

Abstract
The rarity of mixed phenotype acute leukemia (MPAL) has precluded adequate data to incorporate minimal residual disease (MRD) monitoring into therapy. Fluidity in MPAL classification systems further complicates understanding its biology and outcomes; this includes uncertainty surrounding the impact of shifting diagnostic requirements even between iterations of the World Health Organization (WHO) classification. Our primary objective was to address these knowledge gaps. To do so, we analyzed clinicopathologic features, therapy, MRD, and survival in a centrally-reviewed, multi-center cohort of MPAL uniformly diagnosed by the WHO classification and treated with acute lymphoblastic leukemia (ALL) regimens. ALL induction therapy achieved an EOI MRD negative (<0.01%) remission in most patients (70%). EOI MRD positivity was predictive of 5-year EFS (HR=6.00, p<0.001) and OS (HR=9.57, p=0.003). Patients who cleared MRD by EOC had worse survival compared to those EOI MRD negative. In contrast to adults with MPAL, ALL therapy without transplantation was adequate to treat most pediatric patients. Earlier MRD clearance was associated with better treatment success and survival. Prospective trials are now necessary to validate and refine MRD thresholds within the pediatric MPAL population and to identify salvage strategies for those with poor predicted survival.
Introduction
Mixed phenotype acute leukemia (MPAL) is a rare form of leukemia concurrently expressing features of lymphoid and myeloid lineages.1, 2 While this historically resulted in varying approaches to treatment,1-4 emerging data now support beginning therapy with acute lymphoblastic leukemia (ALL) therapy and not more intensive acute myeloid leukemia (AML) therapy.5 However, major gaps in knowledge remain. While monitoring of minimal residual disease (MRD) is standard practice for both AML and ALL to risk-stratify therapy intensity and spare use of hematopoietic stem cell transplantation (HSCT), 6-11 the rarity of MPAL has precluded robust MRD data to support a similar approach. The multinational MPAL registry study AMBI2012 is the only cohort to date to report MRD-related outcomes but includes heterogeneous patient populations, treatment regimens, use of HSCT, and techniques for flow cytometry and MRD.12 No data for MRD from a uniform patient cohort is available to provide a clearer understanding of its association with leukemia biology, relapse, and survival. Interpretation of data is further complicated by differences in international classification systems for MPAL.13 However, even within the World Health Organization (WHO) classification, subtle differences in the criteria set forth in the most recent 2016 iteration (in print 2017) resulted in changes to the population identified as MPAL.14, 15 Defining distinct but overlapping MPAL populations has a profound impact on comparing survival rates among cohorts.13, 16-18 The impact of MRD on survival for patients diagnosed by the most recent WHO2016 definition is therefore similarly unknown. The objectives of this multi-institutional study was to address these knowledge gaps though establishing a large, centrally-reviewed, uniformly diagnosed and similarly treated cohort of children with MPAL from which to analyze biology, disease response by MRD, and survival.
Methods
Cohort generation
Six participating institutions from different regions in the United States identified cases diagnosed locally as MPAL between 2008 and 2016. Data for a distinct cohort of classic pediatric B-ALL cases diagnosed during the same study period was submitted from one participating site to serve as a reference cohort. The study was approved by the institutional review board at each institution. For all patients, data from medical records were obtained for demographics (age, sex, ethnicity), disease characteristics (highest presenting white blood cell [WBC] count, central nervous system involvement [CNS], results for fluorescence in situ hybridization [FISH]/cytogenetics), treatment regimen, and use of HSCT. CNS status (CNS 1, 2, or 3) and leukemia FISH/cytogenetics (adverse, favorable, neutral) were classified using the most recent COG biology stratification AALL08B1.19 Treatment outcomes included initial disease response (end of induction morphology, MRD), relapse, and death.
Central Review Process
The original diagnostic flow cytometry was submitted by sites and reanalyzed by two independent hematopathologists blinded to local diagnosis and clinical outcomes (M.J.O., G.W.). The cases were first evaluated as to whether they met strict criteria for the commonly used 2008 WHO classification of MPAL (WHO2008). Cases meeting the formal definition for WHO2008 MPAL were additionally classified by each hematopathologist for MPAL phenotype based on represented lineages (B+myeloid [B/My], T+myeloid [T/My], B+T [B/T], B+T+myeloid [B/T/My]) and blast population(s) (biphenotypic [Bi-P] = one blast population expressing multiple lineages) or bi-lineal [Bi-L] = two distinct blast populations). All WHO2008 cases were re-evaluated as to whether they met the new, additional criteria for WHO2016 MPAL.15 Following initial review, discordant cases were reviewed by both hematopathologists to achieve internal consensus. A third hematopathologist was available to review cases where consensus could not be reached but was not required. Diagnostic specimens and flow cytometry for the classic B-ALL cohort were centrally reviewed by one hematopathologist (M.J.O.). Due to cautionary verbiage in the WHO2016 classification regarding the adequacy of MPO flow cytometric expression alone to define a case as MPAL,15 the classic B-ALL reference cohort excluded cases of B-ALL with MPO expression to avoid potential overlap. MRD data was not reviewed centrally; MRD analyses were performed either in a Children’s Oncology Group reference laboratory (41/84, 49%) or institutional flow cytometry laboratory (43/84, 51%).
