

Abstract
Because most studies have focused on the intrinsic carcinogenic pathways of tumors, the underlying role of N6-methyladenosine (m6A) methylation in tumor immune microenvironment (TIME) remains elusive. Herein, we systematically explored the correlations of prominent m6A regulators with PD-L1 and immune infiltrates in 769 head and neck squamous cell carcinomas (HNSCCs; The Cancer Genome Atlas [TCGA] cohort, n = 499; GSE65858 cohort, n = 270). The PD-L1 expression evidently associated with m6A regulators. Two molecular subtypes (cluster1/2) were identified by consensus clustering for 15 m6A regulators. The cluster2 preferentially associated with favorable prognosis, upregulated PD-L1 expression, higher immunoscore, and distinct immune cell infiltration. The hallmarks of G2M checkpoint, mTORC1 signaling, and PI3K/AKT/mTOR signaling were remarkably enriched in the cluster1. A prognostic risk score was constructed using seven m6A regulator-associated signatures that represented an independent prognosis factor for HNSCC. Patients with low-risk score exhibited higher immunoscore and upregulated PD-L1 expression than patients with high-risk score. Consistently, m6A regulators showed the same influence on immune modulation and survival in external GSE65858 cohort. Further analysis revealed that m6A regulator-based signatures were implicated in TIME and their copy-number alterations dynamically affected the abundance of tumor-infiltrating immune cells. Collectively, our study elucidated the important role of m6A methylation in TIME of HNSCC. The proposed m6A regulator-based signatures might serve as crucial mediators of TIME in HNSCC, representing promising therapeutic targets in improving immunotherapeutic efficacy.
Keywords: RNA methylation, prognosis, immune infiltrates, PD-L1, TIME, HNSCC
Graphical Abstract
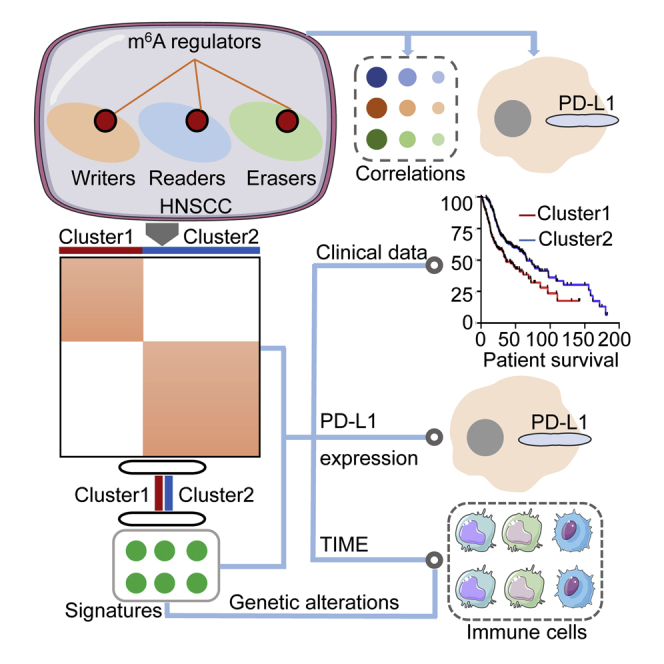
Because most studies focus on tumor-intrinsic oncogenic pathways, potential roles of the m6A methylation in tumor immune microenvironment remain unclear. This study highlights the important role of m6A methylation in immune microenvironment of HNSCC. The proposed m6A regulator-based signatures might serve as promising therapeutic targets for improving immunotherapeutic efficacy.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common malignant tumor in humans worldwide, with approximately 600,000 newly diagnosed cases annually.1,2 More than 50% of diagnosed HNSCC patients present at an advanced stage. The overall 5-year survival rate of HNSCC is ~50%.3 Primary treatments include surgery, radiotherapy, and chemotherapy based on the location and clinical stage of HNSCC, but advanced HNSCC exhibits weak prognosis and limited treatment method. Immunotherapy likely yields preferable therapeutic effects on patients with advanced HNSCC because of its safety and less adverse events. This treatment contributes new insights into clinical treatment management. Since few patients could benefit from immunotherapy, the overall disease control rate and treatment strategies still need to be improved. The imbalance of the immune system has a vital role in the development of HNSCC characterized by an immunosuppressive disease. Patients of HNSCC frequently present low absolute lymphocyte counts, spontaneous apoptosis of cytotoxic T lymphocytes, defects in natural killer (NK) cell activity, and barriers to antigen presentation.4 Numerous cytokines and immunosuppressive cells related to tumor immune escape in the HNSCC microenvironment are also present. Thus, the regulatory mechanism of tumor immune microenvironment (TIME) should be further explored to determine effective biomarkers that precisely predict prognosis and considerably optimize personalized immunotherapy management.
N6-methyladenosine (m6A), the methylation modification at the sixth N atom of adenine, is the most common post-transcriptional modification on mRNA, mediating >60% RNA methylation.5,6 Abnormal m6A methylation levels are closely related to stem cell differentiation, immune response, embryonic development, and microRNA (miRNA) editing; they also play an essential role in the progression of various cancers.7, 8, 9, 10, 11, 12 The epigenetic modification of m6A methylation affects tumor development by modulating the mRNA expression levels of related oncogenes or suppressor genes. The m6A methylation levels in tumors mainly depend on the expression of m6A methylation regulators. The m6A methylation is highly correlated with the expression of intracellular methyltransferases (“writers”) and demethylases (“erasers”), whereas binding proteins (“readers”) bind to m6A methylation sites in performing a series of biological functions.13 The “Writers,” which include methyltransferase like 3 (METTL3), METTL14, WT1-associated protein (WTAP), METTL16, KIAA1429, and RNA-binding motif protein 15 (RBM15), promote m6A RNA methylation.14, 15, 16, 17, 18, 19 The “Erasers,” comprising fat mass- and obesity-associated protein (FTO) and α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5), remove m6A methylation groups from RNA.20 The “Readers,” which include YTH domain-containing 1 (YTHDC1), YTHDC2, YTH N6-methyl-adenosine RNA-binding protein 1 (YTHDF1), YTHDF2, YTHDF3, and heterogeneous nuclear ribonucleoprotein C (HNRNPC), play a specific role by binding to the m6A methylation site.21, 22, 23 The aberrant expression of m6A regulators plays a vital regulatory role in tumor progression, prognosis, and radio-resistance. Chen et al.12 showed that METTL3 upregulates the m6A methylation level of the suppressor gene SOCS2 by regulating the m6A-YTHDF2 pathway to accelerate SOCS2 mRNA degradation. METTL3 overexpression also promotes the proliferation and migration of hepatocellular carcinoma cells, whereas deregulated METTL3 inhibits its growth and metastasis. Nishizawa et al.24 revealed that YTHDF1 expression is dramatically upregulated in patients with colorectal cancer and involved in tumor diameter (p = 0.009), lymph node metastasis (p = 0.044), distant metastasis (p = 0.036), and clinical stage (p = 0.0226). Taketo et al.25 reported that pancreatic cancer cells with downregulated METTL3 expression are sensitive to anticancer drugs and radiotherapy. The study of Zhao et al.26 including thirteen m6A methylation regulators indicated that m6A regulators have the role for predicting prognosis in HNSCC patients. However, the prognosis role of other m6A methylation regulators such as METTL16 and YTHDF3 was not analyzed in HNSCC. In addition, whether m6A methylation regulators have the interface of therapeutic effects or the correlation of PD-L1 has yet to be fully explored.
