

Abstract
Background
Malignant gliomas have disproportionally high morbidity and mortality. Heterozygous mutations in the isocitrate dehydrogenase 1 (IDH1) gene are most common in glioma, resulting in predominantly arginine to histidine substitution at codon 132. Because IDH1R132H requires a wild-type allele to produce (D)-2-hydroxyglutarate for epigenetic reprogramming, loss of IDH1R132H heterozygosity is associated with glioma progression in an IDH1-wildtype-like phenotype. Although previous studies have reported that transgenic IDH1R132H induces the expression of nestin—a neural stem-cell marker, the underlying mechanism remains unclear. Furthermore, this finding seems at odds with better outcome of IDH1R132H glioma because of a negative association of nestin with overall survival.
Methods
Gene expression was compared between IDH1R132H-hemizygous and IDH1R132H-heterozygous glioma cells under adherent and spheroid growth conditions. The results were validated for (D)-2-hydroxyglutarate responsiveness by pharmacologic agents, associations with DNA methylation by bioinformatic analysis, and associations with overall survival. Bisulfite DNA sequencing, chromatin immunoprecipitation, and pharmacological approach were used.
Findings
Neural stem-cell marker genes, including CD44, NES, and PROM1, are generally downregulated in IDH-mutant gliomas and IDH1R132H-heterozygous spheroid growth compared respectively with IDH-wildtype gliomas and IDH1R132H-hemizygous spheroid growth, in agreement with their negative associations with patient outcome. In contrast, CD24 is specifically upregulated and apparently associated with better survival. CD24 and NES expression respond differentially to alteration of (D)-2-hydroxyglutarate levels. CD24 upregulation is associated with histone and DNA demethylation as opposed to hypermethylation in the downregulated genes.
Interpretation
The better outcome of IDH-mutant glioma is orchestrated exquisitely through epigenetic reprogramming that directs bidirectional expression of neural stem-cell marker genes.
Introduction
Gliomas represent 81% of primary brain malignancy and cause significant morbidity and mortality [1]. Whereas glioblastoma (WHO grade IV)—the most common and advanced form of glioma—has a 5-year survival of only 5.5%, WHO grade II and grade III (lower-grade) gliomas—owing to the inevitable recurrence and progression—contribute disproportionately to the high mortality and morbidity [2]. Despite the emergence of novel therapeutics including molecular targeting, the outcome remains dismal.
The human IDH1 gene encodes the cytosolic isocitrate dehydrogenase that catalyzes the conversion of isocitrate and NADP+ to 2-oxoglutarate (aka α-ketoglutarate) and NADPH. Hotspot heterozygous mutations in IDH1 occur in >70% of the lower-grade gliomas and secondary glioblastomas, resulting predominantly in the substitution of arginine 132 with histidine [3]. The mutant enzyme IDH1R132H acquires a neomorphic activity that converts 2-oxoglutarate and NADPH further to (D)-2-hydroxyglutarate (D-2HG) [4,5], thereby leading to NADP+/NADPH imbalance [6,7]. High concentrations of D-2HG inhibit 2-oxoglutarate-dependent histone demethylases and 5-methylcytosine hydroxylases, leading to hypermethylation of lysine residues in histones and CpG islands in DNA [8,9]. It is generally believed that the epigenetic reprogramming through histone and DNA hypermethylation recapitulates the glioma-CpG island methylator phenotype to block cell differentiation and drive IDH-mutant glioma development [[9], [10], [11]].
Interestingly, IDH1R132H neomorphic activity requires the expression of wild-type IDH1; IDH1R132H alone is insufficient to produce D-2HG [[12], [13], [14]]. As such, IDH1R132H-hemizygous glioma cells (resulting from the loss of remaining wild-type allele) show drastic reduction of D-2HG [15]. Loss of IDH1R132H heterozygosity occurs frequently in patient-derived xenografts and ex vivo spheroid cultures and is associated with glioma progression [3]. Compared with IDH1R132H-heterozygous glioma cells, IDH1R132H-hemizygous cells manifest robust anchorage-independent growth and aggressive tumor growth [15,16]. Furthermore, IDH1R132H-hemizygous spheroid growth exhibits significant enrichment of the mesenchymal subtype gene set of glioblastoma and a transcriptomic profile resembling that of IDH1-wildtype glioma [16]. Although uncommon in glioma, loss of IDH1R132H heterozygosity and copy number alterations at the IDH1 locus are associated with glioma recurrence and progression [12,17]. Accordingly, we have proposed that loss of IDH1R132H heterozygosity, but not necessarily IDH1R132H itself, promotes glioma progression [3].
Previous studies showed that transgenic IDH1R132H in contrast to wild-type IDH1 induced nestin expression in immortalized human astrocytes, in correlation with general increases of DNA and histone methylation marks, a key piece of evidence for oncogenic transformation via blocking cell differentiation and adopting a stem-like phenotype [10,18]. Furthermore, treatment of TS603 glioma cells with the IDH1R132H inhibitor AGI-5198 reduced the number of nestin-positive cells, thereby promoting differentiation [9]. These studies suggest the possibility of epigenetic modifications specific for NES (encoding nestin) upregulation in IDH1R132H-heterozygous cells, even though the underlying mechanism remains unclear. Moreover, this finding is apparently at odds with the clinical observation that nestin is an adverse predictor whereas IDH1R132H is a favorable predictor of survival in lower-grade glioma [19,20]. In this study, by taking advantage of the IDH1-wildtype-like phenotype of IDH1R132H hemizygosity [16], we began by comparing nestin expression between IDH1R132H-heterozygous and IDH1R132H-hemizygous cells cultured in adherent and spheroid growth conditions to seek an in-depth understanding of neural stem-cell marker gene expression in relation to IDH1R132H heterozygosity.
