

Abstract
Motivation
Understanding the underlying biological mechanisms and respective interactions of a disease remains an elusive, time consuming and costly task. Computational methodologies that propose pathway/mechanism communities and reveal respective relationships can be of great value as they can help expedite the process of identifying how perturbations in a single pathway can affect other pathways.
Results
We present a random-walks-based methodology called PathWalks, where a walker crosses a pathway-to-pathway network under the guidance of a disease-related map. The latter is a gene network that we construct by integrating multi-source information regarding a specific disease. The most frequent trajectories highlight communities of pathways that are expected to be strongly related to the disease under study.
We apply the PathWalks methodology on Alzheimer's disease and idiopathic pulmonary fibrosis and establish that it can highlight pathways that are also identified by other pathway analysis tools as well as are backed through bibliographic references. More importantly, PathWalks produces additional new pathways that are functionally connected with those already established, giving insight for further experimentation.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Since its introduction more than a century ago, random walks (Pearson, 1905) have been successfully applied to a wide range of sciences including Physics, Chemistry, Biology, Computer Science and Engineering. With its effective algorithmic layout, easy realization and efficiently produced outcomes, the method is still deemed a suitable choice for extracting sub-networks of interest in graph-structures consisting of nodes demonstrating multiple strong connections. The methodology has known weaknesses, such as simply recreating the degree distribution of a graph or getting trapped in highly connected cliques without being able to explore distant neighborhoods. Moreover, random walks entail a finite number of steps and in this respect, if additional neighborhoods are to be explored during the same time period, multiple walkers have to be simultaneously deployed (Ding and Szeto, 2017; Lu et al., 2006). To prevent walker entrapment in strongly connected network regions, restart strategies are used (Chen et al., 2016; Tong et al., 2006). Such strategies allow a walker to discontinue its current course and proceed by following up a different node in the graph. Converging strategies have also been studied, mostly in the context of computer networks where random walks converge according to application-induced probability distributions for visiting nodes (Zhong et al., 2008).
The output from methodologies such as the random walk, heavily depends on the quality of the contained data. In random walks specifically, these data can be integrated in a graph. There is a vast number of online databases offering biological content and an even greater need of parsing and integrating this information (Baxevanis and Bateman, 2015; Navarro et al., 2003; Philippi and Köhler, 2006). The potential knowledge gain could provide researchers the means and tools to extract results that would benefit the health care system by enhancing prevention, diagnosis as well as treatment of maladies.
Computational applications, which allow for fast screening and integration of such biological information, are the prerequisite for speeding up the process of generating quality results. In this respect, tools, such as the PREDICT (Gottlieb et al., 2011), integrate drug information from online databases including DrugBank (Wishart et al., 2018), OMIM (Amberger et al., 2015) and SIDER (Kuhn et al., 2016) to suggest new drug-target indications based on substance similarities. Other models including MutPred (Li et al., 2009) parse protein sequences and provide insights on the mechanisms of diseases. Similar software tools are especially needed in the case of rare diseases as in vivo experiments might not be given the appropriate consideration. The latter could be attributed to the lack of targeted individuals especially if a disease under examination is simply infrequent.
The integration of biological data from different ‘omes’ (e.g. genome, transcriptome and proteome) is essential for bioinformatics applications that yield sophisticated results revolving around pathway analysis, drug repurposing, interaction networks and disease associations. Zachariou et al. (2018) examined the importance of studying disease mechanisms from a multi-omics perspective and proposed a multi-level network for the Alzheimer’s disease (AD). This network was formed by integrating multi-source biological information, such as differentially expressed genes, pathways, single-nucleotide polymorphisms, drugs and microRNAs. Here, genes act as intermediaries between the different layers of the proposed network. Through this methodology, clusters of potential key biological pathways of AD were proposed for further examination.
Community detection algorithms are regularly used to identify meaningful clusters in a graph and have been successfully proposed in the context of social networks for more than a decade now (Clauset et al., 2004; Liakos et al., 2017; Yang and Leskovec, 2012). We have only recently seen the adoption of such techniques in biological settings. In particular, a benchmarking study (Rahiminejad et al., 2019) considered the Louvain method (Blondel et al., 2008) as the best choice in finding protein communities in the protein–protein interaction (PPI) networks of Human and Yeast. While addressing the DREAM challenge, Tripathi et al. (2019) applied their community detection framework in six heterogeneous biological networks (two human PPI, a pathway signaling, a co-expression, a cancer and a homology network) in order to extract core disease communities. More specifically, they showed that overlapping community detection algorithms yield better results for disease module identification, which is justified since a node (e.g. a gene) can participate in multiple diseases at the same time. Wilson et al. (2017) applied community detection algorithms in a gene interaction network and while deploying the Louvain algorithm they sought to identify communities of up to 10 genes that characterize functional and disease pathways.
In this work, we propose a random walk-based methodology on a pathway-to-pathway network and we term this as PathWalks. PathWalks exploits a map that we construct in the form of a synthetic gene network, containing integrated information regarding a disease of interest, as the latter has been presented in Zachariou et al. (2018). We create multi-source integrated information maps regarding AD and idiopathic pulmonary fibrosis (IPF). We use the produced maps to drive random walks on respective pathway-to-pathway networks. Our methodology highlights the most frequently walked candidate pathways and trajectories, identifying pathway communities that are expected to be strongly related to these diseases. The novelty of our approach lies with the exploitation of multi-omics disease-related information that helps drive walks on a functional connectivity network of biological pathways. The approach ultimately highlights key pathways and their functional communities related to the disease of interest.
2 Materials and methods
2.1 The general concept of PathWalks
Our proposed PathWalks methodology integrates random walks and shortest paths computations to walk on a pathway-to-pathway network under the guidance of a synthetic gene network that we construct by integrating a-priori molecular information related to a disease (Zachariou et al., 2018). The PathWalks methodology exploits two main network components related to a disease of interest, which need to be constructed before the execution of the algorithm.
The first component is the multi-source information map; this is a synthetic gene-to-gene network, which represents integrated information (e.g. gene co-expression, physical interactions and miRNA targets) from biological databases in the form of weighted connections. Mathematically, the gene network is represented as a graph (Gg) and described as Gg = (Vg, Eg), where Vg is the set of nodes (genes) and Eg is the set of connections among nodes. The walker performs random walks on the gene network and the visited nodes indicate the walker’s new destination on the PathWalks’ second component; the functional connectivity network of biological pathways.
We construct the pathway-to-pathway network [Gp = (Vp, Ep)], by parsing the biological pathways’ functional connectivity information from KEGG (Kanehisa et al., 2017). Pathways that contain genes already associated with the studied disease, receive higher numeric-value edge scores (i.e. visitation probability). The walker moves on the pathway-to-pathway network according to the instructions given by the map (gene-to-gene network) in order to explore biological pathway relations regarding the disease under examination.
A sorted list of the most visited pathways is generated after a set number of iterations. In order for the algorithm to converge, the two last sorted pathway-visitation lists must have a similarity index above a selected threshold. Finally, the algorithm highlights the most frequently visited edges (i.e. pathway-to-pathway connections) and nodes (pathways), revealing interesting pathway communities, according to the multi-source map. In this study, we explore two use-case scenarios from different disease settings; AD as a neurodegenerative disease and IPF as a fibrotic disease. We show a descriptive diagram of the PathWalks methodology in Figure 1.
Fig. 1.
