

Abstract
TET family enzymes are known for oxidation of the 5-methyl substituent on 5-methylcytosine (5mC) in DNA. 5mC oxidation generates the stable base 5-hydroxymethylcytosine (5hmC), starting an indirect, multi-step process that ends with reversion of 5mC to unmodified cytosine. While probing the nucleobase determinants of 5mC recognition, we discovered that TET enzymes are also proficient as direct N-demethylases of cytosine bases. We find that N-demethylase activity can be readily observed on substrates lacking a 5-methyl group and, remarkably, TET enzymes can be similarly proficient in either oxidation of 5mC or demethylation of N4-methyl substituents. Our results indicate that TET enzymes can act as both direct and indirect demethylases, highlight the active-site plasticity of these Fe(II)/α-ketoglutarate-dependent dioxygenases, and suggest activity on unexplored substrates that could reveal new TET biology.
Keywords: DNA methylation, Enzymes, Epigenetics, TET enzymes
Graphical Abstract
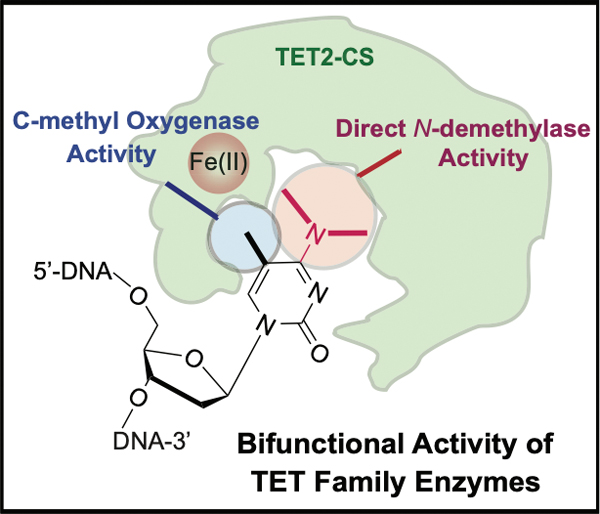
Employing a nucleobase modifier strategy, we discovered that TET family enzymes are robust and direct N-demethylases, and that this activity is comparable to its normal activity in oxidation of 5-methylcytosine. This remarkable N-demethylase activity highlights the plasticity of the TET active site and suggests broader biological roles for TET enzymes beyond impacting DNA methylation dynamics.
5-methylcytosine (5mC) is an epigenetic DNA modification that plays crucial roles in mammalian gene silencing, imprinting, aging, and carcinogenesis.[1] The conversion of 5mC to unmodified cytosine is primarily initiated by Ten-eleven translocation (TET) family dioxygenases, whereby oxidation of 5mC marks a base for demethylation, but does not directly result in loss of the methyl group.[2, 3] This pathway involves TET-mediated oxidation of 5mC to generate 5-hydroxymethylcytosine (5hmC), the predominant product, along with 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC).[4–6] Each of these oxidized 5mC bases (ox-mCs) can promote the passive, replication-dependent 5mC loss by antagonizing the maintenance DNA methyltransferase DNMT1.[7] 5fC and 5caC can also be excised as part of a base excision repair pathway to complete an active, but indirect, demethylation process.[5, 8]
This indirect demethylation of 5mC by TET enzymes stands as a point of contrast to their closely related family members in the larger Fe(II)/ α-ketoglutarate(αKG)-dependent dioxygenase family. In the epigenetic realm, this family more broadly includes histone demethylases of the Jumonji family and AlkB enzymes that act on alkylated nucleobases in DNA/RNA.[9] All family members share the initial catalytic half-reaction, whereby Fe(II) is oxidized to a reactive Fe(IV)-oxo species with concomitant conversion of αKG to succinate (Figure S1). For most of these family members, oxidation occurs on a methyl or alkyl substituent linked to a nitrogen or oxygen heteroatom. Direct dealkylation can then proceed via loss of formaldehyde or other aldehyde products with the heteroatom acting as an electron sink.[10, 11] TET enzymes differ in that the canonical target of oxidation, the 5-methyl group of 5mC, is not linked to a heteroatom, resulting in the stable generation of ox-mCs as part of the multi-step, indirect demethylation pathway.