Materials and methods
Spheroid growth and treatment
BT142 mut/− (ATCC) was used to generate IDH1R132H-heterozygous BT142 mut/IDH1 through a transgene that expresses YFP, P2A, and IDH1, whereas IDH1R132H-hemizygous BT142 mut/YFP* was created similarly with an engineered stop codon at P2A [15]. IDH1R132H-heterozygous (IMA mut/+) and IDH1-deleted IDH1R132H-hemizygous (IMA mut/−) glioma cells were described previously [12]. Adherent and neural spheroid cultures were performed as described previously [15,21]. Briefly, adherent culture was maintained in a complete growth medium consisting of 45% stem cell medium, 45% DMEM, and 10% FBS, whereas spheroid culture used a neurobasal medium supplemented with B-27, 10 ng/mL bFGF, and 20 ng/mL EGF (Invitrogen).
To ensure adequate treatment with epigenetic agents, cells were first grown in adherent culture with 3 μM AGI-5198 (Sigma-Aldrich, St. Louis, MO, USA) in reference to DMSO or 1 mM octyl-(R)-2HG (Sigma-Aldrich) in reference to ethanol for 3 or 5 days. Likewise, 5 μM 5-aza-2′-deoxycytidine (DAC; TCI America, Portland, OR, USA), UNC0642, or UNC1999 (MedChemExpress, Monmouth, NJ, USA) was administered to adherent cells with medium replacement every other day for a total of 5 days in reference to vehicle control. Treated cells were subsequently seeded at a density of 5 × 104 per well in a 48-well plate for spheroid growth, with continued dosing every three days. Unless otherwise specified, spheroid growth was terminated 7 days after seeding for further analyses.
Gene expression
For reverse transcription–quantitative PCR, total RNA was extracted from IDH1R132H-heterozygous and IDH1R132H-hemizygous BT142 cells cultured under spheroid conditions in 3 biological replicates [16] and was converted to cDNA as previously described [15]. Quantitative PCR was performed in quadruplicate with LightScanner Master Mix (BioFire Diagnostics, Salt Lake City, UT, USA) using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). The primer sets are listed in Supplementary Table 1. The annealing temperature was set at 63 °C for 45 cycles. Quantitation cycle (Cq) values were obtained through CFX Manager Software (Bio-Rad) and were normalized by the Cq values of three reference genes: RPL30, YWHAZ, and UBC.
Western blot analysis was performed essentially as described previously [15,22]. Spheroid cultures between 40 and 100 μm were collected with cell sifters. The same membranes were probed with various antibodies with dilutions as follows: 1:500 anti-Nestin (GenScript, Piscataway, NJ, USA), 1:500 anti-IDH1 (Biovision, Milpitas, CA, USA), and 1:500 anti-IDH1R132H (EMD Millipore, Burlington, MA, USA).
Bisulfite sequencing
Bisulfite conversion was conducted with 500 ng of genomic DNA of BT142 cells using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA). The UCSC Genome Browser on Human DEC. 2013 (GRCH38.HG38) Assembly and MethPrimer [23] were used for primer design (Supplementary Table 2). After 30-cycle amplification, PCR products were purified using DNA Clean & Concentrator-5 Kit (Zymo Research) and ligated into pGEM-T vector for bacterial transformation and DNA sequencing.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed essentially as described previously [24]. Three biological replicates of BT142 mut/YFP* and BT142 mut/IDH1 cells in spheroid culture were prepared with 2 × 106 cells per replicate. The antibodies used for immunoprecipitation were anti-H3K4me3 (EMD Millipore), anti-H3K27me3 (Cell Signaling, Danvers, MA, USA), and anti-H3 (Abcam, Cambridge, MA, USA). The levels of NES and CD24 trimethylation at H3K4 and H3K27 were determined with quantitative PCR as described above and were normalized by the Cq values of matched samples immunoprecipitated with the anti-H3 antibody. The primer sets are listed in Supplementary Table 3. Annealing temperatures were set at 63 °C for 45 cycles.
Bioinformatic analysis
The genomic data sets of GSE16011 and The Cancer Genome Atlas (TCGA) Brain Lower Grade Glioma (TCGA-LGG) were acquired as described previously [25,26]. GSE16011 contains 136 cases of IDH-wild-type and 80 cases of IDH1-mutant gliomas of World Health Organization (WHO) grade II to grade IV, and TCGA-LGG contains 53 cases of IDH-wild-type and 233 cases of IDH-mutant gliomas of WHO grade II to grade III. Comparative analyses of gene expression and DNA methylation based on IDH status were performed as described previously [25]. Likewise, Pearson correlations between DNA methylation and gene expression were performed using Prism 8 (GraphPad, San Diego, CA, USA).
Survival
Kaplan–Meier overall survival analysis of the GSE16011 data set was performed using the R2: Genomic Analysis and Visualization Platform (http://r2.amc.nl) with the Kaplan scan. The p values were Bonferroni corrected. Log-rank (Mantel-Cox) tests were performed according to the z-scores of gene expression from the TCGA-LGG data set using Prism 8. Multivariate Cox regressions based on gene expression, sex, age, and tumor grades of the TCGA-LGG data set were performed using OncoLnc (http://www.oncolnc.org) with p values corrected by false-discovery rate (FDR).