Statistical approach
All outcome analyses were restricted to patients beginning treatment with COG ALL therapy for induction and were completed on an “intent to treat” basis. MRD positivity was defined as MRD≥0.01% at all time-points. The primary endpoints for the study were the rate of MRD-positivity at end of induction (EOI) and end of consolidation (EOC) following ALL therapy and their contribution to overall survival (OS) and event free survival (EFS). A logistic regression model was constructed to evaluate demographics, disease, and treatment predictors of EOI MRD positivity (additionally defined as disease progression prior to EOI). Patients missing EOI MRD results (not performed, unavailable, or induction death) were excluded from the analysis of EOI MRD. Induction failure (IF) was defined as disease progression necessitating change in therapy prior to EOI or EOI MRD≥ 5%. Patients with missing FISH/cytogenetics (n=2) were omitted from the survival analyses. The survival times for analyses of OS and EFS were defined as the minimum time between the start of therapy and the qualifying event (all-cause mortality for OS and disease progression, relapse, or death for EFS). Kaplan-Meier survival curves with significance by the log-rank test (LRT) were stratified on treatment and disease characteristics. Survival estimates for each analysis were presented at five years ±standard errors (where data with longer follow-up were sparse, 3-year estimates were substituted and as indicated). Multivariable Cox regression models were constructed using a staged model selection for endpoint-specific models. In this approach, a base model of traditional predictors was established (age, WBC, FISH/cytogenetics). The contribution of other covariates indicated in Table 1 were then assessed in that model and retained at a threshold of p=0.15. In cases when covariates were sparsely distributed, the 95% confidence intervals were calculated using the profile log likelihood function. To examine the effect of EOI MRD and EOC MRD, EFS and OS were defined from time of EOI and EOC, respectively. Patients with events prior to EOI and EOC were excluded from the corresponding analyses. A comparison analysis was also performed incorporating data from the MPAL and B-ALL cohorts and with diagnosis/cohort retained as a covariate in each endpoint-specific multivariable model. The comparison analysis enabled greater statistical power to explore which covariates were potentially predictive of each endpoint after controlling for underlying diagnosis. Unless otherwise stated, all analyses were performed using two-sided tests, with significance level set at p<0.05, and completed using the statistical software Stata (StataCorp 2015, Stata Statistical Software, Release 14, College Station, Tx, USA).
Table 1.
Patient and disease characteristics of entire study cohort
| Characteristic | n | % | |
|---|---|---|---|
| Total cohort | 94 | 100 | |
| Age at diagnosis | <10 years | 43 | 46 |
| ≥10 years | 51 | 54 | |
| Sex | Male | 62 | 66 |
| Female | 32 | 34 | |
| Ethnicity | Hispanic | 52 | 55 |
| Not Hispanic | 40 | 43 | |
| Unknown | 2 | 2 | |
| Presenting WBC (/uL) | <50,000 | 66 | 70 |
| ≥50,000 | 28 | 30 | |
| CNS Status1 | CNS1 | 64 | 68 |
| CNS2 | 26 | 28 | |
| CNS3 | 4 | 4 | |
| Cytogenetic classification1 | Neutral | 67 | 71 |
| Favorable | 16 | 17 | |
| Adverse | 9 | 10 | |
| Unknown | 2 | 2 | |
| MPAL phenotype2 | B/My | 84 | 89 |
| T/My | 9 | 10 | |
| B/T | 1 | 1 | |
| B/T/My | 0 | 0 | |
| MPAL populations3 | Biphenotypic (1 pop.) | 82 | 87 |
| Bilineal (2 pop.) | 12 | 13 | |
| MPO positive4 | Yes | 88 | 94 |
| No | 5 | 5 | |
| Unknown | 1 | 1 | |
| CD19 positive4 | Yes | 85 | 90 |
| No | 9 | 10 | |
| Type of induction regimen | ALL | 87 | 93 |
| AML | 6 | 6 | |
| Hybrid | 1 | 1 | |
| HSCT in CR1 | Yes | 15 | 16 |
| No | 79 | 84 | |
As per AALL08B1.
Phenotype designates lineages involved, B/My = B+Myeloid, T/My = T+Myeloid, B/T= B+T, B/T/My = B+T+Myeloid.
Designates whether lineages are represented on one cell population (Biphenotypic) or two or more distinct populations (bilineal+).
Presence of myeloperoxidase (MPO) or CD19 on the MPAL blasts. MPAL = mixed phenotype acute leukemia. ALL= acute lymphoblastic leukemia, AML = acute myeloid leukemia, “hybrid” = elements of both ALL and AML therapy. CNS = central nervous system involvement, HSCT = hematopoietic stem cell transplantation, CR 1 = first remission.
Results
Patient and disease characteristics
Participating sites diagnosed 112 cases as pediatric MPAL between 2008 and 2016. Of these, 94/112 (83.9%) were confirmed on central review to fulfill strict WHO2008 criteria for MPAL. Presenting clinicopathologic features of MPAL are reported in Table 1. Among the MPAL subtypes, B/myeloid was the most common and comprised 89.4% of cases (84/94). Nine T/myeloid cases and one rare B/T MPAL phenotype were present in the cohort. Similarly, the majority were Bi-P, with only 12 cases (12.8%) presenting as Bi-L. The comparison cohort of routine B-ALL included 248 cases with an expected historical distribution of clinicopathologic features (Supplemental Table 1). Except for initial WBC, presenting features for the pediatric MPAL cohort differed significantly compared to the classic B-ALL cohort.
