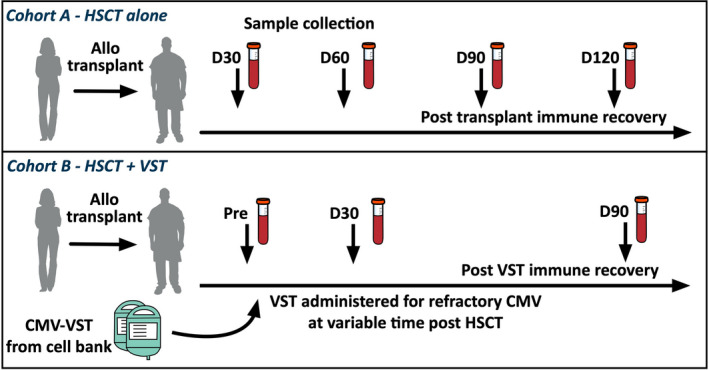
Study schema. Healthy individuals and two cohorts of HSCT recipients had peripheral blood collected for immune profiling.
For the VST cohort, a pre‐CMV‐VST infusion sample was analysed, in addition to samples from day 30 and day 90 post‐infusion. The samples from VST recipients were collected later post‐transplant than those who did not receive VST (median day post‐transplant 128, range 63–255 vs 74.5, range 26–132, respectively; P‐value < 0.0001), reflecting variable timing of trial recruitment.
Immune signatures identified by unsupervised consensus clustering
A mass cytometry panel of 38 metal‐conjugated antibodies was used to gate 72 subsets across the innate and adaptive immune system in up to four sample timepoints per patient over the first 120 days post‐HSCT (Figure 1). To identify global patterns of immune reconstitution, an unsupervised analysis (SC3 algorithm) was performed with all gated subsets (in cells per 109/L) from every sample timepoint. This approach was taken to allow changes over time within individuals and the group as a whole to be included in the unsupervised analysis. The results of this analysis are shown in Figure 2. Initially, this analysis was performed on healthy individuals and HSCT recipients who did not receive VST to describe the immune recovery process that occurs naturally post‐HSCT without the VST intervention (Figure 2a). Three distinct immune profiles were identified. Healthy individuals clustered independently from the HSCT recipients and exhibited higher CD4+ T‐cell subsets (total CD4+, Tconv, Treg, naïve, Tcm, CXCR5+CD45RO+CD4+, integrin B7+CD45ROnegCD4+) and B cells (total B cells and memory B cells) than were seen in HSCT recipients at any timepoint in the observation period.
Figure 2.
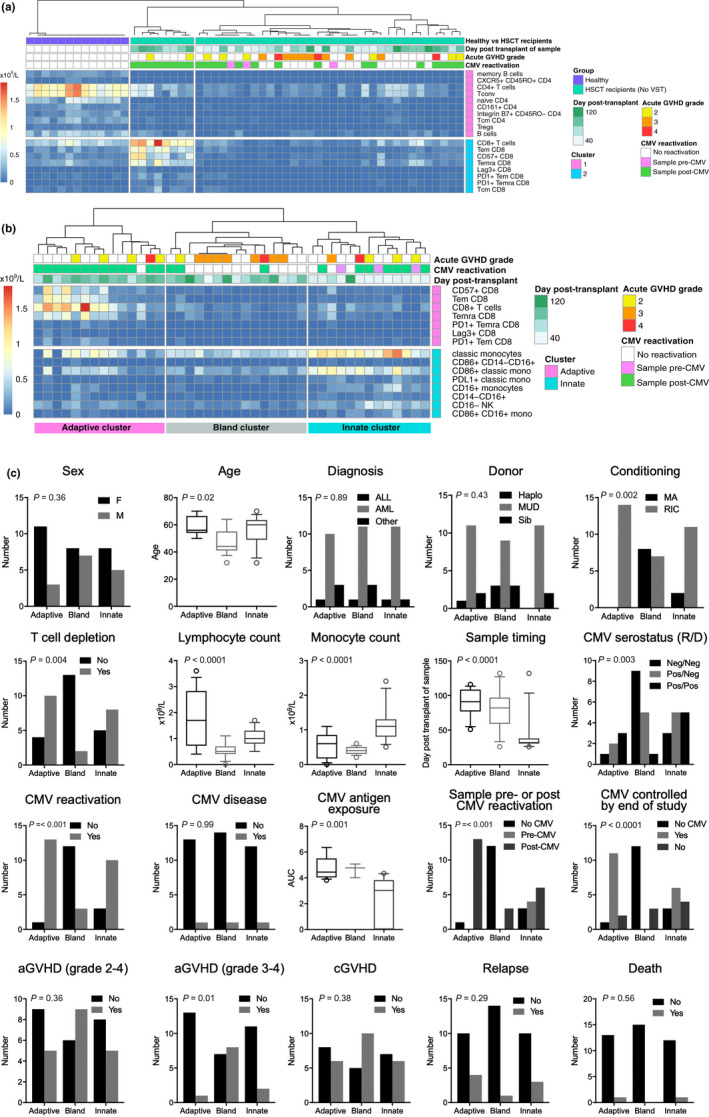
Immune profiles by unsupervised biaxial consensus clustering. (a) In healthy individuals (n = 13; samples collected at steady state) and patients undergoing HSCT without VST (n = 13; up to four timepoints from each patient, day 26 to 132 post‐HSCT). (b) In samples from the patients undergoing HSCT alone (without VST recipients; same samples as in a). Clusters from left to right are designated adaptive, bland and innate, respectively. In the heatmaps in a and b, columns represent samples, and rows represent the cell subsets. All cell subsets from all samples were included in the unsupervised analysis but only subsets contributing significantly to the clustering are visualised in the output. All timepoints were included to allow changes over time to be considered as a variable in subsequent analysis. Clinical annotation in the boxes above the heatmap is shown for reference but was not included in the SC3 analysis. The heatmap colour scale = 0 to > 2 × 109 cells/L. (c) Influence of clinical factors on immune profiles. To assess the impact of patient and transplant characteristics on the immune profile of a sample, univariate analysis was performed. P‐values are based on univariate analysis with Bonferroni correction for multiple tests (α/18). On the x‐axis, the three clusters identified in b are shown. Log10AUC = logarithm of the area under the curve of viral copy number, as a measure of CMV antigen exposure.