Statistical analysis
Unpaired t-tests with Welch's correction were used for comparative analysis of genomic data sets. Quantitative PCR and bisulfite sequencing data were analyzed in unpaired t-tests with Welch's correction or one-way ANOVA using Prism 8. Two-tailed p values were used for statistical significance (ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001).
Results
Pronounced NES upregulation in IDH1R132H-hemizygous but not IDH1R132H-heterozygous spheroids
Our recent study indicates an IDH1-wildtype-like phenotype in IDH1R132H-hemizygous spheroids but not in those with restored IDH1R132H heterozygosity through an IDH1-wildtype transgene [15,16]. Furthermore, the IDH1-wildtype-like phenotype and the enrichment of glioblastoma mesenchymal subtype gene set were seen only in spheroid but not adherent culture [16]. Accordingly, we compared NES expression in relation to IDH1R132H heterozygosity in both culture conditions. In contrast to less than twofold upregulation in adherent IDH1R132H-heterozygous BT142 cells, NES expression was upregulated by 16-fold in IDH1R132H-hemizygous spheroid growth (Fig. 1A). Likewise, nearly 10-fold NES upregulation was seen in spheroid growth of IDH1R132H-hemizygous IMA glioma cells (owing to genetic deletion of wild-type IDH1) [12,15] compared with IDH1R132H-heterozygous spheroid growth (Fig. 1B). Of note, the striking increase of NES expression is specific to the spheroid culture of IDH1R132H-hemizygous cells. Furthermore, nestin protein levels were readily detectable in the spheroid culture of IDH1R132H-hemizygous, but not IDH1R132H-heterozygous, cells (Fig. 1C), which is in agreement with nestin expression in IDH1R132H-hemizygous, but not IDH1R132H-heterozygous, orthotopic xenografts [16]. In keeping with this, there was a conspicuous loss or decrease of IDH1R132H protein levels in IDH1R132H-heterozygous spheroid, but not adherent, growth accompanied by decreased total IDH1 levels. This finding is in agreement with the marked reduction of D-2HG levels in IDH1R132H-heterozygous spheroids and the selection against IDH1R132H expression by an epigenetic mechanism during anchorage-independent growth [15,21]. Of note, glutamate was added to stimulate spheroid growth of IDH1R132H-heterozygous cells [21] for the sake of yielding sufficient cell lysates; however, no obvious effects were observed on NES mRNA levels in the treated cells (Supplementary Fig. 1). Taken together, these results indicate that NES expression in spheroid growth is biologically more relevant than in adherent growth [3,16] and is consistent with stimulated IDH1R132H-hemizygous spheroid growth but inhibited IDH1R132H-heterozygous spheroid growth observed previously [15].
Fig. 1.
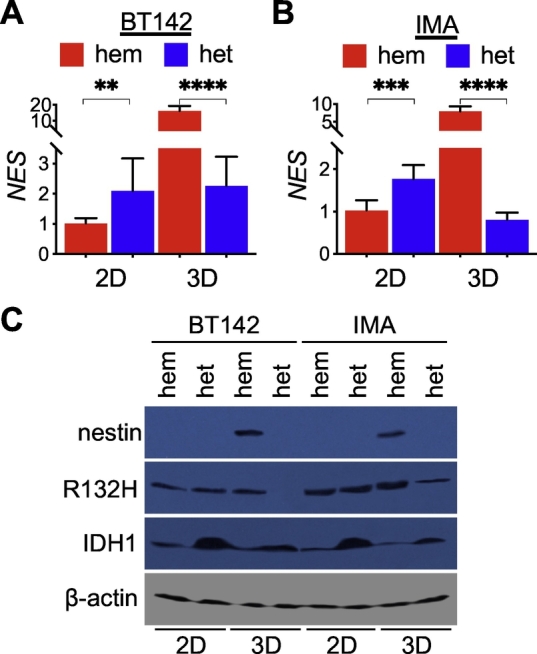
Striking NES upregulation in IDH1R132H-hemizygous spheroids. A and B, Quantitative PCR analysis showed marked increases of NES expression in IDH1R132H-hemizygous (hem) BT142 (A, n = 12) and IMA (B, n = 8) spheroids (3D) in contrast to modest increases in IDH1R132H-heterozygous (het) adherent cells (2D). C. Nestin protein abundance was detected in IDH1R132H-hemizygous, but not IDH1R132H-heterozygous, spheroids. Conspicuous loss or decrease of IDH1R132H protein levels in IDH1R132H-heterozygous spheroids accompanied by decreased total IDH1 levels. Of note, sodium glutamate was used to facilitate spheroid growth of IDH1R132H-heterozygous cells.