Heterogeneity in therapy selection for pediatric MPAL
Among the 94 centrally confirmed MPAL patients, the majority (87/94, 92.6%) received a COG ALL induction regimen, whereas only six received an AML induction regimen; one patient was treated with hybrid therapy. Of note, all ten T-lineage (T/My, B/T) MPAL patients received an ALL induction, as did all CD19-negative cases. Most patients began with a COG high risk (COG HR) induction regimen inclusive of daunorubicin, although nearly a third received standard risk (SR) induction therapy instead. Following induction, nine patients changed from ALL therapy based on the treating physician’s decision. This was not always response-driven as it included three patients who were changed to AML therapy despite being MRD negative at EOI. HSCT in first complete remission (CR1) was performed in only 12 of 87 (13.8%) ALL treated patients. A detailed summary of patient, disease, and treatment characteristics based on initial therapy is outlined in Supplemental Table 2.
Early disease response to ALL induction therapy
Of the 87 patients treated with a COG ALL induction, 93.1% (81/87) successfully achieved a morphologic CR1. MRD was unknown in three patients; One patient suffered a toxic induction death (1/87, 1.1%) and two achieved morphologic CR but did not have EOI MRD performed. Of the 84 patients with known EOI MRD, ALL induction therapy resulted in a MRD-negative CR in 70.2% of cases (59/84). IF was present in only 7.1% (6/84) and consisted of disease progression prior to EOI (n=2) and EOI MRD ≥5% (n=4). On multivariable analysis (Table 2), only the absence of MPO expression was associated with reduced risk for positive MRD, (Odds Ratio [OR] 0.11, 95% confidence interval [95%CI] 0.01–0.87, p=0.036). Neither older age (OR 1.84, 95%CI 0.63–5.33, p=0.26) or high initial WBC (OR 2.54, 95%CI 0.79–8.16, p=0.12) reached significance. T-lineage MPAL (T/My or B/T, n=10/84) was associated with positive EOI MRD on univariable analysis (OR= 4.34, 95%CI 1.10–17.06, p=0.035), but not in the final multivariable model (OR=2.67, 95%CI 0.52–13.54, p=0.237). Cytogenetic abnormalities were not associated with positive EOI MRD. The comparison analysis including data from both MPAL and B-ALL demonstrated similar finding as to those found in the MPAL-only model (Supplemental Table 3).
Table 2.
Multivariable analysis for EOI MRD positivity (≥0.01%) following an ALL induction
| Characteristic1 | n | (%) | Odds Ratio | 95% CI | p-value | |
|---|---|---|---|---|---|---|
| Age at diagnosis | <10 years | 40 | (49) | 1 | -- | 0.258 |
| ≥10 years | 42 | (51) | 1.84 | 0.63, 5.33 | ||
| Presenting WBC (/uL) | <50,000 | 59 | (72) | 1 | -- | 0.118 |
| ≥50,000 | 23 | (28) | 2.54 | 0.79, 8.16 | ||
| Cytogenetic classification2 | Neutral | 60 | (73) | 1 | -- | 0.64 |
| Favorable | 15 | (18) | 0.71 | 0.15, 3.32 | ||
| Adverse | 7 | (9) | 0.42 | 0.05, 3.32 | ||
| Presence of MPO | Negative | 5 | (6) | 1 | -- | 0.036 |
| Positive | 77 | (94) | 0.11 | 0.01, 0.87 | ||
All candidate predictors in Table 1 tested in stepwise multivariable model with endpoint of EOI MRD ≥0.01%, see methods. Age, WBC, cytogenetics included as a priori “base” model irrespective of significance.
As per AALL08B1 criteria; excludes two patients with unknown cytogenetics. WBC = white blood cell count, MPO = myeloperoxidase antigen expression.
Of the 25 patients who remained MRD-positive after an ALL induction, 21 continued with ALL therapy, 14 of whom repeated MRD testing at EOC (MRD was not performed in the remaining seven). For those who were EOI MRD-positive but not IF (i.e. MRD 0.01%−4.9%), 10/12 (83.3%) achieved EOC MRD negativity with ALL therapy. Among the six patients with IF, three of four who changed to AML therapy became EOC MRD-negative; both IF patients who continued with ALL therapy also became EOC MRD-negative. All six EOI MRD-positive T-lineage MPAL patients successfully cleared their MRD by EOC, five with continued ALL therapy alone, while the remaining patient was switched to myeloid therapy at EOI. Notably, the two patients with early disease progression prior to EOI were successfully salvaged with AML therapy without excessive toxicity (<21 days from initial diagnosis). However, of patients receiving AML therapy at any point, 3/12 (25.0%) expired in remission from treatment toxicity as compared to treatment mortality in only 3/81(3.7%) patients receiving continued ALL chemotherapy alone (p=0.005).
MRD-stratified survival for pediatric MPAL
Mean follow-up time for survivors in the WHO2008 MPAL cohort was ~4.1 years (range 0.1–8.8 years). For all WHO2008-confirmed MPAL patients, 5-year EFS and OS were 66.6±5.9% and 83.0±4.3%, respectively. For those beginning with ALL therapy, 5-year EFS and OS were 68.9±6.1% and 85.2±4.3% (Figure 1). In comparison, EFS and OS for the cohort of classic B-ALL were 83.5±2.7% and 93.7±1.8%. For patients surviving to EOI, MRD was predictive of EFS and OS. Patients who were EOI MRD-negative versus EOI MRD positive demonstrated higher 5-year EFS (81.8±6.6% versus 46.8%±12.4%, p<0.001, Figure 2A) and OS (92.7±4.3% versus 75.7±9.5%, p=0.001, Figure 2B). On multivariable analyses for survival (Tables 3 and 4), positive EOI MRD remained highly predictive of EFS (Hazard ratio [HR] 6.00, 95%CI 2.20–16.37, p<0.001) and OS (HR 9.57, 95%CI 1.94–47.20, p=0.003). Kinetics of clearance of blasts influenced survival in the cohort; MPAL patients with delayed clearance of blasts at EOC as compared to EOI experienced poorer EFS (57.1±14.6% versus 91.5±4.1%, p=0.002, Figure 3A) and OS (75.0±12.5% versus 96.2±4.3%, p=0.015, Figure 3B). On multivariable analysis (Tables 3 and 4), as compared to EOI MRD-negative patients, those with delayed clearance of MRD at EOC remained at greater risk for poorer EFS (HR 6.54, 95%CI 1.92, 22.25, global p=0.004) and for poorer OS (HR 9.32, 95%CI 1.45, 59.85, global p=0.028). However, survival analysis for the EOI MRD-positive/EOC MRD-negative group was limited by the unavailability of EOC MRD status in seven EOI MRD-positive patients. Given that the EOI MRD-positive/EOC MRD-unknown group had higher survival (3-year EFS & OS both 83.3±15.2%) than those EOC MRD-negative, survival for EOC MRD-negative patients may be underestimated. The addition of daunorubicin to a COG ALL induction (i.e. COG ALL HR versus SR regimen) was not significantly associated with EFS or OS in our MPAL cohort. The comparison analysis inclusive of MPAL and B-ALL also found an association of MRD with EFS and OS after accounting for disease/cohort (Supplemental Tables 4 and 5).