Most studies have focused on the intrinsic carcinogenic pathways of tumors, but the potential role of m6A methylation in TIME remains unclear. Han et al.27 demonstrated that m6A methylation could prolong new antigen-specific immunity through YTHDF1. The higher levels of CD8+ T and NK cells are evident in YTHDF1-deficient mice than those in wild-type ones, inducing an enhanced antitumor response. This result indicated that YTHDF1 is a vital mediator of tumor immune evasion, emphasizing a potential therapeutic target in improving clinical response to immunotherapy. Consequently, m6A regulators implicated in tumor immunoreactivity pathways may be identified as promising targets in enhancing clinical responses to immunotherapies. Zhao et al.26 showed the interface between m6A regulators and HNSCC prognosis. However, an integrated understanding of m6A RNA methylation in HNSCC, including the interactions between the m6A methylation regulators and TIME, is lacking.
This study aimed to systematically assess the correlations of m6A RNA methylation regulators with prognosis, programmed death ligand 1 (PD-L1), and TIME in HNSCC. Clustering subtypes and risk models for m6A RNA methylation regulators were established to improve prognostic risk stratification and facilitate treatment decision-making for patients with HNSCC. Subsequently, the relationships between clustering subgroups, risk models, PD-L1, immunoscores, and immune cell infiltration were thoroughly analyzed based on the m6A regulator-related signatures to further explore the effect of m6A regulators on TIME. This study also sought to provide insights into the regulatory mechanisms associated with TIME and the strategies for HNSCC immunotherapy.
Results
Expression of m6A RNA Methylation Regulators Was Upregulated in HNSCC
To assess the biological function of m6A regulators in the initiation and development of HNSCC, we systematically investigated the expression patterns of 15 m6A regulatory genes between HNSCC and adjacent normal pairs based on the available The Cancer Genome Atlas (TCGA) dataset. We downloaded the expression profiling datasets for 43 tumor-adjacent normal pairs and analyzed the distinct expression of the selected m6A regulators. The expression levels of m6A regulatory genes in HNSCC and normal tissues were evident (Figures 1A and 1B). The expression levels of “writers” (i.e., KIAA1429, METTL3, RBM15, WTAP, ZC3H13, and METTL16), “readers” (i.e., HNRNPC, YTHDC1, YTHDF1, YTHDF2, and YTHDF3), and “erasers” (ALKBH5 and FTO) were dramatically higher in HNSCC tissues than in normal adjacent tissues (p < 0.01). The expression level of the “reader” YTHDC2 was markedly lower in HNSCC tissues than in normal tissues (p < 0.05). Additionally, no statistically significant difference was evident between the normal and HNSCC tissues regarding the expression level of the “writer” METTL14 (p > 0.05). These results indicated that m6A RNA methylation regulators possessed essential biological roles in HNSCC development.
Figure 1.

Upregulation of m6A RNA Methylation Regulators in HNSCC in TCGA Cohort
(A and B) Heatmap (A) and expression levels (B) of 15 m6A RNA regulators in 43 tumor and adjacent normal pairs. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Significant Correlation of Consensus Clustering for m6A RNA Methylation Regulators with the Characteristics and Survival of Patients with HNSCC
The k = 2 was identified with optimal clustering stability from k = 2 to 9 based on the similarity displayed by the expression levels of m6A regulators and the proportion of ambiguous clustering measure (Figures S1A–S1C). A total of 499 patients with HNSCC were clustered into two subtypes, namely, cluster1 (n = 203) and cluster2 (n = 296), based on the expression levels of the m6A regulators (Figure 2A). The expression of individual m6A methylation regulators was lower in the cluster2 than in the cluster1, especially the expression levels of METTL3 and HNRNPC (Figure 2B). The clinicopathological features between the two subtypes were then compared (Figure 2B). The cluster2 mainly contained female HNSCC. The cluster2 was preferentially associated with a low WHO grade (p < 0.01). The higher immunoscore of the cluster2 was evident than that of the cluster1 (p < 0.05). The overall survival (OS, p < 0.0001) and disease-free survival (DFS, p = 0.009) of the cluster2 were longer than those of the cluster1 (Figures 2C and 2D).
Figure 2.

Differential Clinicopathological Features and Survival of HNSCC in Cluster1/2 Subtypes in TCGA Cohort
(A) Consensus clustering matrix for k = 2. (B) Heatmap and clinicopathologic features of the two clusters (cluster1/2). (C and D) Kaplan-Meier curves of overall survival (OS, C) and disease-free survival (DFS, D) for patients with HNSCC in two clusters (cluster1/2). ∗p < 0.05 and ∗∗p < 0.01.
Our findings suggested that the clustering subtypes defined by m6A regulator expression were closely related to the heterogeneity of patients with HNSCC. To further explore the interaction among m6A regulators, we analyzed the correlations between 15 m6A RNA methylation regulators (Figure S1D). The result showed that the expression levels of YTHDC2, YTHDC1, YTHDF2, METTL14, and RBM15 were positively correlated with one another. The expression levels of KIAA1429, ZC3H13, YTHDF3, and FTO were also positively correlated. The expression level of HNRNPC was positively correlated with the expression levels of METTL3 and WTAP. We further analyzed the gene-expression pattern between the two subtypes by using the principal-component analysis (PCA) method (Figure S1E) and found that the gene-expression profiles between the two subtypes were differentiated well.
Association of PD-L1 with m6A RNA Methylation
To explore the involvement of PD-L1 with m6A RNA methylation, we assessed differential expression in two subtypes and the correlation of PD-L1 with m6A regulators. The expression level of PD-L1 was upregulated in HNSCC tissues compared with normal adjacent tissues (p < 0.05; Figure 3A). The expression level of PD-L1 in the cluster2 was distinctly higher than that in the cluster1 (p < 0.01; Figure 3B). In the TCGA cohort, the expression of PD-L1 had a significantly positive association with METTL3, RBM15, WTAP, HNRNPC, YTHDC2, and YTHDF3 expression levels, whereas a significantly negative correlation was noted with KIAA1429, YTHDF1, and FTO expression levels (Figure 3C). Furthermore, consistent results were obtained in the GSE65858 cohort (Figure 3C).