NES downregulation in IDH-mutant glioma
To test the notion that NES expression in spheroid growth is more relevant to glioma biology, we compared NES expression between IDH-mutant and IDH-wildtype gliomas using two independent genomic data sets: GSE16011 and TCGA-LGG. The mean z-scores of NES transcript in GSE16011 were 0.1790 and −0.3043 (p = 0.0005, 95% CI = −0.7507 to −0.2160) between IDH-wildtype and IDH-mutant samples (Fig. 2A). Furthermore, in agreement with the previous report that nestin is an adverse predictor of lower-grade gliomas [20], NES expression was negatively associated with overall survival (Supplementary Fig. 2A). Moreover, NES upregulation in IDH-wildtype glioma versus IDH-mutant glioma was confirmed in TCGA-LGG: 0.5967 versus −0.1799 (p = 0.0002, 95% CI = −0.7507 to −0.2160) (Fig. 2B). Together, these results not only indicate NES downregulation in IDH-mutant glioma, in agreement with better patient outcome, but also corroborate the biological relevance of spheroid culture to NES expression, which is repressed in IDH1R132H-heterozygous cells but markedly upregulated upon loss of IDH1R132H heterozygosity.
Fig. 2.

Stem-cell marker gene expression in IDH-mutant glioma. A and B, Violin plots showing significant downregulation of CD44, NES, and PROM1 but upregulation of CD24 and SOX2 in IDH-mutant glioma (mIDH) compared with IDH-wildtype (IDH) glioma from the GSE16011 (A) and TCGA-LGG (B) data sets.
CD24 upregulation specifically in IDH-mutant glioma
Our observation that NES expression is downregulated in IDH-mutant glioma compared with IDH-wildtype glioma is apparently at odds with the previous reports that nestin levels were much higher in immortalized human astrocytes transduced with IDH1R132H in reference to wild-type IDH1 [10,18]. To provide further evidence, we sought to examine the expression of additional glioma stem-cell marker genes, including CD24, CD44, PROM1 (prominin 1, aka CD133), and SOX2 (sex-determining region Y-box 2) in patient samples. Interestingly, we observed an extremely significant increase of mean CD24 expression in IDH-mutant glioma of GSE16011; the mean z-scores of CD24 transcript in IDH-wildtype and IDH-mutant samples were −0.2425 and 0.4122, respectively (p < 0.0001, 95% CI = 0.4011 to 0.9082), with a similar finding in TCGA-LGG: −0.2264 and 0.05148 (p = 0.0021, 95% CI = 0.1017 to 0.4541) (Fig. 2A, B). Furthermore, three- and fourfold increases of CD24 expression were detected in IDH1R132H-heterozygous BT142 and IMA spheroid growth compared with IDH1R132H-hemizygous ones (Fig. 3A, B), which is similar to a previous finding of CD24 upregulation in immortalized human astrocytes with long-term IDH1R132H expression in reference to those without [27]. Moreover, we observed significant associations of CD24 upregulation with better overall survival in both GSE16011 and TCGA-LGG (Fig. 3C, D). The result from multivariate Cox regression analysis confirmed the survival benefit of CD24 expression with a significant Cox coefficient of −0.28 (Table 1), which is apparently in contrast to the implication of CD24 in gliomagenesis [27].
Fig. 3.

CD24 upregulation specifically in IDH1R132H-heterozygous glioma cells. A and B. Quantitative PCR revealed significant upregulation of CD24 but downregulation of other specified stem-cell marker genes in both IDH1R132H-heterozygous BT142 (A) and IMA (B) spheroids compared with their IDH1R132H-hemizygous controls (n = 8). C and D, CD24 expression is associated with better overall survival (OS), as shown in Kaplan–Meier survival analysis of the GSE16011 data set with Bonferroni-corrected p = 6.8e−07 (C) and log-rank test of the TCGA-LGG data set (D).
Table 1.
Cox regression analysis of listed genes from the TCGA-LGG data set.

1Listed in an ascending order.
2Insignificant FDR-corrected p-values indicated in gray.
Similar to NES downregulation in IDH-mutant glioma, both CD44 and PROM1 transcript levels were significantly lower in IDH-mutant glioma than in IDH-wildtype glioma (Fig. 2), and in IDH1R132H-heterozygous BT142 and IMA spheroids than in IDH1R132H-hemizygous ones (Fig. 3A, B). Both CD44 and PROM1 were negatively associated with overall survival, with a significant Cox coefficient of 0.40 and 0.26, respectively (Supplementary Fig. 2; Table 1). Oddly, SOX2 transcript levels were significantly higher in IDH-mutant glioma than in IDH-wildtype glioma (Fig. 2) but significantly lower in IDH1R132H-heterozygous BT142 and IMA spheroids than in IDH1R132H-hemizygous ones (Fig. 3A, B). Furthermore, the association of SOX2 expression with overall survival seemed ambiguous (Supplementary Fig. 2; Table 1). Nevertheless, these results support the notion that CD24 is upregulated specifically among the neural stem-cell markers in IDH-mutant glioma whereas the downregulation of CD44, NES, and PROM1 is in agreement with the better patient outcome.