Figure 1: Survival from WHO2008-defined MPAL for those beginning with ALL therapy.
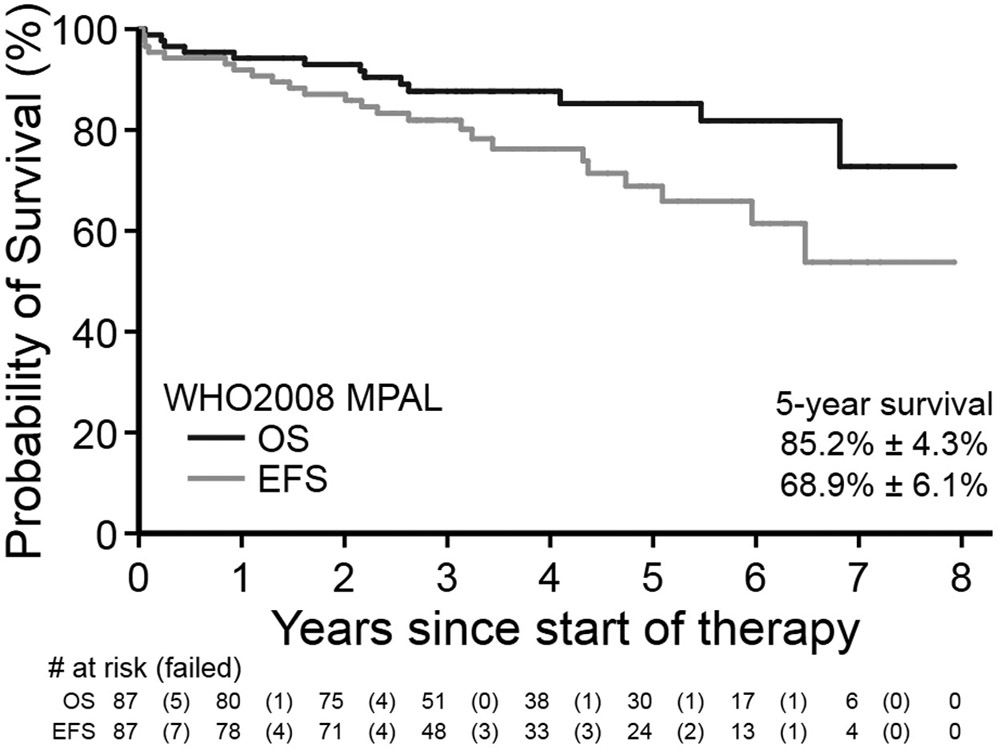
Event-free (A) and overall (B) survival for patients diagnosed with WHO2008 MPAL and beginning therapy with a Children’s Oncology Group ALL induction.
Figure 2: MRD-stratified survival for MPAL following an ALL induction.
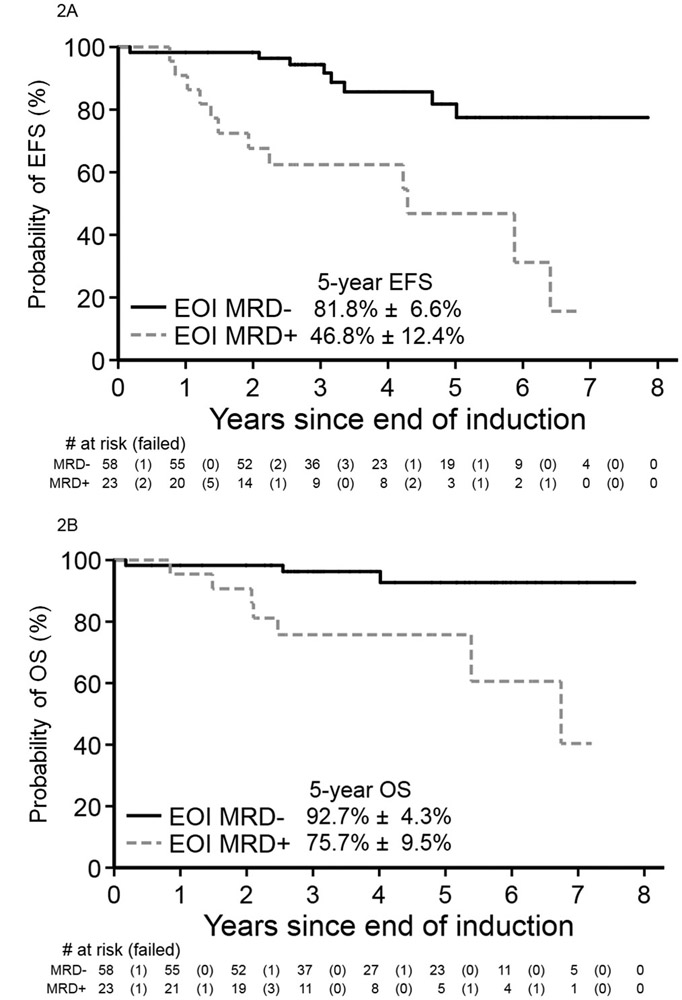
Children with WHO2008 MPAL beginning with a Children’s Oncology Group ALL induction and achieving MRD negativity (<0.01%) experienced significantly higher event-free (A) and overall (B) survival from end of induction than those MRD positive.
