

Abstract
Sample multiplexing using isobaric tagging is a powerful strategy for proteome-wide protein quantification. One major caveat of isobaric tagging is ratio compression that results from the isolation, fragmentation, and quantification of coeluting, near-isobaric peptides, a phenomenon typically referred to as “ion interference”. A robust platform to ensure quality control, optimize parameters, and enable comparisons across samples is essential as new instrumentation and analytical methods evolve. Here, we introduce TKO-iQC, an integrated platform consisting of the Triple Knockout (TKO) yeast digest standard and an automated web-based database search and protein profile visualization application. We highlight two new TKO standards based on the TMTpro reagent (TKOpro9 and TKOpro16) as well as an updated TKO Viewing Tool, TVT2.0. TKO-iQC greatly facilitates the comparison of instrument performance with a straightforward and streamlined workflow.
Keywords: TKO standard, ion interference, TMT, iTRAQ, SPS-MS3, TVT, shiny application
Graphical Abstract

INTRODUCTION
The ability to determine the sequence of a peptide by matching structural peaks in MS2 spectra with predicted b- and y-type fragment ions from a protein database was first introduced by Yates and colleagues in 1993.1 The algorithm they produced was called SEQUEST, and it is still widely used today. Currently, tens of thousands of MS2 spectra are collected and must be matched to peptide sequences during a typical analysis. In this report, we have developed a quality control platform which uses an integrated, SEQUEST-based algorithm as an online database searching and comparison tool.
Isobaric labeling workflows are becoming the method-of-choice for proteome-wide mass spectrometry-based quantitative protein profiling. Sample multiplexing allows simultaneous interrogation of multiple proteomes, thereby increasing throughput, reducing missing values from sampling stochasticity, and improving statistical confidence. As such, controls and multiple experimental conditions can be comprehensively analyzed in a single data set. A major caveat of multiplexing is the coisolation, cofragmentation, and coanalysis of multiple precursor ions, termed “interference”. Here, we present a platform designed to quantify interference using a set of yeast proteome-based standards and companion software. Our Triple KnockOut isobaric tag-based quality control platform, termed TKO-iQC, incorporates two key elements: (1) a highly versatile set of standards that are designed to assess ion interference and (2) an automated, user-friendly, web-based application that permits efficient analysis of these standards. TKO-iQC offers a universal pipeline for data quality assessment across instruments and laboratories.
Previously, we have developed an isobaric tag-based analytical standard, named the Triple KnockOut (TKO), for assessing interference and overall instrument performance. The fundamental basis of this standard is simple, as three Saccharomyces cerevisiae yeast deletions strains are organized with replicates in a multiplexed tandem mass tag (TMT) experiment. In the ideal case of no interference, the TMT signal in the strain with the known deleted protein should be zero. However, TMT signal for channels where the protein is absent is a surrogate for interference. We have described previously the original TKO92 and the unit-resolution TKO6 standards.3 In addition, ThermoScientific has recently released the “Pierce Protein Interference Standard”.4,5 This commercial standard is essentially a TKO9 with the addition of two wildtype yeast-containing channels (establishing a TKO11 standard) and substituting the Δpfk2 deletion strain with Δhis4. We note that Δhis4 has a similar growth rate as Δmet6 and Δura2, and as such is optimal for large-scale processing, despite its caveat of lower abundance and having fewer consistently observed peptides compared to Δpfk2. Here, we expand on these standards leveraging the novel TMTpro16 reagents6 and introduce the TMTpro-series of TKO standards.
The recent introduction of the TMTpro reagent permits higher multiplexing (n = 16) than achievable previously using the classic TMT6/10/11 labels. Using these new reagents, we have designed two standards, namely TKOpro9 and TKOpro16. TKOpro9 is the successor of TKO6, both of which consist of unit-resolved isotopic tags and can be used to evaluate interference of TMT reporter ions measured in the ion trap. While TKO6 was limited to duplicates of each deletion strain, the TKOpro9 uses three additional TMT16pro reporter ions, which mimics the original TKO9 arrangement of 3 × 3 × 3 but is unit resolved. TKOpro16 expands on the TKO11 format by leveraging the 5 additional channels of the TMTpro16 reagent. The availability of 16 channels permits quadruplicates of each deletion strain, and the addition of the wild type strain, to be arranged in a 4 × 4 × 4 × 4 layout.