Figure 3.

Association of PD-L1 with m6A RNA Methylation and the Landscape of Immune Cell Infiltration in HNSCC
(A) PD-L1 upregulation in HNSCC in TCGA cohort. (B) The expression level of PD-L1 in cluster1/2 subtypes in TCGA cohort. (C) The correlation of PD-L1 with m6A methylation regulators in both TCGA and GEO: GSE65858 cohorts. (D) The infiltrating levels of 22 immune cell types in cluster1/2 subtypes in the TCGA cohort. ∗p < 0.05 and ∗∗p < 0.01.
Consensus Clustering for m6A RNA Methylation Regulators Associated with Distinct Immune Cell Infiltration
To investigate the effect of m6A regulators on the TIME of HNSCC, we evaluated the immunoscore and immune infiltrate level between the cluster1 with the upregulated m6A regulator expression and cluster2 with the downregulated m6A regulator expression (Figures 3D and 4). The two clusters revealed a significant difference in immunoscore (Figure 4A). The prognosis of the cluster2 with a higher immunoscore was greater than that of the cluster1 (p < 0.05). Subsequently, the fraction of 22 immune cell types between the two subgroups was analyzed. Cluster1 showed higher infiltration levels of CD4 naive T cell, CD4 memory-activated T cell, and eosinophils (Figures 3D and 4B–4D), whereas cluster2 was more correlated with B memory cells, regulatory T cells (Tregs), NK cells activated, monocytes, macrophages M2, dendritic cells activated, neutrophils, and mast cells infiltration (Figures 3D and 4E and 4F). To elucidate the potential regulatory mechanisms resulting in differences in the TIME between the two subgroups, we performed gene set enrichment analysis (GSEA). The results showed that the malignant hallmarks of tumors, including G2M checkpoint (normalized enrichment score [NES] = 2.511, normalized p < 0.001), mTORC1 signaling (NES = 2.456, normalized p < 0.001), and PI3K/AKT/mTOR signaling (NES = 1.807, normalized p = 0.003), were dynamically correlated with the cluster1 (Figure 4G). Hence, the PI3K/AKT/mTOR signaling pathway might be implicated in the distinct TIME of cluster1/2.
Figure 4.

Distant Immune Cell Infiltration in Two Clusters in TCGA Cohort
(A) Immunoscore in the cluster1/2 subtypes. (B–F) The infiltrating levels of the CD4 native T cells (B), CD4 memory-activated T cells (C), eosinophils (D), neutrophils (E), and dendritic cells resting (F) in two clusters. (G) GSEA showed that G2M checkpoint, mTORC1 signaling, and PI3K/AKT/mTOR signaling are differentially enriched in cluster1. ES, enrichment score; NES, normalized ES; NOM p value, normalized p value. ∗p < 0.05 and ∗∗p < 0.01.
To further verify our hypothesis, we conducted consensus clustering for “erasers” FTO and ALKBH5. The longer OS in the cluster1 was evident than that in the cluster2 (p = 0.0012; Figure 5A). However, cluster1 had lower immunoscore and PD-L1 expression level than cluster2 (Figures 5B and 5C). Moreover, no significant difference of the composition of 22 immune cell types was observed between the two clusters (Figure 5D). Therefore, there was a reverse effect observed with clustering for demethylases on survival and immune infiltration, which further confirmed our hypothesis.
Figure 5.

The Reverse Effect on Survival and Immune Infiltration between the Cluster1/2 Subtypes Generated by Consensus Clustering for Demethylases in TCGA Cohort
(A–C) Kaplan-Meier curves of OS for patients with HNSCC (A), immunoscore (B), and PD-L1 expression level (C) in cluster1/2 subtypes. (D) The infiltrating levels of 22 immune cell types in two clusters (cluster1/2). ∗p < 0.05 and ∗∗p < 0.01.
Construction and Validation of Prognostic Signatures for m6A RNA Methylation Regulators
Then, we explored the prognostic role of m6A regulators in patients with HNSCC. The 499 patients were randomly divided into the TCGA training cohort (351 patients) and validation cohort (148 patients) at a 7:3 ratio. The baseline characteristics among the TCGA training and validation cohorts, including age, gender, T stage, N stage, M stage, grade, and TNM stage, were not statistically different (all p > 0.05; Table S1).
To predict the clinical outcome of m6A regulators in HNSCC patients precisely, we performed the least absolute shrinkage and selection operator (LASSO) regression analysis based on the expression values of 15 m6A regulators in the TCGA training cohort. Seven m6A regulators, namely METTL3, YTHDC2, METTL14, RBM15, YTHDC1, ZC3H13, and HNRNPC, were identified (Figures S2A and S2B). The risk scores of the TCGA training and validation cohorts were calculated using the coefficients obtained by the LASSO algorithm, and the equation is as follows: (risk score = 0.1172 × METTL14 expression level)−(0.0628 × METTL3 expression level) − (0.1163 × YTHDC2 expression level) + (0.0245 × ZC3H13 expression level) − (0.0453 × RBM15 expression level) − (0.0223 × YTHDC1 expression level) + (0.0174 × HNRNPC expression level). Afterward, patients were divided into high- and low-risk groups based on the median risk score. The distribution of the risk scores, OS, OS status, and expression profiles of the seven m6A-regulator-based signatures in TCGA training and validation cohorts is displayed in Figures 6A and 6B. The heatmap results indicated that risky m6A regulators, including METTL3, YTHDC2, RBM15, and YTHDC1, were highly expressed in the high-risk group, whereas the expression levels of protective m6A regulators, including METTL14, ZC3H13, and HNRNPC, were upregulated in the low-risk group. The OS of the low-risk group was longer than that of the high-risk group in the TCGA training and validation cohorts (p < 0.0001, Figures 6C and 6D). To assess the prognostic accuracy of the seven identified risk signatures, we conducted 3- and 5-year receiver operating characteristic (ROC) curve analyses by comparing the respective AUC values. In the TCGA training cohort, the 3- and 5-year AUC values for the seven risk signatures were 0.798 (95% CI: 0.712 to 0.824) and 0.825 (95% CI: 0.768 to 0.873), respectively (Figures S2C and S2D). In the TCGA validation cohort, the 3- and 5-year AUC values for the seven risk signatures were 0.775 (95% CI: 0.709 to 0.816) and 0.814 (95% CI: 0.758 to 0.864), respectively (Figures S2E and S2F). The AUC values showed that the signatures of seven m6A regulators had a favorable discrimination performance for the prognosis of patients with HNSCC. These results suggested that the risk score that were calculated based on the seven risk signatures could accurately predict the prognosis of HNSCC patients.