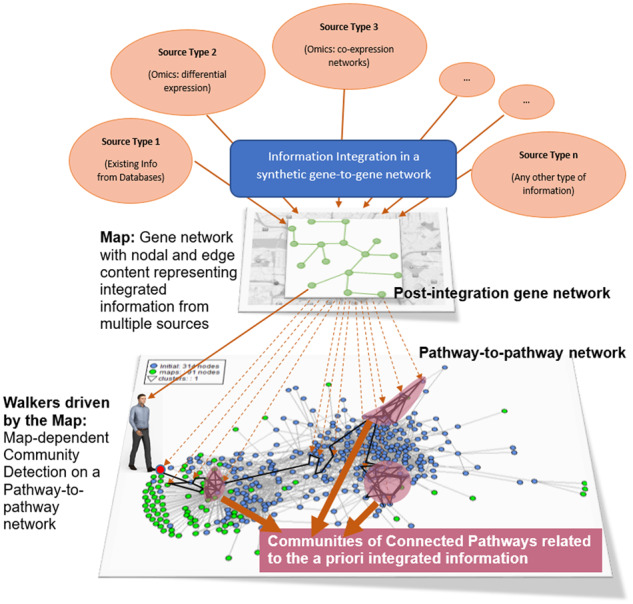
The PathWalks Concept. We integrate multi-source information regarding a disease in a gene map. This gene map guides the walker on a functional connectivity network of biological pathways to identify key pathway communities of the disease
2.2 Multi-source integrated gene map per disease
The first component needed for the execution of PathWalks is the gene map. Here, we create gene maps for the PathWalks algorithm by integrating biological information as described (Zachariou et al., 2018). For both AD and IPF maps, we download genes, drugs, biological pathways and single-nucleotide polymorphisms from Malacards (Espe, 2018). For the AD map, we further include copy-number variations’ information from Malacards, which was missing in the case of IPF. We link drugs of both cases to their gene targets via the DrugBank database. We then extract additional genetic and physical interaction information for each disease’s genes through GeneMANIA’s (Franz et al., 2018) default dataset choices for these two categories. Finally, we map the genes of each disease to miRNAs through MirTarBase (Chou et al., 2018). In the AD use case, we explore additional miRNAs through miRBase (Griffiths-Jones et al., 2007) and TargetScan (Lewis et al., 2005). Following the multi-source integration, we generate gene-to-gene networks to act as guiding maps during the PathWalks execution.
2.3 Pathway-to-pathway reference network
The second component needed for the PathWalks’ execution is the pathways’ network, on which the walker explores pathway relations to highlight sub-networks of disease-related molecular mechanisms. The pathways’ network is an undirected graph of functional connections that we parse from KEGG’s KGML files. A biological pathway in KEGG consists of genes and their molecular interactions, reactions and relations. The nodes in the PathWalks’ pathway-to-pathway network represent biological pathways and an edge connecting two pathways represents a functional link between them. We assign a score on each edge according to the following equation:
where PSiA and PSiB are the pathway scores (PSs) (see below) of the nodes A and B connected with edge i.
The multi-source integration framework combines data across various sources of information into one network and aggregates them into a gene-specific score, based both on the gene characteristic information and on gene–gene integrated inter-relation. We obtain the PS of each pathway by adding the respective participating genes’ specific scores. These specific scores represent the gene’s observed relation to the disease of study. We calculate PSs only for the pathways that we retrieve through Enrichr’s KEGG pathway enrichment analysis (Kuleshov et al., 2016) of the top-100 scored genes of each disease as selected according to the methodology of Zachariou et al. (2018).
2.4 Pathways’ community detection by accumulating guided tours
Following the construction of the gene map and the pathways’ network, we initiate the execution of our proposed algorithm (Fig. 1). At the beginning of the execution, a random gene and a random pathway starting nodes are selected, one for each of the two networks respectively (gene–gene and pathway–pathway). During every iteration, the walker performs a series of steps on the gene-map level and the result assists the walker in deciding its next destination on the pathways’ level. On the genes’ network, the walker moves based on a simple random walk methodology, with a random restart every 50 iterations. In more detail, a random number n is generated in each iteration based on a Cauchy distribution, which indicates the number of steps the walker has to complete on the genes’ level. The walker traverses higher-weighted edges with higher probability via Monte Carlo sampling. The restart parameter prevents the walker from staying trapped inside neighborhoods of high-degree connectivity or bouncing between neighbors with high edge-weight values. Including the starting gene node, the maximum number of genes that can participate in a path in a single iteration is n + 1, in the case where no nodes were visited more than once. The traversed gene nodes indicate the next destination of the random walker on the pathways’ level.
Every pathway receives a + 1 score for each of the selected genes that is included in, normalized by dividing with each pathway’s total number of genes. Through a second Monte Carlo sampling, the next pathway is chosen based on the normalized candidate pathways’ scores. Then, the walker travels the shortest path between the current and the chosen pathway node. In case of multiple shortest paths with the same score, a random one is selected among the options. If no pathways were found containing any of the traversed genes, a new random pathway is sampled and the walker travels there via the shortest path. All of the pathway nodes and all the edges participating in the selected shortest path receive a + 1 on their final score. The resulting list of the top-ranked pathways highlights key molecular mechanisms, according to the genetic map of the disease of interest, while the sorted edge-list result is used for the discovery of pathway communities based on functional relations. The results of the PathWalks algorithm tend to favor nodes with high betweenness score, due to the shortest path usage while pathway-traversing. In order to highlight the most important pathways, we pay special attention to the mostly walked pathways that are not necessarily favored by the network’s topology.
PathWalks convergence criterion is based on the similarity index between the current and the last-sorted list (every a set number steps, 100 in our use cases) of the most visited pathways. If the similarity index between two pathway lists is above a defined threshold, then the walker is allowed to finish the execution. We call this threshold, converging factor of the algorithm. To avoid any random high-similarity result that might occur mid-execution, the variance of the last 10 similarity comparisons is calculated; if the variance is below a certain low threshold (e.g. 0.003), while at the same time the similarity index exceeds the converging factor (e.g. 95% similarity), the execution finishes. The stricter the converging factor and variance thresholds are, the longer the algorithm requires to converge but the resulting pathway communities are less noisy and more related to the disease-related map that guided the walker on the pathway network.
Lastly, the algorithm carries out a Louvain clustering on the re-weighted pathways’ network (i.e. ranked output edge-list) based on igraph’s cluster_louvain function and outputs a text file showing the pathway clusters. We developed the PathWalks software package in R (Ihaka and Gentleman, 1996) and used CRAN’s igraph package (Csardi and Nepusz, 2006) for handling network activity. We show the pseudocode for the PathWalks algorithm in Figure 2. We also plotted network figures (gene, pathway and results) using the Cytoscape tool (Smoot et al., 2011) and provide them as Supplementary Figures and corresponding Cytoscape files in github (https://tinyurl.com/r3psehc).
Fig. 2.

PathWalks Algorithm: outline of input, output and computational steps
3 Results
In this study, we have chosen AD and IPF as our use cases as both are incurable illnesses with sufficient available omics data online. Since these diseases differ significantly in terms of molecular pathology and affected tissues, they furnish a unique opportunity to test PathWalks in two distinct biological subsystems. Furthermore, they are both complex diseases, with AD specifically being a general term including various phenotypes/subphenotypes corresponding to different molecular pathways.
3.1 Pathwalks execution
We run the PathWalks algorithm iteratively until the desired converging similarity and variance output is achieved (see the Materials and methods section 2.4 for more details). For the execution of our two use cases, we set a converging factor of 0.95 and a converging variance of 0.003 (arbitrary values based on a number of initial trials). The similarity indexes and the respective variances are calculated every 100 steps. The algorithm executed 46 800 iterations in the use case of AD and 32 800 in IPF. A faster convergence was achieved for IPF compared to AD (∼2/3 iterations) due to the smaller size of the guiding gene map (∼1/3 connections).
The diagrams of the values of the converging similarity metrics during the execution of PathWalks for the two use-case scenarios are depicted in Figure 3. We use two metrics to manage the algorithm’s convergence: (i) the similarity index, which is calculated every 100 iterations and measures the ordered pathways’ similarity with their previous state and (ii) the variance of these similarity indices, using a sliding window covering at each calculation the 10 last similarity indices. The converging factor and variance designate the exit-thresholds for the two metrics. The combined effect of the converging factor and converging variance impact both the stability and the quality of our results.