The SC3 algorithm draws out only the major differences in data sets, so to identify more subtle differences in immune profiles within the HSCT recipients during natural immune recovery, the analysis was re‐run with the samples from the healthy individuals removed, without the VST cohort included to describe the natural immune recovery process (Figure 2b). Three distinct clusters were identified; Cluster 1 identified samples with increased CD8+ T cells, in particular antigen‐experienced T‐cell subsets (Tem, Temra), immunoinhibitory markers (PD‐1 and Lag3) and the senescence marker CD57. Cluster 3 was characterised by elevated innate subsets, including monocytes (classical, CD86+, PD‐1+, CD16+ monocytes), myeloid cells (CD14negCD16+ and CD86+CD14negCD16+ myeloid cells) and CD16neg NK cells. Finally, an intermediate signature (Cluster 2) was characterised by generally low numbers across all populations that distinguish the innate and adaptive clusters. B cells were not identified as significant features in any of the HSCT clusters. For ease of reference, clusters 1, 2 and 3 will be referred to as adaptive, bland and innate immune signatures, respectively.
CMV and time have the strongest influence on development of immune profiles
To test the relationship between clinical parameters and immune signatures, univariate analysis was performed using samples and clinical data from the HSCT‐alone group and clusters identified by the SC3 analysis (Figure 2c). Lymphocyte and monocyte counts were different between the clusters, with samples present in the adaptive cluster having higher lymphocyte counts (mean 1.8 × 109/L) and samples in the innate cluster having higher monocyte counts (mean 1.2 × 109/L). Both lymphocyte and monocyte counts were low in the bland cluster (means 0.6 and 0.4 × 109/L, respectively). Age, haematological diagnosis and donor type were spread evenly between the clusters, whereas older age, reduced intensity conditioning (RIC) and the use of T‐cell depletion (TCD) in the conditioning regimen were enriched in the adaptive cluster (P = 0.02, 0.002 and 0.004, respectively). Age and RIC are colinear as older transplant recipients are more likely to receive RIC regimens. It is notable that no samples from a patient who received myeloablative conditioning (MAC) had the adaptive immune signature. Sample timing differed significantly between the immune signature clusters (P < 0.0001). A larger proportion of early samples had the innate signature (mean day post‐transplant 42), and the adaptive and bland signatures were enriched for later samples (means 89 and 81, respectively).
Variables related to CMV were strongly associated with differences in immune signatures. CMV‐positive serostatus of donor or recipient, CMV reactivation post‐transplant and samples collected post‐CMV reactivation were all virtually absent in the bland cluster. CMV antigen exposure as measured by area under the curve (log10AUC) of viral copy number was higher in the adaptive and bland cluster, and lower in the innate cluster (means 4.7, 4.6 and 2.4, respectively; P = 0.001) suggesting that the innate signature may be a marker of early antigen exposure. Further, the three samples collected prior to CMV viraemia in those who subsequently reactivated exhibited an innate signature; this may represent an early immune response to subclinical viraemia. Two of the three samples with the bland signature from CMV reactivators were also patients with graft‐versus‐host disease (GVHD), suggesting that these patients were prevented from mounting a mature immune response to CMV by the presence of GVHD or by immune suppression used for treatment. Of the nine CMV reactivators in the HSCT‐alone group, 5 (55%) cleared CMV viraemia by the end of the observation period. Clearance of CMV was associated with the development of the adaptive immune signature (P < 0.0001). CMV tissue disease was seen infrequently in this study (one patient in the HSCT‐alone group and one VST recipient). Samples from the one patient in the HSCT‐alone cohort who developed CMV disease were distributed between the immune profile clusters, as were samples from patients who relapsed or died. Samples from patients with severe acute GVHD were primarily seen in the bland cluster, presumably reflecting the effect of high‐dose corticosteroids and other immunosuppressant medications administered to patients with this condition. Mild acute GVHD (grade 2) did not strongly influence the cluster position of samples.
Characterisation of key immunological changes over time
To describe the evolutionary changes of immune reconstitution after HSCT, a non‐linear fit of the trajectory of each immune subset over time was performed using the loess method on samples from the HSCT‐alone group who did not receive VST (Figure 3). Given the strong influence of CMV seen in the global immunological assessment above, the curves were separated by the occurrence of CMV reactivation at any time during the observation period. To describe the relative changes in all cell subsets, including numerically small ones, this longitudinal analysis was performed on the quantitative data used for SC3 after Z score normalisation. Thus, numeric changes within each cell subset were visualised on the same scale (scale on exemplar plot in Figure 3).
Figure 3.