Figure 6.

Construction and Validation of Prognostic Signatures of m6A Methylation Regulators in TCGA Cohort
(A and B) Distribution of risk score, OS, and OS status and heatmap of the seven prognostic m6A regulator signatures in the TCGA training cohort (A) and TCGA validation cohort (B). (C and D) Kaplan-Meier curves of OS for patients with HNSCC based on the risk score in the TCGA training cohort (C) and TCGA validation cohort (D).
Prognostic Risk Scores Correlated with Grade, Immunoscore, and Radiotherapy in HNSCC
The relationship between risk scores and clinical features was further evaluated. The heatmap demonstrated the expression levels of seven m6A regulators in the high- and low-risk groups in the TCGA training cohort (Figure 7A). The expression levels of METTL3 and YTHDC2 in the high-risk group were typically lower than those of the low-risk group. The expression levels of HNRNPC and ZC3H13 were low in the low-risk group. The difference in terms of clustering subtypes (p < 0.001), grade (p < 0.05), and immunoscore (p < 0.001) between the high- and low-risk groups was significant. We also further examined the relationship between risk score, and subtype, classification, and immunoscore. The risk score of the cluster1 was distinctly higher than that of the cluster2 (p < 0.001, Figure 7B). The risk score increased along with the histological grade increased (p < 0.05, Figure 7C). Compared to the low immunoscore group, the high immunoscore group had lower risk score (p < 0.001, Figure 7D). These findings revealed that the risk score was significantly associated with subtype, grade, and immunoscore in HNSCC patients.
Figure 7.

Prognostic Risk Scores Correlated with Clinicopathological Features and Immunoscore in TCGA Training Cohort
(A) Heatmap and clinicopathologic features of high- and low-risk groups. (B–D) Distribution of risk scores stratified by cluster1/2 (B), grade (C), and immunoscore (D). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
To validate that m6A methylation regulators harbored the same influence on immune modulation and survival in additional HNSCC cases, we selected eligible GEO: GSE65858 dataset from the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65858) as the external validation cohort. Consistent with TCGA analysis, patients with low-risk score had longer OS than patients with high-risk score (p = 0.0025) (Figure 8A). The survival prediction of risk score was evaluated in GEO: GSE65858 cohort by using time-dependent ROC analysis at 3 and 5 years (Figure 8B). Consistently, a higher immunoscore was evident in patients with low-risk score (Figure 8C). Additionally, we found that patients with high-risk score had downregulated PD-L1 expression level than patients with low-risk score in TCGA training, TCGA validation, and independent GSE65858 cohorts (Figures 8D–8F).
Figure 8.

Independent Validation of m6A Regulator-Based Risk Score Model
(A) Kaplan-Meier curves for OS by risk score group in GEO: GSE65858 cohort. (B) Time-dependent ROC curves measuring the predictive value of the risk score in GEO: GSE65858 cohort. (C) Immunoscore by risk score group in GEO: GSE65858 cohort. (D–F) The PD-L1 expression level by risk score group in TCGA training set (D), TCGA validation set (E), and GEO: GSE65858 validation set (F).
Univariate and multivariate Cox regression analyses were conducted in the TCGA training and validation cohorts to determine whether the risk score independently predicted the prognosis of patients with HNSCC. Univariate analysis showed that age (p = 0.002), N stage (p = 0.027), M stage (p = 0.017), TNM stage (p = 0.001), and risk score (p < 0.001) were considerably associated with the OS in the TCGA training cohort (Figure S3A). These factors were then included into multivariate Cox regression analysis, thereby showing that age (p < 0.001), TNM stage (p = 0.009), and risk score (p < 0.001) remained closely correlated with the OS (Figure S3B). In the TCGA validation cohort, univariate Cox analysis revealed that age (p < 0.001), N stage (p = 0.015), TNM stage (p < 0.001), and risk score (p < 0.001) were highly related to the OS (Figure S3C). Similarly, when enrolling these factors into multivariate Cox analysis, the age (p < 0.001), TNM stage (p = 0.001), and risk score (p < 0.001) were still significantly associated with OS (Figure S3D). The results showed that the risk score obtained from seven m6A regulator-based signatures was an independent prognostic factor for HNSCC patients.
The prognostic value of risk score for HNSCC patients subjected to radiotherapy was estimated. We explored whether the application of radiotherapy could influence the predictive performance of the risk score for OS in HNSCC patients. We observed that the OS of patients with radiotherapy in the high- and low-risk groups was superior (Figures 9A–9D), but patients with high-risk score benefited significantly more than those with low-risk score from radiotherapy. Patients with radiotherapy had a favorable survival advantage than patients without radiotherapy in the high-risk group (Figures 9A and 9C). The survival benefit of radiotherapy was relatively not significant in the low-risk group (Figures 9B and 9D). Accordingly, patients with high-risk score were more likely to benefit for survival from radiotherapy than those with low-risk score.
Figure 9.

Considerable Benefit for Survival Was Evident in Patients with HNSCC Subjected to Radiotherapy and High-Risk Score
(A) Kaplan–Meier curves of OS for patients with high-risk score based on the radiotherapy in the training cohort. (B) Kaplan–Meier curves of OS for patients with low-risk score based on the radiotherapy in the training cohort. (C) Kaplan–Meier curves of OS for patients with high-risk score based on the radiotherapy in the validation cohort. (D) Kaplan–Meier curves of OS for patients with low-risk score based on the radiotherapy in the validation cohort.
Effect of Genetic Alterations of the m6A Regulator Signatures on Immune Cell Infiltration
The relationship between the risk score and infiltration levels of six immune cell types was analyzed to estimate the effect of seven m6A regulator-based signatures on the HNSCC immune microenvironment. A significantly negative correlation was observed between the risk score and infiltration levels of the B (p < 0.001), CD4+ T (p = 0.012), and CD8+ T cells (p < 0.001, Figures 10A–10C). The risk score was positively correlated with the infiltration levels of neutrophils, macrophages, and dendritic cells (p < 0.001, Figures 10D–10F). This result confirmed that m6A regulator-based risk signatures were implicated in the HNSCC immune microenvironment.
Figure 10.

Relationships between the Risk Score and Infiltration Abundances of Six Immune Cell Types
(A–F) B cell (A), CD4+ T cell (B), CD8+ T cell (C), neutrophil (D), macrophage (E), and dendritic cell (F).
