

Summary
A SARS-CoV-2 variant carrying the Spike protein amino acid change D614G has become the most prevalent form in the global pandemic. Dynamic tracking of variant frequencies revealed a recurrent pattern of G614 increase at multiple geographic levels: national, regional, and municipal. The shift occurred even in local epidemics where the original D614 form was well established prior to introduction of the G614 variant. The consistency of this pattern was highly statistically significant, suggesting that the G614 variant may have a fitness advantage. We found that the G614 variant grows to a higher titer as pseudotyped virions. In infected individuals, G614 is associated with lower RT-PCR cycle thresholds, suggestive of higher upper respiratory tract viral loads, but not with increased disease severity. These findings illuminate changes important for a mechanistic understanding of the virus and support continuing surveillance of Spike mutations to aid with development of immunological interventions.
Keywords: COVID-19, SARS-CoV-2, diversity, evolution, Spike, antibody, infectivity, neutralization, pseudovirus, PCR cycle threshold
Graphical Abstract
Highlights
-
•
A SARS-CoV-2 variant with Spike G614 has replaced D614 as the dominant pandemic form
-
•
The consistent increase of G614 at regional levels may indicate a fitness advantage
-
•
G614 is associated with lower RT PCR Cts, suggestive of higher viral loads in patients
-
•
The G614 variant grows to higher titers as pseudotyped virions
Korber et al. present evidence that there are now more SARS-CoV-2 viruses circulating in the human population globally that have the G614 form of the Spike protein versus the D614 form that was originally identified from the first human cases in Wuhan, China. Follow-up studies show that patients infected with G614 shed more viral nucleic acid compared with those with D614, and G614-bearing viruses show significantly higher infectious titers in vitro than their D614 counterparts.
Introduction
The past two decades have seen three major pathogenic zoonotic disease outbreaks caused by betacoronaviruses (Cui et al., 2019; de Wit et al., 2016; Liu et al., 2020; Wu et al., 2020). Severe acute respiratory syndrome coronavirus (SARS-CoV) emerged in 2002, infecting ∼8,000 people with a 10% mortality. Middle East respiratory syndrome coronavirus (MERS-CoV) emerged in 2012 with ∼2,300 cases and 35% mortality (Graham and Baric, 2010). The third, SARS-CoV-2, causes the severe respiratory disease coronavirus disease 2019 (COVID-19) (Gorbalenya et al., 2020). First reported in China in December 2019 (Zhou et al., 2020), it rapidly became a pandemic with devastating effects. The June 21, 2020 World Health Organization (WHO) Situation Report records over 8.7 million COVID-19 cases and 460,000 deaths, numbers that increase daily. Humans have no direct immunological experience with SARS-CoV-2, leaving us vulnerable to infection and disease. SARS-CoV-2 is highly transmissible: basic reproduction number, R0,estimates vary between 2.2 and 3.9 (Lv et al., 2020). Estimates of mortality vary regionally between 0.8% and 14.5% (mortality analyses, Johns Hopkins University of Medicine)
Coronaviruses have genetic proofreading mechanisms (Sevajol et al., 2014; Smith et al., 2013), and SARS-CoV-2 sequence diversity is very low (Fauver et al., 2020). Still, natural selection can act upon rare but favorable mutations. By analogy, antigenic drift results in gradual accumulation of mutations by the influenza virus during flu season, and the complex interplay between immunological resistance mutations and the fitness landscape enables antibody resistance to develop across populations (Wu et al., 2020), driving the need to develop new influenza vaccines every few seasons. Longer flu seasons have increased opportunities for selection pressure (Boni et al., 2006). Although SARS-CoV-2 shows evidence of some seasonal waning (Sehra et al., 2020), the persistence of the pandemic may enable accumulation of immunologically relevant mutations in the population even as vaccines are developed. Antigenic drift is seen among the common cold coronaviruses OC43 (Ren et al., 2015; Vijgen et al., 2005) and 229E (Chibo and Birch, 2006) and in SARS-CoV-1 (Guan et al., 2003; Song et al., 2005). Notably, a single SARS-CoV-1 amino acid change, Spike D480A/G in the receptor binding domain (RBD), arose in infected humans and civets and became the dominant variant among 2003/2004 viruses. D480A/G escapes neutralizing antibody 80R, and immune pressure from 80R in vitro could recapitulate emergence of the D480 mutation (Sui et al., 2008). Although there is no evidence yet of antigenic drift for SARS-CoV-2, with extended human-to-human transmission, SARS-CoV-2 could also acquire mutations with fitness advantages and immunological resistance. Attending to this risk now by identifying evolutionary transitions that may be relevant to the fitness or antigenic profile of the virus is important to ensure effectiveness of the vaccines and immunotherapeutic interventions as they advance to the clinic.
In response to the urgent need to develop effective vaccines and antibody-based therapeutic agents against SARS-CoV-2, over 90 vaccine and 50 antibody approaches are currently being explored (Cohen, 2020; Yu et al., 2020). Most target the trimeric Spike protein, which mediates host cell binding and entry and is the major target of neutralizing antibodies (Chen et al., 2020; Yuan et al., 2020). Spike monomers are comprised of an N-terminal S1 subunit that mediates receptor binding and a membrane-proximal S2 subunit that mediates membrane fusion (Hoffmann et al., 2020a; Walls et al., 2020; Wrapp et al., 2020). SARS-CoV-2 and SARS-CoV-1 share ∼79% sequence identity (Lu et al., 2020), and both use angiotensin-converting enzyme 2 (ACE2) as their cellular receptor. Antibody responses to SARS-CoV-1 Spike are complex. In some patients with rapid and high neutralizing antibody responses, an early decline of these responses is associated with increased severity of disease and a higher risk of death (Ho et al., 2005; Liu et al., 2006; Temperton et al., 2005; Zhang et al., 2006). Some antibodies against SARS-CoV-1 Spike mediate antibody-dependent enhancement (ADE) of infection in vitro and exacerbate disease in animal models (Jaume et al., 2011; Wan et al., 2020; Wang et al., 2014; Yip et al., 2016).
Most current SARS-CoV-2 immunogens and testing reagents are based on the Spike protein sequence of the Wuhan reference sequence (Wang et al., 2020), and first-generation antibody therapeutic agents were discovered based on early pandemic infections and evaluated using the Wuhan reference sequence proteins. Alterations of the reference sequence as the virus propagates in human-to-human transmission could potentially alter the viral phenotype and/or the efficacy of immune-based interventions. Therefore, we designed bioinformatics tools to create an “early warning” strategy to evaluate Spike evolution during the pandemic to enable testing of mutations for phenotypic implications and generation of appropriate antibody breadth evaluation panels as vaccines and antibody-based therapeutic agents progress. Phylogenetic analysis of the global sampling of SARS-CoV-2 is being very capably addressed by the Global Initiative for Sharing All Influenza Data (GISAID) database (https://www.gisaid.org/; Elbe and Buckland-Merrett, 2017; Shu and McCauley, 2017) and Nextstrain (https://nextstrain.org; Hadfield et al., 2018). However, in a setting of low genetic diversity like that of SARS-CoV-2, with very few de novo mutational events, phylogenetic methods that use homoplasy to identify positive selection (Crispell et al., 2019) have limited statistical power. Additionally, recombination can add a confounding factor to phylogenetic reconstructions, and recombination is known to play a role in natural coronavirus evolution (Graham and Baric, 2010; Lau et al., 2011; Li et al., 2020; Oong et al., 2017; Rehman et al., 2020), and recombinant sequences (potential sequencing artifacts) have been found among SARS-CoV-2 sequences (De Maio et al., 2020). Given these issues, we developed an alternative indicator of potential positive selection by identifying variants that are recurrently becoming more prevalent in different geographic locations. If increases in relative frequency of a particular variant are observed repeatedly in distinct geographic regions, then that variant becomes a candidate for conferring a selective advantage.
Single amino acid changes are worth monitoring because they can be phenotypically relevant. Among coronaviruses, point mutations have been demonstrated to confer resistance to neutralizing antibodies in MERS-CoV (Tang et al., 2014) and SARS-CoV-1 (Sui et al., 2008; ter Meulen et al., 2006). In the HIV envelope, single amino acid changes are known to alter host species susceptibility (Li et al., 2016), increase expression levels (Asmal et al., 2011), change the viral phenotype from tier 2 to tier 1, cause an overall change in neutralization sensitivity (Gao et al., 2014; LaBranche et al., 2019), and confer complete or nearly complete resistance to classes of neutralizing antibodies (Bricault et al., 2019; Sadjadpour et al., 2013; Zhou et al., 2019).
We developed a bioinformatics pipeline to identify Spike amino acid variants that are increasing in frequency across many geographic regions by monitoring GISAID data. By early April 2020, it was clear that the Spike D614G mutation exhibited this behavior, and G614 has since become the dominant form in the pandemic. We present experimental evidence that the G614 variant is associated with greater infectivity as well as clinical evidence that it is associated with higher viral loads. We continue to monitor other mutations in Spike for frequency shifts at regional and global levels and provide regular updates at a public web site (https://cov.lanl.gov/).
Results
Website Overview
Our analysis pipeline to track SARS-CoV-2 mutations in the COVID-19 pandemic is based on regular updates from the GISAID SARS-CoV-2 sequence database (GISAID acknowledgments are in Table S1). GISAID sequences are generally linked to the location and date of sampling. Our website provides visualizations and summary data that allow regional tracking of SARS-CoV-2 mutations over time. Hundreds of new SARS-CoV-2 sequences are added to GISAID each day, so we have automated steps to create daily working alignments (Kurtz et al., 2004; Figure S1). The analysis presented here is based on a May 29, 2020 download of the GISAID data, when our Spike alignment included 28,576 sequences; updated versions of key figures can recreated at our website (https://cov.lanl.gov). The overall evolutionary rate for SARS-CoV-2 is very low, so we set a low threshold for a Spike mutation to be deemed “of interest,” and we track all sites in Spike where 0.3% of the sequences differ from the Wuhan reference sequence, monitoring them for increasing frequency over time in geographic regions as well as for recurrence in different geographic regions. Here we present results for the first amino acid variant to stand out by these metrics, D614G.
Figure S1.

Schematic of the SARS-CoV-2 Outbreak Sequence Processing Pipeline, Related to Figure 1
The intent of these procedures is to generate, for each of several regions, a set of contiguous codon-aligned sequences, complete in that region, without extensive uncalled bases, large gaps, or regions that are unalignable or highly divergent, in reasonable running time for n > 30,000 ∼30kb viral genomes. This allows daily processing of GISAID data to enable us to track mutations. This process provides the foundational data to enable the generation of Figures 1, 2, 3, and 7.
A. Processing procedures:
1. Download all SARS-CoV-2 sequences from GISAID.org (34,607 as of 2020-05-29). The downloaded sequences are stored in compressed form (via bzip2: https://sourceware.org/git/bzip2.git).
2. Align sequences to the SARS-CoV-2 reference sequence (NC_045512), trim to desired endpoints, and filter for coverage and quality. These steps are incorporated in a single Perl script, ‘align_to_ref.pl’, briefly summarized here: sequences are compressed for identity, then mapped against the given reference sequence using ‘nucmer’ from the ‘MUMmer’ package (Kurtz et al., 2004). The nucmer ‘delta’ file contains locations of matching regions and is parsed and used to, first, partition the sequences into “good” and “bad” subsets, and then to generate alignments from the “good” sequences.
B. Categories of sequences included and excluded from our automated alignments. A series of criteria is used successively to exclude sequences with large internal gaps, excessive five- and three-prime gaps, large numbers of mismatches or ambiguities (> 30) overall, or regions with a high concentration of mismatches or ambiguities (> 10 in any 100 nt subsequence): the counts of these categories of “bad” sequence are shown, for different regional genome alignments.
We then create the following different regional subalignments: CODING-REGIONS2 (“FULL,” from the 5′-most start-codon (orf1ab) to the 3′-most stop-codon (ORF10), NC_045512 bases 266-29,674; SPIKE, the complete surface glycoprotein coding region, bases 21,563-25,384; NEAR-COMPLETE (“NEARCOMP,” the most-commonly-sequenced region of the genome, bases 55-29,836; COMPLETE, matching the NC_045512 sequence from start up to the poly-A tail, bases 1-29,870; 5′ UTR, the five-prime untranslated region, bases 1-265 only. Generally speaking, the smaller the region, the more sequences are included.
Sequences are trimmed to the extent of the reference (with minimum allowed gaps at 5′ and 3′ ends), following which the pairwise alignments are generated from the matching regions, and a multiple sequence alignment is constructed from the pairwise alignments.
3. De-duplicate1. To reduce computational demands, sequences are compressed by identity following trimming to the desired region, by computing a hash value for each sequence (currently the SHA-1 message digest, 160 bits encoded as a 40-character hex string). To prevent the loss of parsimony-informative characters when they occur in identical strings, however, multiple sequences are reduced to a minimum of two occurrences.
4. Codon-align. Gaps are introduced into the entire compressed alignment so that the alignment column containing the last base of each codon has a number divisible by three; this simplifies processing of translations. Code for this procedure is derived from the GeneCutter tool from the LANL HIV database (https://www.hiv.lanl.gov/content/sequence/GENE_CUTTER/cutter.html).
5. Partition (full/spike-only). For subalignments that encompass the spike protein and substantial additional sequence, the spike region is extracted separately, to allow matched comparisons.
6. Build parsimony trees. A brief parsimony search (parsimony ratchet, with 5 replicates) is performed with ‘oblong’ (Goloboff, 2014) This is intended as an efficient clustering procedure rather than an explicit attempt to achieve an accurate phylogenetic reconstruction, but it appears to yield reasonable results in this situation of a very large number of sequences with a very small number of changes, where more complex models may be subject to overfitting. When multiple most-parsimonious trees are found, only the shortest of these (under a p-distance criterion) is retained. Distance scoring is performed with PAUP∗ (Swofford, 2003).
7. Re-duplicate (expand, i.e., uncompress). The original sequence names and occurrence counts are restored to FastA format files and the appropriate leaf taxa added to the parsimony trees.
8. Sort alignment by tree. Sequences in the FastA files are sorted by the expanded tree, allowing patterns of mutation to be discerned by inspection.
9. Mutations of interest can be readily tracked on the trees to resolve whether they are identified in predominantly in single clades or distributed throughout the tree and likely to be recurring (e.g., Figure 7 (sites of interest with low frequency amino acid substitutions) and Figure S6 (Site 614)).
The D614G Variant
Increasing Frequency and Global Distribution
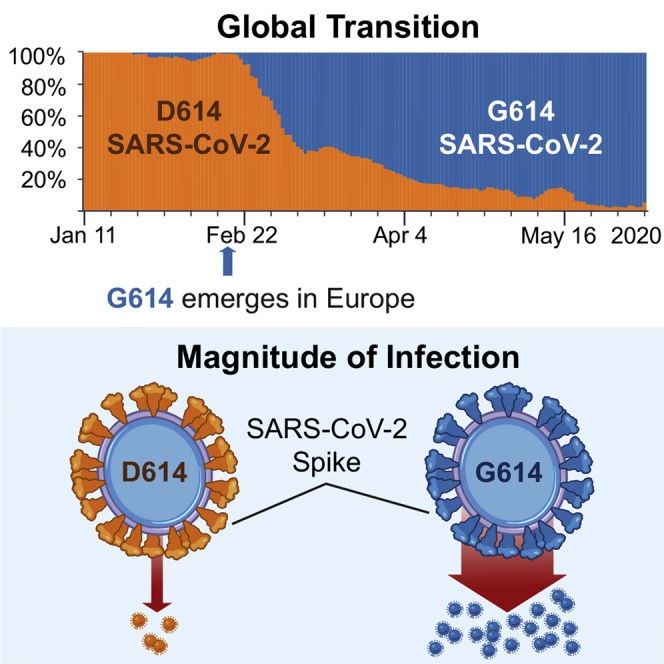
The Spike D614G amino acid change is caused by an A-to-G nucleotide mutation at position 23,403 in the Wuhan reference strain; it was the only site identified in our first Spike variation analysis in early March 2020 that met our threshold criterion. At that time, the G614 form was rare globally but gaining prominence in Europe, and GISAID was also tracking the clade carrying the D614G substitution, designating it the “G clade.” The D614G change is almost always accompanied by three other mutations: a C-to-T mutation in the 5′ UTR (position 241 relative to the Wuhan reference sequence), a silent C-to-T mutation at position 3,037, and a C-to-T mutation at position 14,408 that results in an amino acid change in RNA-dependent RNA polymerase (RdRp P323L). The haplotype comprising these 4 genetically linked mutations is now the globally dominant form. Prior to March 1, 2020, it was found in 10% of 997 global sequences; between March 1 and March 31, 2020, it represented 67% of 14,951 sequences; and between April 1 and May 18, 2020 (the last data point available in our May 29, 2020 sample), it represented 78% of 12,194 sequences. The transition from D614 to G614 occurred asynchronously in different regions throughout the world, beginning in Europe, followed by North America and Oceania and then Asia (Figures 1, 2, 3, S2, and S3).
Figure 1.