Fig. 3.
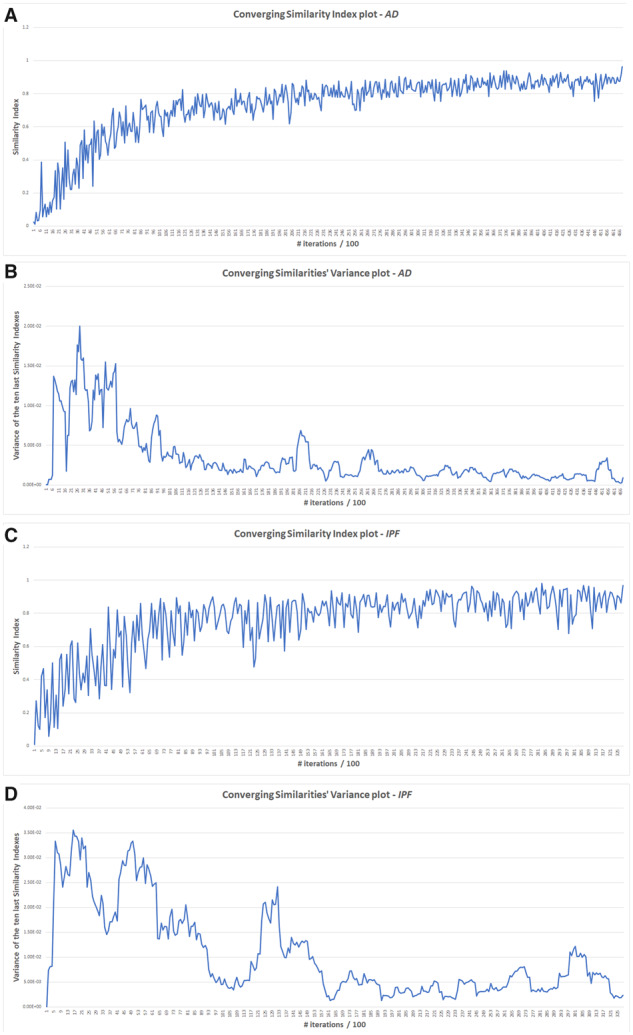
Converging variables’ plots for the AD and IPF cases. We calculate converging similarities every 100 steps and their respective variances for every 10 last observations. (A) Converging similarity index values’ plot of AD. (B) Converging similarity index variance plots of AD. (C) Converging similarity index values’ plot of IPF. (D) Converging similarity index variance plots of IPF
More specifically, the converging factor sets the acceptable level of pathway lists’ similarity of 100 iterations apart and the converging variance is responsible for preventing the algorithm from exiting due to randomly exceeding the selected convergence factor. Figures 3A and C (‘similarity index’ versus ‘100 s of iterations’) depict for both AD and IPF, plateaus due to the algorithm’s convergence. At the same time, the respective converging-variance values shown in Figures 3B and D, decrease. In both use-cases after a small number of iterations, the produced pathway lists consistently include a number of key (top-ranked) pathways. In IPF, the plateau is reached faster than in AD since the IPF gene map is smaller, hence, less pathways are targeted more often.
The quality of the results should be attributed in both the highly as well as the moderately ranked pathways. Regarding the moderately ranked pathways, the respective lists converge when the similarity index has reached the plateau and the similarity variance is reasonably small (i.e. values around 0.005 as seen in Figures 3B and D, with IPF having more fluctuation in its values). Thus, the combination of convergence factor and variance influences both quality and stability of our results. A trade-off exists here: on one hand, a low-converging factor and a high converging-variance achieves fast but only stable calculations regarding the top-ranked pathways. On the other, a combination of high-converging factor and low converging-variance yields a lengthier execution but offers highly stable and qualitative results across the list of pathways, the re-weighted network and the formed communities.
Following the convergence of PathWalks, we obtain the ranked pathways, the edge-list of the re-weighted network of pathways according to the frequency of the walker’s trajectories and the formed pathway clusters in text format. Tables 1–4 present the top-10% ranked pathways and top-10 ranked edge results, while Supplementary Table S1–Tables 1-6 contain, respectively, the ranked pathway, edge-list and cluster entries for the two diseases.
Table 1.
The top-10% ranked pathways (31/319) that are visited in the use case of AD
| Rank | Pathway name | Score |
|---|---|---|
| 1 | Calcium-signaling pathway | 20 739 |
| 2 | Alzheimer’s disease | 17 842 |
| 3 | Apoptosis | 16 673 |
| 4 | MAPK-signaling pathway | 8046 |
| 5 | Serotonergic synapse | 4295 |
| 6 | Pathways in cancer | 3978 |
| 7 | Dopaminergic synapse | 3263 |
| 8 | Metabolic pathways | 3211 |
| 9 | Oxidative phosphorylation | 2535 |
| 10 | Notch-signaling pathway | 2220 |
| 11 | Cocaine addiction | 2092 |
| 12 | Cholesterol metabolism | 1635 |
| 13 | Apoptosis-multiple species | 1617 |
| 14 | Axon guidance | 1414 |
| 15 | Wnt-signaling pathway | 1354 |
| 16 | Bile secretion | 1278 |
| 17 | Cytokine–cytokine receptor interaction | 1263 |
| 18 | TNF-signaling pathway | 1166 |
| 19 | Salivary secretion | 1164 |
| 20 | Prion diseases | 1077 |
| 21 | Neurotrophin-signaling pathway | 1075 |
| 22 | Amyotrophic lateral sclerosis | 1066 |
| 23 | Circadian entrainment | 1056 |
| 24 | Thyroid hormone synthesis | 1051 |
| 25 | Insulin-signaling pathway | 1013 |
| 26 | cAMP-signaling pathway | 996 |
| 27 | Fat digestion and absorption | 979 |
| 28 | Influenza A | 966 |
| 29 | Parkinson disease | 951 |
| 30 | Pancreatic secretion | 928 |
| 31 | Oxytocin-signaling pathway | 922 |
Note: The score denotes the times a pathway participated in the shortest path that was traversed by the random walker.
Table 4.
The top-10 ranked edges walked in the use case of IPF
| Rank | Pathway name 1 | Pathway name 2 | Edge weight |
|---|---|---|---|
| 1 | MAPK-signaling pathway | Toll-like receptor-signaling pathway | 2918 |
| 2 | Pathways in cancer | Cytokine–cytokine receptor interaction | 2539 |
| 3 | Pathways in cancer | MAPK-signaling pathway | 1976 |
| 4 | Toll-like receptor-signaling pathway | Malaria | 1550 |
| 5 | MAPK-signaling pathway | AGE-RAGE-signaling pathway in diabetic complications | 1521 |
| 6 | MAPK-signaling pathway | TNF-signaling pathway | 1463 |
| 7 | MAPK-signaling pathway | TGF-beta-signaling pathway | 1316 |
| 8 | Toll-like receptor-signaling pathway | African trypanosomiasis | 1285 |
| 9 | MAPK-signaling pathway | Melanoma | 1266 |
| 10 | Cytokine–cytokine receptor interaction | Toll-like receptor-signaling pathway | 1252 |
Note: The edge weight denotes the number of times an edge was accessed by the random walker.
Table 2.