Our goal was to provide the proteomics community with a simple, yet useful, standard with dedicated software to investigate ion interference. We have previously developed the TKO Viewing Tool (TVT) for the streamlined analysis of the TKO standard.4 With TVT, an end-user simply uploads a RAW file of a TKO standard into a web browser for which a quick, fully automated database search (based on the SEQUEST/Comet search engine1,7,8) is performed. Here we introduce an updated TVT, referred to as TVT2.0. Changes in TVT2.0 include (1) compatibility with TMTpro16-labeled standards, (2) encrypted links to the results, (3) performance comparisons across multiple experiments, and (4) improved sample tracking. The user is emailed a link to tables and graphics illustrating traditional figures of merit, such as protein and peptide counts, in addition to various ion statistics. TVT2.0 also allows for tracking and comparing these metrics across multiple TKO analyses. Together, TVT2.0 and the TKO standard comprise the TKO-iQC, which is an innovative and scalable platform for the assessment of interference for isobaric tag-based quantitative proteomics.
EXPERIMENTAL SECTION
Samples were processed using the SL-TMT protocol.9 We have included detailed methods for yeast growth, sample processing, data acquisition, and analysis in the Supporting Information. The TKO Viewing Tool can be accessed at http://tko.hms.harvard.edu/ or alternatively at https://wren.hms.harvard.edu/gfy/www/modules/single_standard/. The RAW files are available upon request and can also be downloaded from ProteomeXchange10 with identifier PXD017803.
RESULTS AND DISCUSSION
In this study, we introduce TKO-iQC, an integrated quality control platform for isobaric tag-based quantitative proteomics. The centerpiece of TKO-iQC is the updated TKO Viewing Tool (TVT2.0), which is an automated database search and data visualization application for the TKO standard. TVT2.0 can analyze TKO standards that are labeled with the TMTpro reagents.6 To showcase the TKO-iQC platform, we evaluated the performance of two new TKO standards (TKOpro9 and TKOpro16) across different instruments and data acquisition methods.
TVT2.0 is a web-based application designed to streamline the analysis of the TKO standard, and is an update of the original TVT.4 The TVT2.0 performs an automated, web-based, database search of the TKO standard using Comet.11 Once the standard is searched, the viewer returns tables and plots of traditional figures of merit (e.g., protein and peptide counts, Xcorr values, ion times) as well as the TKO-specific IFI (interference-free index) (Figure 1). The user simply selects the TKO variant used, uploads a RAW file (Figure 1A), and enters an email address. Once the RAW file is uploaded, a predefined database search is initiated.11 A hyperlink is emailed to the user upon completion of the search (~5–15 min depending on server usage). For tracking TKO analyses, a log of the user’s submissions, including date, filename, a link to the data and a unique search identifier (searchID) is appended to the end of each email. This information is useful particularly when comparing files over extended periods of time or across different instruments. Note that as a security feature, each analysis is associated with a specific email address, and an encrypted link to the data is provided in the email for future reference or data sharing (Figure 1B). The link opens the front page of the TKO Viewing Tool Web site (Figure 1C) which provides access to plots and tables displaying the data, in addition to standard figures of merit (Figure 1D). As a quantitative measure, the IFI is calculated for a given TKO protein as the difference from 1 of the ratio of the average TMT signal-to-noise for the TKO protein deficient channels to that of non-TKO channels (Figure 1E). We note that values close to 1, approach no interference, while values which are close to zero are indicative of a high degree of interference. Moreover, a customization feature is available in the TVT2.0 permitting the substitution of any deletion strain into a custom, TKO standard if the sample arrangement fits one of the preassigned formats: 2 × 2 × 2 (e.g., TMT6), 3 × 3 × 3 (e.g., TKO9, TKOpro9), 3 × 3 × 3 × 2 (e.g., TKO11), or 4 × 4 × 4 × 4 (e.g., TKOpro16) (Figure S1, S2). In addition to viewing interference-specific information and performance metrics for a single analysis, the end-user is offered an option to compare results across multiple TKO analyses. Such a feature is useful for assessing method development,12,13 optimizing sample preparation protocols,9,14 or tracking instrument performance.
Figure 1.