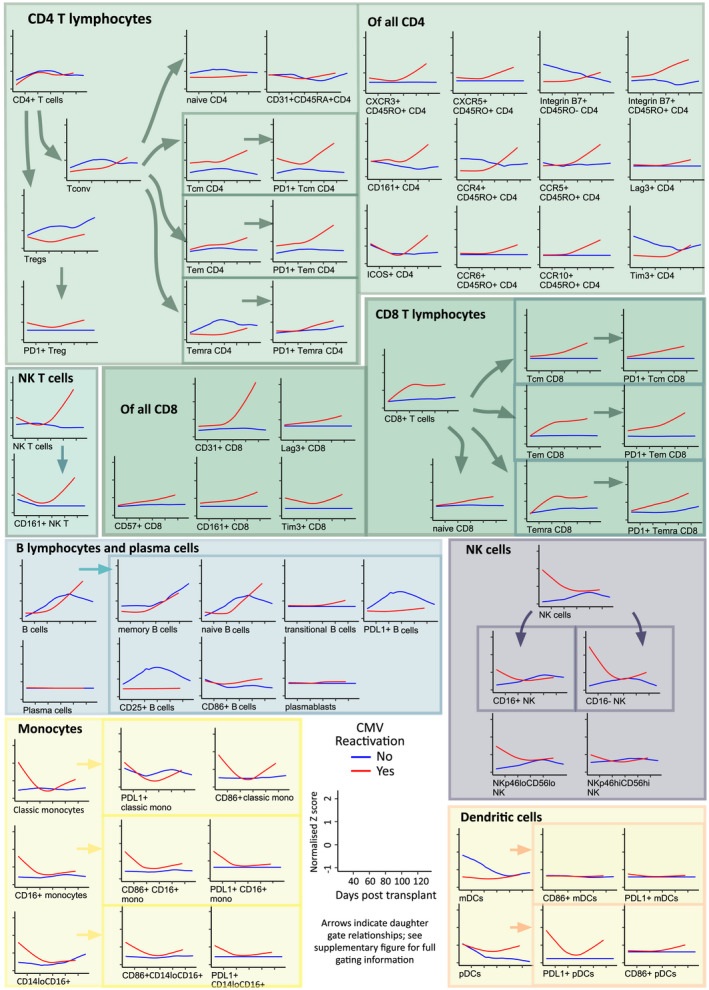
Estimated evolutionary dynamics of immune subsets over time. Scaled population counts (x109/L), z‐score normalised, are shown as smoothed loess curves for the HSCT‐alone cohort over time following transplantation. None of these patients received VST. Those with CMV reactivation are shown in red (n = 9), compared with no CMV reactivation in blue (n = 4). The scale of axes is shown on the exemplar and is uniform in all the plots.
Overall, CMV reactivation resulted in greater dynamic range of the majority of cell subsets. When plotted over time, the early dominance of innate cells (NK, monocytes, myeloid cells) was seen in samples to approximately 60 days post‐transplantation in CMV reactivators. CD16negNK and NKp46loCD56lo NK cells showed the most marked curve separation, although CD16+ NK cells were also elevated in CMV reactivators. Monocytes were elevated early in CMV reactivators, with CD86+, CD16+ and PDL1+ expression. CD14neg myeloid cells were also evident; NKT cells rise later in CMV reactivators.
Later, CD8+ T‐cell subsets are seen to rise in CMV reactivators, primarily in antigen‐experienced subsets (in particular Tem, Temra). Of these, the majority expressed the immunoinhibitory marker PD‐1, consistent with chronic antigenic stimulation. In this analysis, the proportional changes influenced by CMV are seen in quantitatively smaller subsets that are not prominent in the SC3 analysis. In particular, while overall CD4+ T‐cell numbers do not change substantially, some CD4+ T‐cell subsets show marked differences in patients with and without CMV reactivation. Data representation in this manner highlights that even with numerically low recovery of total CD4+ T cells, as was expected for the time period assessed, CMV reactivation can still be seen to influence subset proportions and inhibitory molecule expressing cells. In patients with CMV reactivation, the reconstitution of Tregs was found to be blunted, with CD4+ Tcm and Tem subsets conversely elevated. Co‐expression of PD‐1 (Figure 3) and other inhibitory molecules Lag3 and Tim3 (data not shown) was enriched on memory CD4+ T cells in CMV reactivators. Further, within the CD8+ T cells, CD31+CD8+ T cells showed the greatest curve separation of all the subsets, indicating that CMV‐related T‐cell activation is associated with CD31 expression. CD31 is known to be a regulator of activation, migration and differentiation 21 ; to our knowledge, CD31 expression by CD8+ cells in response to CMV has not previously been reported. In contrast, there was no significant rise in CD31+CD4+CD45RA+ T cells (recent thymic emigrants) within the timeframe of this study, indicating that in this main adult transplant population, the generation of immune effectors via the thymus from graft stem cells was not prominent and supports previous findings that early lymphocyte recovery is via expansion of mature donor T cells infused with the stem cell graft. 22
Clinical success of VST treatment is associated with recovery of adaptive immune response in some recipients
To assess the global immunological trajectory of HSCT patients undergoing treatment with third‐party CMV‐specific VST for treatment of refractory CMV infections, support vector machine (SVM) was used to predict in which immune profile cluster each sample from VST recipients would fall. Results of SVM are shown in Figure 4. The HSCT‐alone patients show a stereotyped transition from innate to adaptive signatures over time, which is not seen in CMV non‐reactivators (Figure 4a–c). Of note, the one patient who developed CMV tissue disease (day 58 post‐transplant) did so after grade 3 hepatic GVHD (day 39 post‐HSCT) which was treated with corticosteroids, anti‐thymocyte globulin and etanercept. CMV was treated successfully with ganciclovir and the colitis resolved. Viraemia resolved after the last study timepoint. This patient had a delayed development of the adaptive signature (Figure 4a grey line, short dashes) which corresponded to the timeframe of resolution of CMV disease.
Figure 4.