The top-10 ranked edges walked in the use case of AD
| Rank | Pathway name 1 | Pathway name 2 | Edge weight |
|---|---|---|---|
| 1 | Alzheimer’s disease | Calcium-signaling pathway | 11 233 |
| 2 | Alzheimer’s disease | Apoptosis | 8829 |
| 3 | Calcium-signaling pathway | Serotonergic synapse | 3850 |
| 4 | Calcium-signaling pathway | Dopaminergic synapse | 2898 |
| 5 | Oxidative phosphorylation | Alzheimer’s disease | 2430 |
| 6 | Oxidative phosphorylation | Metabolic pathways | 2341 |
| 7 | MAPK-signaling pathway | Calcium-signaling pathway | 2271 |
| 8 | Pathways in cancer | Calcium-signaling pathway | 2112 |
| 9 | Dopaminergic synapse | Cocaine addiction | 1956 |
| 10 | Pathways in cancer | Notch-signaling pathway | 1883 |
Note: The edge weight denotes the number of times an edge was accessed by the random walker.
Table 3.
The top-10% ranked pathways (31/319) that are visited in the use case of IPF
| Rank | Pathway name | Score |
|---|---|---|
| 1 | MAPK-signaling pathway | 19 325 |
| 2 | Toll-like receptor-signaling pathway | 6517 |
| 3 | Cytokine–cytokine receptor interaction | 5569 |
| 4 | Pathways in cancer | 3826 |
| 5 | TGF-beta signaling pathway | 2889 |
| 6 | Chemokine-signaling pathway | 2095 |
| 7 | PI3K-Akt-signaling pathway | 1974 |
| 8 | AGE-RAGE-signaling pathway in diabetic complications | 1974 |
| 9 | Malaria | 1903 |
| 10 | TNF-signaling pathway | 1900 |
| 11 | Endocytosis | 1692 |
| 12 | Apoptosis | 1508 |
| 13 | NF-kappa B-signaling pathway | 1471 |
| 14 | African trypanosomiasis | 1466 |
| 15 | Rheumatoid arthritis | 1451 |
| 16 | Melanoma | 1442 |
| 17 | Chagas disease (American trypanosomiasis) | 1298 |
| 18 | Pertussis | 1260 |
| 19 | Gap junction | 1209 |
| 20 | IL-17-signaling pathway | 1194 |
| 21 | Hippo-signaling pathway | 1137 |
| 22 | Calcium-signaling pathway | 1096 |
| 23 | Apelin-signaling pathway | 1060 |
| 24 | Epithelial cell signaling in Helicobacter pylori infection | 1012 |
| 25 | Fluid shear stress and atherosclerosis | 978 |
| 26 | Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 953 |
| 27 | Osteoclast differentiation | 949 |
| 28 | Inflammatory bowel disease | 949 |
| 29 | Gastric cancer | 944 |
| 30 | EGFR tyrosine kinase inhibitor resistance | 938 |
| 31 | Adherens junction | 931 |
Note: The score denotes the times a pathway participated in the shortest path that was traversed by the random walker.
3.2 Comparisons and validation
In this section, we compare our PathWalks results with other approaches regarding pathway analysis for AD and IPF. Our goal is to discover which pathways are commonly highlighted among various methods, as a baseline validation approach for the outcomes of our approach and designate entries exclusively highlighted by PathWalks.
PathWalks implements shortest path traversing on the biological pathways’ network level. Due to the network’s topology and the assigned edge weights, certain pathway nodes are consistently highlighted in the results. We perform a PathWalks execution with random biological pathway selection at each iteration (without gene-map guidance) to identify these topology-favored nodes that are not necessarily highlighted due to their association with each use-case disease. For this random-PathWalks experiment, we use our functional connectivity network of biological pathways and assign edge weights equal to the number of common genes between two pathways. We show the top-10% of the topology-favored nodes in Table 5 and provide the respective total lists of ranked pathways, re-weighted network and formed clusters in the Supplementary Table S1-Tables 7-9. We first compare the top-10% ranked pathway lists among the respective IPF and AD PathWalks and the random-PathWalks experiments to identify which pathways are re-ranked due to direct association with the biological map and which mostly due to the topology. We then compare the top-10% PathWalks results (31 pathways) with the respective top-31 significant results from other pathway analysis tools to evaluate our results.
Table 5.
The top-10% ranked pathways (31/319) that are visited in a random-PathWalks execution
| Rank | Pathway name | Score |
|---|---|---|
| 1 | Metabolic pathways | 1 135 390 |
| 2 | Oxidative phosphorylation | 907 950 |
| 3 | PI3K-Akt-signaling pathway | 684 541 |
| 4 | Non-alcoholic fatty liver disease | 523 109 |
| 5 | MAPK-signaling pathway | 472 571 |
| 6 | Pathways in cancer | 457 037 |
| 7 | Calcium-signaling pathway | 342 037 |
| 8 | Apoptosis | 301 799 |
| 9 | Thermogenesis | 171 517 |
| 10 | cAMP-signaling pathway | 161 076 |
| 11 | Alzheimer’s disease | 158 931 |
| 12 | Focal adhesion | 112 460 |
| 13 | Influenza A | 108 816 |
| 14 | Toll-like receptor-signaling pathway | 104 478 |
| 15 | Wnt-signaling pathway | 92 442 |
| 16 | Regulation of actin cytoskeleton | 88 624 |
| 17 | Human papillomavirus infection | 87 482 |
| 18 | Retrograde endocannabinoid signaling | 66 385 |
| 19 | Pancreatic secretion | 62 218 |
| 20 | Dopaminergic synapse | 61 069 |
| 21 | Antigen processing and presentation | 60 265 |
| 22 | Colorectal cancer | 57 818 |
| 23 | Epstein–Barr virus infection | 54 034 |
| 24 | Glutamatergic synapse | 49 419 |
| 25 | JAK-STAT-signaling pathway | 47 463 |
| 26 | Human T-cell leukemia virus 1 infection | 47 009 |
| 27 | Phospholipase D-signaling pathway | 46 703 |
| 28 | Viral carcinogenesis | 46 195 |
| 29 | RNA transport | 44 420 |
| 30 | Citrate cycle (TCA cycle) | 44 015 |
| 31 | Herpes simplex virus 1 infection | 43 873 |
Note: The pathways’ network initial edge weights denote the number of common genes between two pathways.
Figures 4 and 5 show Venn diagrams of the top-10% topology-favored pathways with the respective results from AD and IPF. PathWalks brings 19 pathways to the top of the results of AD and 25 of IPF due to the integrated biological information rather than due to the topology. ‘Serotonergic synapse’ and ‘Notch signaling’ pathways are the first two entries highlighted directly by AD’s gene map. ‘Cytokine–cytokine receptor interaction’, ‘TGF-beta signaling’ and ‘Chemokine signaling’ pathways are the top-3 IPF related results with direct biological connection to the disease. Nevertheless, we do not necessarily consider topology-favored nodes as true-negative entries. Topology-favored nodes either contain functional connections with multiple biological pathways (high-degree value) or connect distinct functional sub-networks (high betweenness value). Therefore, perturbations in the functional connectivity network potentially affect these nodes indirectly. However, we observe that several of the topology-favored pathways decrease in rank for non-relevant diseases. For example, the ‘Oxidative phosphorylation’ pathway is ranked second in the random-PathWalks example and ninth in the AD use case, but only 162nd in the use case of IPF. All top-31 pathway lists of PathWalks, GeneTrail3, Enrichr, EnrichNet and random PathWalks can be found in Supplementary Table S2.
Fig. 4.
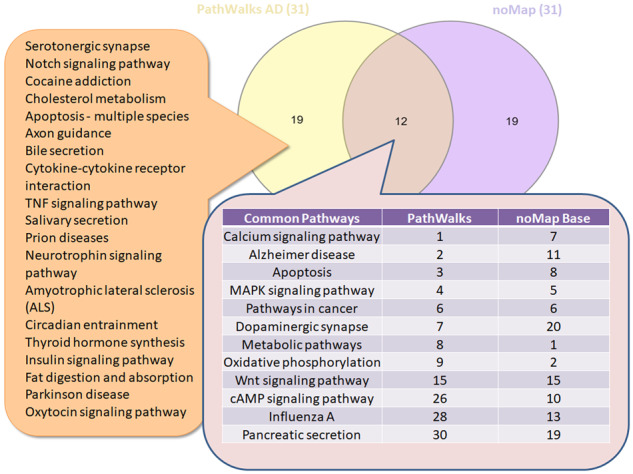
Venn diagram between the top-10% AD PathWalks and random PathWalks (no gene map) results. In the intersection, we observe the respective ranks of the 12 common pathways for each execution while on the left list, we depict the 19 pathways highlighted by PathWalks due to their direct association with the integrated AD gene map
Fig. 5.