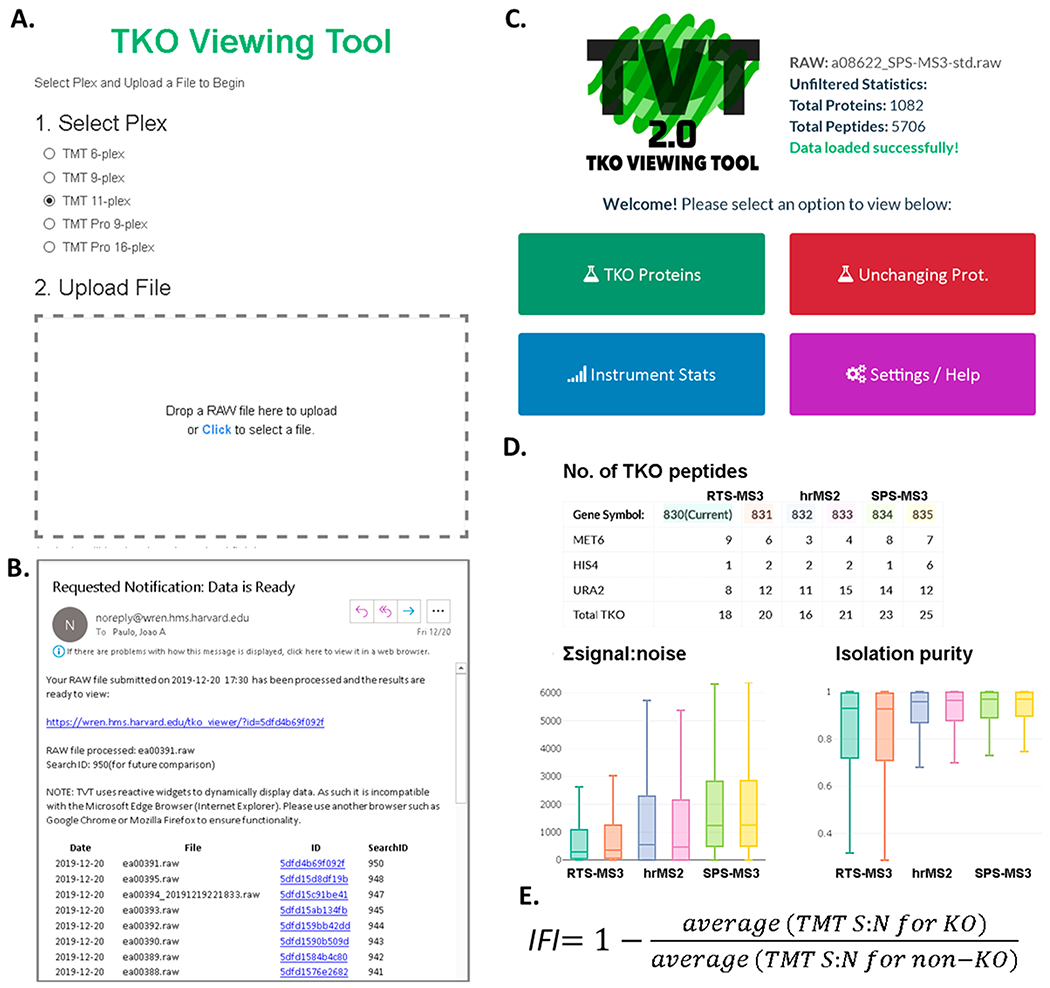
TKO Viewing Tool. (A) The TVT2.0 uploader can be assessed by navigating to http://tko.hms.harvard.edu (Chrome and Firefox web browsers are recommended). The TKO variant used must be selected. The RAW file is then dragged directly into the web browser to initiate an automated, preconfigured database search, as was done in the original TVT application.4 (B) The user will receive an email with a link to this analysis as well as a unique identifier (search ID) and a list of previous jobs. (C) Following the provided link in the email, search data can be retrieved via intuitive dropdown menus allowing for quality control assessment of the submitted analysis and comparison with previous analyses. (D) We highlight a new feature of TVT2.0 that allows for comparison of multiple previous analyses (we recommend up to 12). The input for these comparisons are “SearchIDs” as shown in panel B. Additional example comparisons are illustrated in Supplemental Figure 3. (E) We highlight the equation used to calculate the IFI.