Recent work has demonstrated that TET enzymes can tolerate substrates beyond 5mC in DNA, including RNA or alternative 5-modified substrates, such as 5-ethylcytosine.[12–16] Observing this promiscuity prompted us to further explore the features that determine if a nucleobase is recognized and oxidized. Prior work using structure-guided biochemistry and MD simulation both have suggested a role for Asn1387 of human TET2 in recognition of the N4 amino group of 5mC (Figure 1A).[17–19] Specifically, alteration of Asn1387 via an N1387A mutation led to loss of activity on 5mC in DNA.[17] Given that substrate recognition could therefore hinge on discrimination of the Watson-Crick face of cytosine, we adopted an orthogonal nucleobase modification approach to probe this possibility.
Figure 1.
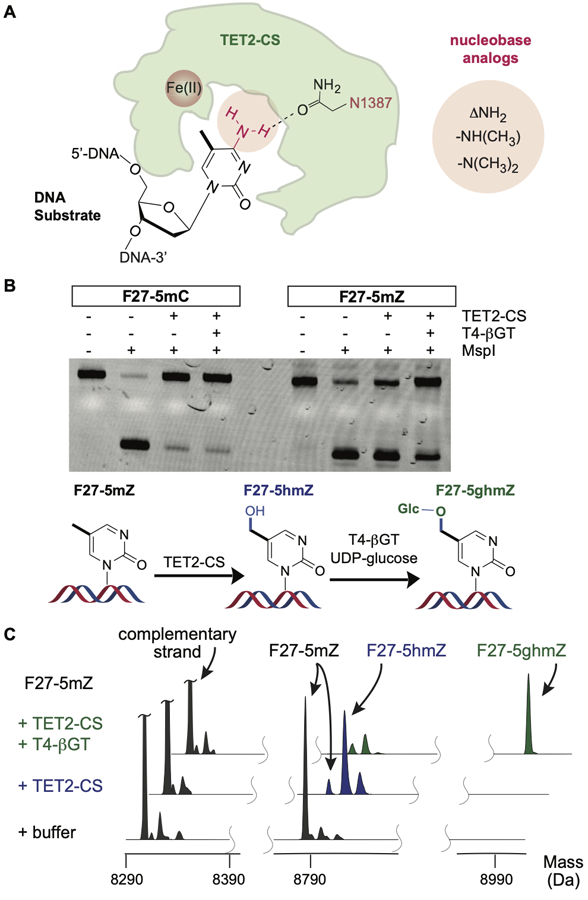
The role of N4 of 5mC in TET-mediated oxidation. (A) The interaction between 5mC and Asn1387 of TET2 is postulated to be critical to reactivity. The N4 interaction can be probed by N4-removal (ΔNH2), as in 5-methylzebularine (5mZ), or via methyl substitution. (B) A 27-mer FAM-labeled substrate with 5mC or 5mZ (200 nM) was reacted with or without TET2 -CS (1.6 µM) in the presence or absence of T4-βGT and the products analyzed on a denaturing gel after cleavage with MspI. While the substrates can be cleaved, modified products are partially or fully protected from MspI cleavage. (C) Spectral traces from ESI-MS analysis of reaction products were normalized to the complementary strand. The product containing 5-glucosyl-hmZ is denoted by F27–5ghmZ.
For our nucleobase modification approach, we aimed to make targeted alterations to the 5mC substrate (Figure 1A). We first used solid phase synthesis to generate matched fluorescein-labeled 27mer DNA substrates containing a central CXGG sequence, where X was either 5mC (F27–5mC) or 5-methylzebularine (F27–5mZ), a nucleobase lacking the exocyclic NH2, which would remove the Asn1387 H-bond partner. To simultaneously probe the consequences of alternative disruptions, we also pursued N4 modification. To this end, we synthesized a convertible dT phosphoramidite,[20] and introduced this base in a matched substrate sequence by solid phase synthesis. Upon reaction with appropriate amines, the base was converted, yielding DNA containing either 4-N-methyl-5-methylcytosine or 4,4-N,N-dimethyl-5-methylcytosine (F27–4m5mC and F27–4dm5mC, respectively) (Figure S2).
Next, to explore the reactivity of these nucleobase analogues in DNA, the four DNA oligonucleotides were hybridized to a complementary strand containing unmodified cytosine bases, incubated with purified human TET2 (TET2-CS) and subjected to further analysis. We first focused on a comparison between F27–5mC and F27–5mZ. For 5mC, TET-mediated oxidation can be tracked using coupling enzymes. While MspI can cleave a CXGG sequence when X is 5mC, this cleavage is blocked either by oxidation of X to 5caC or oxidation to 5hmC followed by its glucosylation with T4 phage-derived β-glucosyltransferase (βGT) (Figure 1B).[21] To probe 5mZ reactivity, we first validated that unreacted F27–5mZ, like F27–5mC, can also be cleaved by MspI. Upon reaction with TET2, however, the DNA becomes partially protected from cleavage by MspI, and protection is further enhanced by including βGT (Figure 1B). To validate the generation of 5-hydroxymethylzebularine (5hmZ), and its glucosylation by βGT, we analyzed the reaction products using electrospray ionization mass spectrometry (ESI-MS) on the intact DNA and found efficient depletion of starting material, conversion to F27–5hmZ, and its subsequent glucosylation (Figure 1C).