The Global Transition from the Original D614 Form to the G614 Variant
For a Figure360 author presentation of this figure, see https://doi.org/10.1016/j.cell.2020.06.043#mmc4.
(A) Changes in the global distribution of the relative frequencies of the D614 (orange) and G614 (blue) variants in 2 time frames. Circle size indicates the relative sampling within each map. Through March 1, 2020, the G614 variant was rare outside of Europe, but by the end of March 2020 it had increased in frequency worldwide. These data are explored regionally in Figure 2 (Europe), Figure S2 (North America), and Figure S3 (Australia and Asia).
(B) Paired bar charts compare the fraction of sequences with D614 and with G614 for two time periods separated by a 2-week gap. The first time period (left bar) includes all sequences up to the onset day (see main text). The second time period (right bar) includes all sequences acquired at least 2 weeks after the onset date. All regions are shown that met the minimal threshold criteria for inclusion (see main text) with a significant shift in frequency (two-sided Fisher’s exact test, p < 0.05). Four hierarchical geographic levels are split out based on GISAID naming conventions.
(C) Running weekly average counts of sampled sequences exhibiting the D614 (orange) and G614 (blue) variants on different continents between January 12 and May 12, 2020. The measure of interest is the relative frequency over time. The shape of the overall curve just reflects sample availability; sequencing was more limited earlier in the epidemic (hence the left-hand tail), and there is a time lag between viral sampling and sequence availability in GISAID (hence the right-hand tail).
Weekly running count plots were generated with Python Matplotlib (Hunter, 2007); all elements of this figure are frequently updated at https://cov.lanl.gov/.
Figure 2.

The Transition from D614 to G614 in Europe
(A) Maps of relative D614 and G614 frequencies in Europe in 2 time frames.
(B) Weekly running counts of G614 illustrating the timing of its spread in Europe. The legend for Figure 1 explains how to read these figures. Some nations essentially had G614 epidemics when sampling began, but even in these cases, small traces of D614 found early were soon lost (e.g., France and Italy). The Italian epidemic started with the D614 clade, but Italy had the first sampled case of the full G614 haplotype and had shifted to all G614 samples prior to March 1, 2020 (Figure S5). European nations that began with a mixture of D614 and G614 most clearly reveal the frequency shifts (e.g., Germany, Spain, and the United Kingdom). The United Kingdom is richly sampled and so is subdivided into smaller regions (England, Wales, and Scotland) and then further divided to display two well-sampled English cities. Even in settings with very well-established D614 epidemics (e.g., Wales and Nottingham; see also Figures S2 and S3), G614 becomes prevalent soon after its appearance. The increase in G614 frequency often continues well after stay-at-home orders are in place (pink line) and past the subsequent 2-week incubation period (pink transparent box). The figures shown here can be recreated with contemporary data from GISAID at the http://cov.lanl.gov/ website. UK stay-at-home order dates were based on the date of the national proclamation, and others were documented on the web.
Figure 3.

Modeling the Daily Fraction of the G614 Variant as a Function of Time in Local Regions Using Isotonic Regression
(A) Analysis summaries for all of the level 3 and 4 regional subdivisions from GISAID data (Figure 1) that have at least 5 each of D614 and G614 variants and that are sampled on at least 14 days. We report the number of each variant, the number of days with test results, and the number of days spanning the first and the last reported tests. the p values are for two one-sided tests, comparing the null hypothesis of no consistent changes in relative frequency over time with positive pressure (the fraction of the G614 variant increasing with time) or negative pressure (the fraction of the G614 variant decreasing with time). Across all regions with sufficient data, binomial p values against the null that increases and decreases are equally likely to indicate that the consistency of increasing G614 is highly significant. California has increasing and decreasing patterns with low p values; this can happen when different time windows support opposing patterns. The G614 decreasing time window in California was driven by sampling from Santa Clara county, a rare region that has retained the D614 form (Figure S4). In the May 29, 2020 dataset used here, Santa Clara county was sampled later in May than any other region in California, so the California G614 frequency dips at this last available time point. When Santa Clara county is removed from the California sample, the pattern of increasing levels of G614 is restored (red asterisk).
(B) Three examples of cities, plotting the daily fraction of G614 as a function of time and accompanied by plots of running weekly counts. The dot size is proportional to the number of sequences sampled that day. The staircase line is the maximum likelihood estimate under the constraint that the logarithm of the odds ratio is non-decreasing. Two typical examples are shown, highlighted in blue (Sydney and Cambridge), and one exception is shown, highlighted in orange (Yakima). Yakima had a brief sampling window enriched for G614 early in the sampling period, but otherwise G614 maintained a low frequency.
Summaries and plots for all regional data at levels 2–4 (included country) are included in Data S1.
Figure S2.

The Increasing Frequency of the D614G Variant over Time in North America, Related to Figure 1
Maps of the relative frequencies of D614 and G614 in North America in two different time windows. B. Weekly running counts of G614 illustrating the timing of its spread in North America. This figure complements Figures 2 and S3, and Figure 1 has details about how to read these figures. When a particular stay-at-home order date was known for a state or county it is shown as a pink line, followed by a light pink block indicating the maximum two-week incubation time. Different counties in California had different stay-at-home order dates (Mar. 16-19) so are not highlighted, but more detail can be seen regarding California in Figure S4. The decline in D614 frequency often continues well after the stay-at-home orders were initiated, and sometimes beyond the 14-day maximum incubation period, when serial reintroduction of the G614 would be unlikely. On the right, Washington State is shown, with details from two heavily sampled counties, Snohomish and King. Both counties had well-established ongoing D614 epidemics when G614 variants were introduced, undoubtedly by travelers. Washington state’s stay-at-home order was initiated March 24. At this time there were 1170 confirmed cases in King County, and 614 confirmed cases in Snohomish County. (Confirmed COVID19 case count data from: COVID-19 Data Repository by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University). Testing was limited, and so this is lower bound on actual cases. Of the sequences sampled by March 24, 95% from King County (153/161) and 100% from Snohomish County (33/33) were the original D614 form (Part B, details at https://cov.lanl.gov/). By mid-April, D614 was rarely sampled. Whatever the geographic origin of the G614 variants that entered these counties, and whether one or if multiple G614 variants were introduced, the rapid expansion of G614 variants occurred in the framework of well-established local D614 variant epidemics. Santa Clara county is one of the two exceptions to the pattern of D614 decline in Figure 1B: details are provided in Figures S4A and S4C.
Figure S3.

The Increasing Frequency of the D614G Variant over Time in Australia and Asia, Related to Figures 1 and 2
This figure complements Figure 2 and Figure S2, and Figure 1 has details about how to read these figures. The plot representing national sampling in Australia is on the left, with two regional subsets of the data on the right. In each case a local epidemic started with the D614 variant, and despite being well established, the G614 variant soon dominates the sampling. Only limited recent sampling from Asia is currently available in GISAID; to include more samples on the map the 10-day period between March 11-20, is shown rather than the period between March 21-30; even the limited sampling mid-March the supports the repeated pattern of a shift to G614. The Asian epidemic was overwhelmingly D614 through February, and despite this, G614 repeatedly becomes prominent in sampling by mid-March.
We developed two statistical approaches to assess the consistency and significance of the D614-to-G614 transition. In general, to observe a significant change in the frequency of variants in a geographic region, three requirements must be met. First, both variants must at some point be co-circulating in the geographic area. Second, there must be sampling over an adequate duration to observe a change in frequency. Third, enough samples must be available for adequate statistical power to detect a difference. Both of our approaches enable us to systematically extract all GISIAD local and regional data that meet these three requirements.
Our first approach requires that there be an “onset,” defined as the first day where the cumulative number of sequences reached 15 and both forms were represented at least 3 times; we further require that there be at least 15 sequences available at least 2 weeks after onset. Each geographic region that meets these criteria is extracted separately based on the hierarchical geographic/political levels designated in GISAID (Figure 1B). A two-sided Fisher’s exact test compares the counts in the pre-onset period with the counts after the 2-week delay period and provides a p value against the null hypothesis that the fraction of D614 versus G614 sequences did not change. All regions that met the above criteria and that showed a significant change in either direction (p < 0.05) are included. Almost all shifted toward increasing G614 frequencies: 5 of 5 continents, 16 of 17 countries (two-sided binomial p value of 0.00027), 16 of 16 regions (p = 0.00003), and 11 of 12 counties and cities (p = 0.0063).
In Figure 2 (Europe), Figure S2 (North America), and Figure S3 (Australia and Asia), we break down the relationships shown in Figure 1B in detail. The G614 variant increased in frequency even in regions where D614 was the clearly dominant form of a well-established local epidemic when G614 entered the population. Examples of this scenario include Wales, Nottingham, and Spain (Figure 2); Snohomish county and King county (Figure S2); and New South Wales, China, Japan, Hong Kong, and Thailand (Figure S3). Although introduction of a new variant might sometimes result in emergence of the new form because of stochastic effects or serial re-introductions or apparent emergence because of sampling biases, the consistency of the shift to G614 across regions is striking. The increase in G614 often continued after national stay-at-home orders were implemented and, in some cases, beyond the 2-week maximum incubation period.
We found two exceptions to the pattern of increasing G614 frequency in Figure 1B; details regarding these cases are shown in Figure S4. The first is Iceland. Changes in sampling strategy during a regional molecular epidemiology survey conducted through the month of March 2020 might explain this exception (Gudbjartsson et al., 2020). In early March 2020, only high-risk people were sampled, the majority being travelers from countries in Europe where G614 dominated. In mid-March 2020, screening began to include the local population; this coincided with the appearance of the D614 variant in the sequence dataset. The second exception is Santa Clara county, one of the most heavily sampled regions in California (Deng et al., 2020). The D614 variant dominates sequences from the Santa Clara Department of Public Health (DPH) to date; the G614 variant was apparently not established in that community. In contrast, a smaller set of Santa Clara county sequences, sampled from mid-March to early April 2020, were specifically noted to be from Stanford; the Stanford samples had a mixture of both forms co-circulating (Figure S4), suggesting that the two communities in Santa Clara County are effectively distinct. A June 19, 2020 GISAID update for several California counties is provided in Figure S4C, and the G614 form is present in the most recent Santa Clara DPH samples.
Figure S4.