The effects of somatic cell copy number alternations (CNAs) of the m6A regulator-based signatures on immune cell infiltration were further analyzed to elucidate the underlying mechanisms by which the risk score was associated with different immune cell infiltrations. The CNAs of the identified m6A regulators signatures, including arm-level deletion and arm-level gain, significantly affected the infiltration levels of B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells in HNSCC (Figure 11). These results demonstrated that m6A regulators had pivotal regulatory effects on the TIME for HNSCC patients.
Figure 11.

Effect of the Genetic Alterations of m6A Regulator-Relevant Signature on the Immune Cell Infiltration
(A–F) METTL3 (A), YTHDC1 (B), METTL14 (C), YTHDC2 (D), RBM15 (E), and HNRNPC (F). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Discussion
The m6A methylation is the most common form of mRNA modification that plays a crucial role in post-transcriptional regulation.28 The dysregulation of m6A methylation regulatory protein triggers downstream RNA metabolism disorder and is involved in the progression of various tumors.10,29, 30, 31, 32 However, the m6A modification level of specific m6A regulators that serve as a tumor inhibitor or promoter is unknown. For example, ALKBH5 plays a distinct role in different tumor types. Kwok et al.33 reported that the deletion of the ALKBH5 expression is an unfavorable prognostic indicator of acute myeloid leukemia and is associated with TP53 mutation. Zhang et al.10 showed that the ALKBH5 expression is upregulated in gliomas, and ALKBH5 enhances the FOXM1 expression level by increasing the stability of transcription products and promoting the proliferation and invasion of glioma. The diverse functions of m6A regulators involved in different tumor types suggested that the regulation of m6A methylation modification levels is overwhelmingly complex. Furthermore, since most studies have focused on the intrinsic carcinogenic pathways of tumors, further research focusing on m6A regulators is warranted to elucidate the potential regulatory mechanism of m6A methylation in TIME. At present, the effects of m6A RNA methylation on the TIME of HNSCC have yet to be fully analyzed.
In this study, the expression patterns, prognostic values, and effects on the TIME of m6A regulators in HNSCC were demonstrated. The expression of YTHDC2 significantly decreased in HNSCC tissues, whereas the METTL14 expression was not significant between cancer and adjacent normal tissues. The expression levels of other thirteen m6A regulators and PD-L1 were higher in the HNSCC tissues than in the adjacent normal tissues. Analyses revealed unanticipated associations of PD-L1 with m6A regulators. We identified two subtypes of HNSCC, that is, cluster1 and cluster2, by consensus clustering for m6A regulators. The cluster1/2 subtype affected the prognosis and different clinicopathological features of HNSCC and was closely related to PD-L1, immunoscore, and immune cell infiltration levels. We also derived seven prognostic risk signatures from m6A regulators, which effectively stratified the OS of HNSCC patients into high- and low-risk groups in TCGA and GEO cohorts. The high- and low-risk groups were also associated with distinct clustering subtypes, PD-L1 expression level, and immunoscore. Among these risk signatures, METTL3 functions as an oncogene in liver cancer and acute leukemia.6,12 METTL3 and METTL14 serve as suppressor genes in cervical cancer and breast cancer, respectively.6,34 RBM15 emerges as an oncogene in leukemia.35 ZC3H13 functions as a suppressor gene in colorectal cancer, but Panahi et al.36,37 showed that ZC3H13 acts as an oncogenic protein. HNRNPC is an oncogene in breast cancer, but it is a suppressor gene in liver cancer and malignant glioma.38, 39, 40 These findings revealed that the dysregulation of specific m6A regulators served as distinct functions in various kinds of cancer.
We characterized the effects of differential m6A methylation modification on different HNSCC subtypes by clustering m6A regulators. The patients in the cluster2 showed a high histological grade. Similarly, the cluster2 had a preferred survival, including OS and DFS, compared with that of the cluster1. The difference in the TIME between cluster1/2 subtypes was significant. The PD-L1 expression level and immunoscore of the cluster2 with favorable prognosis were significantly higher than that of the cluster1. There is a significant survival difference between the two clusters, which may be related to the more important role of the higher immunoscore and PD-L1 expression level in cluster2. This finding was consistent with the results of a previous study, which revealed that the survival of patients with high immunoscore is longer than that of patients with low immunoscore.3 Further analysis showed that the infiltration levels of CD4 naive T cells, CD4 memory-activated T cells, and eosinophils in the cluster1 were higher than those in the cluster2. Conversely, the infiltration levels of B memory cells, Tregs, NK cells activated, monocytes, macrophages M2, neutrophil, and mast cell levels in the cluster2 were higher than those in the cluster1. Moreover, there was a reverse effect observed with “eraser” clustering on survival and immune infiltration. The GSEA results indicated that the malignant functional features of the tumor, including mTORC1 signaling and PI3K/AKT/mTOR signaling, were significantly enriched in the cluster1. Liu et al.41 showed that a decrease in m6A RNA methylation levels regulates the AKT signaling pathway in endometrial cancer. Zhou et al.42 disclosed that downregulated METTL3 expression is associated with the mTOR signaling pathway in renal cell carcinoma. Similarly, Li et al.43 found that the decreased METTL3 expression level triggers increased PI3K, AKT, and mTOR expression levels, which are correlated with poor prognosis in patients with renal cell carcinoma. These studies have suggested that the mRNA of the PI3K/AKT/mTOR signaling pathway molecule may serve as targets for m6A methylation modification. Therefore, we hypothesized that the m6A methylation modification and the PI3K/AKT/mTOR signaling pathway are jointly involved in the regulation of the differential TIME between the two subgroups in HNSCC.
The prognostic value of the m6A regulators-relevant signatures was evaluated in patients with HNSCC and validated in the TCGA cohort and external GSE65858 cohort, respectively. The risk score obtained from seven risk signatures effectively stratified the patients with HNSCC into high- and low-risk groups. The OS of the patients in high-risk group was shorter than that of the patients in low-risk group in the TCGA training and validation cohorts. Consistent results were also obtained in independent GEO: GSE65858 validation cohort. The risk score of the cluster2 was significantly lower than those of the cluster1. The survival of patients with high immunoscore was greater in the low-risk group than in the high-risk group. This observation was consistent with previous results. Furthermore, patients in low-risk group had higher PD-L1 expression level than in high-risk group. Univariate and multivariate Cox regression analyses showed that the risk score was an independent prognostic factor for HNSCC patients. The risk score calculated with m6A regulator-based signatures predicted the sensitivity of HNSCC patients to radiotherapy. Patients with high-risk scores benefited significantly more from radiotherapy than those with low-risk scores. Wang et al.34 indicated that the expression levels of METTL3 and METTL14 significantly decrease in 286 cervical cancer tissues, whereas the FTO and ALKBH5 expression levels dynamically increase; clinical data analysis suggested that patients with m6A hypomethylation have short DFS and OS but have a high recurrence rate (p < 0.01). Zhou et al.44 discovered that the FTO expression level significantly increases in patients with cervical squamous cell carcinoma, resulting in resistance to chemoradiotherapy probably because FTO reduces the m6A methylation level of certain genes and then affects ERCC1 expression. Consequently, the identification of m6A regulator-associated risk signatures can accurately predict the prognosis of patients with HNSCC, thereby promoting the choice of individually therapeutic strategies and expanding insights into the advancement of therapeutic approaches.