Differential regulation of CD24 and NES expression by D-2HG
Previously, we showed a marked decrease of D-2HG in IDH1R132H-hemizygous cells compared with IDH1R132H-heterozygous cells [15]. Therefore, we sought to ascertain whether octyl-(R)-2HG—a membrane-permeant precursor form of D-2HG—could alter CD24 and NES expression in IDH1R132H-hemizygous cells. Indeed, such treatment led to an approximately fivefold increase of CD24 expression but an eightfold decrease of NES expression in IDH1R132H-hemizygous BT142 spheroids (Fig. 4A) along with nearly fourfold inhibition of spheroid growth (Supplementary Fig. 3A). In IDH1R132H-heterozygous spheroids, however, such treatment had no effect on CD24 expression but further decreased NES expression by tenfold (Fig. 4B) along with moderate inhibition of spheroid growth (Supplementary Fig. 3A). Conversely, treatment of IDH1R132H-heterozygous spheroids with AGI-5198—a potent IDH1R132H inhibitor [9]—resulted in 70% decrease in CD24 expression, but a 14-fold increase of NES expression along with nearly threefold increase of spheroid growth (Supplementary Fig. 3B), in contrast to modest effects on IDH1R132H-hemizygous spheroids (Fig. 4C, D). Consistently, AGI-5198 increased CD44 expression in IDH1R132H-heterozygous spheroids, whereas octyl-(R)-2HG inhibited CD44 and PROM1 expression in both types of spheroids. Similar results were obtained in IMA spheroids (Supplementary Figs. 3 and 4). Thus, our finding not only supports differential regulation of CD24 and NES in IDH1R132H-heterozygous cells but also suggests a distinct epigenetic mechanism underlying CD24 upregulation.
Fig. 4.

Differential regulation of CD24 and NES by D-2HG in BT142 spheroid growth. A and B, Octyl-(R)-2HG treatment stimulated CD24 expression in IDH1R132H-hemizygous spheroids (A, hem + 2HG) but inhibited NES expression in both IDH1R132H-hemizygous and IDH1R132H-heterozygous spheroids (B, het + 2HG) in reference to vehicle treatment (+ethanol). C and D, In contrast to modest effects in IDH1R132H-hemizygous spheroids (C, hem + AGI5198), AGI-5198 treatment stimulated NES expression but inhibited CD24 expression in IDH1R132H-heterozygous spheroids (D, het + AGI5198) in reference to vehicle treatment (+DMSO). Gene expression was assayed with quantitative PCR (n = 8).
Correlation of DNA methylation with neural stem-cell marker gene expression
IDH-mutant glioma is known to be associated with genome-wide DNA hypermethylation owing to D-2HG inhibition of the TET 5-methylcytosine hydroxylases, even though DNA hypomethylation also occurs to a lesser extent [28,29]. To provide evidence for an association of gene expression with epigenetic modification, we compared DNA methylation of neural stem-cell marker genes between IDH-mutant and IDH-wildtype gliomas using the TCGA-LGG data set. Whereas the mean values of DNA methylation for CD44, NES, PROM1, and SOX2 in IDH-wildtype glioma were in a descending order (0.5980, 0.3635, 0.1381, and 0.09519), significant increases of DNA methylation (0.9023, 0.5389, 0.4503, and 0.04857) were evident in IDH-mutant glioma (Fig. 5A). Of note, SOX2 remained essentially hypomethylated, with a minute difference in the mean values between the two glioma types. Furthermore, we observed significant inverse correlations between DNA methylation and gene expression in IDH-mutant glioma, albeit with PROM1 showing relatively weak Pearson R2 (Fig. 5B). In IDH-wildtype glioma, however, only CD44 and NES showed extremely significant correlations. These results suggest that heterozygous IDH1R132H induces the downregulation of neural stem-cell marker genes through epigenetic reprogramming.
Fig. 5.

Inverse correlations of DNA methylation with glioma stem-cell marker gene expression. A, Comparative analysis of the TCGA-LGG data set showing significantly higher median levels of DNA methylation in CD44, NES, and PROM1 but lower in SOX2 in IDH-mutant (mIDH) glioma compared with IDH-wildtype (IDH) glioma. B, Pearson correlations showing inverse correlations between DNA methylation and gene expression in IDH-mutant glioma (circles), as indicated by p and R2 values, and also in IDH-wildtype glioma (triangles).
Distinctive epigenetic modifications at the CD24 locus
To gain insight into epigenetic modification of CD24, which was unavailable from the TCGA-LGG data set, we performed bisulfite sequencing of CD24 in reference to NES with genomic DNA extracted from IDH1R132H-heterozygous and IDH1R132H-hemizygous BT142 cells. Within the CD24 promoter region examined, we observed an average of 28% CpG methylation in IDH1R132H-hemizygous cells versus 6% in IDH1R132H-heterozygous cells—a more than fourfold decrease of DNA methylation (Fig. 6A, B). In contrast, the NES promoter region showed a greater than eightfold increase in DNA methylation in IDH1R132H-heterozygous cells. This result suggests that the upregulation of CD24 in IDH-mutant glioma is associated with a decrease of DNA methylation.
Fig. 6.

Differential epigenetic modifications between CD24 and NES in IDH1R132H-heterozygous cells. A, Bisulfite sequencing results showing lower levels of CpG methylation (filled circles) in CD24 but higher levels in NES in IDH1R132H-heterozygous cells than in IDH1R132H-hemizygous cells. B, Quantitative analysis of the bisulfite sequencing results. C, Chromatin immunoprecipitation of BT142 spheroids revealed an opposing trend of H3K27me3 (n = 9) between CD24 and NES, presented in a log2 ratio of IDH1R132H-heterozygous cells over IDH1R132H-hemizygous cells.
Moreover, we assessed specific changes in histone methylation at the gene locus of CD24 in reference to NES using the transcriptional activation mark—trimethylation of histone 3 lysine 4 (H3K4me3)—and the repression mark—trimethylation of histone 3 lysine 27 (H3K27me3) [30]. Chromatin immunoprecipitation revealed that IDH1R132H-heterozygous spheroid growth exhibited a 6-fold decrease of H3K27me3 at the CD24 locus while a ~5.5-fold increase at the NES locus compared with IDH1R132H-hemizygous spheroid growth (Fig. 6C). Interestingly, H3K4me3 was increased for both genes, suggesting the possibility of bivalent histone modifications [31]. Taken together, these results support the notion that heterozygous IDH1R132H induces epigenetic modifications specific for CD24 upregulation, whereas loss of IDH1R132H heterozygosity alters epigenetic modifications to promote IDH1-wildtype-like gene expression.