Two Exceptions to the Pattern of Increasing Frequency of the G614 Variant over Time, from Figure 1B, Related to Figure 1
A. Details regarding Santa Clara county, the only exceptional pattern at the county/city level in Figure 1B. Many samples from the Santa Clara County Department of Public Heath (DPH) were obtained from March into May, and D614 has steadily dominated the local epidemic among those samples. The subset of Santa Clara county samples specifically labeled “Stanford,” however, were sampled over a few weeks mid-March through early April, and have a mixture of both the G614 and D614 forms. These distinct patterns suggest relatively little mixing between the two local epidemics. Why Santa Clara county DPH samples should maintain the original form is unknown, but one possibility is that they may represent a relatively isolated community that had limited exposure to the G614 form, and G614 may not have had the opportunity to become established in this community – though this may be changing, see Part C. The local stay-at-home orders were initiated relatively early, March 16, 2020. B. Details regarding Iceland, the only country with an exceptional pattern from Figure 1B. All Icelandic samples are from Reykjavik, and only G614 variants were initially observed there, with a modest but stable introduction of the original form D614 in mid-March. This atypical pattern might be explained by local sampling. The Icelanders conducted a detailed study of their early epidemic (Gudbjartsson et al., 2020), and all early March samples were collected from high risk travelers from Europe and people in contact with people who were ill; the majority of the traveler samples from early March were from people coming in from Italy and Austria, and G614 dominated both regions. On March 13, they began to sequence samples from local population screening, and on March 15, more travelers from the UK and USA with mixed G614/D614 infections began to be sampled in the high-risk group, and those events were coincident with the appearance of D614. C. Updated data regarding California from the June 19, 2020 GISAID sampling. Most of the analysis in this paper was undertaken using the May 29, 2020 GISAID download, but as California was an interesting outlier, and more recent sampling conducted while the paper was under review was informative, we have included some additional plots from California data that were available at the time of our final response to review, on June 19th. Informative examples from well-sampled local regions are shown. Stay-at-home order dates are shown as a pink line, followed by a light pink block indicating the maximum two-week incubation time. N indicates the number of available sequences. Overall California, and specifically, San Diego and San Joaquin, show a clear shift from D614 to G614. The transition for San Joaquin was well after the stay-at-home orders and incubation period had passed. San Francisco shows a trend toward G614. Santa Clara DPH, which was essentially all D614 in our May 29th GISAID download, had 7 G614 forms sampled in late May that were evident in our June 19th GISAID download. Ventura is an example of a setting that was essentially all G614 when it began to be sampled significantly in early April, so a transition cannot be tracked; i.e., we cannot differentiate in such cases whether the local epidemic originated as a G614 epidemic, or whether it went through a transition from D614 to G614 prior to sampling. The figures in Parts A, B, and C can be recreated with more current data at http://cov.lanl.govcontent/index.
Our second statistical approach to evaluating the significance of the D614-to-G614 transition (Figure 3) uses the time series data in GISAID more fully. Here we extracted all regional data from GISAID that had a minimum of 5 sequences representing each of the D614 and the G614 variants and at least 14 days of sampling. We then modeled the daily fraction of G614 as a function of time using isotonic regression, testing the null hypothesis that this fraction does not change over time (i.e., it remains roughly flat over time with equally likely random fluctuations of increase or decrease). We then separately tested the null against two alternative hypotheses: that the fraction of G614 increases or that it decreases. Figure 3A shows separate p values for all subcountries/states and counties/cities that met the minimal criteria. 30 of 31 subcountries/states with a significant change in frequency were increasing in G614; a binomial test indicates that G614 increases are highly significantly enriched (p = 2.98e–09). This was also found in 17 of 19 counties/cites (p = 0.0007). Figure 3B shows examples for 3 cities, plotting the daily fraction of G614 as a function of time. Country summaries (similar to Figure 3A) and plots for all regions (similar to Figure 3B) are included in Data S1.
Origins of the D614G 4-Base Haplotype
The earliest examples of sequences carrying parts of the 4-mutation haplotype that characterizes the D614G GISAID G clade were found in China and Germany in late January 2020, and they carried 3 of the 4 mutations that define the clade, lacking only the RdRp P323L substitution (Figure S5D). This may be an ancestral form of the G clade. One early Wuhan sequence and one early Thai sequence had the D614G change but not the other 3 mutations (Figure S5D); these may have arisen independently. The earliest sequence we detected that carried all 4 mutations was sampled in Italy on February 20, 2020 (Figure S5D). Within days, this haplotype was sampled in many countries in Europe.
Figure S5.

Relationships among the Earliest Examples of the G Clade and Other Early Epidemic Samples, Related to Figures 1 and 2
Weekly running counts of the earliest forms of the virus carrying G614. B. The GISAID G clade is based on a 4 base haplotype that distinguishes it from the original Wuhan form. Part B shows the tallies of all of the variants among the 4 base mutations that are the foundation of the G clade haplotype, versus the bases found in the original Wuhan reference strain, and highlights of some of the earliest identified sequences bearing these mutations. We first consider just the 3 mutations that are in coding regions, to enable using an alignment that contains a larger set of sequences. One is in the RdRp protein (nucleotide C14408T resulting in a P323L amino acid change), one in Spike (nucleotide A23403G resulting in the D614G amino acid change) and one is silent (C3037T). The other mutation is in the 5′ UTR (C241T), and tallies based on all 4 positions are done separately, as they are based on an alignment with fewer sequences. The earliest examples of a partial haplotype, TTCG, are found in Germany and China (Parts A and B). This form was present in Shanghai but never expanded (Part A). A cluster of infections that all carried this form were identified in Germany, but they did not expand and were subsequently replaced with the original Wuhan form, only to be replaced again by sequences that carry the full 4 base haplotype variant TTTG. The first example in our alignments (see Figure S1) found to carry the full 4 base haplotype was sampled in Italy on Feb. 20. The first cases in Italy were of the original Wuhan form CCCA form, but by the end of February TTTG was the only form sampled in Italy, and it is the TTTG form that has come the dominate the pandemic. Of note, the TTCG form did not expand, and it lacked the RdRp P323L change, raising the possibility that the P323L change may contribute to a selective advantage of the haplotype. TTTG and CCCA are almost always linked in SARS-CoV-2 genomes, > 99.9% of the time (Part B). The number of cases where the clade haplotype is disrupted, and the form of the disruption, are all noted in part B (not including ambiguous base calls). Some cases of a disrupted haplotype may be due to recombination events and not de novo mutations. Given the fact that these two forms are were co-circulating in many communities throughout the spring of 2020, and the fact that disruptions in the 4 base pattern are rare, suggests that recombination is overall relatively rare among pandemic sequences.
Structural Implications of the Spike D614G Change
D614 is located on the surface of the Spike protein protomer, where it can form contacts with the neighboring protomer (Figure 4A). Cryoelectron microscopy (cryo-EM) structures (Walls et al., 2020; Wrapp et al., 2020) indicate that the side chains of D614 and T859 of the neighboring protomer (Figure 4B) form a between-protomer hydrogen bond, bringing together a residue from the S1 unit of one protomer and a residue of the S2 unit of the other protomer (Figure 4C). The change to G614 would eliminate this side-chain hydrogen bond, possibly increasing main-chain flexibility and altering between-protomer interactions. In addition, this substitution could modulate glycosylation at the nearby N616 site, influence the dynamics of the spatially proximal fusion peptide (Figure 4D) of the neighboring protomer, or have other effects.
Figure 4.

Structural Mapping of Amino Acid Changes and Clusters of Variation in the Spike Protein
(A) Sites including Spike 614 and those noted in Figure 7 mapped onto S1 and S2 units of the Spike protein (PDB: 6VSB). S1 and S2 are defined based on the furin cleavage site (protomer 1: S1, dark blue; S2, light blue; protomer 2: S1, dark green; S2, light green; protomer 3: gray). The RBD of protomer 1 is in the “up” position for engagement with the ACE2 receptor. Sites of interest are indicated by red balls, and variable clusters are labeled in red. The missing RBD residues at the ACE2 interface are shown in (D).
(B) Proximity of D614 (red) to an N-linked glycosylation sequon of its own protomer (blue) and to residues T859 and Q853 of the neighboring protomer (green) are shown. Black dashed lines indicate possible hydrogen bond formation. Dotted lines indicate the structurally unresolved region of the fusion peptide connecting to Q853.
(C) Schematic representation of potential protomer-protomer interactions shown in (B), in which D614 (red) from the S1 unit of one protomer (blue) is brought close to T859 from the S2 unit of the neighboring protomer.
(D) Sites of interest (red, residues 475–483) near the RBD (blue)-ACE2 (yellow) binding interface. The interfacial region is shown as a molecular surface (PDB: 6M17).
(E) Variable cluster 936–940 (red) in the HR1 region of S2. These residues occur in a region that undergoes conformational transition during fusion; pre-fusion (PDB: 6VSB) and post-fusion (PDB: 6LXT) conformations of HR1 are shown (left and right).
G614 Is Associated with Potentially Higher Viral Loads in COVID-19 Patients but Not with Disease Severity
SARS-CoV-2 sequences from 999 individuals presenting with COVID-19 disease at the Sheffield Teaching Hospitals NHS Foundation Trust were available and linked to clinical data. The Sheffield data include age, sex, date of sampling, hospitalization status (defined as outpatient [OP], inpatient [IP], requiring hospitalization, or admittance to the intensive care unit [ICU]), and the cycle threshold (Ct) for a positive signal in E-gene based RT-PCR. The Ct is used here as a surrogate for relative viral loads; lower Ct values indicate higher viral loads (Corman et al., 2020), but not all viral nucleic acids represent infectious viral particles. RT-PCR methods changed during the course of the study because of limited availability of testing kits. The first method involved nucleic acid extraction and the second method heat treatment (Fomsgaard and Rosenstierne, 2020). A generalized linear model (GLM) used to predict the PCR Ct based on the RT-PCR method, sex, age, and D614G status showed only the RT-PCR method (p < 2e−16) and D614G status (p = 0.037) to be statistically significant (Figure 5A). Lower Ct values were observed in G614 infections. While our paper was in revision, G614-variant association with low Ct values in vivo (Figure 5) was reported independently by two other groups (Lorenzo-Redondo et al., 2020; Wagner et al., 2020) in preprints that have not yet been peer reviewed.
Figure 5.

Clinical Status and D614G Associations Based on 999 Subjects with COVID-19 and Linked Sequence and Clinical Data Were Sampled in Sheffield, England
(A) G614 was associated with a lower cycle threshold (Ct) required for detection; lower values are indicative of higher viral loads. The PCR method was changed partway through April 2020 because of shortages of nucleic acid extraction kits (Fomsgaard and Rosenstierne, 2020). The Ct levels for the two PCR methods (nucleic acid extraction versus simple heat inactivation) differ, and so we used a GLM to evaluate the statistical effect of D614G across methods.
(B) D614G status was not statistically associated with hospitalization status (outpatient [OP], inpatient [IP], or ICU) as a marker of disease severity, but age was highly correlated. The number of counts in each category is noted in the top right corner of each graph. See the main text and STAR Methods for statistical details.
We found no significant association between D614G status and disease severity as measured by hospitalization outcomes. A comparison of D614G status and hospitalization (combining IP and ICU) was not significant (p = 0.66, Fisher’s exact test), although comparing ICU admission with IP and OP did have borderline significance (p = 0.047) (Figure 5B). Regression analysis reinforced the result that G614 status was not associated with greater levels of hospitalization but that higher age (Dowd et al., 2020; Promislow, 2020), male sex (Conti and Younes, 2020; Promislow, 2020) and higher Ct values (lower viral loads) were highly predictive of hospitalization. Further analysis showed that viral load was not masking a potential D614G status effect on hospitalization (STAR Methods). Univariate analysis also found highly significant associations between age and male sex and hospitalization (STAR Methods).
G614 Is Associated with Higher Infectious Titers of Spike-Pseudotyped Virus
We quantified the infectious titers of pseudotyped single-cycle vesicular stomatitis virus (VSV) and lentiviral particles displaying D614 or G614 SARS-CoV2 Spike protein. For the VSV and lentiviral pseudotypes, G614-bearing viruses had significantly higher infectious titers (2.6- to 9.3-fold increase) than their D614 counterparts; this was confirmed in multiple cell types (Figures 6A–6C). Similar results, reported recently in a preprint that has not yet been peer reviewed, also suggest that G614 increases Spike stability and membrane incorporation (Zhang et al., 2020).
Figure 6.