Table 3.
Multivariable analysis for event-free survival from EOI and EOC
| Characteristic1 | n | (%) | Event-free Survival from EOI | Event-free Survival from EOC | |||||
|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | p-value | HR | 95% CI | p-value | ||||
| Age at diagnosis | <10 years | 39 | (49) | 1 | -- | 0.278 | 1 | -- | 0.346 |
| ≥10 years | 41 | (51) | 1.69 | 0.65, 4.38 | 1.59 | 0.60, 4.22 | |||
| Initial WBC (/uL) | <50,000 | 57 | (71) | 1 | -- | 0.419 | 1 | -- | 0.384 |
| ≥50,000 | 23 | (29) | 0.65 | 0.23, 1.87 | 0.61 | 0.19, 1.91 | |||
| Cytogenetic classification2 | Neutral | 59 | (74) | 1 | -- | 0.092 | 1 | -- | 0.099 |
| Favorable | 15 | (19) | 0.18 | 0.02, 1.47 | 0.19 | 0.02, 1.48 | |||
| Adverse | 6 | (7) | 2.18 | 0.42, 11.22 | 2.12 | 0.41, 10.90 | |||
| EOI MRD3 | Negative | 57 | (71) | 1 | -- | <0.001 | -- | -- | -- |
| Positive | 23 | (29) | 6.00 | 2.20, 16.37 | -- | -- | -- | ||
| EOI/EOC MRD3 | Neg/Neg | 57 | (71) | -- | -- | -- | 1 | -- | 0.004 |
| Pos/Neg | 13 | (16) | -- | -- | -- | 6.54 | 1.92, 22.25 | ||
| Pos/Pos | 3 | (4) | -- | -- | -- | 9.01 | 1.51, 53.86 | ||
| Pos/Unk. | 7 | (9) | -- | -- | -- | 5.06 | 1.45, 17.70 | ||
All candidate predictors in Table 1 tested in stepwise multivariable model with endpoint of OS, see methods. Age, WBC, cytogenetics included as a priori “base” model irrespective of significance.
As per AALL08B1 criteria; excludes two patients with unknown cytogenetics.
MRD considered positive ≥0.01% at either time-point. HR = hazard ratio, EOI = end of induction, EOC = end of consolidation WBC = white blood cell count
Table 4.
Multivariable analysis for overall survival from EOI and EOC
| Characteristic1 | n | (%) | Overall Survival from EOI | Overall Survival from EOC | |||||
|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | p-value | HR | 95% CI | p-value | ||||
| Age at diagnosis | <10 years | 39 | (49) | 1 | -- | 0.917 | 1 | -- | 0.893 |
| ≥10 years | 41 | (51) | 1.1 | 0.19, 6.25 | 1.13 | 0.19, 6.55 | |||
| Initial WBC (/uL) | <50,000 | 57 | (71) | 1 | -- | 0.372 | 1 | -- | 0.396 |
| ≥50,000 | 23 | (29) | 0.44 | 0.07, 2.69 | 0.45 | 0.07, 2.91 | |||
| Cytogenetic classification2 | Neutral | 59 | (74) | 1 | -- | 0.163 | 1 | -- | 0.165 |
| Favorable | 15 | (19) | 0.54 | 0.05, 5.37 | 0.53 | 0.05, 5.38 | |||
| Adverse | 6 | (7) | 7.61 | 1.02, 56.64 | 7.68 | 1.01, 58.16 | |||
| Induction therapy3 | COG SR | 26 | (32) | 1 | -- | 0.561 | 1 | -- | 0.567 |
| COG HR | 54 | (68) | 2.05 | 0.18, 22.75 | 2.05 | 0.18, 23.47 | |||
| HSCT in CR14 | No | 69 | (89) | 1 | -- | 0.674 | 1 | -- | 0.712 |
| Yes | 11 | (14) | 1.44 | 0.26, 7.87 | 1.4 | 0.24, 8.15 | |||
| EOI MRD5 | Negative | 57 | (71) | 1 | -- | 0.003 | -- | -- | -- |
| Positive | 23 | (29) | 9.57 | 1.94, 47.20 | -- | -- | |||
| EOI/EOC MRD5 | neg/neg | 57 | (71) | -- | -- | -- | 1 | -- | 0.028 |
| pos/neg | 13 | (16) | -- | -- | -- | 9.32 | 1.45, 59.85 | ||
| pos/pos | 3 | (4) | -- | -- | -- | 10.03 | 0.64, 157.42 | ||
| pos/unk. | 7 | (9) | -- | -- | -- | 9.94 | 1.51, 65.47 | ||
All candidate predictors in Table 1 tested in stepwise multivariable model with endpoint of OS, see methods. Age, WBC, cytogenetics included as a priori “base” model irrespective of significance.