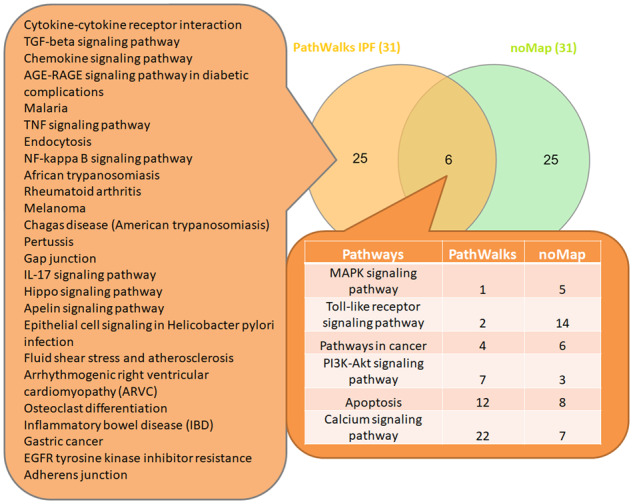
Venn diagram between the top-10% IPF PathWalks and random PathWalks (no gene map) results. In the intersection, we observe the respective ranks of the 6 common pathways for each execution while on the left list, we depict the 25 pathways highlighted by PathWalks due to their direct association with the integrated IPF gene map
To evaluate our findings, we compare our PathWalks results with those derived from pathway analysis tools including GeneTrail3 (Backes et al., 2007), Enrichr and EnrichNet. We feed as input to these tools the gene nodes of each map. Subsequently, we establish common highlighted pathway entries between PathWalks and the tools in discussion. This exercise partially helps validate our PathWalks-derived results and constitutes a common pathway analysis technique. For example, Glaab et al. (2012) have successfully used the intersection of the results of the enrichment analysis tools SAM-GS (Dinu et al., 2007) and GAGE (Luo et al., 2009) while testing for the confidence of their EnrichNet tool’s pathway analysis results. PathWalks also exclusively highlights several biological pathways not necessarily favored by the topology. Furthermore, the key value-added of PathWalks compared to prior pathway analysis approaches is that it yields functional connections among pathways as well as proposes pathway clusters. In Figures 6 and 7, we provide the Venn diagrams of the top-10% highlighted pathways from each tool, for AD and IPF, respectively.
Fig. 6.
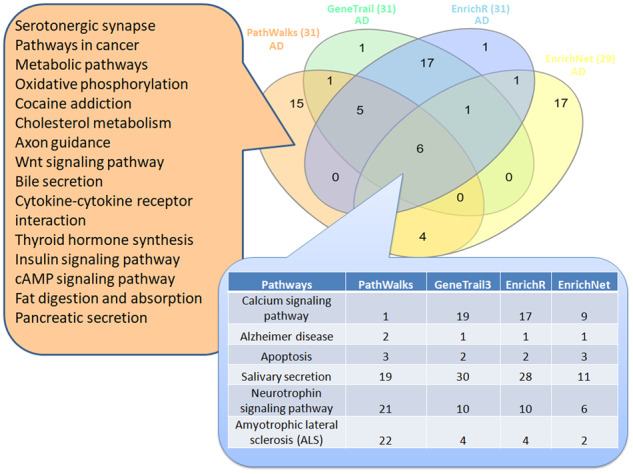
Venn diagram among the top-31 results from PathWalks and the respective significant pathways produced by other pathway analysis tools for the use case of AD. We note that, EnrichNet returned only 29 significant pathway results. In the intersection among all four tools, we observe the respective pathway ranks. On the left, we show the 15 exclusive pathways highlighted by PathWalks in its top-31 results
Fig. 7.
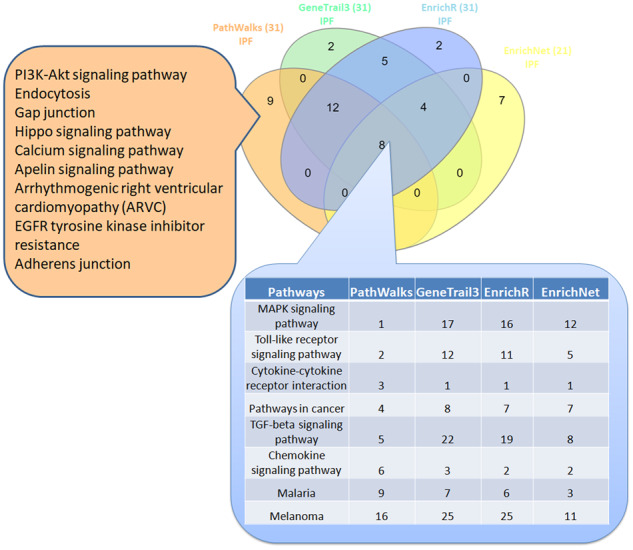
Venn diagram among the top-31 results from PathWalks and the respective significant pathways of other pathway analysis tools for the use case of IPF. We note that, EnrichNet returned only 21 significant pathway results. In the intersection among all four tools, we observe the respective pathway ranks. On the left, we show the nine exclusive pathways highlighted by PathWalks in its top-31 results
In the AD use case, 15 terms are ranked exclusively in PathWalks, 6 of which are favored by the network’s topology. The remaining nine top-ranked candidates, some of which are interestingly ranked very low in a random-PathWalks execution (Supplementary Table S1-Table 7), include pathways, such as ‘Serotonergic synapse’, ‘Cholesterol metabolism’, ‘Bile secretion’ and ‘Axon guidance’. In the IPF use case, the top-9 terms are exclusively produced by PathWalks, seven of which are not favored by the topology including ‘Endocytosis’, ‘Gap junction’, ‘Hippo signaling’ and ‘Apelin signaling’ pathways.
Validating pathway analysis methodologies is an invariably challenging task since ground truths and gold standards are often unavailable. Yu et al. (2017) discuss these difficulties and present a model, which can evaluate a pathway analysis methodology based on the consistency of its results on smaller subsets of a main gene expression dataset. However, such an approach can only be followed when parsing gene expression datasets. In our case that entails gathering of multi-omics data from various sources, we choose to validate our PathWalks results by comparing them with the results from other tools, similar to Glaab’s approach (Glaab et al., 2012). Furthermore, we identify corroborating bibliographic evidence to further ascertain the effectiveness of PathWalks mechanisms in AD and IPF. Without doubt, there is no single best approach in pathway analysis or in validating its results. Although common indications provided by several tools offer a baseline for validating results, one should keep in mind that every individual tool contributes its own incremental value-added through its own unique produced outcome(s).
4 Discussion
Our methodology combines random walks and network-based integration to detect key disease-related pathway clusters in the use cases of AD and IPF. In AD, the two most visited pathways are ‘Calcium signaling pathway’ (ranked seventh in random PathWalks) and as expected, the ‘Alzheimer disease’ pathway (ranked 11th in random PathWalks), which includes a set of known components and interactions related to the AD pathology.