Viral Infectivity and D614G Associations
(A) A recombinant VSV pseudotyped with the G614 Spike grows to a higher titer than D614 Spike in Vero, 293T-ACE2, and 293T-ACE2-TMPRSS2 cells, as measured in terms of focus-forming units (ffu). ∗∗∗∗p < 0.0001 by Student’s t test in pairwise comparisons. Experiments were repeated twice, each time in triplicate. Using a GLM to assess viral infectivity of the D614 and G614 variants across cell types and to account for repeat experiments, we found that the G614 variant had an average 3-fold higher infectious titer than D614 and that this difference was highly significant (p = 9 × 10−11) (STAR Methods).
(B and C) Recombinant lentiviruses pseudotyped with the G614 Spike were more infectious than corresponding D614 S-pseudotyped viruses in (B) 293T/ACE2 (6.5-fold increase) and (C) TZM-bl/ACE2 cells (2.8-fold increase, p < 0.0001). Relative luminescence units (RLUs) of Luc reporter gene expression (Naldini et al., 1996) were standardized to the p24 content of the pseudoviruses (p24 content of pseudoviruses for 293T/ACE2 cells: D614 = 269 ng/mL, G614 = 255 ng/mL; p24 content of pseudoviruses for TZM-bl/ACE2 cells, D614 = 680 ng/mL, G614 = 605 ng/mL). Background RLUs were measured in wells that received cells but no pseudovirus.
(D and E) Convalescent serum from six individuals in San Diego (Donors A–F) can neutralize D614-bearing (orange) and G614-bearing (blue) VSV pseudoviruses. Percent relative infection is plotted versus log polyclonal antibody concentration. Error bars indicate the mean ± SD of two biological replicates, each having two technical replicates. In (D), the sensitivity to each form is shown seperately for each sera. In (E) the responses to all sera are combined in one graph, and two negative control normal human sera are indicated in grey.
TMPRSS2, a type-II transmembrane serine protease, cleaves the viral Spike after receptor binding to enhance entry of MERS-CoV, SARS-CoV, and SARS-CoV-2 (Hoffmann et al., 2020b; Kleine-Weber et al., 2018; Matsuyama et al., 2020; Millet and Whittaker, 2014; Park et al., 2016; Shulla et al., 2011; Zang et al., 2020). Spike 614 is in a pocket adjacent to the fusion peptide near the expected TMPRSS2 cleavage site, suggesting that there could be differences in the propensity and/or requirement for TMPRSS2 of the G614 variant. To test this hypothesis, we infected 293T cells stably expressing the ACE2 receptor in the presence or absence of TMPRSS2 and quantified the titer of infectious virus. We found similar fold changes in titers between D614 and G614 regardless of TMPRSS2 expression (Figure 6A). Hence, entry of G614-bearing viruses into 293T-ACE2 cells compared with D614-bearing viruses is not enhanced by TMPRSS2. Further studies are required to determine whether the G614 variant shows increased titers in lung cells, which may recapitulate native protease expression levels more faithfully, and to determine whether this variant increases the fitness of authentic SARS-CoV-2.
We also tested whether the D614G variations would be similarly neutralized by a polyclonal antibody. Convalescent sera of six San Diego residents, likely infected in early to mid-March 2020, when D614 and G614 were circulating, demonstrate equivalent or better neutralization of a G614-bearing pseudovirus compared with a D614-bearing pseudovirus (Figures 6D and 6E). Although we do not know with which virus each of these individuals were infected, these initial data suggest that, despite increased fitness in cell culture, G614-bearing virions are not intrinsically more resistant to neutralization by convalescent sera.
Additional Sites of Interest in the Spike Gene with Rare Mutations
Spike has very few mutations overall. A small set has reached 0.3% or more of the global population sample, the threshold for automatic tracking at the https://cov.lanl.gov website (Figures 7A and 7B; details are provided in Table S2). Regions in the alignment where entropy is relatively high compared with the rest of Spike (i.e., local clusters of rare mutations) are also tracked (Table S2). Genetic mutations of interest are mapped as amino acid changes onto a Spike structure (Figure 4). The mutation resulting in the signal peptide L5F change recurs many times in the tree and is stably maintained in about 0.6% of the global GISAID data. There are several clusters of mutations in the region of the Spike gene encoding the N-terminal domain (NTD) and RBD that are potential targets for neutralizing antibodies (Chen et al., 2017; Zhou et al., 2019; Sui et al., 2008; Tang et al., 2014; ter Meulen et al., 2006). The RBD cluster (positives 475–483) spans two positions, 475 and 476, that are located within 4 Å of bound ACE2 (Figure 4D; Yan et al., 2020). The fusion peptide contains a cluster of amino acid changes between 826–839; this cluster is highlighted in Figure 7 to illustrate our web-based tools for tracking variation (Figures 7A–7C). The fusion core of HR1 (Xia et al., 2020), next to the helix break in pre-fusion Spike, also contains a cluster of amino acid changes between 936-940 (Figure 4E). The motif SXSS (937–940) may enhance the association of helices (Dawson et al., 2002; Salamango and Johnson, 2015). The cytoplasmic tail of Spike also contains a site of interest, P1263L.
Figure 7.

Tracking Variation in Spike
(A) Spike sites of interest (with a minimum frequency of 0.3% variant amino acids) are mapped onto a parsimony tree (for D614G; Figure S6). L5F recurs throughout the tree and is often clustered in small local clades. A829T is found in a single lineage. Other sites of interest cluster in a main lineage but are occasionally found in other parts of the tree in distant geographic regions and, thus, likely to recur at a low level. Build parsimony trees. A brief parsimony search (parsimony ratchet with 5 replicates) is performed with “oblong” (Goloboff, 2014). This is intended as an efficient clustering procedure rather than an explicit attempt to achieve accurate phylogenetic reconstruction, but it appears to yield reasonable results in this situation of a very large number of sequences with a very small number of changes, where more complex models may be subject to overfitting. When multiple most-parsimonious trees are found, only the shortest of these (under a p-distance criterion) is retained. Distance scoring is performed with PAUP∗ (Swofford, 2003).
(B) The global frequency of amino acid variants in sites of interest and the place where they are most commonly found. Such information could be useful if a vaccine or antibody is intended for use in a geographic region with a commonly circulating variant, so it could be experimentally evaluated prior to testing the planned intervention.
(C) Examples of exploratory plots showing A829T in Thailand and D839Y in New Zealand. Such plots for any variant in any region can be readily created at https://cov.lanl.gov/ to enable monitoring local frequency changes.
(D) Contiguous regions of relatively high entropy in the Spike alignment, indicative of local clusters of amino acid variation in the protein. The fusion-peptide cluster is used as example. It spans two sites of interest, labeled in blue and purple in (B) and (C). The alignment is created with AnalyzeAlign. Contemporary versions of these figures can be created at https://cov.lanl.gov/. Care should be taken to try to avoid systematic sequencing errors and processing artifacts among rare variants (for example, see Figure S7; De Maio et al., 2020; Freeman et al., 2020).
Discussion
Our data show that, over the course of 1 month, the variant carrying the D614G Spike mutation became the globally dominant form of SARS-CoV-2. Phylogenetic tracking of SARS-CoV-2 variants at Nextstrain reveals complex webs of evolutionary and geographical relationships (https://nextstrain.org; Hadfield et al., 2018); travelers globally dispersed G614 variants and likely would have introduced and reintroduced G614 variants into different locations. Still, D614 prevalent epidemics were very well established in many locations when G614 first began to appear (see Figure S2 for examples). The mutation that causes the D614G amino change is transmitted as part of a conserved haplotype defined by 4 mutations that almost always track together (Figures S5 and S6). The pattern of increasing G614 frequency within many different populations where D614 and G614 were co-circulating is highly significant, suggesting that G614 may be under positive selection (Figures 1B and 3). We also found G614 to be associated with higher levels of viral nucleic acid in the upper respiratory tract in human patients (Figure 5), suggestive of higher viral loads, and with higher infectivity in multiple pseudotyping assays (Figure 6).
Figure S6.