We showcase the TVT2.0 using three examples, each examining the performance of the TKOpro standards across different instruments and data acquisition methods. First, we used the TKOpro16 (Figure 2A) to investigate three common data acquisition methods for isobaric tag-based experiments, specifically: real-time search (RTS)-MS3,12,15 high-resolution MS2 (hrMS2), and traditional SPS-MS316,17 (Figure 2B). We analyzed two replicate samples using a 45 min data acquisition method on an Orbitrap Fusion Tribrid mass spectrometer. We designed the TKO standard such that each deletion strain is an abundant protein (e.g., Met6, His4, Ura2). In doing so, we are confident that peptides from TKO proteins are identified in every analysis, which is important when using short gradients. As such, we recommend 45 min methods to minimize instrument time spent on quantity control. The data illustrated that the interference-free index (IFI) was highest for RTS-MS3 followed by SPS-MS3, with hrMS2 having the most interference (Figure 2C).15 Moreover, we compared the number of proteins and peptides quantified by each analytical method (Figure 2D) and confirmed that with short gradients, these complex standards revealed similar numbers of proteins and peptides quantified for RTS-MS3 and hrMS2, with SPS-MS3 trailing behind. These data support that RTS-MS3 achieves a good balance with respect to low interference and high protein/peptide quantification.12,15 Figure S3 provides additional tables and plots of these data generated directly from the TVT2.0 instrument comparison web interface.
Figure 2.

Example analysis using the TKOpro16 standard. (A) TKOpro16 standard is composed of four replicates each of deletion strains (Δmet6, Δhis4, and Δura2) as well as wildtype yeast. Each strain/replicate is labeled with the specified TMTpro16 reagent. (B) Schematic of three acquisition methods investigated: real time search (RTS)-MS3, high-resolution HCD-MS2 (hrMS2), and synchronous precursor selection (SPS)-MS3. We highlight the mass analyzer for each stage of m/z measurement. (C) Bar plot of the interference-free index (IFI) for duplicates of the acquisition methods outlined in (B). (D) Combination plot indicating the number of proteins (circle) and peptides (bars) for the acquisition methods outlined in (B). OT, Orbitrap; IT, ion trap.
Next, we used the TKOpro9 (Figure 3A) to compare MS3-based data acquisition using the Orbitrap (OT-MS3) or ion trap (IT-MS3) mass analyzer (Figure 3B). Unlike the TKOpro16 standard, the TKOpro9 lacks 13C and 15N isotopologue mass differences, as the labels are all unit resolved. As such, high resolution (>45000) mass analysis is not required for reporter-based quantification. Isobaric tags that are separated by approximately one Dalton can be analyzed by an ion trap for TMT-based reporter ion quantification, thereby permitting faster scan speeds and shorter duty cycles, as demonstrated previously.18 We analyzed three replicates samples using a 45 min liquid chromatography gradient on a FAIMS-equipped Orbitrap Eclipse Tribrid mass spectrometer. Our data illustrated that the interference-free index (IFI) was lower for data analyzed with OT-MS3 than IT-MS3 (Figure 3C), yet the number of proteins and peptides were higher for IT-MS3 than OT-MS3 (Figure 3D). These data support that IT-MS3 may be a viable alternative to OT-MS3, but trades depth for quantitative accuracy, analogous to how hrMS2 behaves when compared to SPS-MS3 (Figure 2).
Figure 3.

Example analysis using the TKOpro9 standard. (A) The TKOpro9 standard is composed of three replicates of deletion strains (Δmet6, Δhis4, and Δura2) each labeled with the specified TMTpro reagent and separated by unit resolution. (B) Schematic of two acquisition methods investigated: the classic Orbitrap-based SPS-MS3 (OT-MS3) and the alternative ion trap-based SPS-MS3 (IT-MS3). We highlight the mass analyzer for each stage of m/z measurement. (C) Bar plot of interference-free index (IFI) for duplicates of the acquisition methods outlined in (B). (D) Combination plot indicating the number of proteins (circle) and peptides (bars) for the acquisition methods outlined in (B). OT, Orbitrap; IT, ion trap.