The ‘Calcium signaling pathway’ has the strongest connection to the ‘Alzheimer disease’ pathway based on the most walked edges of the pathways network. Calcium plays a major role in the normal function of the cells. Deregulation of calcium signaling has been implicated in many neurodegenerative diseases including AD (Mattson and Chan, 2003; Supnet and Bezprozvanny, 2010; Woods and Padmanabhan, 2012). Alteration in calcium homeostasis has been found to lead to elevated levels of resting calcium in AD animal models (Alzheimer’s Association Calcium Hypothesis Workgroup, 2017). Calcium overload has also been correlated with disrupted neuronal structure and function (Kuchibhotla et al., 2008). Recent efforts investigate the calcium dysregulation in order to find additional pathogenic mechanisms and new treatment methods for AD (Alvarez et al., 2020; Dave and Jha, 2020; Galla et al., 2020). Several therapeutic drugs that currently target plasma Ca2+ channels have received good efficacy on in vitro and in vivo AD models. A number of such drugs either have been already approved by the Food and Drug Administration for AD treatment or are in clinical trials (Tong et al., 2018).
The ‘Apoptosis’ pathway is directly linked to the ‘Alzheimer disease’ pathway and ranked third. The ‘Alzheimer disease’ pathway is also indirectly linked, through the ‘Calcium signaling pathway’, via frequently traversed edges to other high-rank pathways, such as the ‘Serotonergic synapse’, ‘Dopaminergic Synapse’ and ‘MAPK signaling’. ‘MAPK signaling pathway’ is ranked fourth in AD. The persistent activation of mitogen-activated protein kinases (MAPKs) is thought to play a key role in neurodegeneration, including AD, through mediating hyper-phosphorylation of neuronal proteins, eventually causing neuronal death (Fadaka et al., 2017).
The ‘Serotonergic synapse’ pathway is distinctly produced by PathWalks and ranked fifth. The serotonergic system has an important role in memory, cognitive process and learning. Moreover, it has been found to be impaired in AD, where extensive serotonergic denervation is observed (Butzlaff and Ponimaskin, 2016). Serotonergic markers, specifically 5-HT receptors, are affected by AD-associated neurodegeneration. Recent studies suggest the examination of all markers and related signaling pathways of the serotonergic system in order to discover novel treatment and methods for AD (Lennon et al., 2019).
The ‘Dopaminergic synapse’ pathway is ranked seventh by PathWalks (22nd by GeneTrail3, 21st by Enrichr and 20th by random PathWalks). A deficit in the dopaminergic system has also been observed in AD, with the loss of that dopaminergic neurons in the ventral tegmental area during the early (pre-plaque) stages of AD(Nobili et al., 2017). Furthermore, the dopaminergic system has been intensively studied as a key neurotransmitter involved with emotion and cognition (Nardone et al., 2014). New findings on the relation of dopamine neurons in AD start to emerge as well (Krashia et al., 2019; Pan et al., 2019). Both dopaminergic and serotonergic can be associated to AD through the calcium pathway. For example, a T-type calcium channel enhancer (known as SAK3) was shown to boost serotonin and dopamine in the hippocampus of both naive and amyloid precursor protein knock-in mice (Wang et al., 2018).
We also observe highly ranked edges connecting ‘Metabolism’ to ‘Alzheimer disease’ pathways, through the ‘Oxidative phosphorylation’. Both the hypometabolism and oxidative stress have been implicated as key contributors in initiation and progression for the synapse vulnerability in AD (Mosconi et al., 2008).
‘Pathways in Cancer’ is also associated with AD and connects to the ‘Calcium signaling pathway’. Interestingly certain types of cancers, such as lung cancer, have been found to be anti-correlated with the occurrence of neurodegenerative diseases, such as AD, although both types of diseases are associated to aging (Sánchez-Valle et al., 2017).
Moreover, we identify ‘Cholesterol metabolism’ (rank 12) and ‘Bile secretion’ (rank 16) as uniquely produced pathways by our PathWalks analysis. Cholesterol is particularly important in the brain since it is a major component of cell membranes, and consequently, altered cholesterol metabolism may contribute to AD development (Gamba et al., 2019). Bile acids are the end-products of cholesterol metabolism produced by human and gut microbiome co-metabolism and appear to play a role in the central nervous system. Recent studies suggest that microbiota influence pathological features of AD including amyloid-β deposition and neuroinflammation. These efforts urge additional research into the role that cholesterol and bile acid pathways play in AD pathology (Chang et al., 2017; MahmoudianDehkordi et al., 2019; Nho et al., 2019).
The PathWalks exclusively highlighted pathway ‘cAMP signaling’ and the pathway ‘Oxytocin signaling’ (common among PathWalks, GeneTrail3 and Enrichr), are not yet associated with AD. We suggest that further research should be pursued regarding these pathways to potentially discover novel perturbed mechanisms of AD.
In the use case of IPF, we identify the ‘MAPK signaling pathway’ to be top-ranked, based on the walker’s visitation frequency. ‘MAPK signaling pathway’ has received high betweenness and degree scores, but is linked to other highlighted pathways of IPF and hence might be a key intermediate functional node in the pathogenesis of IPF. In a relevant study (Antoniou et al., 2010), a significant overexpression in the Braf oncogene, a key gene in the MAPK pathway, was observed in IPF versus a control group. In another study (Yoshida et al., 2002), three MAP kinases (ERK, JNK and p38 MAPK) were suggested to be involved in the regulation of lung inflammation and injury in IPF. Additionally, we have suggested in our previous computational drug repurposing study on IPF (Karatzas et al., 2017) that the MAPK-signaling pathway plays a key role in the transition of early stage IPF toward a more advanced stage.
The second highest ranked pathway in IPF, directly connected to the ‘MAPK signaling’ is the ‘Toll-like receptor signaling’ pathway. Recent Toll-like receptor studies related to IPF suggest promising genes as therapeutic targets. TLR7, TLR9 and TLR2 mRNA expressions were found to be significantly increased in IPF compared to control subjects, even though TLR9 protein expression was lower in IPF than controls (Samara et al., 2012). TLR9 has also been shown to drive the fibrosis progression in IPF in another study (Hogaboam et al., 2012). A TLR3 polymorphism, namely TLR3 L412F, has also been linked to a more aggressive and profibrotic disease phenotype in IPF (O’Dwyer et al., 2013). In a regulatory network, an edge would be directed from the ‘Toll-like receptor signaling pathway’ toward the MAPK one, as TLR signaling leads to the activation of MAPKs in mammals through the sequential recruitment of the adapter molecule MyD88 and the serine-threonine kinase IRAK (Hemmi et al., 2002). In turn, the activated MAPKs (ERKs, JNKs and p38 proteins) regulate cellular mechanisms associated with inflammatory responses as well as cell proliferation and survival (Li et al., 2010) and so MAPKs are key components in the pathogenesis of IPF.
‘Cytokine-cytokine receptor interaction’ is the third ranked pathway, which has also been suggested by our previous study (Karatzas et al., 2017) to play a key role in all stages of the IPF disease. The important role of cytokines as therapeutic targets in IPF has also been emphasized (Coker and Laurent, 1998). Bouros et al. (2017) recently proposed the tumor necrosis factor-like cytokine 1A (TL1A), as a novel fibrogenic factor. Specifically, they found upregulated mRNA and protein levels of TL1A in subepithelial lung myofibroblasts that were treated either with pro-inflammatory factors or bronchoalveolar lavage fluid from IPF patients.
‘Pathways in cancer’ is the next pathway result in rank. IPF is known to have many similar alterations and behaviors to cancer biology (Vancheri et al., 2010). The second and third most traversed edges link the ‘Cytokine–cytokine receptor interaction’ pathway to the ‘Pathways in cancer’, which is then linked to the ‘MAPK signaling’ pathway. Yong and colleagues presented information about p38 MAPK being a key player in cellular processes that are related to inflammation and cancer. p38 MAPK can activate both anti-inflammatory and pro-inflammatory cytokines. p38 MAPK inhibitors have been tested as potential therapeutic drugs against inflammatory diseases and cancer but with numerous side effects (Yong et al., 2009).