Distribution of A23403G (D614G) Mutation and Other Mutations on an Approximate Phylogenetic Tree Using Parsimony, Related to Figure 7
This tree the same as the tree shown in Figure 6A, but highlights complementary information: the G614 substitution, and patterns of bases that underly the clades. It is based on the “FULL” alignment of 17,760 sequences, from the June 2 alignment, described in the pipeline in Figure S1, from the beginning of (orf1ab) to the last stop-codon (ORF10), NC_045512 bases 266-29,674). The outer element is a radial presentation of a full-coding-region parsimony tree; branches are colored by the global region of origin for each virus isolate. The inner element is a radial bar chart showing the identity of common mutations (any of the top 20 single-nucleotide mutations from the June 2 alignment), so that sectors of the tree containing a particular mutation at high frequency are subtended by an inner colored arc; mutations not in the top 20 are presented together in gray. The tree is rooted on a reference sequence derived from the original Wuhan isolates (GenBank accession number NC_045512), at the 3 o’clock position. Branch ends representing sequence isolates bearing the D614G change are decorated with a gray square; sectors of the tree containing that mutation are subtended by a dark blue arc in the inner element; other mutations are denoted by different colors. As an example, in this tree, the region from approximately 12:30 to 3 o’clock represents GISAID’s “GR” clade, defined both by mutations we are tracking in this paper that carry the G614 variant (the GISAID G clade, defined by mutations A23403G, C14408T, C3037T, and a mutation in the 5′ UTR (C241T, not shown here), and an additional 3-position polymorphism: G28881A + G28882A + G28883C. These base substitutions are contiguous and result two amino acid changes, including N-G204R, hence GISAID’s “GR clade” name. Close examination of this triplet in sequences from the Sheffield dataset suggests the mutations are not a sequencing artifact. The outer phylogenetic tree was computed using oblong (see STAR Methods), and plotted with the APE package in R. The inner element is a bar chart plotted with polar coordinates using the gglot2 package in R. The frequency of the GR clade appears to be increased in the UK and Europe as a subset of the regional G clade expansion, given that both carry G614D.
Given that most G614 variants belong to the G clade lineage, phylogenetic methods that depend on recurrence of mutational events for their signal are poorly powered to resolve whether D614G is under positive selection. The GISAID data, however, provided the opportunity to look into the relationships among the SARS-CoV-2 variants in the context of time and geography, enabling us to track the increase in frequency of G614 as an early indicator of possible positive selection. This approach is potentially subject to founder effects and sampling biases, and so we generally view this strategy as simply an early indicator of an amino acid change that should be monitored further and tested. The G614 variant stood out, however, in our early detection framework for several reasons. First was the consistency of increase across geographic regions, which was highly significantly non-random (Figures 1B and 3). Second, if the two forms were equally likely to propagate, then one would expect the D614 form to persist in many locations where the G614 form was introduced into the ongoing well-established D614 epidemics. Instead, we found that, even in such cases, G614 increased (Figures 1, 2, 3, S2, and S3). Third, the increase in G614 frequency often continued well after national stay-at-home orders were in place, when serial reseeding from travelers was likely to be reduced significantly (Figures 2, S2, and S3).
Our global tracking data show that the G614 variant in Spike has spread faster than D614. We interpret this to mean that the virus is likely to be more infectious, a hypothesis consistent with the higher infectivity observed with G614 Spike-pseudotyped viruses we observed in vitro (Figure 6) and the G614 variant association with higher patient Ct values, indicative of potentially higher in vivo viral loads (Figure 5). Interestingly, we did not find evidence of G614 effects on disease severity; i.e., it was not significantly associated with hospitalization status. However, an association between the G614 variant and higher fatality rates has been reported in a comparison of mortality rates across countries, although this kind of analysis can be complicated by different availability of testing and care in different nations (Becerra-Flores and Cardozo, 2020).
Although higher infectiousness of the G614 variant may fully account for its rapid spread and persistence, other factors should also be considered. These include epidemiological factors because viral spread also depends on whom it infects, and epidemiological influences can also cause changes in genotype frequency to mimic evolutionary pressures. In all likelihood, a combination of evolutionary selection for G614 and the founder’s effects of being introduced into highly mobile and connected populations may have together contributed, in part, to its rise. The G-clade mutations in the 5′ UTR or in the RdRP protein might also have effects. In addition, there could be immunological consequences resulting from the G614 change in Spike. The G614 variant is sensitive to neutralization by polyclonal convalescent serum (Figure 5), which is encouraging in terms of immune interventions, but it will be important to determine whether the D614 and G614 forms of SARS-CoV-2 are differentially sensitive to neutralization by vaccine-elicited antibodies or by antibodies produced in response to infection with either form of the virus. Also, if the G614 variant is indeed more infectious than the D614 form (Figure 6), then it may require higher antibody levels for protection by vaccines or antibody therapeutic agents than the D614 form. Antibodies against an immunodominant linear epitope spanning Spike 614 in SARS-CoV-1 were associated with ADE activity (Wang et al., 2016), and so it is possible that this mutation may affect ADE.
Tracking mutations in the Spike gene has been our primary focus to date because of its relevance to vaccine and antibody-based therapy strategies currently under development. Such interventions take months to years to develop. For the sake of efficiency, contemporary variation should be factored in during development to ensure that the interventions will be effective against circulating variants when they are eventually deployed. To this end, we built a data analysis pipeline to enable exploration of potentially interesting mutations on SARS-CoV-2 sequences. The analysis is updated daily as the data become available through GISAID, enabling experimentalists to make use of the most current data available to inform vaccine development, reagents for evaluating antibody response, and experimental design. The speed with which the G614 variant became the dominant form globally suggests a need for continued vigilance.
Limitations of Study
Shifts in frequency toward the G614 variant in any given geographic region could, in principle, result from founder effects or sampling biases; it was the consistency of this pattern across regions where both forms of the virus were initially co-circulating that led us to suggest that the G614 form might be transmitted more readily because of an intrinsic fitness advantage; however, systematic biases across many regions could affect the levels of significance we observed. The lack of association between G614 and hospitalization we report may miss effects on disease severity that are more subtle than we can detect. The experimental approach taken here to acquire laboratory evidence of increased fitness of the D614G mutation is based on two different pseudovirus models of infection in established cell lines. The extent to which this model faithfully recapitulates wild-type virus infection in natural target cells of the respiratory system is still being determined, and our laboratory experiments do not directly address the biology and mechanics of natural transmission. Infectiousness and transmissibility are not always synonymous, and more studies are needed to determine whether the D614G mutation actually led to an increase in number of infections and not just higher viral loads during infection. We encourage others to study this phenomenon in greater detail with a wild-type virus in natural infection and varied target cells (Hou et al., 2020) and in relevant animal models. Finally, the neutralization assays performed were based on sera from SARS-CoV-2 infected individuals with an unknown D614G status. Thus, although they show that the G614 variants are neutralization sensitive, more work is needed to resolve whether the potency of neutralization is affected when the variant that initiated the immune response differs from the test variant or when monoclonal antibodies are used.
Consortia
The members of the Sheffield COVID-19 Genomics Group are Adrienne Angyal, Rebecca L. Brown, Laura Carrilero, Katie Cooke, Alison Cope, Thushan I. de Silva, Mark Dunning, Cariad M. Evans, Luke R. Green, Danielle C. Groves, Hailey Hornsby, Katie J. Johnson, Sokratis Karitois, Alexander J. Keeley, Katjusa Koler, Benjamin B. Lindsey, Matthew D. Parker, Paul J. Parsons, David G. Partridge, Mohammad Raza, Sarah Rowland-Jones, Nikki Smith, Rachel M. Tucker, Dennis Wang, and Matthew D. Wyles.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Polyclonal human sera | This study | N/A |
| Bacterial and Virus Strains | ||
| VSV-ΔG-GFP | Karafast | Cat# EH1020 |
| rVSV-SARS-CoV-2 | This study | N/A |
| rVSV-SARS-CoV-2 – D614G | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fugene 6 | Promega | Cat# E2692 |
| TransIT-LT1 | Mirus | Cat# MIR 2304 |
| PFA | Electron Microscopy Sciences | Cat# 15710 |
| Hoechst | ThermoFisher Scientific | Cat# 62249 |
| SuperScript IV (50rxn | ThermoFisher Scientific | Cat# 18090050 |
| dNTP mix (10mM each) | ThermoFisher Scientific | Cat# R0192 |
| Random hexamers (50uM) | ThermoFisher Scientific | Cat# N8080127 |
| RNase OUT | ThermoFisher Scientific | Cat# 10777019 |
| Q5 High-fidelity polymerase | New England Biolabs | Cat# M0491S |
| Critical Commercial Assays | ||
| Promega Luciferase Assay System | Promega | Cat# E1501 |
| Britelite Plus Reporter Gene Assay System | Perkin-Elmer Part | Cat# 6066769 |
| MagnaPure96 extraction platform | Roche Diagnostics Ltd, Burgess Hill, UK | Cat# 06 543 588 001 |
| SensiFASTTM Probe No-ROX One-Step Real-time PCR kit | Bioline | Cat# BIO-76001 |
| Ligation sequencing kit | Oxford Nanopore | Cat# SQK-LSK109 |
| Native barcoding expansion kit 13-24 | Oxford Nanopore | Cat# EXP-NBD114 |
| Native barcoding expansion kit1-12 | Oxford Nanopore | Cat# EXP-NBD104 |
| Flow cell priming kit | Oxford Nanopore | Cat# EXP-FLP002 |
| Flow cells R9.4.1 48pk | Oxford Nanopore | Cat# FLO-MIN106D |
| Flow cell wash kit | Oxford Nanopore | Cat# EXP-WSH003 |
| SFP Expansion kit | Oxford Nanopore | Cat# EXP-SFB001 |
| Next Ultra II library prep kit (illumina) | New England Biolabs | Cat# E7645L |
| Quick ligation module | New England Biolabs | Cat# E6056L |
| HIV-1 p24 ELISA | Perkin-Elmer | Cat# NEK050B |
| Deposited Data | ||
| COVID-19 Data Repository by the Center for Systems Science and Engineering (CSSE) | Johns Hopkins University | https://github.com/CSSEGISandData/COVID-19/blob/master/csse_covid_19_data/csse_covid_19_time_series/time_series_covid19_confirmed_US.csv |
| GISAID | Elbe and Buckland-Merrett, 2017; Shu and McCauley, 2017 | https://www.gisaid.org |
| STAY-AT-HOME ORDERS IN EUROPE | https://www.sidley.com/-/media/uploads/stay-at-home-tracker_europe.pdf?la=en | |
| Experimental Models: Cell Lines | ||
| HEK293T/17 cells | ATCC | Cat# CRL-11268 |
| 293T/ACE2.MF | Dr. Michael Farzan and Huihui Mu | N/A |
| TZM-bl/ACE2.MF | Dr. Michael Farzan and Huihui Mu | N/A |
| 293T | ATCC | Cat# CRL-3216 |
| Vero | ATCC | Cat# CCL-81 |
| 293T-Ace2 | This study | N/A |
| 293T-Ace2-TMPRSS2 | This study | N/A |
| Oligonucleotides | ||
| Primer pool1 and Primer Pool2 for sequencing (98 oligonucleotides | Artic Network | https://artic.network/ncov-2019 |
| Recombinant DNA | ||
| VRC7480 (Spike plasmid) | Drs. Barney Graham and Kizzmekia Corbett | N/A |
| VRC7480.D614G (Spike plasmid) | This study | N/A |
| pCMV ΔR8.2 (lentiviral backbone) | Drs. Barney Graham and Kizzmekia Corbett | N/A |
| pHR’ CMV Luc (luciferase reporter) | Drs. Barney Graham and Kizzmekia Corbett; Naldini et al., 1996 | N/A |
| pSG3ΔEnv | Drs. Beatrice Hahn and Feng Gao | N/A |
| Empty vector: phCMV3 | Genlantis | Cat# P003300 |
| pCAGGS-VSV-G | Kerfast | Cat# EH1017 |
| phCMV3-SARS-CoV-2 | This study, Spike cloned from synthetic, codon optimized DNA | N/A |
| phCMV3-SARS-CoV-2 – D614G | This study, generated through site-directed mutagenesis | N/A |
| Software and Algorithms | ||
| R | The R Foundation for Statistical Computing | http://www.R-project.org |
| Nanopolish | © Ontario Institute for Cancer Research 2015 MPL liscense | https://github.com/jts/nanopolish |
| R packages: phangorn (version 2.5.5), ggplot2 (version 3.3.0), beeswarm (version 0.2.0), tidyverse (version 1.3.0), ape (version 5.3), lme4 (version 1.1.21) | The R Foundation for Statistical Computing | https://cran.r-project.org/ |
| data.table (version 1.12.8) | Matt Dowle | https://github.com/Rdatatable/data.table |
| Aliview | Anders Larsson | https://ormbunkar.se/aliview/ |
| cgam (version 1.14) | Xiyue Liao, Mary C. Meyer | https://www.jstatsoft.org/htaccess.php?volume=089&type=i&issue=05 |
| ARTIC network protocol (accessed the 19th of April) | ARTIC network | https://artic.network/ncov-2019 |
| Python Matplotlib A 2D Graphics Environment v 3.2.2 | Hunter, 2007 | https://matplotlib.org |
| The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC | Schrödinger | https://pymol.org/2/ |
| Oblong | Goloboff, 2014 | http://www.lillo.org.ar/phylogeny/oblong/ |
| Los Alamos National Lab HIV Database: Analyze Align and Entropy | Los Alamos National Lab | http://cov.lanl.govcontent/index |
| Los Alamos National Lab SARS-CoV-2 Analysis Pipeline: SARS-CoV-2 map, Relative Frequency Change by Geographical Region, Rainbow Tree | This study | http://cov.lanl.govcontent/index |
| PAUP | David Swofford | https://paup.phylosolutions.com |
| GraphPad Prism 8 | GraphPad Software, Inc | https://www.graphpad.com |
| Other | ||
| Published primers and probes for the SARS-CoV-2 E-gene RT- qPCR SARS-CoV-2 | Corman et al., 2020 | N/A |
| ABI Thermal Cycler | Applied Biosystems, Foster City, United States | Cat# 4375305 |
| CellInsight CX5 High Content Screening Platform | ThermoFisher Scientific | Cat# CX51110 |
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Bette Korber (btk@lanl.gov).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All sequence data used here are available from The Global Initiative for Sharing All Influenza Data (GISAID), at https://www.gisaid.org/. The user agreement for GISAID does not permit redistribution of sequences. Other data have been deposited to Mendeley Data: https://doi.org/10.17632/hn3h9gdrgj.1.
Web-based tools to recreate much of the analyses provided in this paper but based on contemporary GISAID data downloads are available at https://cov.lanl.gov/.
Code to create the alignments as described in Figure S1 and to perform the Isotonic regression analysis in Figure 3 will be available through https://cov.lanl.gov, at also GitHub, once permission from our funders is obtained.
Experimental Model and Subject Details
Human Subjects
999 individuals presenting with active COVID-19 disease were sampled for SARS CoV-2 sequencing at Sheffield Teaching Hospitals NHS Foundation Trust, UK using samples collected for routine clinical diagnostic use. This work was performed under approval by the Public Health England Research Ethics and Governance Group for the COVID-19 Genomics UK consortium (R&D NR0195). SARS-CoV-2 sequences were generated using samples taken for routine clinical diagnostic use from 999 individuals presenting with active COVID-19 disease: 593 female, 399 male, 6 no gender specified; ages 15-103 (median 55) years.
Method Details
Detection and Sequencing of Sars-Cov-2 Isolates from Clinical Samples
Samples for PCR detection of SARS-CoV-2 (Figure 5A) were all obtained from either throat or combined nose/throat swabs. Nucleic acid was extracted from 200μl of sample on MagnaPure96 extraction platform (Roche Diagnostics Ltd, Burgess Hill, UK). SARS-CoV-2 RNA was detected using primers and probes targeting the E gene and the RdRp genes for routine clinical diagnostic purposes, with thermocycling and fluorescence detection on ABI Thermal Cycler (Applied Biosystems, Foster City, United States) using previously described primer and probe sets (Corman et al., 2020). Nucleic acid from positive cases underwent long-read whole genome sequencing (Oxford Nanopore Technologies (ONT), Oxford, UK) using the ARTIC network protocol (accessed the 19th of April; https://artic.network/ncov-2019) Following base calling, data were demultiplexed using ONT Guppy using a high accuracy model. Reads were filtered based on quality and length (400 to 700bp), then mapped to the Wuhan reference genome and primer sites trimmed. Reads were then downsampled to 200x coverage in each direction. Variants were called using nanopolish (https://github.com/jts/nanopolish) and used to determine changes from the reference. Consensus sequences were constructed using reference and variants called.
Pseudotyped Virus Infectivity
VSV System
Plasmids for full-length SARS-Cov-2 Spike were generated from synthetic codon-optimized DNA (Wuhan-Hu-1 isolate, GenBank: MN908947.3) through sub-cloning into the pHCMV3 expression vector, with a stop codon included prior to the HA tag. The D614G variant was generated by site-directed mutagenesis. Positive clones were fully sequenced to ensure that no additional mutations were introduced.
Lentiviruses for stable cell line production were generated by seeding 293T cells at a density of 1x106 cells/well in a 6-well dish. Once the cells reached confluency, they were transfected with 2ug pCaggs-VSV-G, 2ug of lentiviral packaging vector pSPAX2, and 2ug of lentiviral expression plasmid pCW62 encoding ACE2-V5 and the puromycin resistance gene (pCW62-ACE2.V5-PuroR) or TMPRSS2-FLAG and the blasticidin resistance gene (pCW62-TMPRSS2.FLAG-BlastR) using Trans-IT transfection reagent according to manufacturer’s instructions. 24 hours post-transfection, media was replaced with fresh DMEM containing 10% FBS and 20mM HEPES. 48 hours post-transfection, supernatants were collected and filtered using a 0.45um syringe filter (VWR Catalog #28200-026).
293T-ACE2 cells were generated by seeding 293T cells at a density of 1x106 cells/well in a 6-well dish. At confluency, cells were transduced with 100uL of ACE2.V5-PuroR lentivirus. 48 hours post-transduction, cells were placed under 5ug/ml puromycin. 293T-ACE2+TMPRSS2 cells were generated by seeding 293T-ACE2 cells at a density of 1x106 cells/well in a 6-well dish. At confluency, cells were transduced with 100uL of TMPRSS2.FLAG-BlastR lentivirus. 48 hours post-transduction, cells were placed under 10ug/ml blasticidin selection.