As per AALL08B1 criteria; excludes two patients with unknown cytogenetics.
COG SR/HR = Children’s Oncology Group three drug (steroid, vincristine, asparaginase) or four drug induction (+daunorubicin) for NCI/Rome Standard Risk or High Risk ALL
Hematopoetic stem cell transplantation (HSCT) in first remission (CR1).
MRD considered positive ≥0.01% at either time-point. EOI = end of induction, EOC = end of consolidation WBC = white blood cell count.
Figure 3: MRD kinetics during ALL therapy on survival from MPAL.

For children with WHO2008 MPAL receiving a Children’s Oncology Group ALL induction and consolidation, those achieving earlier MRD negativity (<0.01%) at end of induction experienced higher event-free (A) and overall (B) survival from end of consolidation than those patients becoming MRD negative only after consolidation. Patients with unknown end consolidation MRD are not shown.
Kaplan-Meier survival curves of EFS and OS for patients beginning with ALL therapy and stratified based on HSCT vs no HSCT, MPAL phenotype, and MPAL lineage are included in the supplementary material (Supplementary Figures 1-3). Although 32.0% (8/25) EOI MRD-positive patients underwent HSCT, 6.8% (4/59) of EOI MRD-negative patients still proceeded to HSCT. For those becoming EOC MRD negative with ALL therapy alone, most patients completed chemotherapy without HSCT (10/14, 71.4%). Survival remained excellent for EOI MRD-negative MPAL patients treated with ALL chemotherapy alone without HSCT with 5-year EFS and OS from EOI of 80.1±6.8% and 92.1±4.6%.
Influence of diagnostic criteria for MPAL on survival
Insufficient flow cytometry was performed routinely by sites to fully classify all cases diagnosed prior to publication of the WHO2016 update. Upon central review, 21/94 (22.3%) cases were confirmed to fully meet the additional WHO2016 criteria for heterogeneity of concurrent antigen expression. Of note, this group excluded patients defined by MPO antigen expression alone. For those fulfilling WHO2016 criteria, 15/21 (71%) patients began therapy with an ALL induction regimen. While limited numbers precluded analysis of therapy type for WHO2016 MPAL, this sub-cohort meeting the WHO2016 classification demonstrated a 3-year EFS and OS of 64.8±10.8% and 69.9±10.3% (Supplemental Figure 4). Evaluation of MRD status demonstrated 3-year EFS and OS for EOI MRD-negative patients was 78.8±13.4% and 90.0±9.5%, as compared to those who were MRD-positive where EFS and OS was 63.5±16.9% and 63.5±16.9%, respectively (Figure 4A and 4B).
Figure 4: Association of MRD with event free and overall survival for MPAL defined by WHO2016 update.

Event-free (A) and overall survival (B) for the subset of MPAL patients fulfilling the WHO2016 update with known end of induction MRD (n=19).
Discussion
Over the past two decades, MRD testing has become an integral part of risk-stratification in both ALL and AML therapy.6-10 However, data for MRD’s predictive value in pediatric MPAL is limited to one publication, the recently published iBFM-AMBI2012 study.12 Predictive thresholds for MRD are disease- and regimen-specific; applying MRD results from iBFM-AMBI2012 to clinical practice is complicated by its inclusion of a myriad of international patients and treatment regimens. In our multi-center, centrally reviewed cohort, patients were uniformly defined using the WHO MPAL classification and treated exclusively with COG ALL induction and consolidation therapy prior to MRD assessment. With this approach, we found MRD to be highly predictive of relapse and death. To our knowledge, this is also the first description for the potential impact of MRD kinetics on MPAL survival. Our data confirmed recent reports suggesting MPAL patients respond well to ALL-directed therapy and demonstrated a subset of early-responding patients have excellent survival from ALL therapy alone without HSCT consolidation.
In our contemporary cohort, only a minority of MPAL patients began therapy with an AML induction. This represents a marked shift from a historically more even selection of AML or ALL regimens as initial therapy for MPAL.5, 20, 21 Most patients in our study responded well to ALL therapy and achieved a MRD-negative complete remission. Analysis of clinical features at diagnosis of MPAL did not reveal a predictor of EOI MRD positivity, although trends were present for patients at higher age, with elevated WBC, and/or T-lineage MPAL. Interestingly, expression of MPO was significantly associated with reduced risk for EOI MRD. Although the number of MPO-negative cases in our cohort were very few, the recent iBFM-AMBI2012 also showed a similar trend with MPO positivity predictive of better EFS in patients treated with ALL therapy (HR 0.27, 95% CI 0.09–0.83, p=0.022).12 Absence of MPO likely represents monocytic differentiation of MPAL blasts and, in the context of a common stem cell precursor in MPAL, may indicate differences in MPAL biology reflected by this phenotype. Patients with T-lineage MPAL appear to be more likely to clear MRD by the end of consolidation, similar to the typical pattern seen in T-ALL.22 A recent study by Alexander et. al. characterizing the genetic basis of pediatric MPAL showed that the two principal subtypes of MPAL, T/myeloid and B/myeloid, overlap but are genetically distinct.23 Similar findings have been replicated in adult MPAL.24, 25 Understanding the relationship of the biology, phenotypes, clinical presentation, and chemotherapy with MPAL MRD and survival will be crucial to refine future approaches to risk-stratification. Further study is necessary to define clinical and biologic risk factors to provide context for MRD predictive thresholds.