The fifth ranked pathway ‘TGF-beta signaling’ is also known to be linked not only with IPF but with fibrotic diseases in general (Rosenbloom et al., 2017) and it is one of the key drivers in fibrogenesis (Meng et al., 2016). The sixth ranked pathway ‘Chemokine signaling’ has been also shown to contribute to the pathogenesis of interstitial lung diseases including IPF via mechanisms, such as the regulation of vascular modeling and the mediation of the traffic of bone marrow derived progenitor cells to the lungs (Mehrad and Strieter, 2010).
A number of PathWalks results for IPF are neither highlighted by the benchmark tools we explore in our analysis nor by the random (no-map) PathWalks execution. The pathway of ‘Endocytosis’, which is directly connected to ‘Cytokine–cytokine receptor interaction’, is ranked 11th, but there is little evidence in bibliography associating this pathway with IPF. Specifically, Hsu et al. (2011) show that IPF and Systemic Sclerosis-Pulmonary Fibrosis share enriched functional groups regarding genes involved in caveolin-mediated endocytosis. Caveolins are a family of plasma membrane proteins, which form caves that are involved in receptor-independent endocytosis (Williams and Lisanti, 2004). In another study, Shi and Sottile (2008) suggest a possibility that IPF patients may have perturbations in extracellular matrix endocytosis due to caveolin-1 turnover of the fibronectin matrix.
Similarly, the ‘Apelin signaling’ pathway, which is directly connected to ‘MAPK signaling’, ranked 23rd and was uniquely produced by PathWalks. Apelin is an endogenous ligand that binds to the G-protein-coupled receptor, is expressed in multiple tissues and organ systems and is implicated in various physiological processes (Tatemoto et al., 1998). There is no bibliographic evidence directly associating this pathway with IPF. Hence, both ‘Apelin signaling’ and ‘Endocytosis’ pathways should be further explored for potential contribution to the fibrogenesis of IPF patients.
Without a doubt, a limitation in pathway analysis is the fact that there is often no ground truth to validate the identified pathways apart from comparing results with those derived with other tools, looking into the literature and carrying out wet lab experiments. Nevertheless, PathWalks has yielded promising results for AD and IPF as the pathway-to-pathway network and the gene map significantly assist with their biological information.
Funding
E.K. is a PhD student in the National and Kapodistrian University of Athens. His doctoral thesis was funded by the State Scholarships Foundation (IKY) scholarship, under the Action ‘Strengthening Human Resources, Education and Lifelong Learning’, 2014–2020; co-funded by the European Social Fund (ESF) and the Greek State [MIS-5000432]. M.M.B. is a post-doctoral researcher in the Democritus University of Thrace. Her post-doctoral research was funded by the State Scholarships Foundation (IKY) scholarship; co-financed by Greece and the European Union (European Social Fund- ESF) through the Operational Programme «Human Resources Development, Education and Lifelong Learning» in the context of the project ‘Reinforcement of Post-doctoral Researchers - 2nd Cycle’ [MIS-5033021], implemented by the State Scholarships Foundation (ΙΚΥ). M.Z., G.M. and A.O. hold post-doctoral research fellow positions funded by the European Commission Research Executive Agency Grant BIORISE [number 669026], under the Spreading Excellence, Widening Participation, Science with and for Society Framework. G.M.S. holds the Bioinformatics European Research Area (ERA) Chair Position funded by the European Commission Research Executive Agency (REA) Grant BIORISE [number 669026], under the Spreading Excellence, Widening Participation, Science with and for Society Framework.
Conflict of Interest: none declared.
Supplementary Material
References
- Alvarez J. et al. (2020) The role of Ca2+ signaling in aging and neurodegeneration: insights from Caenorhabditis elegans models. Cells, 9, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer’s Association Calcium Hypothesis Workgroup (2017) Calcium hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement., 13, 178. [DOI] [PubMed] [Google Scholar]
- Amberger J.S. et al. (2015) OMIM. org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res., 43, D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou K.M. et al. (2010) Expression analysis of Akt and MAPK signaling pathways in lung tissue of patients with idiopathic pulmonary fibrosis (IPF). J. Recept. Signal Transduct. Res., 30, 262–269. [DOI] [PubMed] [Google Scholar]
- Backes C. et al. (2007) GeneTrail—advanced gene set enrichment analysis. Nucleic Acids Res., 35, W186–W192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxevanis A.D., Bateman A. (2015) The importance of biological databases in biological discovery. Curr. Protoc. Bioinformatics, 50, 1.1.1–1.1.8. [DOI] [PubMed] [Google Scholar]
- Blondel V.D. et al. (2008) Fast unfolding of communities in large networks. J. Stat. Mech., 2008, P10008. [Google Scholar]
- Bouros E. et al. (2017) Lung fibrosis-associated soluble mediators and bronchoalveolar lavage from idiopathic pulmonary fibrosis patients promote the expression of fibrogenic factors in subepithelial lung myofibroblasts. Pulm. Pharmacol. Ther., 46, 78–87. [DOI] [PubMed] [Google Scholar]
- Butzlaff M., Ponimaskin E. (2016) The role of serotonin receptors in Alzheimer’s disease. Opera Med. Physiol., 2, 77–86. [Google Scholar]
- Chang T.-Y. et al. (2017) Cellular cholesterol homeostasis and Alzheimer’s disease. J. Lipid Res., 58, 2239–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. et al. (2016) IRWRLDA: improved random walk with restart for lncRNA-disease association prediction. Oncotarget, 7, 57919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.-H. et al. (2018) miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res., 46, D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauset A. et al. (2004) Finding community structure in very large networks. Phys. Rev. E, 70, 066111. [DOI] [PubMed] [Google Scholar]
- Coker R., Laurent G. (1998) Pulmonary fibrosis: cytokines in the balance. Eur. Respir. J., 11, 1218–1221. [DOI] [PubMed] [Google Scholar]
- Csardi G., Nepusz T. (2006) The igraph software package for complex network research. InterJ Complex Syst., 1695, 1–9. [Google Scholar]
- Dave D.D., Jha B.K. (2020) 3D mathematical modeling of calcium signaling in Alzheimer’s disease. Netw. Model. Anal. Health Inform. Bioinform., 9, 1. [Google Scholar]
- Ding M.C., Szeto K.Y. (2017) Selection of random walkers that optimizes the global mean first-passage time for search in complex networks. Procedia Comput. Sci., 108, 2423–2427. [Google Scholar]
- Dinu I. et al. (2007) Improving gene set analysis of microarray data by SAM-GS. BMC Bioinformatics, 8, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espe S. (2018) MalaCards: the human disease database. J. Med. Libr. Assoc., 106, 140. [Google Scholar]
- Fadaka A.O. et al. (2017) Role of p38 MAPK signaling in neurodegenerative diseases: a mechanistic perspective. Ann. Neurodegener. Dis., 2, 1026. [Google Scholar]
- Franz M. et al. (2018) GeneMANIA update 2018. Nucleic Acids Res., 46, W60–W64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galla L. et al. (2020) Intracellular calcium dysregulation by the Alzheimer’s disease-linked protein presenilin 2. Int. J. Mol. Sci., 21, 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba P. et al. (2019) A crosstalk between brain cholesterol oxidation and glucose metabolism in Alzheimer’s disease. Front. Neurosci., 13, 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaab E. et al. (2012) EnrichNet: network-based gene set enrichment analysis. Bioinformatics, 28, i451–i457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb A. et al. (2011) PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol., 7, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S. et al. (2007) miRBase: tools for microRNA genomics. Nucleic Acids Res., 36, D154–D158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H. et al. (2002) Small anti-viral compounds activate immune cells via the TLR7 MyD88–dependent signaling pathway. Nat. Immunol., 3, 196–200. [DOI] [PubMed] [Google Scholar]
- Hogaboam C.M. et al. (2012) Epigenetic mechanisms through which toll-like receptor–9 drives idiopathic pulmonary fibrosis progression. Proc. Am. Thorac. Soc., 9, 172–176. [DOI] [PubMed] [Google Scholar]
- Hsu E. et al. (2011) Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum., 63, 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R., Gentleman R. (1996) R: a language for data analysis and graphics. J. Comput. Graph. Stat., 5, 299–314. [Google Scholar]
- Kanehisa M. et al. (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res., 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatzas E. et al. (2017) Drug repurposing in idiopathic pulmonary fibrosis filtered by a bioinformatics-derived composite score. Sci. Rep., 7, 12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashia P. et al. (2019) Unifying hypothesis of dopamine neuron loss in neurodegenerative diseases: focusing on Alzheimer’s disease. Front. Mol. Neurosci., 12, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla K.V. et al. (2008) Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron, 59, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M. et al. (2016) The SIDER database of drugs and side effects. Nucleic Acids Res., 44, D1075–D1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V. et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res., 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon J. et al. (2019) C-15 serotonergic system activity and BPSD in Alzheimer’s disease pathogenesis: a systematic review. Arch, Clin. Neuropsych., 34, 1044–1044. [Google Scholar]
- Lewis B.P. et al. (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell, 120, 15–20. [DOI] [PubMed] [Google Scholar]
- Li B. et al. (2009) Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics, 25, 2744–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. (2010) Toll-like receptor signaling in cell proliferation and survival. Cytokine, 49, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liakos P. et al. (2017) COEUS: community detection via seed-set expansion on graph streams. In: 2017 IEEE International Conference on Big Data (Big Data). pp. 676–685. IEEE, Boston, USA.