Recombinant SARS-CoV-2-pseduotyped VSV-ΔG-GFP were generated by transfecting 293T cells with phCMV3 expressing the indicated version of codon-optimized SARS-CoV-2 Spike using TransIT according to the manufacturer’s instructions. At 24 hr post-transfection, the medium was removed, and cells were infected with rVSV-G pseudotyped ΔG-GFP parent virus (VSV-G∗ΔG-GFP) at MOI = 2 for 2 hours with rocking. The virus was then removed, and the cells were washed twice with OPTI-MEM containing 2% FBS (OPTI-2) before fresh OPTI-2 was added. Supernatants containing rVSV-SARS-2 were removed 24 hours post-infection and clarified by centrifugation.
Viral titrations were performed by seeding cells in 96-well plates at a density sufficient to produce a monolayer at the time of infection. Then, 10-fold serial dilutions of pseudovirus were made and added to cells in triplicate wells. Infection was allowed to proceed for 12-16 hr at 37°C. The cells were then fixed with 4% PFA, washed two times with 1xPBS and stained with Hoescht (1ug/mL in PBS). After two additional washes with PBS, pseudovirus titers were quantified as the number of fluorescent forming units (ffu/mL) using a CellInsight CX5 imager (ThermoScientific) and automated enumeration of cells expressing GFP.
Lentiviral System
Additional assessments of corresponding D614 and G614 Spike pseudotyped viruses were performed by using lentiviral vectors and infection in 293T/ACE2.MF and TZM-bl/ACE2.MF cells (both cell lines kindly provided by Drs. Mike Farzan and Huihui Mu at Scripps). Cells were maintained in DMEM containing 10% FBS, 1% Pen Strep and 3 ug/ml puromycin. An expression plasmid encoding codon-optimized full-length spike of the Wuhan-1 strain (VRC7480), was provided by Drs. Barney Graham and Kizzmekia Corbett at the Vaccine Research Center, National Institutes of Health (USA). The D614G amino acid change was introduced into VRC7480 by site-directed mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit from Agilent Technologies (Catalog # 210518). The mutation was confirmed by full-length spike gene sequencing. Pseudovirions were produced in HEK293T/17 cells (ATCC cat. no. CRL-11268) by transfection using Fugene 6 (Promega Cat#E2692). Pseudovirions for 293T/ACE2 infection were produced by co-transfection with a lentiviral backbone (pCMV ΔR8.2) and firefly luciferase reporter gene (pHR’ CMV Luc) (Naldini et al., 1996). Pseudovirions for TZM-bl/ACE2 infection were produced by co-transfection with the Env-deficient lentiviral backbone pSG3ΔEnv (kindly provided by Drs Beatrice Hahn and Feng Gao). Culture supernatants from transfections were clarified of cells by low-speed centrifugation and filtration (0.45 μm filter) and used immediately for infection in 96-well culture plates. 293T/ACE2.MF cells were preseeded at 5,000 cells per well in 96-well black/white culture plates (Perkin-Elmer Catalog # 6005060) one day prior to infection. Sixteen wells were inoculated with 50 ul of a 1:10-dilution of each pseudovirus and incubated for three days. Luminescence was measured using the Promega Luciferase Assay System (Catalog # E1501). For infection of TZM-bl/ACE2.MF cells, 10,000 freshly trypsinized cells were added to 16 wells of a 96-well clear culture plate (Fisher Scientific) and inoculated with undiluted pseudovirus. Luminescence was measured after 2 days in a solid black plate using the Britelite Plus Reporter Gene Assay System (Perkin-Elmer). Luminescence in both assays was measured using a PerkinElmer Life Sciences, Model Victor2 luminometer. HIV-1 p24 content (produced by the backbone vectors) was quantified using the Alliance p24 ELISA Kit (PerkinElmer Health Sciences, Cat# NEK050B001KT). Reported relative luminescence units (RLUs) were adjusted for p24 content.
Neutralization Assay
Pre-titrated amounts of rVSV-SARS-CoV-2 (D614 or G614 variant) were incubated with serially diluted human sera at 37°C for 1 hr before addition to confluent Vero monolayers in 96-well plates. Infection proceeded for 12-16 hr at 37°C in 5% CO2 before cells were fixed in 4% paraformaldehyde and stained with 1ug/mL Hoescht. Cells were imaged using a CellInsight CX5 imager and infection was quantitated by automated enumeration of total cells and those expressing GFP. Infection was normalized to the percent cells infected with rVSV-SARS-CoV-2 incubated with normal human sera. Data are presented as the relative neutralization for each concentration of sera.
Data Pipeline
Background and General Approach
The Global Initiative for Sharing All Influenza Data (GISAID) (Elbe and Buckland-Merrett, 2017; Shu and McCauley, 2017) has been coordinating SARS-CoV-2 genome sequence submissions and making data available for download since early in the pandemic. At time of this writing, hundreds of sequences were being added every day. These sequences result from extraordinary efforts by a wide variety of institutions and individuals: while an invaluable resource, but are mixed in quality. The complete sequence download includes a large number of partial sequences, with variable coverage, and extensive ‘N’ runs in many sequences. To assemble a high-quality dataset for mutational analysis, we constructed a data pipeline using some off-the-shelf bioinformatic tools and a small amount of custom code.
From theSARS-CoV-2 sequences available from GISAID, we derived a “clean” codon-aligned dataset comprising near-complete viral genomes, without large insertions or deletions (“indels”) or runs of undetermined or ambiguous bases. For convenience in mutation assessment, we generated a codon-based nucleotide multiple sequence alignment, and extracted translations of each reading frame, from which we generated lists of mutations. The cleaning process was in general a process of deletion, with alignment of retained sequences; the following criteria were used to exclude sequences:
-
1
Fragmented matching (> 20 nt gap in match to reference)
-
2
Gaps at 5′ or 3′ end (> 3 nt)
-
3
High numbers of mismatched nucleotides (> 20), ‘N’ or other ambiguous IUPAC codes.
-
4
Regions with concentrated ambiguity calls: > 10 in any 50 nt window)
Any sequence matching any of the above criteria was excluded in its entirety.
Sequence Mapping and Alignment
Sequences were mapped to a reference (bases 266:29674 of GenBank entry NC_045512; i.e., the first base of the ORF1ab start codon to the last base of the ORF10 stop codon) using “nucmer” from the MUMmer package (version 3.23; Kurtz et al., 2004). The nucmer output “delta” file was parsed directly using custom Perl code to partition sequences into the various exclusion categories (Sequence Mapping Table) and to construct a multiple sequence alignment (MSA). The MSA was refined using code derived from the Los Alamos HIV database “Gene Cutter” tool code base. At this stage, alignment columns comprising an insertion of a single “N” in a single sequence (generating a frameshift) were deleted, and gaps were shifted to conform with codon boundaries.
Using the initial “good-sequence” alignment, a low-effort parsimony tree was constructed. Initially, trees were built using PAUP∗ (Swofford, 2003) with a single replicate heuristic search using stepwise random sequence addition; subsequently, a parsimony ratchet was added; currently, oblong (Goloboff, 2014) is used. Sequences in the alignment were sorted vertically to correspond to the (ladderized) tree, and reference-sequence reading frames were added. See Figure S1 for a pipeline schematic.
Data partitioning and phylogenetic trees
Alignments were made and trees inferred for three distinct data partitions, the longer the alignments, the fewer sequences the sequences (Figure S1.) The full genome tree was used for Figure 7. Trees were inferred by either of two methods: 1. neighbor-joining using a p-distance criterion, (Swofford, 2003) or 2. parsimony heuristic search using a version of the parsimony ratchet (Goloboff, 2014), the general conclusions in Figure 7 were substantiated in both; the parsimony tree is shown.
Global Maps
The Covid-19 pie chart map is generated by overlaying Leaflet (a JavaScript library for interactive maps) pie charts on maps provided by OpenStreetMap. The interface is presented using rocker/shiny, a Docker for Shiny Server.
Quantification and Statistical Analysis
Systematic Regional Analysis of D614/G614 Frequencies
To observe a significant change in the frequency of two SARS-CoV-2 variants in a geographic region, three minimal requirements must be met. Both variants must have been introduced into an area and be co-circulating, data must be sampled for a long enough period to observe a change in frequency, and there must be enough data to be powered adequately to detect a difference.
We use the bioinformatic approaches described above to extract from GISAID all the politically defined geographic regions within the data that met these criteria, to track changes in frequency in a systematic way using all available data. The political/geographical regions we use are strictly hierarchically segmented based on the naming conventions used in GISAID. GISAID data is labeled such that the geographic source is noted first as a continent or Oceania; we call this Level 1. Level 2 is the country of origin of a sample. Level 3 are subcountries and states, and although occasionally level 3 includes a major city in a small country. For this purpose, England, Scotland, and Wales are considered sub-countries of the United Kingdom, and assigned level 3; the sampling in the UK has been the most extensive globally to date. Level 4, is the county or city of origin. The levels are strictly hierarchical, and within a given level, the geographical regions do not overlap. In some cases (e.g., Nepal_Kathmandu and Nepal, Greece_Athens and Greece, Italy_Veneto_Verona and Italy_Veneto, or Iceland_Reykjavik and Iceland) the sampling in a sub-level exactly matches the sampling in the corresponding upper level, in which case the sub-level is not presented. Levels 3 and 4 are not always available, and the day of sampling is also not always available.
The statistical strategies we use are then applied separately in each country, region or city, and we do not assume that outbreaks in each political subdivision are independent and identically distributed. Instead, our model assumption is that the individuals we test within a region are independent. This assumption may fail if there are sampling biases in a region that change over a given period of time. The G614 form is part of the G clade haplotype that is introduced by travelers, as we discuss in the text, and it is rare for it to arise independently. Our null hypothesis is that the observed shifts in frequency are random nondirectional drift. We have taken two statistical approaches to test this.
Fisher’s exact comparison
For this comparison, we used a two-sided Fisher’s exact test to compare the G614 and D614 counts in the pre-onset and the post-delay periods, as described in the text, and provides a p value against the null hypothesis that the fraction of D614 and G614 sequences did not change. To be included in the analysis, 15 sequences were required pre-onset, with a mixture of D614 and G614 present such that the rarer form was present at least 3 times; we also required a minimum of 15 sequences be sampled at least 2 weeks later, to create a post-delay set. Only regions for which p < 0.05 are considered, based on a two sided-test. We then use a binomial test to evaluate the null hypothesis that in regions where we saw significant change in sampling frequency over time, the shift was as likely to be an increase or a decrease in G614 across geographic regions. This analysis is presented in Figure 1B.
Isotonic Regression
Isotonic regression forms the basis of a one-sided test of the hypothesis for positive selection based on fitting the indicator that the typed strain is G as a logistic regression in which the logarithm of the odds ratio is a non-decreasing function of time. We use the residual deviance of the fitted model as our test statistics. To be included in this analysis, a region was required to have at least 5 sequences each of D614 and G614, and a minimum of 14 sampling days of data available. While we have a composite null hypothesis (the log-odds ratio is non-increasing), assuming that the log-odds ratio remains constant over time leads to tests that have largest power. While the classical chi-square approximation does not hold, we can sample from the constant log-odds ratio by permuting the vector of variant labels, and refitting the isotonic logistic regression. We performed 400 randomizations of the data in each region. Hence the lowest p value we can obtain is 0.0025. The reverse hypothesis, namely than the fraction of G variant decreases with time is also tested by fitting a non-increasing function of time. The isotonic logistic regression was done using R and the cgam package. We applied the bionomial test across regions with a significant change in one direction, as we did for the Fisher's test results. This analysis is presented in Figure 3 and Data S1.
Clinical Data and Modeling
Baseline Comparisons of Clinical Parameters
Univariate analysis showed no associations between the age of individuals and their D614 (median 54.8, IR 39.4-77) or G614 status (median 54.6 (38.7-72.8) (Wilcoxon rank sum p = 0.37), nor with D614 and G614 and sex (Fisher’s exact p = 0.32). Comparing hospitalization and age, the median (IR) are: for all hospitalized, (IP+OCU), 74 years (59-83); for all OP, 44 (32-54), Wilcoxon p < 2.2e-16.
67% of males were hospitalized, versus 33% of females (Fisher’s exact p = p value < 2.2e-16).
Modeling PCR Ct
Two PCR Ct methods were used as a surrogate for estimating in vivo viral load in the upper respiratory tract, switching methods in mid-April due a shortage of kits. The first method involved nucleic acid extraction; the second method, heat treatment (Fomsgaard and Rosenstierne, 2020).
To assess the impact of available clinical parameters on viral load as measured by PCR Cts, we used a linear model, predicting Ct from PCR method, Sex, Age and D614G variant. This revealed that only the PCR method and the D614G variant were statistically significant. A negative coefficient for the G variant indicated that patients infected by the latter have, on average, a higher viral load, but that that viral load is not impacted by neither age nor sex.
The results from the smaller model are:
Coefficients:
Estimate Std. Error t value Pr(> |t|)
(Intercept) 24.301 0.3166 76.757 < 2e-16 ∗∗∗
G614 −0.7763 0.3718 −2.088 0.037 ∗
Method_2 3.1979 0.3658 8.743 < 2e-16
Results comparing D614G status for the two methods were also evaluated independently, and the first method showed a significant association between lower Ct values and presence of G614 (Wilcoxon p = 0.033), but the second method, with many fewer samples, did not reach significance.
Predicting Hospitalization
The simple Fisher’s exact test analysis in Figure 5 indicates that the D614G status is not predictive of hospitalization, even though it is predictive of viral load. We can make a first analysis to predict hospitalization from viral load, gender, age and D614G status:
Coefficients:
Estimate Std. Error z value Pr(> |z|)
(Intercept) −7.548823 0.624270 −12.092 < 2e-16 ∗∗∗
G614 0.112038 0.214107 0.523 0.600779
Male 1.490789 0.181695 8.205 2.31e-16 ∗∗∗
Age 0.089444 0.005664 15.791 < 2e-16 ∗∗∗
CT 0.069376 0.018243 3.803 0.000143 ∗∗∗
Method_2 −0.358397 0.218856 −1.638 0.101506
As somewhat expected, the D614G status is not statistically significant, even though viral load is, but the coefficient goes in the opposite direction than we would have intuited: a lower viral load is predictive of a higher probability of hospitalization. Sex (Male) and Age both increase the probability of hospitalization.
Predicting Hospitalization, revisited
Although the above analysis indicates that aa614G does not predict hospitalization directly, it does predict viral load and viral load predicts hospitalization; so there is a concern that aa614G might affect hospitalization, but that this effect is “masked” by the viral load. To explore this hypothesis, we “unmask” the aa614G by using the residuals from the regression of Ct on extraction method and D614G status to get a second predictive model for hospitalization:
Coefficients:
Estimate Std. Error z value Pr(> |z|)
(Intercept) −5.889991 0.393950 −14.951 < 2e-16 ∗∗∗
G614 0.029858 0.209349 0.143 0.886587
Corrected Ct 0.069276 0.018225 3.801 0.000144 ∗∗∗
Male 1.490690 0.181584 8.209 2.22e-16 ∗∗∗
Age 0.089714 0.005661 15.849 < 2e-16 ∗∗∗
In these regression analyses, the estimated coefficients for age, sex and viral load (corrected or not for method and strain) remain mostly unchanged, and strain still does not have an effect.
All other comparisons were not significant. All coding was done using R. Results of these analysis are presented in the main text and in Figure 5.
Modeling pseudotype virus infectivity
We used a log-normal generalized linear model (GLM) to test whether the G614 variant grew to higher titers than the wild-type D614 virus in Vero, 293T-ACE2 and 293T-ACE2-TMPRSS2 cell lines. The full experiment was repeated twice, each time in triplicate, and the 2 experimental repeats were considered random effects. Viral variant and cell line were considered as fixed effects. On average, across all cell lines, G614 grows to about a 3-fold (2.95) higher titer than D614 (p = 9x10−11). A significant interaction was found between viral variant and cell line (p = 0.002), indicating that the relative increase of G614 compared to D614 was significantly different across cell lines (p = 0.002).
Results of these analysis are presented in Figure 6A.
Sequence quality control
We discovered a sequencing processing error that gave rise to what appeared at first to be a mutation of interest at position 943 (24389 A > C and 24390 C > G) in Spike that was evident in sequences from Belgium. It was frequent enough to be a site of interest, and was tracked. We contacted the group in Belgium, the source of the data, who were already aware of the issue, concurred with our interpretation, and they had been in touch with GISAID with a request to remove the problematic sequences.
We identified the issue with this site as part of another study using a method to detect systematic sequencing errors (Freeman et al., 2020); we are interrogating the quality of available sequencing
data and these positions were highlighted as suspect. We interrogated these positions in the raw sequencing data from Sheffield, and although these two variants are not present in the final consensus sequence from any of the Sheffield isolates, the raw, untrimmed bam files show their presence in only one of the amplicons covering the site (Figure S7A and S7B). We noticed that in fact this position is to the left of the 5′ primer of amplicon 81 in what we believe to be an adaptor sequence. Comparison of the Wuhan reference and the adaptor sequence reveals similarity around this position:
Figure S7.