A tumor microenvironment that formed in a process of dynamic changes is regulated by a variety of immunosuppressive signals. Tumor microenvironment plays an essential regulatory role in tumorigenesis, and its heterogeneity can lead to multiple dimensions, including patient prognosis and therapeutic response.45, 46, 47, 48 Tumor-infiltrating lymphocytes and immunoscores are associated with the prognosis of HNSCC and predict the efficacy of radiotherapy and chemotherapy.49,50 However, the underlying mechanism of immune infiltration against tumor response in HNSCC remains unclear. HNSCC is an immunosuppressive disease that induces a variety of immunosuppressive cells. At present, the mediation of the m6A regulators on TIME in HNSCC is still limited. In this study, the risk score based on the seven risk signatures of m6A regulator was significantly correlated with the PD-L1 expression level and immune cells infiltration. Risk scores were negatively correlated with the infiltration levels of B, CD4+ T, and CD8+ T cells but were positively correlated with the infiltration levels of neutrophils, macrophages, and dendritic cells. Li et al.8 reported that the loss of METTL3 or METT14 triggers the disorders of proliferation and differentiation in T cells, which in turn reduce the sensitivity of interleukin-7 (IL-7) in vivo. Han et al.27 showed that the infiltration levels of CD8+ T and NK cells increase in YTHDF1-deficient mouse tumors, thereby enhancing the cross-expression of tumor antigens and cross-priming of CD8+ T cells in vivo. These findings suggested that m6A RNA methylation regulators are involved in TIME regulation to some extent. Our study also revealed that the CNAs of m6A regulators significantly affected the immune cell infiltration level in HNSCC, providing insight into the TIME.
Undeniably, there are several limitations in our study. First, extrapolation of our results was substantiated in both TCGA and GSE65858 cohort. The proposed risk score model and interactions between the TIME and m6A regulators was not subjected to external verification due to the lack of sufficiently available data in our own cohort. Consequently, further external validation in the multicenter cohorts is needed to be carried out to test the findings. Additionally, the regulatory mechanism of m6A regulators in TIME is warranted to be further investigated to reshape the TIME and improve the precision immunotherapy of HNSCC.
In conclusion, this study systematically evaluated prognostic value, the correlation of PD-L1, role in the TIME, and potential regulatory mechanisms of m6A RNA regulators in HNSCC. The upregulated PD-L1 was correlated with m6A methylation. Two HNSCC subtypes (cluster1/2) were determined via the consensus clustering for m6A regulators that stratified the prognosis of patients with HNSCC and simultaneously presented the significantly different TIME. The risk score developed from seven m6A regulator-based signatures was an independent prognostic indicator of patients with HNSCC. Patients with high-risk score likely benefited more from radiotherapy. The m6A regulator-based risk signatures were significantly associated with the immune cell infiltration levels of patients with HNSCC. The m6A RNA methylation might be involved in the regulation of HNSCC immune microenvironment in synergy with the PI3K/AKT/mTOR signaling pathway. Therefore, identifying m6A regulators related to molecular pathways affecting tumor immune responses and further studying its regulatory mechanisms might provide promising targets for improving the responsiveness of HNSCC to immunotherapy.
Materials and Methods
Datasets
The RNA-seq transcriptome data of patients with HNSCC and the corresponding clinical data were downloaded from the TCGA data portal (https://portal.gdc.cancer.gov/). For RNA-seq data, level 3 mRNA expression profiles integrated by the Illumina HiSeq RNASeqV2 system were derived from TCGA. The data of 528 HNSCC samples and 43 adjacent normal tissues were downloaded on June 22, 2019. The following inclusion criteria were used: (1) histologically confirmed HNSCC and (2) simultaneously available information on mRNA expression profile data and OS. Lastly, 499 patients with HNSCC and with the corresponding clinicopathological information, including age, gender, TNM staging, and grade were enrolled for further analysis. A total of 499 patients with HNSCC were randomly assigned into a training cohort (351 patients) and a validation cohort (148 patients) at a 7:3 ratio by using the caret package. The baseline clinicopathological features of the two cohorts are summarized in Table S1. The GEO: GSE65858 dataset from GEO database was used as the external validation cohort. The expression profiling of 270 HNSCC patients and survival information were extracted from GEO: GSE65858 dataset.51
m6A RNA Methylation Regulator Detection
According to previously published literature, 16 m6A RNA methylation regulators were collected.6,13,33 A total of 15 m6A methylation regulators were identified on the basis of available mRNA expression data of HNSCC from TCGA. Next, the differential expression of the 15 m6A regulators was determined in the tumor tissues versus adjacent normal pairs.
Bioinformatics Analysis
To functionally elucidate the biological characteristics of the m6A regulators in HNSCC, we employed the “ConsensusClusterPlus” package (1,000 iterations and resample rate of 80%, http://www.bioconductor.org/) to classify the patients with HNSCC into different subtypes. The PCA was performed using the R package for R v3.6.0 to assess gene-expression patterns among distinct HNSCC subtypes. The GSEA was conducted in the Hallmark gene set “h.all.v6.2.symbols.gmt” of MSigDB by using the JAVA program to illustrate the difference in survival among different HNSCC subtypes. The algorithm of random sampling was 1,000 permutations. An enrichment pathway between two subtypes was determined by utilizing the false discovery rate of < 0.05 and the NES.
The immunoscore for each patient was calculated with the ESTIMATE algorithm through the R “estimate package.”52 The fraction of 22 immune cell types for each contained sample was yielded through cell type identification by estimating relative subsets of RNA transcripts (CIBERSORT; https://cibersort.stanford.edu/). The algorithm of 1,000 permutations was adopted. Only samples with a CIBERSORT p of < 0.05 were included to perform the subsequent analysis of comparing differential immune infiltration levels between the subgroups grouped by clustering subtypes and risk scores.
The prognostic risk signatures of 15 m6A regulatory genes were established using the LASSO regression analysis in the TCGA training cohort.53 Signatures were screened by selecting the optimal penalty parameter λ correlated with the minimum 10-fold cross-validation. The coefficients obtained from the LASSO regression algorithm were used to yield the following risk score equation: risk score = sum of coefficients × m6A regulator expression level. According to this equation, the risk score of each patient was separately calculated in the training and validation cohorts. Subsequently, the patients were divided into high- and low-risk groups, and the median value of the risk score was set as the cut-off point.