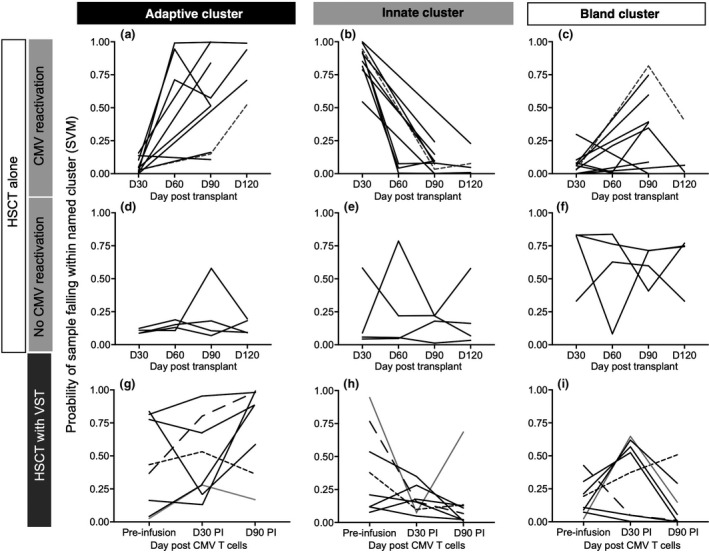
Immune signatures in patients who received VST. A support vector machine (SVM) was used to calculate the probability of a sample collected from patients pre‐ and post‐VST infusion falling within the immune signatures defined by the SC3 algorithm. The similarity of each sample to the clusters defined in SC3 is expressed by the probability of a sample falling in a given cluster (y‐axis). HSCT alone (teaching set) is shown in (a–f). A stereotyped pattern of innate signature progressing to adaptive signature over time is seen in CMV reactivators. One case of CMV disease and grade 3 hepatic GVHD is shown in (a–c) (grey line, short dashes). GVHD treatment delays development of adaptive immune signature, and the bland signature is seen at the time of treatment for GVHD (c). CMV colitis resolved at the time that the adaptive signature is seen (a). (g–i) Patients who received HSCT and VST. Samples pre‐VST infusion showed heterogeneity with three of eight patients having attained the adaptive immune signature at the time of VST infusion (g) despite all having high CMV antigen exposure (Table 1) and ample time develop an appropriate immune response. Six of eight patients had developed an adaptive signature by day 90 post‐infusion. Patient 1 (line with short dashes) did not develop an adaptive signature at any time. Patient 2 (line with long dashes) had an innate signature that progressed to adaptive signature. Patient 25 (grey line) had a cord blood transplant and did not develop an adaptive signature by day 90 post‐infusion but was able to control CMV.
In contrast, prior to VST infusion, despite a high level of CMV antigen exposure (median log10AUC at the first study timepoint 2.4 for HSCT‐alone vs 5.7 for VST recipients; P = 0.001) and a longer time frame in which to develop the appropriate immunological response, five of eight patients on the VST trial had not developed the adaptive immune signature (Figure 4g). However, by the end of study, six of the seven patients who received VST and controlled CMV had developed the adaptive immune signature. Of the patients with favorable clinical outcome, one patient (Patient 25) did not exhibit the adaptive immune signature by the end of this study despite experiencing virological control. This patient was the only study participant to receive a cord blood transplant, which is associated with delay in immune reconstitution. 23 The one patient in the VST group (Patient 1) who did not control CMV post‐VST treatment did not develop the adaptive immune profile at any point in the study period.
ViSNE was used to visualise changes in two CMV‐VST recipients with diverse clinical outcomes (Figure 5). Patient 1 failed to control CMV despite lengthy periods of antiviral treatment and 3 VST infusions and eventually died of presumed CMV encephalitis. Patient 2 controlled CMV post‐VST infusion and did not require further antiviral pharmacotherapy. Patient 2 still had an innate signature at the time of study entry despite 27 days of antiviral therapy. This patient is then seen to progress from an innate to an adaptive immune signature after VST, whereas Patient 1 did not. Figure 5 illustrates global hypocellularity in Patient 1 compared to Patient 2 pre‐infusion. Patient 2 demonstrated a rapid recovery of CD4+ T cells, CD8+ T cells and B cells at day 30 post‐infusion that was sustained thereafter. Patient 1 had sustained hypocellularity without CD4+ or CD8+ T‐cell recovery, as well as loss of the small NK cell population seen pre‐infusion.
Figure 5.
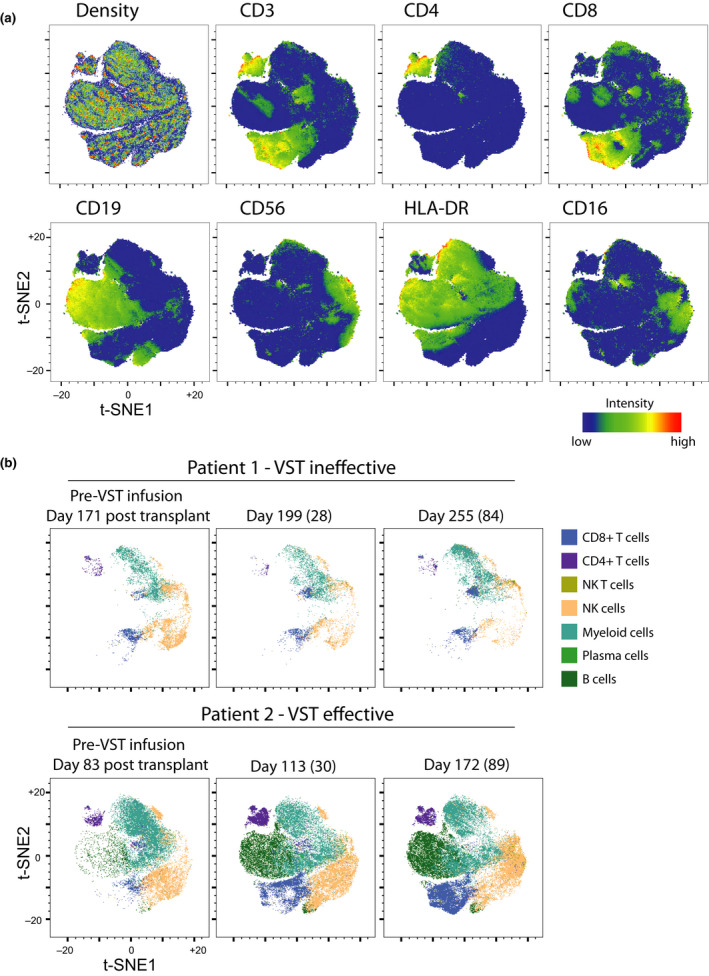
A viSNE was constructed to illustrate the differences in immunological features between two VST recipients with diverse outcomes. Patient 1 did not control CMV despite maximal pharmacologic therapy and 3 VST infusions and died of CMV encephalitis. Patient 2 had failed pharmacologic therapy but CMV viraemia was controlled after VST infusion. (a) Density plot, CD3, CD4, CD8, CD19, CD56, HLA‐DR and CD16 intensity of signal are shown across combined analysis. (b) Tracking of viSNE space occupancy comparing manually gated subsets from a time series of patients 1 and 2 who had divergent clinical outcomes following VST treatment. Day post‐transplant is shown, with day post‐VST infusion in parentheses.