Investigation of S943P, Related to Figure 7
A. IGV plots showing bam files from nanopore sequencing data of amplicons produced by the Artic network protocol. Raw data from amplicon 81 contains a portion of adaptor sequence which is homologous to the reference genome, apart from the C variants which lead to a S943P mutation call. This region is therefore included in variant calling if location-based trimming is not carried out. Subsequent panels show that this region is soft clipped when trimming adapters and primers and is therefore not available for variant calling. B. Base frequencies at position 24389 in 23 samples from the Sheffield data show that C is present in half of the reads in the raw data, but is absent from trimmed and primer trimmed data. This figure is also associated with the STAR Methods.
Nanopore adaptor sequence:
CAGCACCTT
The Wuhan reference sequence:
CAGCAAGTT
In our validation set, we see a C present at around 50% of called bases at both these positions in raw data but this region is trimmed by the ARTIC pipeline and is therefore not used to call variants and contribute to the final consensus sequence. Although it is evident in amplicon 81, in this region, there is no evidence for these variants in the data from amplicon 80, which also covers these positions. We include a figure (Figure S7) to explain our finding.
In summary this is an error that has arisen due to a combination of improper trimming of adaptor and primer regions from raw sequencing reads before downstream analysis, and the coincidental homology between the nanopore adaptor sequence and the Wuhan reference genome in this region. This is included here as a cautionary note; resolving rare biological mutations and sequencing error will be an important balance going forward in terms of interpretation of rare mutations (De Maio et al., 2020). A recurrent amino acid change like L5F (Figure 7) could potentially result from a recurrent sequencing or sequence processing error (De Maio et al., 2020), or alternatively, it may be of particular interest if it is naturally recurring homoplasy.
Additional Resources
Current data updates, analytical results, and webtools: https://cov.lanl.gov
Acknowledgments
We thank Andrew McMichael, Sarah Rowland-Jones, and Xiao-Ning Xu for bringing together the clinical and theory teams. We thank Anthony West for pointing out the 5′ UTR G-clade mutation; George Ellison for suggestions regarding clinical data analyses; Barbara Imperiali for insights regarding the structural implications of the D614G change; and Rachael Mansbach, Srirupa Chakraborty, and Kien Nguyen for sharing preliminary MD data. We thank Davey Smith and Stephen Rawlings of UCSD and Alessandro Sette, Jennifer M. Dan, and Shane Crotty of LJI for survivor sera and Sharon Schendel for manuscript edits. We acknowledge Barney Graham, Kizzmekia Corbett, Nicole Doria-Rose, Adrian McDermott, and John Mascola at the Vaccine Research Center, NIH for reagents and assistance with the lentivirus-based SARS-CoV-2 infection assay and Elize Domin for technical support. The Sheffield COVID-19 Genomics Group is part of the COG-UK CONSORTIUM, supported by the Medical Research Council (MRC), part of UK Research & Innovation (UKRI), the National Institute of Health Research (NIHR), and Genome Research Limited, operating as the Wellcome Sanger Institute. T.I.d.S. is supported by a Wellcome Trust intermediate clinical fellowship (110058/Z/15/Z). M.P. was funded by the NIHR Sheffield Biomedical Research Centre (BRC). B.K., E.E.G., T.B., N.H., W.M.F., H.Y., and W.A. were supported by LANL LDRD projects 20200554ECR and 20200706ER and through NIH NIAID, DHHS Interagency Agreement AAI12007-001-00000. D.S. acknowledges support from the John and Mary Tu Foundation and the San Diego CFAR AI036214. A.S. and S.C. acknowledge support from NIH NIAID AI42742 (Cooperative Centers for Human Immunology). E.O.S. acknowledges support from CoVIC INV-006133 of the COVID-19 Therapeutics Accelerator, supported by the Bill and Melinda Gates Foundation, Mastercard, Wellcome and private philanthropic support, the Overton family, and a FastGrant from Emergent Ventures in aid of COVID-19 research. We gratefully acknowledge the team at GISAID for creating SARS-CoV-2 global database and the many people who provided sequence data (Table S1).
Author Contributions
Conceptualization, B.K. and D.C.M.; Methodology, B.K., W.M.F., J.T., N.H., E.O.S., and D.C.M.; Software, W.M.F., J.T., H.Y., W.A., N.H., E.E.G., T.B., T.M.F., M.D.P., and B.K.; Validation, E.O.S., D.C.M., J.T., B.K., B.F., and N.H.; Formal Analysis, B.K., J.T., N.H., W.M.F., S.G., M.D.P., T.M.F., D.G.P., C.M.E., T.I.d.S., T.B., and E.E.G.; Investigation, E.O.S., D.C.M., K.M.H., C.M.E., D.G.P., L.G.P., H.T., A.M.-W., S.P.W., C.C.L., and T.I.d.S.; Writing – Original Draft, B.K., W.M.F., S.G., D.C.M., and E.O.S; Writing – Review & Editing, T.I.d.S., C.C.L., E.E.G., N.H., H.Y., and T.B.; Visualization, B.K., E.O.S., J.T., N.H., W.M.F., E.E.G., and S.G.; Supervision, B.K., D.C.M., E.O.S., T.I.d.S., and S.P.W.; Funding Acquisition, B.K., E.E.G., D.C.M., S.G., E.O.S., and T.I.d.S.
Declaration of Interests
The authors declare no competing interests.
Published: July 3, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.06.043.
Contributor Information
Bette Korber, Email: btk@lanl.gov.
Sheffield COVID-19 Genomics Group:
Adrienne Angyal, Rebecca L. Brown, Laura Carrilero, Luke R. Green, Danielle C. Groves, Katie J. Johnson, Alexander J. Keeley, Benjamin B. Lindsey, Paul J. Parsons, Mohammad Raza, Sarah Rowland-Jones, Nikki Smith, Rachel M. Tucker, Dennis Wang, and Matthew D. Wyles
Supplemental Information
This is a spreadsheet with tabs including the following data: i) Sites of interest: A listing of all sequences with amino acid changes in sites of interest in Spike (those that reach > 0.3% of the global population, or if in ACE2 contact regions, > 0.1% of the population). ii) D614G: A separate tab for D614G variants. iii) Spike Variation: A tally of all amino acid variants and the overall entropy for each position in Spike, as well as the entropy for each 10 amino acid contiguous stretch across the protein, is listed (https://www.hiv.lanl.gov/content/sequence/ENTROPY/entropy.html). This table includes annotation of the RDB location and ACE2 contact sites. Regular updates of all information in Table S2 can be found at http://cov.lanl.govcontent/index.
References
- Asmal M., Hellmann I., Liu W., Keele B.F., Perelson A.S., Bhattacharya T., Gnanakaran S., Daniels M., Haynes B.F., Korber B.T. A signature in HIV-1 envelope leader peptide associated with transition from acute to chronic infection impacts envelope processing and infectivity. PLoS ONE. 2011;6:e23673. doi: 10.1371/journal.pone.0023673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra-Flores M., Cardozo T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int. J. Clin. Pract. 2020 doi: 10.1111/ijcp.13525. Published online May 6, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni M.F., Gog J.R., Andreasen V., Feldman M.W. Epidemic dynamics and antigenic evolution in a single season of influenza A. Proc. Biol. Sci. 2006;273:1307–1316. doi: 10.1098/rspb.2006.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricault C.A., Yusim K., Seaman M.S., Yoon H., Theiler J., Giorgi E.E., Wagh K., Theiler M., Hraber P., Macke J.P. HIV-1 Neutralizing Antibody Signatures and Application to Epitope-Targeted Vaccine Design. Cell Host Microbe. 2019;26:296. doi: 10.1016/j.chom.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.H., Hotez P.J., Bottazzi M.E. Potential for developing a SARS-CoV receptor-binding domain (RBD) recombinant protein as a heterologous human vaccine against coronavirus infectious disease (COVID)-19. Hum. Vaccin. Immunother. 2020;16:1239–1242. doi: 10.1080/21645515.2020.1740560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Lu S., Jia H., Deng Y., Zhou J., Huang B., Yu Y., Lan J., Wang W., Lou Y. A novel neutralizing monoclonal antibody targeting the N-terminal domain of the MERS-CoV spike protein. Emerging Microbes and Infections. 2017;6:e37. doi: 10.1038/emi.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibo D., Birch C. Analysis of human coronavirus 229E spike and nucleoprotein genes demonstrates genetic drift between chronologically distinct strains. J. Gen. Virol. 2006;87:1203–1208. doi: 10.1099/vir.0.81662-0. [DOI] [PubMed] [Google Scholar]
- Cohen J. COVID-19 shot protects monkeys. Science. 2020;368:456–457. doi: 10.1126/science.368.6490.456. [DOI] [PubMed] [Google Scholar]
- Conti P., Younes A. Coronavirus COV-19/SARS-CoV-2 affects women less than men: clinical response to viral infection. J. Biol. Regul. Homeost. Agents. 2020;34 doi: 10.23812/Editorial-Conti-3. [DOI] [PubMed] [Google Scholar]
- Corman V.M., Landt O., Kaiser M., Molenkamp R., Meijer A., Chu D.K., Bleicker T., Brünink S., Schneider J., Schmidt M.L. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25:2000045. doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispell J., Balaz D., Gordon S.V. HomoplasyFinder: a simple tool to identify homoplasies on a phylogeny. Microb. Genom. 2019;5 doi: 10.1099/mgen.0.000245. e000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson J.P., Weinger J.S., Engelman D.M. Motifs of serine and threonine can drive association of transmembrane helices. J. Mol. Biol. 2002;316:799–805. doi: 10.1006/jmbi.2001.5353. [DOI] [PubMed] [Google Scholar]
- De Maio N., Walker C., Borges R., Weilguny L., Slodkowicz G., Goldman N. Issues with SARS-CoV-2 sequencing data. 2020. https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473
- de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X., Gu W., Federman S., du Plessis L., Pybus O.G., Faria N., Wang C., Yu G., Bushnell B., Pan C.Y. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science. 2020:eabb9263. doi: 10.1126/science.abb9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd J.B., Andriano L., Brazel D.M., Rotondi V., Block P., Ding X., Liu Y., Mills M.C. Demographic science aids in understanding the spread and fatality rates of COVID-19. Proc. Natl. Acad. Sci. USA. 2020;117:9696–9698. doi: 10.1073/pnas.2004911117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauver J.R., Petrone M.E., Hodcroft E.B., Shioda K., Ehrlich H.Y., Watts A.G., Vogels C.B.F., Brito A.F., Alpert T., Muyombwe A. Coast-to-Coast Spread of SARS-CoV-2 during the Early Epidemic in the United States. Cell. 2020;181:990–996.e5. doi: 10.1016/j.cell.2020.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomsgaard A.S., Rosenstierne M.W. An alternative workflow for molecular detection of SARS-CoV-2 - escape from the NA extraction kit-shortage, Copenhagen, Denmark, March 2020. Euro Surveill. 2020;25:2000398. doi: 10.2807/1560-7917.ES.2020.25.14.2000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman T.M., Wang D., Harris J., Genomics England Research Consortium Genomic loci susceptible to systematic sequencing bias in clinical whole genomes. Genome Res. 2020;30:415–426. doi: 10.1101/gr.255349.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F., Bonsignori M., Liao H.X., Kumar A., Xia S.M., Lu X., Cai F., Hwang K.K., Song H., Zhou T. Cooperation of B cell lineages in induction of HIV-1-broadly neutralizing antibodies. Cell. 2014;158:481–491. doi: 10.1016/j.cell.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloboff P.A. Oblong, a program to analyse phylogenomic data sets with millions of characters, requiring negligible amounts of RAM. Cladistics. 2014;30:273–281. doi: 10.1111/cla.12056. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E., Baker S.C., Baric R.S., de Groot R.J., Drosten C., Gulyaeva A.A., Haagmans B.L., Lauber C., Leontovich A.M., Neuman B.W., Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.L., Baric R.S. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J. Virol. 2010;84:3134–3146. doi: 10.1128/JVI.01394-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Gudbjartsson D.F., Helgason A., Jonsson H., Magnusson O.T., Melsted P., Norddahl G.L., Saemundsdottir J., Sigurdsson A., Sulem P., Agustsdottir A.B. Spread of SARS-CoV-2 in the Icelandic Population. N. Engl. J. Med. 2020;382:2302–2315. doi: 10.1056/NEJMoa2006100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M.-S., Chen W.-J., Chen H.-Y., Lin S.-F., Wang M.-C., Di J., Lu Y.-T., Liu C.-L., Chang S.-C., Chao C.-L. Neutralizing antibody response and SARS severity. Emerg. Infect. Dis. 2005;11:1730–1737. doi: 10.3201/eid1111.040659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.J., Okuda K., Edwards C.E., Martinez D.R., Asakura T., Dinnon K.H., III, Kato T., Lee R.E., Yount B.L., Mascenik T.M. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell. 2020 doi: 10.1016/j.cell.2020.05.042. Published online May 27, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- Jaume M., Yip M.S., Cheung C.Y., Leung H.L., Li P.H., Kien F., Dutry I., Callendret B., Escriou N., Altmeyer R. Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH- and cysteine protease-independent FcγR pathway. J. Virol. 