The effect of CNAs of the m6A regulators on immune cell infiltration levels was evaluated by applying the Tumor Immune Estimation Resource (TIMER, https://cistrome.shinyapps.io/timer/) that consisted of six immune cell types (i.e., B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells). The GISTIC 2.0 data were utilized in the TIMER.
Statistical Analysis
Statistical tests were carried out using R version 3.6.0, SPSS 24.0 (IBM, NY, USA) and GraphPad Prism 8.0. The expression levels of the m6A RNA methylation regulators were compared with the Mann-Whitney U test in cancer tissues versus normal tissues. Student’s t test and one-way ANOVA were used to separately perform the group comparisons of two subgroups and more than two subgroups. Categorical variables were compared with chi-square tests in the training and validation cohorts. Survival curves were generated using the Kaplan-Meier method, and the difference between groups was compared with the log rank test. Subtypes, clinicopathological features, risk scores, PD-L1, and immune infiltration levels were subjected to correlation analysis by using a Pearson correlation test. Univariate and multivariate analyses were conducted using Cox regression models to determine the independent prognostic value of the risk scores integrated other clinical features. The predictive efficiency of the m6A regulator-relevant signatures for 3- and 5-year OS was estimated using the ROC curves. p < 0.05 indicated statistical significance.
Author Contributions
L.Y. conceived, designed, and supervised the study. L.Y. and G.W. drafted the manuscript. L.Y., G.W., L.G., X.Z., and P.H. collected the data. L.Y., G.W., and L.G. performed all data analysis. All authors reviewed and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors would like to thank the TCGA, GEO, and TIMER databases for the availability of the data and the perseverance of Alan.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.06.001.
Supplemental Information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X.M., Song L.J., Shen J., Yue H., Han Y.Q., Yang C.L., Liu S.Y., Deng J.W., Jiang Y., Fu G.H., Shen W.W. Prognostic and predictive values of immune infiltrate in patients with head and neck squamous cell carcinoma. Hum. Pathol. 2018;82:104–112. doi: 10.1016/j.humpath.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 4.Ferris R.L., Whiteside T.L., Ferrone S. Immune escape associated with functional defects in antigen-processing machinery in head and neck cancer. Clin. Cancer Res. 2006;12:3890–3895. doi: 10.1158/1078-0432.CCR-05-2750. [DOI] [PubMed] [Google Scholar]
- 5.Du K., Zhang L., Lee T., Sun T. m6A RNA Methylation Controls Neural Development and Is Involved in Human Diseases. Mol. Neurobiol. 2019;56:1596–1606. doi: 10.1007/s12035-018-1138-1. [DOI] [PubMed] [Google Scholar]
- 6.Wang S., Sun C., Li J., Zhang E., Ma Z., Xu W., Li H., Qiu M., Xu Y., Xia W. Roles of RNA methylation by means of N6-methyladenosine (m6A) in human cancers. Cancer Lett. 2017;408:112–120. doi: 10.1016/j.canlet.2017.08.030. [DOI] [PubMed] [Google Scholar]
- 7.Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A.A., Kol N., Salmon-Divon M., Hershkovitz V., Peer E., Mor N., Manor Y.S. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 8.Li H.B., Tong J., Zhu S., Batista P.J., Duffy E.E., Zhao J., Bailis W., Cao G., Kroehling L., Chen Y. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548:338–342. doi: 10.1038/nature23450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen T., Hao Y.J., Zhang Y., Li M.M., Wang M., Han W., Wu Y., Lv Y., Hao J., Wang L. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16:289–301. doi: 10.1016/j.stem.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 10.Zhang S., Zhao B.S., Zhou A., Lin K., Zheng S., Lu Z., Chen Y., Sulman E.P., Xie K., Bögler O. m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell. 2017;31:591–606. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui Q., Shi H., Ye P., Li L., Qu Q., Sun G., Sun G., Lu Z., Huang Y., Yang C.G. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017;18:2622–2634. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen M., Wei L., Law C.T., Tsang F.H., Shen J., Cheng C.L., Tsang L.H., Ho D.W., Chiu D.K., Lee J.M. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–2270. doi: 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y., Hsu P.J., Chen Y.S., Yang Y.G. Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616–624. doi: 10.1038/s41422-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bokar J.A., Shambaugh M.E., Polayes D., Matera A.G., Rottman F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3:1233–1247. [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y., Li Y., Toth J.I., Petroski M.D., Zhang Z., Zhao J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ping X.L., Sun B.F., Wang L., Xiao W., Yang X., Wang W.J., Adhikari S., Shi Y., Lv Y., Chen Y.S. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horiuchi K., Kawamura T., Iwanari H., Ohashi R., Naito M., Kodama T., Hamakubo T. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J. Biol. Chem. 2013;288:33292–33302. doi: 10.1074/jbc.M113.500397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pendleton K.E., Chen B., Liu K., Hunter O.V., Xie Y., Tu B.P., Conrad N.K. The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell. 2017;169:824–835. doi: 10.1016/j.cell.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartz S., Mumbach M.R., Jovanovic M., Wang T., Maciag K., Bushkin G.G., Mertins P., Ter-Ovanesyan D., Habib N., Cacchiarelli D. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8:284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Y., Yan J., Li Q., Li J., Gong S., Zhou H., Gan J., Jiang H., Jia G.F., Luo C., Yang C.G. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–384. doi: 10.1093/nar/gku1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer K.D., Jaffrey S.R. Rethinking m6A Readers, Writers, and Erasers. Annu. Rev. Cell Dev. Biol. 2017;33:319–342. doi: 10.1146/annurev-cellbio-100616-060758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu N., Dai Q., Zheng G., He C., Parisien M., Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishizawa Y., Konno M., Asai A., Koseki J., Kawamoto K., Miyoshi N., Takahashi H., Nishida N., Haraguchi N., Sakai D. Oncogene c-Myc promotes epitranscriptome m6A reader YTHDF1 expression in colorectal cancer. Oncotarget. 2017;9:7476–7486. doi: 10.18632/oncotarget.23554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taketo K., Konno M., Asai A., Koseki J., Toratani M., Satoh T., Doki Y., Mori M., Ishii H., Ogawa K. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int. J. Oncol. 2018;52:621–629. doi: 10.3892/ijo.2017.4219. [DOI] [PubMed] [Google Scholar]