Questions also remain how best to integrate MRD into therapy. In our multicenter cohort, treatment for several patients was decided by the treating physician irrespective of MRD results; some EOI MRD negative patients were switched from ALL to AML therapy while a few EOI MRD positive patients continued with ALL therapy. While we found a survival advantage for early MRD negativity at EOI, OS for those with delayed MRD clearance by EOC remained >70%. Most EOI MRD positive patients successfully cleared their disease with continued ALL therapy. This raises the question as to whether a switch to AML or other intensive therapy should be considered earlier in therapy than EOC. Similarly, the role for HSCT in CR1 from a first therapy attempt remains uncertain. Patients undergoing HSCT in our cohort experienced a trend for worse survival, but this population was likely highly selected from those with poorer disease response and/or features considered “higher risk” by the provider. For instance, irrespective of therapy type, of the 15 cases who were MRD negative by EOC, five nonetheless underwent HSCT, thus suggesting the treating providers considered these cases as higher risk for relapse despite clearance of MRD. While data supports the recommendation for HSCT in adults with MPAL,21, 26-28 we found no clear benefit from HSCT consolidation in CR1 in our cohort even for these potentially “higher risk” patients. The recent iBFM-AMBI2012 study had a similar conclusion regarding the absence of a clear benefit for HSCT in CR1 for children with MPAL.12 Prospective study is necessary to identify populations likely to be failed by frontline ALL therapy and to conclusively determine potential benefit from HSCT in poorly responding patients.
Further complicating our understanding of MRD risk-stratification in MPAL is the heterogeneous populations defined by different classification systems (i.e. EGIL, WHO2008, and WHO2016). Most recently, because limited data exists on the biology and outcomes of MPAL defined by MPO alone, the WHO2016 update excludes cases with subjective “weak” MPO expression.14, 15 Our previous studies have examined the clinical implications of defining MPAL by MPO expression.17, 18 It is therefore notable that the presenting hematopathology of the MPAL cohort, as defined by WHO2008 criteria for MPO, differed significantly from the reference B-ALL cohort. We would also note that use of the WHO2016 criteria for MPAL resulted in markedly fewer cases being confirmed as MPAL from use of historical, routinely tested antibody panels. This was primarily due to the new criterion for concurrent antigen heterogeneity in leukemia blasts. Complete and harmonized testing panels might have resulted in more cases meeting the WHO2016 MPAL criteria. The more comprehensive testing necessary to establish this defining characteristic marks a change from the initial intention of the WHO2008 classification to utilize smaller, streamlined, highly lineage -specific antibody panels. The sub-population classified by the WHO2016 criteria appeared to have poorer survival than the parent WHO2008 cohort. It is unclear if differences in survival were inherent to a higher-risk population defined by WHO2016’s reliance on additional myeloid antigen expression or from more patients receiving potentially sub-optimal non-ALL therapy. Despite small numbers, a trend remained present for the predictive value of MRD in stratifying WHO2016-defined MPAL.
This study clearly identified MRD positivity as a poor prognostic factor specific to children with MPAL treated with ALL regimens. However, one limitation of this study is the lack of central review of MRD; MPAL MRD is challenging to interpret and prospective validation with central review is an essential next step to understanding its predictive value. We must also acknowledge the relatively small numbers of T/My MPAL cases included in the cohort. Given the distinct clinical and biologic differences of T/My and B/My MPAL, caution should be used before extrapolating results of this study to T/My MPAL. For B/My MPAL, since most of the EOI MRD-positive patients continued on ALL therapy and attained MRD negative remission by end of consolidation, it remains unclear when intensified therapy may be beneficial to prevent relapse. Similarly, the role for HSCT in pediatric MPAL remains unknown. It therefore remains challenging to balance intensification of therapy to potentially prevent relapse with inherent risk for acute and long-term treatment toxicity, and even toxic mortality, as seen in our cohort. However, MPAL patients who remain MRD-positive at the end of consolidation have a poorer prognosis and may warrant intensification of therapy, HSCT, or alternative cellular, immune, or molecular-therapy approaches. Planned prospective validation of MRD disease burden and incidence of relapse will further guide these challenging treatment decisions.
Supplementary Material
Acknowledgements
Data was collated using the REDCAP database supported by NIH/NCATS UL1TR001855 and UL1TR000130. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Potential Competing Interests
M.J.O. is currently employed at Caris Life Sciences. There are no other conflicts of interest to report.