Unlike with the N1387A TET2 mutant, where 5mC oxidation was almost completely deficient,[17] we readily observed 5mZ oxidation by TET2-CS. This result suggests that there is no obligate requirement for an H-bond between Asn1387 of TET2 and the 5mC exocyclic amine. The absence of this interaction, however, slows down oxidation, and while we detected 5hmZ, higher-order oxidation was not readily observed. No products consistent with 5-carboxyl-dZ were observed by ESI-MS, and while 5-formyl-dZ would be difficult to differentiate by ESI-MS alone, no new product was detected upon incubation with an aldehyde reactive probe (Figure S3). Notably, in step-wise oxidation, 5mC is a favored substrate compared to 5hmC and 5fC.[19] The predominant formation of 5hmZ from 5mZ implies that the 5-hydroxymethyl group might be similarly less reactive in absence of a stabilizing H-bond from N1387 to N4, leading to an exaggerated deficit in iterative oxidation. We posit that the N1387A mutant may have either disrupted the nearby His1904-cytosine N3 interaction or be structurally destabilizing.
Having shown a tolerance to loss of the H-bonding partner, we next turned our attention to the F27–4m5mC and F27–4dm5mC substrates, which could also alter or prevent H-bond formation. As these substrates were not readily amenable to the conventional MspI-based assay, we first used ESI-MS to explore product formation. Upon reaction with TET2 we noted multiple new products with both substrates, including species reduced in mass relative to the starting material (Figure S4). This surprising result lead us to degrade the DNA to individual nucleosides and characterize by LC-MS/MS. For F27–4m5mC, we noted 4m5mC consumption, with detectable generation of species consistent with 4-N-methyl versions of 5hmC, 5fC, and 5caC. To our surprise, we also detected 5caC itself, a product consistent with N-demethylation, despite complete absence of 5mC in the starting substrate (Figure S5). F27–4dm5mC was similarly consumed, and yielded a product mixture nearly identical to that associated with F27–4m5mC (Figure S5). The results were consistent with initial mono-N-demethylation, followed by a mixture of oxidations of the 5-methyl substituent and N-demethylation.
The observation of concurrent 5-methyl oxidation and N-demethylation led us to further consider the nucleobase determinants of TET-mediated oxidation. To more directly probe N-demethylation, we designed two additional matched substrates lacking the 5-methyl group. Using solid phase synthesis and a convertible nucleoside strategy, we generated substrates containing 4-N-methyl-dC (F27–4mC) and a 4,4-N,N-dimethyl-dC (F27–4dmC) (Figure S6A). The associated duplexes were reacted with TET2-CS and analyzed by ESI-MS of the intact DNA (Figure 2A/2B). Under conditions identical to those used for F27–5mC oxidation, N-demethylase activity could be readily observed. F27–4mC was ~30% demethylated to generate F27-C, while F27–4dmC was almost completely mono-demethylated and ~30% di-demethylated. Thus, TET2 has direct N-demethylase activity and this activity is independent of the presence of 5-methyl group.
Figure 2.