- Lu T. et al. (2006) Statistics of cellular signal transduction as a race to the nucleus by multiple random walkers in compartment/phosphorylation space. Proc. Natl. Acad. Sci. USA, 103, 16752–16757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W. et al. (2009) GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics, 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MahmoudianDehkordi S. et al. ; for the Alzheimer's Disease Neuroimaging Initiative and the Alzheimer Disease Metabolomics Consortium. (2019) Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease—an emerging role for gut microbiome. Alzheimers Dement., 15, 76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M.P., Chan S.L. (2003) Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium, 34, 385–397. [DOI] [PubMed] [Google Scholar]
- Mehrad B., Strieter R.M. (2010) CXC chemokine signaling in interstitial lung diseases In: Handbook of Cell Signaling. Elsevier, Academic Press, Cambridge, Massachusetts. ; 2907–2911. [Google Scholar]
- Meng X-m. et al. (2016) TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol., 12, 325–338. [DOI] [PubMed] [Google Scholar]
- Mosconi L. et al. (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. NY Acad. Sci., 1147, 180–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardone R. et al. (2014) Dopamine differently modulates central cholinergic circuits in patients with Alzheimer disease and CADASIL. J. Neural Transm., 121, 1313–1320. [DOI] [PubMed] [Google Scholar]
- Navarro J.D. et al. (2003) From biological databases to platforms for biomedical discovery. Trends Biotechnol., 21, 263–268. [DOI] [PubMed] [Google Scholar]
- Nho K. et al. ; for the Alzheimer's Disease Neuroimaging Initiative and the Alzheimer Disease Metabolomics Consortium. (2019) Altered bile acid profile in mild cognitive impairment and Alzheimer’s disease: relationship to neuroimaging and CSF biomarkers. Alzheimers Dement., 15, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobili A. et al. (2017) Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat. Commun., 8, 14727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dwyer D.N. et al. (2013) The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med., 188, 1442–1450. [DOI] [PubMed] [Google Scholar]
- Pan X. et al. (2019) Dopamine and dopamine receptors in Alzheimer’s disease: a systematic review and network meta-analysis. Front. Aging Neurosci., 11, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson K. (1905) The problem of the random walk. Nature, 72, 342–342. [Google Scholar]
- Philippi S., Köhler J. (2006) Addressing the problems with life-science databases for traditional uses and systems biology. Nat. Rev. Genet., 7, 482–488. [DOI] [PubMed] [Google Scholar]
- Rahiminejad S. et al. (2019) Topological and functional comparison of community detection algorithms in biological networks. BMC Bioinformatics, 20, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom J. et al. (2017) Human fibrotic diseases: current challenges in fibrosis research In: Rittié L. Fibrosis. Springer, Humana Press, New York; pp. 1–23. [DOI] [PubMed] [Google Scholar]
- Samara K.D. et al. (2012) Expression profiles of Toll-like receptors in non-small cell lung cancer and idiopathic pulmonary fibrosis. Int. J. Oncol., 40, 1397–1404. [DOI] [PubMed] [Google Scholar]
- Sánchez-Valle J. et al. (2017) A molecular hypothesis to explain direct and inverse co-morbidities between Alzheimer’s Disease, Glioblastoma and Lung cancer. Sci. Rep., 7, 4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F., Sottile J. (2008) Caveolin-1-dependent β1 integrin endocytosis is a critical regulator of fibronectin turnover. J. Cell Sci., 121, 2360–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot M.E. et al. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics, 27, 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supnet C., Bezprozvanny I. (2010) Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer’s disease. J. Alzheimers Dis., 20, S487–S498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatemoto K. et al. (1998) Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun., 251, 471–476. [DOI] [PubMed] [Google Scholar]
- Tong B.C.-K. et al. (2018) Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta Mol. Cell Res., 1865, 1745–1760. [DOI] [PubMed] [Google Scholar]
- Tong H. et al. (2006) Fast random walk with restart and its applications. In: Sixth International Conference on Data Mining (ICDM’06). pp. 613–622. IEEE, Hong Kong, China.
- Tripathi B. et al. (2019) Adapting community detection algorithms for disease module identification in heterogeneous biological networks. Front. Genet., 10, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancheri C. et al. (2010) Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur. Respir. J., 35, 496–504. [DOI] [PubMed] [Google Scholar]
- Wang S. et al. (2018) T-type calcium channel enhancer SAK3 promotes dopamine and serotonin releases in the hippocampus in naive and amyloid precursor protein knock-in mice. PLoS One, 13, e0206986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T.M., Lisanti M.P. (2004) The caveolin proteins. Genome Biol., 5, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S.J. et al. (2017) Discovery of functional and disease pathways by community detection in protein-protein interaction networks. Pac. Symp. Biocomput., 22, 336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S. et al. (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res., 46, D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods N.K., Padmanabhan J. (2012) Neuronal calcium signaling and Alzheimer’s disease In: Islam M. Calcium Signaling. Advances in Experimental Medicine and Biology, Springer, Dordrecht. pp. 1193–1217. [DOI] [PubMed] [Google Scholar]
- Yang J., Leskovec J. (2012) Community-affiliation graph model for overlapping network community detection. In: 2012 IEEE 12th International Conference on Data Mining. pp. 1170–1175. IEEE, Brussels, Belgium.
- Yong H.-Y. et al. (2009) The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin. Inv. Drugs, 18, 1893–1905. [DOI] [PubMed] [Google Scholar]
- Yoshida K. et al. (2002) MAP kinase activation and apoptosis in lung tissues from patients with idiopathic pulmonary fibrosis. J. Pathol., 198, 388–396. [DOI] [PubMed] [Google Scholar]
- Yu C. et al. (2017) A strategy for evaluating pathway analysis methods. BMC Bioinformatics, 18, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariou M. et al. (2018) Integrating multi-source information on a single network to detect disease-related clusters of molecular mechanisms. J. Proteom., 188, 15–29. [DOI] [PubMed] [Google Scholar]
- Zhong M. et al. (2008) The convergence-guaranteed random walk and its applications in peer-to-peer networks. IEEE Trans. Comput., 57, 619–633. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.