Discussion
We have developed a method for assessing multiple immune cell subsets using a single peripheral blood sample, and a new analysis strategy utilising existing analysis tools. This comprehensive immunophenotyping panel captures the diversity of immune subpopulations expected in peripheral blood from a single PBMC sample per timepoint making it feasible for applications to human studies of immunotherapy.
We have used a systems approach to assess immune reconstitution in HSCT recipients with and without VST administration to demonstrate the natural process of immune reconstitution, to explore mechanisms for failure of CMV control and the clinical effects of VST therapy. We have identified distinct immune signatures, using unsupervised analysis, that are associated with clinical parameters in patients undergoing HSCT. These show the strong influence of CMV in determining the pattern of immune reconstitution, with two distinct immune signatures (innate and adaptive) associated with the early and late immune responses to CMV, respectively, and that are absent in patients without CMV reactivation.
Cytomegalovirus resulted in elevation of NK, monocyte and myeloid populations early post‐reactivation, which transitioned to elevation in CD8+ antigen‐experienced T‐cell subsets later in the observation period. While overall CD4+ T‐cell numbers remained static, memory subsets with PD‐1 expression rose in CMV reactivators, although these changes were numerically much smaller than those observed in CD8+ subsets. Tregs rose in patients without CMV reactivation. Patients with CMV reactivation who experienced severe acute GVHD did not develop the adaptive immune signature, indicating that GVHD and its treatment impair the natural immune recovery process.
The canonical pattern of immune cell reconstitution after HSCT involves the early recovery of innate immune cells followed by later reconstitution of adaptive immune cells. The early CMV signature with a predominance of monocyte and NK cells observed here may be reflective of a rapid innate immune response to the early phases of viral reactivation. CMV reactivation post‐HSCT is known to drive NK cell reconstitution and maturation, 24 , 25 , 26 which was clearly seen in this study. Monocytes are a known site of CMV latency and may also serve as vehicles for viral dissemination; however, few studies have performed detailed characterisation of the monocyte compartment in the early immune response to CMV after HSCT. To our knowledge, this study presents the most detailed picture of monocyte subset reconstitution in the context of CMV reactivation post‐HSCT. We have previously shown that latent CMV drives infected cells into an anergic‐like monocyte state. 27 It is not clear whether the increase in monocytes seen early in reactivating patients in the current study was present prior to reactivation or caused by the effect of early viral reactivation, because of the set time of sample collection (day 30) which was not prior to CMV reactivation in the majority of cases. We intend to explore this question prospectively in a larger study with the first sample timepoint collected earlier than day 30, prior to CMV reactivation.
Adoptive cellular therapy with antigen‐specific VST can lead to viraemia control in patients with treatment‐resistant CMV. 10 , 14 , 17 In the present analysis, some cases of refractory CMV were associated with a failure to establish the appropriate adaptive immune signature in the timeframe observed in natural immune recovery, despite substantial antigen stimulation. VST infusion is able to induce the development of the adaptive, CD8+ T‐cell‐dominated immune signature in some patients in whom clinical recovery is observed. In contrast, the one patient who never controlled CMV viraemia despite administration of VST did not develop the adaptive immune signature at any point. We hypothesise that the adaptive immune signature represents the immunological manifestation of control of viraemia, and therefore of successful VST therapy, although this will need confirmation in a larger study. It is possible that failure to mount an adequate cellular immune response in some patients is the basis for the elevated mortality in CMV reactivators as a whole, 1 and also for the increase in co‐infection in some patients. 28 , 29 In this study, a large proportion of HSCT recipients underwent RIC regimens. It is not possible to draw conclusions about the origin of the expanded T cells (recipient, original transplant donor or the VST third‐party donor). This aspect will be addressed in future work.
This study has several limitations. Certain outcomes such as CMV tissue disease, relapse or death are infrequent, preventing conclusions being drawn about predictive immune signatures for these outcomes. We have concentrated only on the first few months post‐transplant even though immune reconstitution is known to develop over the first 12 months or longer. The observations made of the recipients who underwent HSCT plus VST should be approached with caution as this was not a randomised study and the numbers in the VST group were small. It should be noted, however, that there have been no randomised studies of VST for immune reconstitution published to date. Our study has two prospectively enrolled cohorts and provides a methodological framework for larger upcoming studies. In future work, the immune profiling approach described here will be used to assess immune signatures prospectively in a randomised trial that is due to begin recruitment in 2020. Predictive immune signature(s) may be identified that allow the identification of patients who are likely to recover without intervention, those who are likely to fail standard antiviral therapy and those likely to respond to VST infusion, and this may inform rational use of VST therapy.
Methods
Study subjects and samples – HSCT and VST trial treatment
The study schema is shown in Figure 1. A single steady‐state peripheral blood sample was collected from healthy volunteers. Four peripheral blood samples were collected from patients undergoing allogeneic HSCT without VST on days 30, 60, 90 and 120 post‐HSCT. These patients were recruited sequentially and were treated with conditioning regimens, transplant procedures and post‐transplant care according to standard institutional practice. From the samples collected for HSCT recipients enrolled on a phase I trial of third‐party VST, three timepoints were assessed: pre‐VST infusion, day 30 and day 90 post‐VST infusion. The timing post‐transplant of sample collection was determined by the day of trial enrolment. The trial conduct and clinical results have been previously reported. 14 In brief, patients received partially HLA‐matched third‐party donor‐derived CMV‐specific VST from a cell bank 11 after CMV reactivation and subsequent failure of at least 2 weeks of anti‐CMV pharmacotherapy to control viraemia. Conditioning, immunosuppression and supportive care were administered according to institutional practice. CMV viraemia was defined as any positive PCR result in the blood [Roche COBAS® AmpliPrep/COBAS® TaqMan (Roche Diagnostics, Branchburg, NJ)]. CMV antigen exposure was quantified by the logarithm of the area under the curve (log10AUC) of CMV PCR quantitation in the blood in copies/mL. 28
The study was approved by the institutional ethics committees of the participating institutions. Informed consent was obtained from all study participants prior to enrolment in accordance with the Declaration of Helsinki.