Complex effects of epigenetic inhibitors on neural stem-cell marker gene expression
To test the functional relevance of epigenetic modifications to gene expression, we employed inhibitors of DNA methyltransferase and histone demethylases in IDH1R132H-heterozygous cells. Treatment with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine resulted in a general downregulation of glioma stem-cell marker genes in comparison with the vehicle controls (Fig. 7A). This finding apparently differs from increased tissue factor (F3) expression after 5-aza-2′-deoxycytidine treatment [32] but may support the previous finding that treatment with 5-azacytidine effectively results in glioma regression [33]. Treatment with the histone methyltransferases G9a/GLP inhibitor UNC0642, which has been shown to reverse histone H3K9 methylation and restore Atm expression [34], yielded a modest increase in NES expression but downregulation of the other genes (Fig. 7B). However, treatment with the histone methyltransferase EZH2/EZH1 inhibitor UNC1999, which removes H3K27me3, resulted in a reversal of gene expression—CD44 and NES upregulation but CD24 and PROM1 downregulation (Fig. 7C). Although the results indicate differential mechanisms of epigenetic regulation in IDH-mutant glioma, the precise mechanism for individual genes requires further investigation.
Fig. 7.

Effects of epigenetic inhibitors on stem-cell marker gene expression in IDH1R132H-heterozygous BT142 spheroids. Quantitative PCR analysis showed general inhibition of gene expression by 5-aza-2′-deoxycytidine (DAC, A) and UNC0642 except for NES (B), and stimulation of CD44 and NES but inhibition of CD24 and PROM1 by UNC1999 (C) in reference to DMSO treatment. One-way ANOVA was performed by comparing with vehicle treatment.
Discussion
The transgenic IDH1R132H effect on nestin expression was a key piece of evidence supporting the notion that IDH1R132H induces oncogenic transformation through epigenetic changes that block neural differentiation and adopt a stem-like phenotype [9,10,18]. However, our analyses of patient data and DNA and histone modifications indicate NES downregulation in IDH-mutant gliomas and IDH1R132H-heterozygous spheroid growth compared respectively with IDH-wildtype gliomas and IDH1R132H-hemizygous spheroid growth. Furthermore, NES expression is well correlated with epigenetic modifications, supporting epigenetic inhibition of NES expression in IDH-mutant glioma. The NES downregulation is in agreement with glioma patient outcome, which is associated negatively with nestin but positively with IDH1R132H [6,19,20]. Moreover, the general downregulation of neural stem-cell marker genes in IDH-mutant glioma compared with IDH-wildtype glioma is consistent with the inhibition of anchorage-independent growth of IDH1R132H-heterozygous cells [15,16] and the retardation of glioma growth [16,35,36]. The modest increase of NES expression in adherent IDH1R132H-heterozygous cells compared with IDH1R132H-hemizygous cells, however, might explain the previous finding [9,10,18] presumably obtained under similar growth conditions. In light of our recent studies that anchorage-independent growth is more relevant to IDH1R132H glioma biology [15,16], we conclude therefore that heterozygous IDH1R132H epigenetically suppresses NES expression.
Of note, despite NES downregulation in IDH-mutant glioma, our studies also suggest that nestin is nonessential to anchorage-independent growth and tumor growth. We provided evidence here that although extracellular glutamate stimulated IDH1R132H-heterozygous cells for spheroid growth, no NES upregulation was detected at the transcript and protein levels. Likewise, in orthotopic transplantation models, nestin was expressed in tumors derived from IDH1R132H-hemizygous, but not IDH1R132H-heterozygous, glioma cells [16]. Thus, these findings not only are consistent with the term of stem-cell marker but, more importantly, support the notion of a tissue-specific role for glutamate in IDH1R132H glioma biology [3,16,21].
Interestingly, heterozygous IDH1R132H induced CD24 upregulation in stark contrast to the general downregulation of glioma stem-cell marker genes. CD24—a glycosylphosphatidylinositol-anchored molecule—is a marker of neural cell lineage tumors [37] and has been implicated in IDH1R132H gliomagenesis [27]. Likewise, CD24 expression is generally associated with cancer progression and poor survival including glioblastoma [38,39]. Furthermore, CD24 has been identified as a novel ‘don't eat me’ signal of cancer cells through the interaction with Siglec-10 expressed in tumor-associated macrophages [40]. Surprisingly, we observed an association of CD24 expression with better survival in both single- and multi-variate analyses. Of note, intracellular CD24 expression is required for the inactivation of remnant activity of mutant TP53, and high-level CD24 expression is associated with TP53 mutations in TCGA lower-grade glioma [41]. Moreover, CD24 expresses differential isoforms according to the tissue of origin, differentiation status, and post-translational modifications [37]. Therefore, further studies are warranted to unravel the molecular intricacies between TP53 mutations, CD24 upregulation, and CD24 isoforms.