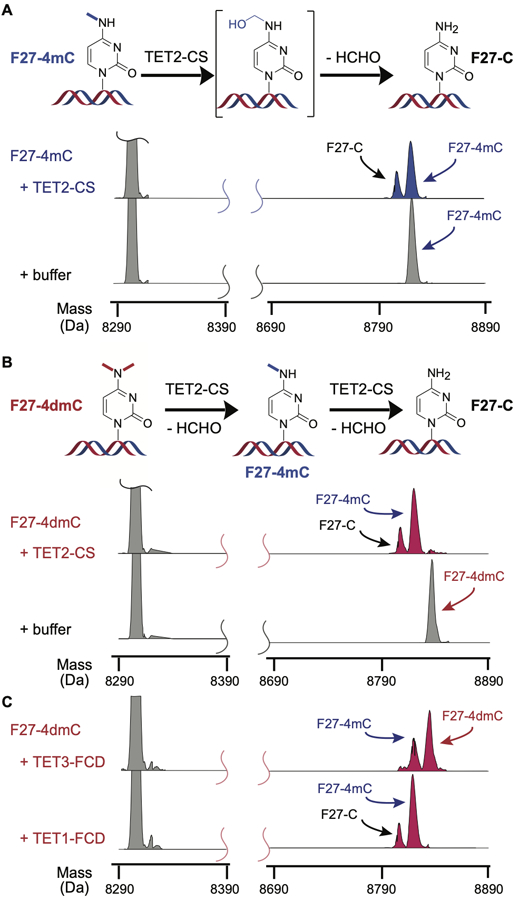
TET family enzymes are robust N-demethylases. (A) The F27–4mC substrate (200 nM) was reacted with or without TET2-CS (1.6 µM) and products were analyzed by ESI-MS with masses corresponding to substrates and potential products noted. A plausible mechanism for N-demethylation involving oxidation followed by deformylation is also shown. (B) The F27–4dmC substrate was reacted with TET2-CS or buffer and analyzed by ESI-MS. A plausible mechanism for di-demethylation involving iterative oxidation and deformylation is shown. (C) The F27–4dmC was reacted with TET1-FCD or TET3-FCD under conditions identical to the TET2-CS reaction and buffer only controls above and analyzed by ESI-MS.
To examine if the novel N-demethylation activity is specific to TET2 or a more general feature of TET enzymes, we next reacted F27–4dmC with the purified catalytic domains from either human TET1 or human TET3, and analyzed reaction products by ESI-MS. When reacted under similar conditions, TET1 showed complete mono-demethylation and partial di-demethylation to a comparable extent relative to TET2-CS (Figure 2C). TET3 also showed N-demethylation activity, with partial mono-demethylation that is consistent with prior observations that TET3 is globally reduced in activity relative to TET1 and TET2.[13] Most notably, these observations demonstrate that the capacity for N-demethylation is intrinsic to the entire human TET enzyme family.
The observation that TET2 can qualitatively act in either 5-methyl oxidation or direct N4-demethylation prompted us consider the comparative efficiency of these distinct reactions. For the F27–5mC and F27–5mZ substrates, the βGT/MspI-coupled assay provides a quantitative assay for substrate reactivity. Unlike these substrates, we found that F27–4dmC is unable to be cleaved by MspI (Figure S6B), likely due to poor duplex DNA formation at the 4dmC base. However, MspI was proficient in cleaving F27–4mC, which therefore permits analysis of mono-demethylation of F27–4dmC via MspI cleavage. To compare the relative reactivity, we next incubated a fixed concentration of each substrate with serial dilutions of TET2-CS, and analyzed using the appropriate format of the MspI-coupled assay (Figure 3A). Using this approach, oxidation of 5mC and mono-demethylation of 4dmC proceeded at comparable efficiency to one another (Figure 3B), with a small quantitative preference for N-demethylation over 5mC oxidation. The fact that different methyl groups on the cytosine nucleobase can be targeted to a similar extent suggests a degree of active site plasticity that has been underappreciated in TET2. Using a similar assay format, we also compared the relative reactivity of F27–5mZ. Unlike F27–4dmC, reactivity is ~4-fold reduced relative to the canonical F27–5mC substrate (Figure 3B). Although reduced in efficiency, the oxidation of 5mZ again points to a notable degree of active site tolerance in TET2.
Figure 3.