Cell collection and storage
Peripheral blood mononuclear cells (PBMCs) from healthy individuals and transplant recipients were collected, processed and stored according to previously published methods. 14 PBMCs were isolated by Ficoll‐Paque (GE Healthcare, Uppsala, Sweden) centrifugation, cryopreserved in 10% dimethyl sulfoxide, 20% foetal calf serum (FCS) and phosphate‐buffered saline (PBS) and stored in vapour phase liquid nitrogen. Samples were thawed in batches and washed with RMPI1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat‐inactivated foetal calf serum (FCS) and benzonase.
Mass cytometry
Panel design
A panel of 38 metal‐tagged monoclonal antibodies, a biotin‐tagged antibody and a PE‐tagged antibody, was used for analysis of PBMCs. A list of antibodies and corresponding metal tags is provided in Supplementary table 1. The panel allowed quantification of 72 gated populations expressing specific markers of interest which encompass immune cell activation, co‐stimulation capacity and functionality. The gating strategy (shown in Supplementary figure 1A and B) assigned lineage determination to 95.6 + 0.3% of live CD45+ cells (median ± SEM) as shown in Supplementary figure 1C. It would be possible to identify novel subsets using this dataset but is beyond the scope of this manuscript.
Mass cytometry experiments
All antibodies were validated, pre‐titred and supplied in per‐test amounts by the Ramaciotti Facility for Human Systems Biology Mass Cytometry Reagent Bank. Reagent bank antibodies were either purchased from Fluidigm (San Francisco, CA, USA) in pre‐conjugated form or unlabelled antibodies were purchased in a carrier‐protein‐free format and conjugated by the Ramaciotti Facility for Human Systems Biology with the indicated respective metal isotope using the MaxPAR antibody conjugation kit (Fluidigm) according to the manufacturer's recommended protocol. For live/dead cell distinction, PBMCs were stained with 1.25 µm cisplatin in PBS for 3 min at room temperature and quenched with RMPI1640 supplemented with 10% heat‐inactivated FCS. Cells were incubated for 30 min initially with anti‐CD45 antibodies conjugated to various metals which facilitated barcoding of two samples together, as well as fluorochrome and biotin‐conjugated antibodies as indicated in the cocktail shown in Supplementary table 1. Following two washes with FACS buffer (PBS with 5% FCS), differentially barcoded samples were combined and incubated with the remaining metal‐conjugated antibodies targeting surface antigens. Following wash with FACS buffer, cells were fixed and permeabilised with FoxP3 buffer kit (eBioscience, San Diego, CA, USA) according to manufacturer's recommendations, and stained with FoxP3 antibody for 45 min at 4°C. Cells were washed and fixed overnight in 4% paraformaldehyde containing DNA intercalator (0.125 μm iridium‐191/193; Fluidigm). After multiple washes with FACS buffer and MilliQ water, cells were diluted to 800 000 cells/mL in MilliQ water containing 1:10 diluted EQ beads (Fluidigm) and filtered through a 35 μm nylon mesh. Cells were acquired at a rate of 200–400 cells/s using a CyTOF 2 Helios upgraded mass cytometer (Fluidigm).
Mass cytometry analysis
All FCS files were normalised using the MATLAB (MathWorks, Natick, MA, USA) normaliser and concurrently run EQ beads 30 and subsequently analysed using FlowJo X 10.0.7r2 software (FlowJo, LLC, Ashland, OR, USA). Samples were pre‐gated on DNA+, live, CD45+ cells and exported for further analysis (Supplementary figure 1A). Anticipating cytopenias in the early post‐transplant samples, acquisition of a minimum number of 1000 live CD45+ cells was needed to be included for further analysis. To ensure significant cells were sampled to be featured in the analysis of each cell subset, a cut‐off was set such that a minimum of 100 cells in the parental gate was required for a sub‐gate to be counted; otherwise, the value was returned as a zero. On these criteria, some samples from HSCT recipients with extremely low mononuclear cell counts were excluded from further analysis.
Scaling of lymphocyte and monocyte counts
In order to factor in the large numeric changes in lymphocytes and monocytes seen over the first few months post‐transplant, lymphocyte and monocyte subpopulation counts were scaled based on counts measured on the automated full blood count analyser on the day of sample collection and expressed as cells per 109/L.
For lymphocyte subpopulations, the scaling factor xl was calculated to be
where L S is lymphocyte count from blood (×109/L as determined by full blood analyser) and L C is the lymphocyte count (lymphocytes as a proportion of total live immune cells) in the CyTOF sample.
The monocyte subpopulations scaling factor xm was calculated to be
where M S is the monocyte count from blood (×109/L) and M C is monocyte count (monocytes as a proportion of total live immune cells) in the CyTOF sample.
Thereafter, all lymphocyte subpopulation counts (frequencies of live immune cells) were multiplied by xl, and all monocyte subpopulation counts were multiplied by xm.
The lymphocyte populations added together to calculate L C were as follows: B cells, CD19+ CD20neg, CD14neg CD16+, CD14neg, CD16neg, NK cells and CD3+ cells. The monocyte populations added together to calculate MC were as follows: CD16+ monocytes and classical monocytes.
Quality control
Batch consistency
Samples were stained and acquired in six experimental batches. To ensure no bias was introduced into the analysis, each batch had fair representation of healthy control and patient samples. For each patient, all timepoints were analysed in the same batch and barcoded together in pairs. To assess consistency between batches, analysis was repeated for six of the 13 healthy control samples across different batches. Upon applying the gating strategy outlined in Supplementary figure 1A and B, each control sample showed comparable population frequencies when stained, acquired and analysed independently in two batches (see Supplementary figure 2A). Furthermore, t‐SNE plots generated for normalised count and proportion data (see next section) showed good mixing of batches across the plots (see Supplementary figure 2B and C), demonstrating the reproducibility of the results over repeated measures.