- 26.Zhao X., Cui L. Development and validation of a m6A RNA methylation regulators-based signature for predicting the prognosis of head and neck squamous cell carcinoma. Am. J. Cancer Res. 2019;9:2156–2169. [PMC free article] [PubMed] [Google Scholar]
- 27.Han D., Liu J., Chen C., Dong L., Liu Y., Chang R., Huang X., Liu Y., Wang J., Dougherty U. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature. 2019;566:270–274. doi: 10.1038/s41586-019-0916-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He C. Grand challenge commentary: RNA epigenetics? Nat. Chem. Biol. 2010;6:863–865. doi: 10.1038/nchembio.482. [DOI] [PubMed] [Google Scholar]
- 29.Roignant J.Y., Soller M. m6A in mRNA: An Ancient Mechanism for Fine-Tuning Gene Expression. Trends Genet. 2017;33:380–390. doi: 10.1016/j.tig.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Ma J.Z., Yang F., Zhou C.C., Liu F., Yuan J.H., Wang F., Wang T.T., Xu Q.G., Zhou W.P., Sun S.H. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6 -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529–543. doi: 10.1002/hep.28885. [DOI] [PubMed] [Google Scholar]
- 31.Li Z., Weng H., Su R., Weng X., Zuo Z., Li C., Huang H., Nachtergaele S., Dong L., Hu C. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31:127–141. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin S., Choe J., Du P., Triboulet R., Gregory R.I. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell. 2016;62:335–345. doi: 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwok C.T., Marshall A.D., Rasko J.E., Wong J.J. Genetic alterations of m6A regulators predict poorer survival in acute myeloid leukemia. J. Hematol. Oncol. 2017;10:39. doi: 10.1186/s13045-017-0410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X., Li Z., Kong B., Song C., Cong J., Hou J., Wang S. Reduced m6A mRNA methylation is correlated with the progression of human cervical cancer. Oncotarget. 2017;8:98918–98930. doi: 10.18632/oncotarget.22041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raffel G.D., Mercher T., Shigematsu H., Williams I.R., Cullen D.E., Akashi K., Bernard O.A., Gilliland D.G. Ott1(Rbm15) has pleiotropic roles in hematopoietic development. Proc. Natl. Acad. Sci. USA. 2007;104:6001–6006. doi: 10.1073/pnas.0609041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu D., Zhou J., Zhao J., Jiang G., Zhang X., Zhang Y., Dong M. ZC3H13 suppresses colorectal cancer proliferation and invasion via inactivating Ras-ERK signaling. J. Cell. Physiol. 2019;234:8899–8907. doi: 10.1002/jcp.27551. [DOI] [PubMed] [Google Scholar]
- 37.Panahi Y., Darvishi B., Ghanei M., Jowzi N., Beiraghdar F., Varnamkhasti B.S. Molecular mechanisms of curcumins suppressing effects on tumorigenesis, angiogenesis and metastasis, focusing on NF-κB pathway. Cytokine Growth Factor Rev. 2016;28:21–29. doi: 10.1016/j.cytogfr.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 38.Wu Y., Zhao W., Liu Y., Tan X., Li X., Zou Q., Xiao Z., Xu H., Wang Y., Yang X. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. EMBO J. 2018;37:e99017. doi: 10.15252/embj.201899017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park Y.M., Hwang S.J., Masuda K., Choi K.M., Jeong M.R., Nam D.H., Gorospe M., Kim H.H. Heterogeneous nuclear ribonucleoprotein C1/C2 controls the metastatic potential of glioblastoma by regulating PDCD4. Mol. Cell. Biol. 2012;32:4237–4244. doi: 10.1128/MCB.00443-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun W., Xing B., Sun Y., Du X., Lu M., Hao C., Lu Z., Mi W., Wu S., Wei H. Proteome analysis of hepatocellular carcinoma by two-dimensional difference gel electrophoresis: novel protein markers in hepatocellular carcinoma tissues. Mol. Cell. Proteomics. 2007;6:1798–1808. doi: 10.1074/mcp.M600449-MCP200. [DOI] [PubMed] [Google Scholar]
- 41.Liu J., Eckert M.A., Harada B.T., Liu S.M., Lu Z., Yu K., Tienda S.M., Chryplewicz A., Zhu A.C., Yang Y. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018;20:1074–1083. doi: 10.1038/s41556-018-0174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou J., Wang J., Hong B., Ma K., Xie H., Li L., Zhang K., Zhou B., Cai L., Gong K. Gene signatures and prognostic values of m6A regulators in clear cell renal cell carcinoma - a retrospective study using TCGA database. Aging (Albany NY) 2019;11:1633–1647. doi: 10.18632/aging.101856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li X., Tang J., Huang W., Wang F., Li P., Qin C., Qin Z., Zou Q., Wei J., Hua L. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8:96103–96116. doi: 10.18632/oncotarget.21726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou S., Bai Z.L., Xia D., Zhao Z.J., Zhao R., Wang Y.Y., Zhe H. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting β-catenin through mRNA demethylation. Mol. Carcinog. 2018;57:590–597. doi: 10.1002/mc.22782. [DOI] [PubMed] [Google Scholar]
- 45.Fridman W.H., Pagès F., Sautès-Fridman C., Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 46.Galon J., Mlecnik B., Bindea G., Angell H.K., Berger A., Lagorce C., Lugli A., Zlobec I., Hartmann A., Bifulco C. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014;232:199–209. doi: 10.1002/path.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hui L., Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015;368:7–13. doi: 10.1016/j.canlet.2015.07.039. [DOI] [PubMed] [Google Scholar]
- 48.Whiteside T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uppaluri R., Dunn G.P., Lewis J.S., Jr. Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in head and neck cancers. Cancer Immun. 2008;8:16. [PMC free article] [PubMed] [Google Scholar]
- 50.Balermpas P., Michel Y., Wagenblast J., Seitz O., Weiss C., Rödel F., Rödel C., Fokas E. Tumour-infiltrating lymphocytes predict response to definitive chemoradiotherapy in head and neck cancer. Br. J. Cancer. 2014;110:501–509. doi: 10.1038/bjc.2013.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wichmann G., Rosolowski M., Krohn K., Kreuz M., Boehm A., Reiche A., Scharrer U., Halama D., Bertolini J., Bauer U., Leipzig Head and Neck Group (LHNG) The role of HPV RNA transcription, immune response-related gene expression and disruptive TP53 mutations in diagnostic and prognostic profiling of head and neck cancer. Int. J. Cancer. 2015;137:2846–2857. doi: 10.1002/ijc.29649. [DOI] [PubMed] [Google Scholar]
- 52.Yoshihara K., Shahmoradgoli M., Martínez E., Vegesna R., Kim H., Torres-Garcia W., Treviño V., Shen H., Laird P.W., Levine D.A. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bøvelstad H.M., Nygård S., Størvold H.L., Aldrin M., Borgan Ø., Frigessi A., Lingjaerde O.C. Predicting survival from microarray data--a comparative study. Bioinformatics. 2007;23:2080–2087. doi: 10.1093/bioinformatics/btm305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.