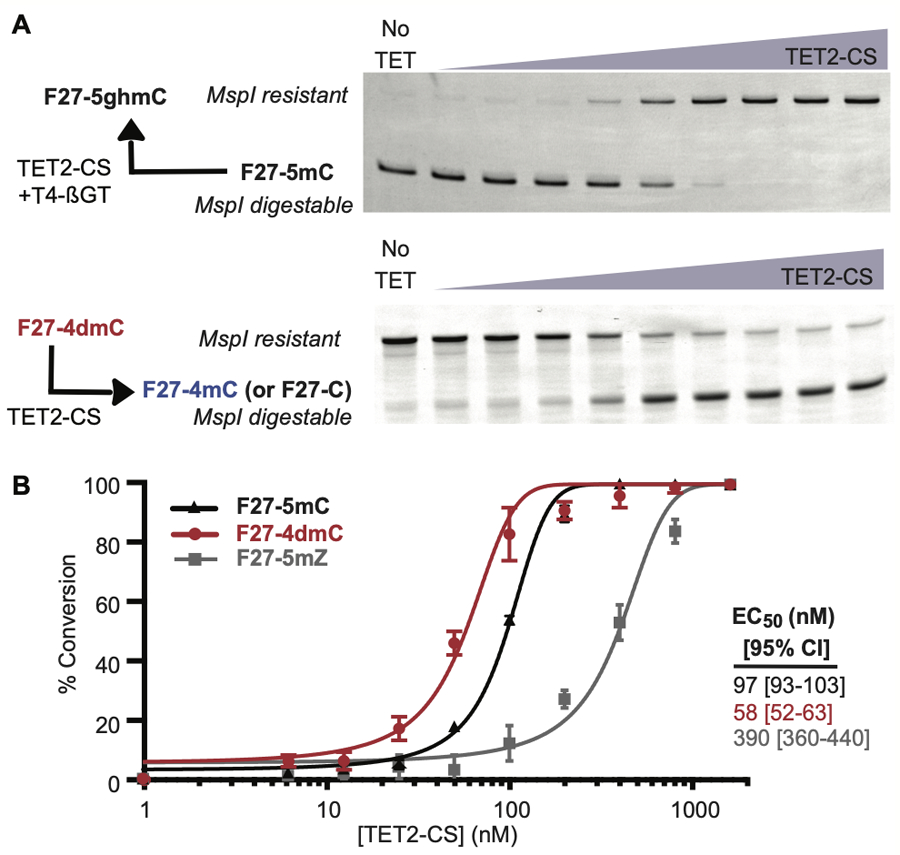
TET2 direct N-demethylation activity is comparable to 5mC oxidation activity. (A) Product formation can be detected by either protection from MspI cleavage, for F27–5mC, or susceptibility to cleavage for F27–4dmC. Fixed concentrations of F27–4dmC, F27–5mC and F27–5mZ substrates (200 nM) were co-incubated with serial dilutions of TET2-CS (6 nM to 1.6 μM), then subjected to the appropriate MspI-based assay. Representative product gels, imaged for FAM fluorescence, are shown. (B) Percent conversion of F27–5mC, F27–5dmC, and F27–5mZ are shown as a function of TET2-CS concentration. Error bars represent standard deviation from three independent replicates and the best fit sigmoidal curve to aggregated data is shown. The EC50 value derived from this fit represents the amount of enzyme required to convert half of the substrate, with 95% confidence interval (CI) noted.
The surprising proficiency of TET as a bifunctional DNA dioxygenase has potential biological and mechanistic implications. From the biological angle, our results provide grounds for asking about an expanded suite of potential cellular substrates. As TET enzymes are both proficient on RNA and also found in organisms without 5mC in DNA,[13, 22] modified RNA may be a relevant substrate in some settings. While the most common RNA modification N6-methyladenine (6mA) does not appear to be a TET substrate (Figure S7), N4 methylation is a physiologically relevant RNA modification that could be a viable TET substrate.[23] As 4mC is also found in the DNA of diverse bacteria, our findings also hold implications for the use of TET enzymes in novel sequencing approaches aimed at localizing various modifications.[24]
From the mechanistic angle, our study offers new insights into TET-mediated oxidation. For many oxidation reactions in metabolism, high specificity is required to differentiate between closely related metabolites, while others, such as cytochrome P450 enzymes, exhibit broad substrate tolerance.[25] Our results suggest that in the Fe(II)/αKG dioxygenase family, DNA/RNA modifying enzymes are likely to inhabit a middle territory.[10] After the generation of a highly reactive Fe(IV)-oxo intermediate, it is likely that any well-placed C-H bond can be targeted to complete the oxidative cycle (Figure S1). Consistent with this suggestion, AlkB family members, which typically act on N- or O-alkylated DNA,[10] have also recently been shown to potentially oxidize 5mC, albeit far less efficiently than substrates such as 3-methylcytosine.[26] The fact that TET enzymes can be highly proficient in alternative activities highlights the plasticity of the TET active site, and suggests a need to broaden exploration of the biological roles for these nucleobase modifying enzymes.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R01-GM118501 and R01-HG010646 to R.M.K.). The 6mA-containing oligonucleotide was a generous gift from Kathy Fange Liu. T.W. is supported by a Cambridge Epigenetix Fellowship.
Footnotes
No competing financial interests have been declared.
Supporting information for this article is given via a link at the end of the document.