Statistical analyses
Clustering using SC3
Unsupervised hierarchical clustering was performed with the SC3 R package based on filtered cell population numbers using all samples that passed QC from the patients who did not receive VST. The SC3 algorithm generates a consensus score resulting from the integration of three similarity metrics commonly utilised for calculating sample distances in hierarchical clustering (Euclidian distance, Pearson's and Spearman's correlation). The number of clusters was chosen to optimise the stability of each cluster. Finally, population counts that were associated with the chosen clustering were extracted (AUC > 0.65, P < 0.05). Using SC3 functionalities, each sample in the heat map was annotated with the associated clinical information.
Support vector machine (SVM)
The probability of a sample from the VST group falling within an immune signature cluster was calculated with SVM utilising a linear kernel. Clustering was predicted based on SVM trained on samples from the HSCT‐alone group (N = 42) using as input only features extracted from SC3 analysis. The accuracy of the SVM classifier was assessed using 5‐fold cross validation (Acc = 0.83). As comparison, another SVM classifier was trained using all cell populations. The accuracy of the classifier decreases to 0.74, therefore validating the importance of the features extracted from the SC3 analysis.
Clinical information, demographics, baseline clinical characteristics, transplantation procedures and post‐transplant outcomes were compared between HSCT‐alone and VST recipients. For categorical variables, the chi‐square test, Fisher's exact test or one‐way ANOVA was used as appropriate. The 2‐sample Student's t‐test was used for normally distributed continuous variables and the Mann–Whitney U‐test for skewed continuous variables. P‐value < 0.05 was considered significant when comparing the distribution of clinical variables between patient groups. To assess the influence of clinical factors on immune profile clusters generated by SC3, univariate regression was performed. The Bonferroni method was used to correct for multiple comparisons (α = 18). P < 0.0028 was the threshold for statistical significance. Statistical analysis was performed using IBM SPSS for Mac version 24.0.0 (IBM, New York, NY, USA) and Prism 7.0b for Mac (GraphPad Software Inc., La Jolla, CA, USA) and R. The fit of the trajectories for immune subsets over time was performed in R using loess curve fitting technique using degree = 1, span = 0.75 and Tukey's biweight function. The visualised t‐distributed stochastic neighbour embedding (ViSNE) algorithm (implemented in FlowJo as a plugin) was utilised to perform dimensionality reduction and visualisation of live immune subsets across samples. 20 , 31 Cells were sampled without replacement from each file relative to density of cells in blood (×109/L) and combined for analysis. The markers used for clustering were CCR10, CD3, CD4, CD8, CD11c, CD14, CD16, CD19, CD20, CD25, CD27, CD45RA, CD45RO, CD56, CD62L, CD86, CD127, CD161, FoxP3 and HLADR. The resulting t‐SNE plots were visualised by marker expression using the FlowJo colour map axis function, with patient time series comparisons visualised with an overlay of manually gated subsets.
Conflict of interest
EB reports advisory board membership with Abbvie, Novartis, Astellas and MSD. DG reports advisory board membership with Abbvie, Gilead and Novartis. DG reports research funding from Haemalogix. EB, DG and LC report patents in the field of adoptive cell therapy manufacture.
Author contributions
Helen M McGuire: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Simone Rizzetto: Conceptualization; Data curation; Formal analysis; Methodology; Resources; Software; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Barbara P Withers: Data curation; Investigation; Project administration; Writing‐review & editing. Leighton E Clancy: Data curation; Investigation; Writing‐review & editing. Selmir Avdic: Data curation; Formal analysis; Supervision; Writing‐review & editing. Lauren Stern: Formal analysis; Writing‐original draft; Writing‐review & editing. Ellis Patrick: Formal analysis; Visualization. Barbara Fazekas de St Groth: Formal analysis; Supervision; Writing‐review & editing. Barry Slobedman: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Visualization; Writing‐original draft; Writing‐review & editing. David J Gottlieb: Conceptualization; Funding acquisition; Project administration; Resources; Supervision; Writing‐review & editing. Fabio Luciani: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Resources; Software; Supervision; Validation; Visualization; Writing‐review & editing. Emily Blyth: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing.
Supporting information
Supplementary Material
Acknowledgments
This project received funding support from the Australian National Health and Medical Research Council (NHMRC) (GNT1121643 and GNT1032431), Australian Leukaemia Foundation, Cancer Council of NSW, Royal College of Pathologists of Australasia and NSW Cancer Institute. EB is a NSW Cancer Institute Early Career Fellow and former NHMRC post‐doctoral fellow (GNT1089398). LS is supported by an Australian Postgraduate Award. HMM is currently supported by the International Society for the Advancement of Cytometry (ISAC) Marylou Ingram Scholars Program, received Sydney Medical School Kickstart grant funding support and was previously an NHMRC post‐doctoral fellow (GNT1037298). BS and EB received Sydney Medical School BioMed‐Connect grant funding. FL is a NHMRC Career Development Fellow (GNT1128417).
The trial was registered at www.clinicaltrials.gov (#NCT02779439) and www.anzctr.org.au (#ACTRN12613000603718).