Finally, we compared data acquired using the new TKOpro16 standard with the classic TKO11 using TVT2.0 (Figure S4A). We note that the deletions strains used in these standards were identical. Here, we analyzed three replicates of ~0.5 μg standard over a 45 min liquid chromatography gradient using a Q-Exactive HF-X mass spectrometer. We note that, as expected, both standards generated consistent IFI measurements across the three replicates (Figure S4B). As these were, in fact, completely different standards that were prepared months apart and used different labeling chemistries, the number of proteins and peptides quantified may be expected to differ. However, we observed only a slight difference in the number of peptides per protein when comparing the two standards (Figure S4C), as also observed previously.19 These findings support that the data obtained from the two TKO variants will result in similar conclusions and that the TVT2.0-based data analysis and visualization is compatible with both TMT- and TMTpro-based TKO standards.
TKO-iQC streamlines and consolidates database searching and visualization of all currently available TKO standards. In addition to the standards described herein, we stress that the format of the TKO standard may also serve as a template for alternative versions of the standard that should also be compatible with TVT2.0. For example, some end-users may be concerned that the selection of very abundant deleted proteins in the TKO standard does not pose enough of a challenge if sensitivity, as well as interference, is to be assayed. One can choose different (e.g., low-abundant) proteins as the TKO proteins, while an alternative option would be to add a background of TMT-labeled human peptides (equivalent in all channels). The TVT2.0 application was coded to be easily adapted to standards well beyond those described herein. For TVT2.0 we chose open-source Comet as our search engine to replace SEQUEST due to similarity in results.11,20 However, we presume that applications with similar functionality of TVT may be designed using Andromeda,21 MS-GF+,22 or other search engines. We also anticipate that standards equivalent to the TKO, but for post-translational modifications, such as phosphorylation or ubiquitylation, will be available and the analysis there of could be based on the TVT2.0 infrastructure. As such, we expect that TKO-iQC will remain an innovative and scalable platform for the assessment of ion interference as computational and technological improvements emerge.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Gygi Lab at Harvard Medical School for invaluable discussion. We also thank members of ThermoFisher Scientific, particularly John Rogers, Rosa Viner, Jae Choi, Ryan Bomgarden, and Aaron Robitaille, for valuable discussions and early access to the commercially available Pierce Yeast TKO11-plex interference standard. This work was funded in part by NIH/NIGMS grants R01 GM132129 (J.A.P.) and GM97645 (S.P.G.). J.N.P. was supported by CONACyT (CVU 289937) from Mexico.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.0c00029.
Supplemental Methods: Extended sample preparation and mass spectrometry data analysis. Figure S1: Composition and assembly of TKOpro standards. Figure S2: TVT2.0 output customization. Figure S3: Example output generated by the TVT2.0 of TKO16pro data acquired from three acquisition strategies. Figure S4: Comparison of TKOpro16 and TKO11 standards (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jasms.0c00029
The authors declare no competing financial interest.
Contributor Information
Jeremy P. Gygi, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States
Ramin Rad, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
Jose Navarrete-Perea, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
Simon Younesi, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
Steven P. Gygi, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
Joao A. Paulo, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
REFERENCES
- (1).Eng JK; McCormack AL; Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5 (11), 976–89. [DOI] [PubMed] [Google Scholar]