Mechanistically, we provided evidence that IDH1R132H-heterozygous cells not only responded to AGI-5198 by decreasing CD24 expression while increasing NES expression but also exhibited DNA hypomethylation at the CD24 locus, consistent with the previous reporting of the existence of DNA hypomethylation despite genome-wide DNA hypermethylation in IDH-mutant glioma [28,29]. Conversely, octyl-(R)-2HG treatment specifically increased CD24 expression in IDH1R132H-hemizygous cells. Furthermore, the divergent trend of DNA methylation and gene expression between CD24 and NES is in good agreement with the levels of transcriptional repressive H3K27me3—significantly lower in CD24 but much higher in NES. Further investigations are required, however, to determine whether the H3K27me3 level is key to the differential expression of CD24 and NES because the H3K27me3 inhibitor UNC1999 increased the expression of NES and CD44 but, unexpectedly, decreased the expression of CD24 in IDH1R132H-heterozygous cells. Moreover, previous studies showed that the accumulation of H3K9me3—another histone-repressive mark—was particularly noticeable in IDH-mutant cells [18] and the use of H3K9me3 inhibitor UNC0642 reduced H3K9 methylation and increased Atm expression [34]. The consequence of UNC0642 treatment in this study, however, was gene downregulation in general, suggesting the complexity of epigenetic regulation in IDH-mutant cells. An integrated genomic approach is expected to reveal how differential gene expression in response to these treatments correlates with epigenetic changes of these neural stem cell marker genes. Nevertheless, our results support the notion that CD24 is upregulated specifically in IDH-mutant glioma in association with epigenetic modifications.
A relatively small number of genes are known to be markedly upregulated and biologically relevant to IDH1R132H glioma. GLUD2 (glutamate dehydrogenase 2) is upregulated in IDH-mutant glioma to alleviate IDH1 mutation-induced metabolic stress and promote glioma growth [42,43]. Interestingly, GLUD2 upregulation also has positive correlation with patient survival (Table 1; data not shown). PDGFRA is another example of aberrant upregulation via DNA hypermethylation that compromises the binding of methylation-sensitive insulator protein CTCF for gene activation [44], but whether PDGFRA upregulation promotes IDH-mutant gliomagenesis requires further investigation, especially considering its association with better overall survival [25] and has a negative Cox coefficient in multivariate analysis (Table 1). Moreover, ATM (ataxia telangiectasia mutated) upregulation was reported in a mouse model of IDH1R132H glioma with Trp53 and Atrx mutations [45], but Atm downregulation was shown in a myeloid lineage-specific conditional Idh1-mutated mouse model [34]. Nonetheless, the prognostic value of ATM expression in glioma patients seems less clear (Table 1; data not shown).
In sum, we presented evidence for the general downregulation of neural stem-cell marker genes including nestin in IDH-mutant glioma but markedly upregulated upon loss of IDH1R132H heterozygosity. However, CD24 is specifically upregulated in IDH-mutant glioma in association with reduced DNA methylation and histone-repressive mark. Further studies are warranted to determine the underlying mechanism and biological role of CD24 expression in IDH-mutant glioma.
Declaration of competing interest
None.
Acknowledgments
Acknowledgments
The authors thank Luming Zhou and Carl T. Wittwer for the assistance in quantitative PCR analysis and Kristin Kraus for editorial assistance.
Funding sources
This work was supported in part by NIH R21NS108065 (LEH), NIH R21HG009181 (MBC), and NIH R01CA188520 (SB).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.tranon.2020.100819.
Appendix A. Supplementary data
Supplementary material
References
- 1.Ostrom Q.T., Bauchet L., Davis F.G. The epidemiology of glioma in adults: a “state of the science” review. Neuro-Oncol. 2014;16:896–913. doi: 10.1093/neuonc/nou087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wen P.Y., Kesari S. Malignant gliomas in adults. N. Engl. J. Med. 2008;359:492–507. doi: 10.1056/nejmra0708126. [DOI] [PubMed] [Google Scholar]
- 3.Huang L.E. Friend or foe—IDH1 mutations in glioma 10 years on. Carcinogenesis. 2019;40:1299–1307. doi: 10.1093/carcin/bgz134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang L., White D.W., Gross S. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ward P.S., Patel J., Wise D.R. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan H., Parsons D.W., Jin G. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009;360:765–773. doi: 10.1056/nejmoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang X., Fu X., Liu Y. Blockade of glutathione metabolism in IDH1-mutated glioma. Mol. Cancer Ther. 2019;19:221–230. doi: 10.1158/1535-7163.mct-19-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu W., Yang H., Liu Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rohle D., Popovici-Muller J., Palaskas N. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turcan S., Rohle D., Goenka A. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang F., Travins J., DeLaBarre B. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 12.Jin G., Reitman Z.J., Duncan C.G. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Research. 2013;73:496–501. doi: 10.1158/0008-5472.can-12-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward P.S., Lu C., Cross J.R. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J. Biol. Chem. 2013;288:3804–3815. doi: 10.1074/jbc.m112.435495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han S., Liu Y., Cai S.J. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Brit J Cancer. 2020;122:1580–1589. doi: 10.1038/s41416-020-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tiburcio P.D.B., Xiao B., Berg S. Functional requirement of a wild-type allele for mutant IDH1 to suppress anchorage-independent growth through redox homeostasis. Acta Neuropathol. 2018;135:285–298. doi: 10.1007/s00401-017-1800-0. [DOI] [PubMed] [Google Scholar]