References
- 1. Green ML, Leisenring W, Xie H et al Cytomegalovirus viral load and mortality after haemopoietic stem cell transplantation in the era of pre‐emptive therapy: a retrospective cohort study. Lancet Haematol 2016; 3: e119–e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walter EA, Greenberg PD, Gilbert MJ et al Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T‐cell clones from the donor. N Engl J Med 1995; 333: 1038–1044. [DOI] [PubMed] [Google Scholar]
- 3. Heslop HE, Ng C, Li C et al Long‐term restoration of immunity against Epstein‐Barr virus infection by adoptive transfer of gene‐modified virus‐specific T lymphocytes. Nat Med 1996; 2: 551–555. [DOI] [PubMed] [Google Scholar]
- 4. Peggs KS, Thomson K, Samuel E et al Directly selected cytomegalovirus‐reactive donor T cells confer rapid and safe systemic reconstitution of virus‐specific immunity following stem cell transplantation. Clin Infect Dis 2011; 52: 49–57. [DOI] [PubMed] [Google Scholar]
- 5. Blyth E, Clancy L, Simms R et al Donor‐derived CMV specific T‐cells reduce the requirement for CMV‐directed pharmacotherapy after allogeneic stem cell transplantation. Blood 2013; 121: 3745–3758. [DOI] [PubMed] [Google Scholar]
- 6. Feucht J, Opherk K, Lang P et al Adoptive T‐cell therapy with hexon‐specific Th1 cells as a treatment of refractory adenovirus infection after HSCT. Blood 2015; 125: 1986–1994. [DOI] [PubMed] [Google Scholar]
- 7. Vickers MA, Wilkie GM, Robinson N et al Establishment and operation of a Good Manufacturing Practice‐compliant allogeneic Epstein‐Barr virus (EBV)‐specific cytotoxic cell bank for the treatment of EBV‐associated lymphoproliferative disease. Br J Haematol 2014; 167: 402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wilkie GM, Taylor C, Jones MM et al Establishment and characterization of a bank of cytotoxic T lymphocytes for immunotherapy of epstein‐barr virus‐associated diseases. J Immunother 2004; 27: 309–316. [DOI] [PubMed] [Google Scholar]
- 9. Haque T, Wilkie GM, Taylor C et al Treatment of Epstein‐Barr‐virus‐positive post‐transplantation lymphoproliferative disease with partly HLA‐matched allogeneic cytotoxic T cells. Lancet 2002; 360: 436–442. [DOI] [PubMed] [Google Scholar]
- 10. Leen AM, Bollard CM, Mendizabal AM et al Multicenter study of banked third‐party virus‐specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood 2013; 121: 5113–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Withers B, Clancy L, Burgess J et al Establishment and operation of a third‐party virus‐specific T cell bank within an allogeneic stem cell transplant program. Biol Blood Marrow Transplant 2018; 24: 2433–2442. [DOI] [PubMed] [Google Scholar]
- 12. Neuenhahn M, Albrecht J, Odendahl M et al Transfer of minimally manipulated CMV‐specific T cells from stem cell or third‐party donors to treat CMV infection after allo‐HSCT. Leukemia 2017; 31: 2161–2171. [DOI] [PubMed] [Google Scholar]
- 13. Aguayo‐Hiraldo PI, Arasaratnam RJ, Tzannou I et al Characterizing the cellular immune response to parainfluenza virus 3. J Infect 2017; 216: 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Withers B, Blyth E, Clancy LE et al Long‐term control of recurrent or refractory viral infections after allogeneic HSCT with third‐party virus‐specific T cells. Blood Adv 2017; 1: 2193–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doubrovina ES, Oflaz‐Sozmen B, Prockop SE et al Adoptive immunotherapy with unselected or EBV‐specific T cells for biopsy‐proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012; 119: 2644–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barker JN, Doubrovina ES, Sauter C et al Successful treatment of EBV‐associated posttransplantation lymphoma after cord blood transplantation using third‐party EBV‐specific cytotoxic T lymphocytes. Blood 2010; 116: 5045–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tzannou I, Papadopoulou A, Naik S et al Off‐the‐shelf virus‐specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, Epstein‐Barr virus, and adenovirus infections after allogeneic hematopoietic stem‐cell transplantation. J Clin Oncol 2017; 35: 3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lakshmikanth T, Olin A, Chen Y et al Mass cytometry and topological data analysis reveal immune parameters associated with complications after allogeneic stem cell transplantation. Cell Rep 2017; 20: 2238–2250. [DOI] [PubMed] [Google Scholar]
- 19. Stern L, McGuire H, Avdic S et al Mass cytometry for the assessment of immune reconstitution after hematopoietic stem cell transplantation. Front Immunol 2018; 9: 1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hartmann FJ, Babdor J, Gherardini PF et al Comprehensive immune monitoring of clinical trials to advance human immunotherapy. Cell Rep 2019; 28: 819–831.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marelli‐Berg FM, Clement M, Mauro C, Caligiuri G. An immunologist's guide to CD31 function in T‐cells. J Cell Sci 2013; 126: 2343–2352. [DOI] [PubMed] [Google Scholar]
- 22. Fry TJ, Mackall CL. Immune reconstitution following hematopoietic progenitor cell transplantation: challenges for the future. Bone Marrow Transplant 2005; 35: S53–S57. [DOI] [PubMed] [Google Scholar]
- 23. Castillo N, García‐Cadenas I, Barba P et al Early and long‐term impaired T lymphocyte immune reconstitution after cord blood transplantation with antithymocyte globulin. Biol Blood Marrow Transplant 2017; 23: 491–497. [DOI] [PubMed] [Google Scholar]
- 24. Foley B, Cooley S, Verneris MR et al Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 2012; 119: 2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muccio L, Bertaina A, Falco M et al Analysis of memory‐like natural killer cells in human cytomegalovirus‐infected children undergoing αβ+T and B cell‐depleted hematopoietic stem cell transplantation for hematological malignancies. Haematologica 2016; 101: 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drylewicz J, Schellens IMM, Gaiser R et al Rapid reconstitution of CD4 T cells and NK cells protects against CMV‐reactivation after allogeneic stem cell transplantation. J Transl Med 2016; 14: 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shnayder M, Nachshon A, Rozman B et al Single cell analysis reveals human cytomegalovirus drives latently infected cells towards an anergic‐like monocyte state. Elife 2020; 9: e52168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hill JA, Mayer BT, Xie H et al Kinetics of double‐stranded DNA viremia after allogeneic hematopoietic cell transplantation. Clin Infect Dis. 2018; 66: 368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hill JA, Mayer BT, Xie H et al The cumulative burden of double‐stranded DNA virus detection after allogeneic HCT is associated with increased mortality. Blood 2017; 129: 2316–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Finck R, Simonds EF, Jager A et al Normalization of mass cytometry data with bead standards. Cytometry Part A 2013; 83: 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. E‐aD Amir, Davis KL, Tadmor MD et al viSNE enables visualization of high dimensional single‐cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 2013; 31: 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material