2011;85:10582–10597. doi: 10.1128/JVI.00671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleine-Weber H., Elzayat M.T., Hoffmann M., Pöhlmann S. Functional analysis of potential cleavage sites in the MERS-coronavirus spike protein. Sci. Rep. 2018;8:16597. doi: 10.1038/s41598-018-34859-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S., Phillippy A., Delcher A.L., Smoot M., Shumway M., Antonescu C., Salzberg S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBranche C.C., Henderson R., Hsu A., Behrens S., Chen X., Zhou T., Wiehe K., Saunders K.O., Alam S.M., Bonsignori M. Neutralization-guided design of HIV-1 envelope trimers with high affinity for the unmutated common ancestor of CH235 lineage CD4bs broadly neutralizing antibodies. PLoS Pathog. 2019;15:e1008026. doi: 10.1371/journal.ppat.1008026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Lee P., Tsang A.K., Yip C.C., Tse H., Lee R.A., So L.Y., Lau Y.L., Chan K.H., Woo P.C. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J. Virol. 2011;85:11325–11337. doi: 10.1128/JVI.05512-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Wang S., Kong R., Ding W., Lee F.H., Parker Z., Kim E., Learn G.H., Hahn P., Policicchio B. Envelope residue 375 substitutions in simian-human immunodeficiency viruses enhance CD4 binding and replication in rhesus macaques. Proc. Natl. Acad. Sci. USA. 2016;113:E3413–E3422. doi: 10.1073/pnas.1606636113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Giorgi E.E., Marichannegowda M.H., Foley B., Xiao C., Kong X.-P., Chen Y., Gnanakaran S., Korber B., Gao F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Science Advances. 2020;6:eabb9153. doi: 10.1126/sciadv.abb9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Fontanet A., Zhang P.-H., Zhan L., Xin Z.-T., Baril L., Tang F., Lv H., Cao W.-C. Two-year prospective study of the humoral immune response of patients with severe acute respiratory syndrome. J. Infect. Dis. 2006;193:792–795. doi: 10.1086/500469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Yang Y., Zhang C., Huang F., Wang F., Yuan J., Wang Z., Li J., Li J., Feng C. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020;63:364–374. doi: 10.1007/s11427-020-1643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo-Redondo R., Nam H.H., Roberts S.C., Simons L.M., Jennings L.J., Qi C., Achenbach C.J., Hauser A.R., Ison M.G., Hultquist J.F. A Unique Clade of SARS-CoV-2 Viruses is Associated with Lower Viral Loads in Patient Upper Airways. medRxiv. 2020 doi: 10.1101/2020.2005.2019.20107144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv M., Luo X., Estill J., Liu Y., Ren M., Wang J., Wang Q., Zhao S., Wang X., Yang S., On Behalf Of The Covid-Evidence And Recommendations Working Group Coronavirus disease (COVID-19): a scoping review. Euro Surveill. 2020;25:2000125. doi: 10.2807/1560-7917.ES.2020.25.15.2000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Nao N., Shirato K., Kawase M., Saito S., Takayama I., Nagata N., Sekizuka T., Katoh H., Kato F. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA. 2020;117:7001–7003. doi: 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet J.K., Whittaker G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA. 2014;111:15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L., Blömer U., Gage F.H., Trono D., Verma I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oong X.Y., Ng K.T., Takebe Y., Ng L.J., Chan K.G., Chook J.B., Kamarulzaman A., Tee K.K. Identification and evolutionary dynamics of two novel human coronavirus OC43 genotypes associated with acute respiratory infections: phylogenetic, spatiotemporal and transmission network analyses. Emerg. Microbes Infect. 2017;6:e3. doi: 10.1038/emi.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.E., Li K., Barlan A., Fehr A.R., Perlman S., McCray P.B., Jr., Gallagher T. Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc. Natl. Acad. Sci. USA. 2016;113:12262–12267. doi: 10.1073/pnas.1608147113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promislow D.E.L. A geroscience perspective on COVID-19 mortality. J. Gerontol. A Biol. Sci. Med. Sci. 2020 doi: 10.1093/gerona/glaa094. Published online April 17, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman S., Sharique L., Ihsan A., Liu Q. Evolutionary Trajectory for the Emergence of Novel Coronavirus SARS-CoV-2. Pathogens. 2020;9:240. doi: 10.3390/pathogens9030240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren L., Zhang Y., Li J., Xiao Y., Zhang J., Wang Y., Chen L., Paranhos-Baccalà G., Wang J. Genetic drift of human coronavirus OC43 spike gene during adaptive evolution. Sci. Rep. 2015;5:11451. doi: 10.1038/srep11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadjadpour R., Donau O.K., Shingai M., Buckler-White A., Kao S., Strebel K., Nishimura Y., Martin M.A. Emergence of gp120 V3 variants confers neutralization resistance in an R5 simian-human immunodeficiency virus-infected macaque elite neutralizer that targets the N332 glycan of the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 2013;87:8798–8804. doi: 10.1128/JVI.00878-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamango D.J., Johnson M.C. Characterizing the Murine Leukemia Virus Envelope Glycoprotein Membrane-Spanning Domain for Its Roles in Interface Alignment and Fusogenicity. J. Virol. 2015;89:12492–12500. doi: 10.1128/JVI.01901-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehra S.T., Salciccioli J.D., Wiebe D.J., Fundin S., Baker J.F. Maximum Daily Temperature, Precipitation, Ultra-Violet Light and Rates of Transmission of SARS-Cov-2 in the United States. Clin. Infect. Dis. 2020:ciaa681. doi: 10.1093/cid/ciaa681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevajol M., Subissi L., Decroly E., Canard B., Imbert I. Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Res. 2014;194:90–99. doi: 10.1016/j.virusres.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y., McCauley J. GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017;22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulla A., Heald-Sargent T., Subramanya G., Zhao J., Perlman S., Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011;85:873–882. doi: 10.1128/JVI.02062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.C., Blanc H., Surdel M.C., Vignuzzi M., Denison M.R. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog. 2013;9:e1003565. doi: 10.1371/journal.ppat.1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.D., Tu C.C., Zhang G.W., Wang S.Y., Zheng K., Lei L.C., Chen Q.X., Gao Y.W., Zhou H.Q., Xiang H. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. USA. 2005;102:2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui J., Aird D.R., Tamin A., Murakami A., Yan M., Yammanuru A., Jing H., Kan B., Liu X., Zhu Q. Broadening of neutralization activity to directly block a dominant antibody-driven SARS-coronavirus evolution pathway. PLoS Pathog. 2008;4:e1000197. doi: 10.1371/journal.ppat.1000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford D.L. PAUP∗ Phylogenetic Analysis Using Parsimony. Sinauer Associates; 2003. PAUP∗ {Phylogenetic Analysis Using Parsimony (and Other Methods)], Version 4. [Google Scholar]
- Tang X.-C., Agnihothram S.S., Jiao Y., Stanhope J., Graham R.L., Peterson E.C., Avnir Y., Tallarico A.S.C., Sheehan J., Zhu Q. Identification of human neutralizing antibodies against MERS-CoV and their role in virus adaptive evolution. Proc. Natl. Acad. Sci. USA. 2014;111:E2018–E2026. doi: 10.1073/pnas.1402074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temperton N.J., Chan P.K., Simmons G., Zambon M.C., Tedder R.S., Takeuchi Y., Weiss R.A. Longitudinally profiling neutralizing antibody response to SARS coronavirus with pseudotypes. Emerg. Infect. Dis. 2005;11:411–416. doi: 10.3201/eid1103.040906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Meulen J., van den Brink E.N., Poon L.L., Marissen W.E., Leung C.S., Cox F., Cheung C.Y., Bakker A.Q., Bogaards J.A., van Deventer E. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 2006;3:e237. doi: 10.1371/journal.pmed.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgen L., Keyaerts E., Lemey P., Moës E., Li S., Vandamme A.-M., Van Ranst M. Circulation of genetically distinct contemporary human coronavirus OC43 strains. Virology. 2005;337:85–92. doi: 10.1016/j.virol.2005.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner C., Roychoudhury P., Hadfield J., Hodcroft E., Lee J., Moncla L., Muller N., Behrens C., Huang M.-L., Mathias P. Comparing viral load and clinical outcomes in Washington State across D614G mutation in spike protein of SARS-CoV-2. 2020. https://github.com/blab/ncov-D614G
- Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y., Shang J., Sun S., Tai W., Chen J., Geng Q., He L., Chen Y., Wu J., Shi Z. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J. Virol. 2020;94:e02015–e02019. doi: 10.1128/JVI.02015-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.-F., Tseng S.-P., Yen C.-H., Yang J.-Y., Tsao C.-H., Shen C.-W., Chen K.-H., Liu F.-T., Liu W.-T., Chen Y.-M.A., Huang J.C. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem. Biophys. Res. Commun. 2014;451:208–214. doi: 10.1016/j.bbrc.2014.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Zhang L., Kuwahara K., Li L., Liu Z., Li T., Zhu H., Liu J., Xu Y., Xie J. Immunodominant SARS Coronavirus Epitopes in Humans Elicited both Enhancing and Neutralizing Effects on Infection in Non-human Primates. ACS Infect. Dis. 2016;2:361–376. doi: 10.1021/acsinfecdis.6b00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Liu Z., Chen Z., Huang X., Xu M., He T., Zhang Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020;92:667–674. doi: 10.1002/jmv.25762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A., Peng Y., Huang B., Ding X., Wang X., Niu P., Meng J., Zhu Z., Zhang Z., Wang J. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe. 2020;27:325–328. doi: 10.1016/j.chom.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip M.S., Leung H.L., Li P.H., Cheung C.Y., Dutry I., Li D., Daëron M., Bruzzone R., Peiris J.S., Jaume M. Antibody-dependent enhancement of SARS coronavirus infection and its role in the pathogenesis of SARS. Hong Kong Med. J. 2016;22(3, Suppl 4):25–31. [PubMed] [Google Scholar]
- Yu J., Tostanoski L.H., Peter L., Mercado N.B., McMahan K., Mahrokhian S.H., Nkolola J.P., Liu J., Li Z., Chandrashekar A. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science. 2020:eabc6284. doi: 10.1126/science.abc6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M., Wu N.C., Zhu X., Lee C.-C.D., So R.T.Y., Lv H., Mok C.K.P., Wilson I.A. A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV. Science. 2020:eabb7269. doi: 10.1126/science.abb7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang R., Gomez Castro M.F., McCune B.T., Zeng Q., Rothlauf P.W., Sonnek N.M., Liu Z., Brulois K.F., Wang X., Greenberg H.B. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.abc3582. eabc3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Zhang F., Yu W., He T., Yu J., Yi C.E., Ba L., Li W., Farzan M., Chen Z. Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J. Med. Virol. 2006;78:1–8. doi: 10.1002/jmv.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Jackson C.B., Mou H., Ojha A., Rangarajan E.S., Izard T., Farzan M., Choe H. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. 2020. https://www.scripps.edu/news-and-events/press-room/2020/20200611-choe-farzan-sars-cov-2-spike-protein.html
- Zhou P., Wang H., Fang M., Li Y., Wang H., Shi S., Li Z., Wu J., Han X., Shi X. Broadly resistant HIV-1 against CD4-binding site neutralizing antibodies. PLoS Pathog. 2019;15:e1007819. doi: 10.1371/journal.ppat.1007819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This is a spreadsheet with tabs including the following data: i) Sites of interest: A listing of all sequences with amino acid changes in sites of interest in Spike (those that reach > 0.3% of the global population, or if in ACE2 contact regions, > 0.1% of the population). ii) D614G: A separate tab for D614G variants. iii) Spike Variation: A tally of all amino acid variants and the overall entropy for each position in Spike, as well as the entropy for each 10 amino acid contiguous stretch across the protein, is listed (https://www.hiv.lanl.gov/content/sequence/ENTROPY/entropy.html). This table includes annotation of the RDB location and ACE2 contact sites. Regular updates of all information in Table S2 can be found at http://cov.lanl.govcontent/index.
Data Availability Statement
All sequence data used here are available from The Global Initiative for Sharing All Influenza Data (GISAID), at https://www.gisaid.org/. The user agreement for GISAID does not permit redistribution of sequences. Other data have been deposited to Mendeley Data: https://doi.org/10.17632/hn3h9gdrgj.1.
Web-based tools to recreate much of the analyses provided in this paper but based on contemporary GISAID data downloads are available at https://cov.lanl.gov/.
Code to create the alignments as described in Figure S1 and to perform the Isotonic regression analysis in Figure 3 will be available through https://cov.lanl.gov, at also GitHub, once permission from our funders is obtained.