- 16.Tiburcio P.D.B., Gillespie D.L., Jensen R.L., Huang L.E. Extracellular glutamate and IDH1R132H inhibitor promote glioma growth by boosting redox potential. J. Neuro-Oncol. 2020;146:427–437. doi: 10.1007/s11060-019-03359-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazor T., Chesnelong C., Pankov A. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc. Natl. Acad. Sci. 2017;114:10743–10748. doi: 10.1073/pnas.1708914114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu C., Ward P.S., Kapoor G.S. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanson M., Marie Y., Paris S. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J. Clin. Oncol. 2009;27:4150–4154. doi: 10.1200/jco.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- 20.Hatanpaa K.J., Hu T., Vemireddy V. High expression of the stem cell marker nestin is an adverse prognostic factor in WHO grade II-III astrocytomas and oligoastrocytomas. J. Neuro-oncol. 2014;117:183–189. doi: 10.1007/s11060-014-1376-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tiburcio P.D.B., Xiao B., Chai Y. IDH1R132H is intrinsically tumor-suppressive but functionally attenuated by the glutamate-rich cerebral environment. Oncotarget. 2018;9:35100–35,113. doi: 10.18632/oncotarget.26203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi H., Gillespie D.L., Berg S. Intermittent induction of HIF-1α produces lasting effects on malignant progression independent of its continued expression. PLOS One. 2015;10 doi: 10.1371/journal.pone.0125125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L.-C., Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 24.Bhaskara S., Knutson S.K., Jiang G. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436–447. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang L.E., Cohen A.L., Colman H. IGFBP2 expression predicts IDH-mutant glioma patient survival. Oncotarget. 2016;8:191–202. doi: 10.18632/oncotarget.13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karsy M., Guan J., Huang L.E. Prognostic role of mitochondrial pyruvate carrier in isocitrate dehydrogenase-mutant glioma. J. Neurosurg. 2018;130:56–66. doi: 10.3171/2017.9.jns172036. [DOI] [PubMed] [Google Scholar]
- 27.Turcan S., Makarov V., Taranda J. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018;50:62–72. doi: 10.1038/s41588-017-0001-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duncan C.G., Barwick B.G., Jin G. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012;22:2339–2355. doi: 10.1101/gr.132738.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei S., Wang J., Oyinlade O. Heterozygous IDH1R132H/WT created by “single base editing” inhibits human astroglial cell growth by downregulating YAP. Oncogene. 2018;37:5160–5174. doi: 10.1038/s41388-018-0334-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007;8:829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- 31.Vastenhouw N.L., Schier A.F. Bivalent histone modifications in early embryogenesis. Curr. Opin. Cell Biol. 2012;24:374–386. doi: 10.1016/j.ceb.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Unruh D., Mirkov S., Wray B. Methylation-dependent tissue factor suppression contributes to the reduced malignancy of IDH1-mutant gliomas. Clin. Cancer Res. 2018;25:747–759. doi: 10.1158/1078-0432.ccr-18-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borodovsky A., Salmasi V., Turcan S. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget. 2013;4:1737–1747. doi: 10.18632/oncotarget.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoue S., Li W.Y., Tseng A. Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell. 2016;30:337–348. doi: 10.1016/j.ccell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anido J., Sáez-Borderías A., Gonzàlez-Juncà A. TGF-β receptor inhibitors target the CD44high/Id1high glioma-initiating cell population in human glioblastoma. Cancer Cell. 2010;18:655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 36.Singh S.K., Hawkins C., Clarke I.D. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 37.Poncet C., Frances V., Gristina R. CD24, a glycosylphosphatidylinositol-anchored molecule, is transiently expressed during the development of human central nervous system and is a marker of human neural cell lineage tumors. Acta Neuropathol. 1996;91:400–408. doi: 10.1007/s004010050442. [DOI] [PubMed] [Google Scholar]
- 38.Fukushima T., Tezuka T., Shimomura T. Silencing of insulin-like growth factor-binding protein-2 in human glioblastoma cells reduces both invasiveness and expression of progression-associated gene CD24. J. Biol. Chem. 2007;282:18634–18,644. doi: 10.1074/jbc.m609567200. [DOI] [PubMed] [Google Scholar]
- 39.Gilliam D.T., Menon V., Bretz N.P., Pruszak J. The CD24 surface antigen in neural development and disease. Neurobiol. Dis. 2017;99:133–144. doi: 10.1016/j.nbd.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 40.Barkal A.A., Brewer R.E., Markovic M. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572:392–396. doi: 10.1038/s41586-019-1456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L., Liu R., Ye P. Intracellular CD24 disrupts the ARF–NPM interaction and enables mutational and viral oncogene-mediated p53 inactivation. Nat. Commun. 2015;6:5909. doi: 10.1038/ncomms6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen R., Nishimura M.C., Kharbanda S. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. P Natl Acad Sci Usa. 2014;111:14217–14222. doi: 10.1073/pnas.1409653111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waitkus M.S., Pirozzi C.J., Moure C.J. Adaptive evolution of the GDH2 allosteric domain promotes gliomagenesis by resolving IDH1R132H induced metabolic liabilities. Cancer Res. 2018;78:36–50. doi: 10.1158/0008-5472.can-17-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flavahan W.A., Drier Y., Liau B.B. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2015;529:110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Núñez F.J., Mendez F.M., Kadiyala P. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019;11:eaaq1427. doi: 10.1126/scitranslmed.aaq1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material