References
- 1.Weinberg OK, Arber DA. Mixed-phenotype acute leukemia: historical overview and a new definition. Leukemia 2010. November; 24(11): 1844–1851. [DOI] [PubMed] [Google Scholar]
- 2.Bene MC, Porwit A. Acute leukemias of ambiguous lineage. Semin Diagn Pathol 2012. February; 29(1): 12–18. [DOI] [PubMed] [Google Scholar]
- 3.Steensma DP. Oddballs: acute leukemias of mixed phenotype and ambiguous origin. Hematol Oncol Clin North Am 2011. December; 25(6): 1235–1253. [DOI] [PubMed] [Google Scholar]
- 4.Wolach O, Stone RM. Mixed-phenotype acute leukemia: current challenges in diagnosis and therapy. Curr Opin Hematol 2017. March; 24(2): 139–145. [DOI] [PubMed] [Google Scholar]
- 5.Maruffi M, Sposto R, Oberley MJ, Kysh L, Orgel E. Therapy for children and adults with mixed phenotype acute leukemia: a systematic review and meta-analysis. Leukemia 2018. July; 32(7): 1515–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Dongen JJ, Seriu T, Panzer-Grumayer ER, Biondi A, Pongers-Willemse MJ, Corral L, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 1998. November 28; 352(9142): 1731–1738. [DOI] [PubMed] [Google Scholar]
- 7.Ciudad J, San Miguel JF, Lopez-Berges MC, Vidriales B, Valverde B, Ocqueteau M, et al. Prognostic value of immunophenotypic detection of minimal residual disease in acute lymphoblastic leukemia. J Clin Oncol 1998. December; 16(12): 3774–3781. [DOI] [PubMed] [Google Scholar]
- 8.Coustan-Smith E, Sancho J, Hancock ML, Boyett JM, Behm FG, Raimondi SC, et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood 2000. October 15; 96(8): 2691–2696. [PubMed] [Google Scholar]
- 9.San Miguel JF, Vidriales MB, Lopez-Berges C, Diaz-Mediavilla J, Gutierrez N, Canizo C, et al. Early immunophenotypical evaluation of minimal residual disease in acute myeloid leukemia identifies different patient risk groups and may contribute to postinduction treatment stratification. Blood 2001. September 15; 98(6): 1746–1751. [DOI] [PubMed] [Google Scholar]
- 10.Rubnitz JE, Inaba H, Dahl G, Ribeiro RC, Bowman WP, Taub J, et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol 2010. June; 11(6): 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pulsipher MA, Peters C, Pui CH. High-risk pediatric acute lymphoblastic leukemia: to transplant or not to transplant? Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2011. January; 17(1 Suppl): S137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hrusak O, de Haas V, Stancikova J, Vakrmanova B, Janotova I, Mejstrikova E, et al. International cooperative study identifies treatment strategy in childhood ambiguous lineage leukemia. Blood 2018. July 19; 132(3): 264–276. [DOI] [PubMed] [Google Scholar]
- 13.Weinberg OK, Seetharam M, Ren L, Alizadeh A, Arber DA. Mixed phenotype acute leukemia: A study of 61 cases using World Health Organization and European Group for the Immunological Classification of Leukaemias criteria. American journal of clinical pathology 2014. December; 142(6): 803–808. [DOI] [PubMed] [Google Scholar]
- 14.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016. May 19; 127(20): 2391–2405. [DOI] [PubMed] [Google Scholar]
- 15.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised Fourth Edition, vol. 2 IARC/WHO Press: Lyon, France, 2017, 585pp. [Google Scholar]
- 16.Pomerantz A, Rodriguez-Rodriguez S, Demichelis-Gomez R, Barrera-Lumbreras G, Barrales-Benitez O, Lopez-Karpovitch X, et al. Mixed-phenotype acute leukemia: suboptimal treatment when the 2008/2016 WHO classification is used. Blood Res 2016. December; 51(4): 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oberley MJ, Li S, Orgel E, Phei Wee C, Hagiya A, O’Gorman MRG. Clinical Significance of Isolated Myeloperoxidase Expression in Pediatric B-Lymphoblastic Leukemia. American journal of clinical pathology 2017. April 1; 147(4): 374–381. [DOI] [PubMed] [Google Scholar]
- 18.Raikar SS, Park SI, Leong T, Jaye DL, Keller FG, Horan JT, et al. Isolated myeloperoxidase expression in pediatric B/myeloid mixed phenotype acute leukemia is linked with better survival. Blood 2018. February 1; 131(5): 573–577. [DOI] [PubMed] [Google Scholar]
- 19.Hunger SP, Loh ML, Whitlock JA, Winick NJ, Carroll WL, Devidas M, et al. Children’s Oncology Group’s 2013 blueprint for research: acute lymphoblastic leukemia. Pediatr Blood Cancer 2013. June; 60(6): 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matutes E, Pickl WF, Van’t Veer M, Morilla R, Swansbury J, Strobl H, et al. Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood 2011. March 17; 117(11): 3163–3171. [DOI] [PubMed] [Google Scholar]
- 21.Gerr H, Zimmermann M, Schrappe M, Dworzak M, Ludwig WD, Bradtke J, et al. Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol 2010. April; 149(1): 84–92. [DOI] [PubMed] [Google Scholar]
- 22.Schrappe M, Valsecchi MG, Bartram CR, Schrauder A, Panzer-Grumayer R, Moricke A, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood 2011. August 25; 118(8): 2077–2084. [DOI] [PubMed] [Google Scholar]
- 23.Alexander TB, Gu Z, Iacobucci I, Dickerson K, Choi JK, Xu B, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018. 2018/October/01; 562(7727): 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morita K, Wang F, Patel K, Bueso-Ramos CE, Zahr AA, Gumbs C, et al. Genomic landscape of adult mixed phenotype acute leukemia (MPAL). Journal of Clinical Oncology 2017; 35(15_suppl): 7023–7023. [Google Scholar]
- 25.Takahashi K, Wang F, Morita K, Yan Y, Hu P, Zhao P, et al. Integrative genomic analysis of adult mixed phenotype acute leukemia delineates lineage associated molecular subtypes. Nature Communications 2018. 2018/July/10; 9(1): 2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolach O, Stone RM. How I treat mixed-phenotype acute leukemia. Blood 2015. April 16; 125(16): 2477–2485. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu H, Saitoh T, Machida S, Kako S, Doki N, Mori T, et al. Allogeneic hematopoietic stem cell transplantation for adult patients with mixed phenotype acute leukemia: results of a matched-pair analysis. Eur J Haematol 2015. November; 95(5): 455–460. [DOI] [PubMed] [Google Scholar]
- 28.Munker R, Labopin M, Esteve J, Schmid C, Mohty M, Nagler A. Mixed phenotype acute leukemia: outcomes with allogeneic stem cell transplantation. A retrospective study from the Acute Leukemia Working Party of the EBMT. Haematologica 2017. December; 102(12): 2134–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.