- (2).Paulo JA; O’Connell’ JD; Gygi SP A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J. Am. Soc. Mass Spectrom. 2016, 27 (10), 1620–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Paulo JA; Navarrete-Perea J; Guha Thakurta S; Gygi SP TKO6: A Peptide Standard To Assess Interference for Unit-Resolved Isobaric Labeling Platforms. J. Proteome Res. 2019, 18 (1), 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gygi JP; Yu Q; Navarrete-Perea J; Rad R; Gygi SP; Paulo JA Web-Based Search Tool for Visualizing Instrument Performance Using the Triple Knockout (TKO) Proteome Standard. J. Proteome Res. 2019, 18 (2), 687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Myers SA; Klaeger S; Satpathy S; Viner R; Choi J; Rogers J; Clauser K; Udeshi ND; Carr SA Evaluation of Advanced Precursor Determination for Tandem Mass Tag (TMT)-Based Quantitative Proteomics across Instrument Platforms. J. Proteome Res. 2019, 18 (1), 542–547. [DOI] [PubMed] [Google Scholar]
- (6).Li J; Van Vranken JG; Pontano Vaites L; Schweppe DK; Huttlin EL; Etienne C; Nandhikonda P; Viner R; Robitaille AM; Thompson AH; Kuhn K; Pike I; Bomgarden RD; Rogers JC; Gygi SP; Paulo JA TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 2020, DOI: 10.1038/s41592-020-0781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).MacCoss MJ; Wu CC; Yates JR 3rd Probability-based validation of protein identifications using a modified SEQUEST algorithm. Anal. Chem 2002, 74 (21), 5593–9. [DOI] [PubMed] [Google Scholar]
- (8).Yates JR 3rd Pivotal role of computers and software in mass spectrometry - SEQUEST and 20 years of tandem MS database searching. J. Am. Soc. Mass Spectrom. 2015, 26 (11), 1804–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Navarrete-Perea J; Yu Q; Gygi SP; Paulo JA Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res. 2018, 17 (6), 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Jarnuczak AF; Vizcaino JA Using the PRIDE Database and ProteomeXchange for Submitting and Accessing Public Proteomics Datasets. Curr. Protoc Bioinformatics 2017, 59, 13311–133112. [DOI] [PubMed] [Google Scholar]
- (11).Eng JK; Jahan TA; Hoopmann MR Comet: an open-source MS/MS sequence database search tool. Proteomics 2013, 13 (1), 22–4. [DOI] [PubMed] [Google Scholar]
- (12).Schweppe DK; Eng JK; Bailey D; Rad R; Yu Q; Navarrete-Perea J; Huttlin EL; Erickson BK; Paulo JA; Gygi SP Full-featured, real-time database searching platform enables fast and accurate multiplexed quantitative proteomics. J. Proteome Res 2020, DOI: 10.1021/acs.jproteome.9b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schweppe DK; Prasad S; Belford MW; Navarrete-Perea J; Bailey DJ; Huguet R; Jedrychowski MP; Rad R; McAlister G; Abbatiello SE; Woulters ER; Zabrouskov V; Dunyach JJ; Paulo JA; Gygi SP Characterization and Optimization of Multiplexed Quantitative Analyses Using High-Field Asymmetric-Waveform Ion Mobility Mass Spectrometry. Anal. Chem 2019, 91 (6), 4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Paulo JA; Navarrete-Perea J; Gygi SP Multiplexed proteome profiling of carbon source perturbations in two yeast species with SL-SP3-TMT. J. Proteomics 2020, 210, 103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Erickson BK; Mintseris J; Schweppe DK; Navarrete-Perea J; Erickson AR; Nusinow DP; Paulo JA; Gygi SP Active Instrument Engagement Combined with a Real-Time Database Search for Improved Performance of Sample Multiplexing Workflows. J. Proteome Res 2019, 18 (3), 1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).McAlister GC; Nusinow DP; Jedrychowski MP; Wuhr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 2014, 86 (14), 7150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ting L; Rad R; Gygi SP; Haas W MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8 (11), 937–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Liu JM; Sweredoski MJ; Hess S Improved 6-Plex Tandem Mass Tags Quantification Throughput Using a Linear Ion Trap-High-Energy Collision Induced Dissociation MS(3) Scan. Anal. Chem 2016, 88 (15), 7471–5. [DOI] [PubMed] [Google Scholar]
- (19).Li J; Van Vranken J; Pontano Vaites L; Schweppe D; Huttlin EL; Etienne C; Nandhikonda P; Viner R; Robitaille A; Thompson A; Kuhn K; Pike I; Bomgarden R; Rogers J; Gygi SP; Paulo JA TMTpro16plex Isobaric Labeling Reagents: Attaining New Sample Multiplexing Heights at Proteome-scale Depths in Quantitative Proteomics. Nat. Methods 2020, x DOI: 10.1038/s41592-020-0781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Eng JK; Hoopmann MR; Jahan TA; Egertson JD; Noble WS; MacCoss MJ A deeper look into Comet–implementation and features. J. Am. Soc. Mass Spectrom. 2015, 26 (11), 1865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cox J; Neuhauser N; Michalski A; Scheltema RA; Olsen JV; Mann M Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10 (4), 1794–805. [DOI] [PubMed] [Google Scholar]
- (22).Kim S; Pevzner PA MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun 2014, 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.