Institute and/or researcher Twitter usernames: @kohlilab
References
- [1].Schubeler D, Nature 2015, 517, 321–326. [DOI] [PubMed] [Google Scholar]
- [2].Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A, Science 2009, 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kohli RM, Zhang Y, Nature 2013, 502, 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pfaffeneder T, Hackner B, Truss M, Munzel M, Muller M, Deiml CA, Hagemeier C, Carell T, Angew. Chem. Int. Ed Engl 2011, 50, 7008–7012. [DOI] [PubMed] [Google Scholar]
- [5].He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL, Science 2011, 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y, Science 2011, 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Seiler CL, Fernandez J, Koerperich Z, Andersen MP, Kotandeniya D, Nguyen ME, Sham YY, Tretyakova NY, Biochemistry 2018, 57, 6061–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Maiti A, Drohat AC, J. Biol. Chem 2011, 286, 35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ, Annu. Rev. Biochem, 2018, 87, 585–620. [DOI] [PubMed] [Google Scholar]
- [10].Fedeles BI, Singh V, Delaney JC, Li D, Essigmann JM, J. Biol. Chem, 2015, 290, 20734–20742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, Smemo S, Dai Q, Bailey KA, Nobrega MA, Han KL, Cui Q, He C, Nat. Commun 2013, 4, 1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pfaffeneder T, Spada F, Wagner M, Brandmayr C, Laube SK, Eisen D, Truss M, Steinbacher J, Hackner B, Kotljarova O, Schuermann D, Michalakis S, Kosmatchev O, Schiesser S, Steigenberger B, Raddaoui N, Kashiwazaki G, Muller U, Spruijt CG, Vermeulen M, Leonhardt H, Schar P, Muller M, Carell T, Nat. Chem. Biol 2014, 10, 574–581. [DOI] [PubMed] [Google Scholar]
- [13].DeNizio JE, Liu MY, Leddin EM, Cisneros GA, Kohli RM, Biochemistry 2019, 58, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kavoosi S, Sudhamalla B, Dey D, Shriver K, Arora S, Sappa S, Islam K, Chemical Science 2019, 10, 10550–10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ghanty U, DeNizio JE, Liu MY, Kohli RM, J. Am. Chem. Soc 2018, 140, 17329–17332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fu L, Guerrero CR, Zhong N, Amato NJ, Liu Y, Liu S, Cai Q, Ji D, Jin SG, Niedernhofer LJ, Pfeifer GP, Xu GL, Wang Y, J. Am. Chem. Soc 2014, 136, 11582–11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, Shi YG, Zhu J, Wang P, Xu Y, Cell 2013, 155, 1545–1555. [DOI] [PubMed] [Google Scholar]
- [18].Liu MY, Torabifard H, Crawford DJ, DeNizio JE, Cao XJ, Garcia BA, Cisneros GA, Kohli RM, Nat. Chem. Biol 2017, 13, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hu L, Lu J, Cheng J, Rao Q, Li Z, Hou H, Lou Z, Zhang L, Li W, Gong W, Liu M, Sun C, Yin X, Li J, Tan X, Wang P, Wang Y, Fang D, Cui Q, Yang P, He C, Jiang H, Luo C, Xu Y, Nature 2015, 527, 118–122. [DOI] [PubMed] [Google Scholar]
- [20].Saudi M, Zmurko J, Kaptein S, Rozenski J, Neyts J, Van Aerschot A, Eur. J. Med. Chem 2017, 126, 101–109. [DOI] [PubMed] [Google Scholar]
- [21].Liu MY, DeNizio JE, Kohli RM, Methods Enzymol 2016, 573, 365–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, Deplus R, Calonne E, Hassabi B, Putmans P, Awe S, Wetzel C, Kreher J, Soin R, Creppe C, Limbach PA, Gueydan C, Kruys V, Brehm A, Minakhina S, Defrance M, Steward R, Fuks F, Science 2016, 351, 282–285. [DOI] [PubMed] [Google Scholar]
- [23].Van Haute L, Hendrick AG, D’Souza AR, Powell CA, Rebelo-Guiomar P, Harbour ME, Ding S, Fearnley IM, Andrews B, Minczuk M, Nucleic Acids Res 2019, 47, 10267–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yu M, Ji L, Neumann DA, Chung DH, Groom J, Westpheling J, He C, Schmitz RJ, Nucleic Acids Res 2015, 43, e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jung ST, Lauchli R, Arnold FH, Curr. Opin. Biotechnol 2011, 22, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bian K, Lenz SAP, Tang Q, Chen F, Qi R, Jost M, Drennan CL, Essigmann JM, Wetmore SD, Li D, Nucleic Acids Res 2019, 47, 5522–5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.