

Abstract
This is a short paper on new ways to think about the structure and function of small heat shock proteins (sHSPs), perhaps the most enigmatic family among protein chaperones. The goal is to incorporate new observations regarding the disordered regions of small heat shock proteins (sHSPs) into the large body of structural information on the conserved structural alpha-crystallin domains (ACD) that define the sHSP family. Disordered regions (N-terminal region and C-terminal region or NTR and CTR, respectively) represent over 50% of the sHSP sequence space in the human genome and are refractory to traditional structural biology approaches, posing a roadblock on the path towards a mechanistic understanding of how sHSPs function. A model in which an ACD dimer serves as a template that presents three grooves into which other proteins or other segments of sHSPs can bind is presented. Short segments within the disordered regions are observed to bind into the ACD grooves. There are more binding segments than there are grooves, and each binding event is weak and transient, creating a dynamic equilibrium of tethered and untethered disordered regions. The ability of an NTR to be in dynamic equilibrium between tethered/sequestered and untethered states suggests several mechanistic alternatives that need not be mutually exclusive. New ways of thinking about (and approaching) the intrinsic properties of sHSPs may finally allow the veil of enigma to be removed from sHSPs.
Keywords: Small heat shock proteins, Disorder, Binding grooves
Preamble
This short paper is not a scholarly treatise on small heat shock protein (sHSP) structure. It is one side of a conversation about how to think about the structure and function of these enigmatic and often frustrating proteins. In the more than 15 years I have been studying sHSPs, I have often felt that the more we learn, the less we understand. My goal here is to offer food for thought and fodder for future investigations with the goal of finally lifting the veil of enigma that has surrounded this class of proteins since their discovery. In line with the informal and conversational format, I have kept citations to a minimum (mainly those that have appeared since the most recent reviews were written) and point readers to two excellent review articles for more details (Haslbeck et al. 2019; Janowska et al. 2019). Apologies to those whose work has not been specifically cited here.
Small heat shock proteins are enigmatic
The family of ATP-independent protein chaperones known as small heat shock proteins remains among the most enigmatic of all chaperones. Following the dogma that structure can reveal function, laboratories worldwide have expended considerable effort to define the structure(s) of sHSPs. The current state of knowledge in this regard has been reviewed recently (Haslbeck et al. 2019; Janowska et al. 2019). In a nutshell, we know a lot about the central conserved structural domain, called the α-crystallin domain (ACD), and precious little about the flanking regions, which are highly flexible and do not adopt unique structures. These regions (N-terminal region and C-terminal region or NTR and CTR, respectively) represent over 50% of the sHSP sequence space in the human genome and have been refractory to traditional structural biology approaches, posing a roadblock on the path towards a mechanistic understanding of how sHSPs function. What has made sHSPs so intractable is not their high disorder content per se but the dynamic assemblies they form that can simultaneously contain different numbers of subunits. Conditions such as temperature and pH or modifications such as phosphorylation alter both the oligomeric landscape and the chaperone activity, raising the question of what sHSP species are the most functionally relevant. Their remarkable plasticity is likely central to the ability of sHSPs to act as general housekeeping chaperones and as first responders under conditions of cellular stress. But the same plasticity has been a bane to structural biologists and biochemists. Here, I attempt to synthesize emerging and available information in hopes of bypassing or, perhaps, even dismantling the roadblock that impedes understanding sHSP structure and function.
ACDs are groove presentation templates
Over two dozen structures of human sHSPs are available in the public Protein Data Bank, representing six of the ten paralogs (HSPB1, HSPB2, HSPB3, HSPB4, HSPB5, and HSPB6). All the depositions include atomic-level information for the entire ACD, and some include small amounts of additional bits from NTRs and/or CTRs. The first important takeaway from the structures is the remarkable similarity of the ACD structures, regardless of whether the structures were solved from protein crystals (X-ray diffraction) or proteins in solution (NMR). With only a few exceptions (see below), all ACD structures from human sHSPs display these key features:
Each ACD adopts an IgG-like β-sandwich structure, and two ACDs come together to form the ACD dimer (Fig. 1a).
The ACD dimer has a large 6-stranded sheet formed by β-strands 4, 5, and 6 + 7 of one subunit and β-strands 6 + 7, 5, and 4 of the second subunit, starting from the outside edge and working towards the center (Fig. 1a). This “bottom” sheet presents a fairly featureless (i.e., no obvious grooves) solvent-exposed surface.
Two copies of a smaller 3-stranded sheet sit atop the bottom sheet: starting from the outside edge, the strands are β8, β9, and β3 (Fig. 1a, b).
- Each ACD dimer contains three grooves (Fig. 1b).
- One long central groove (often referred to as either the β3-groove or the dimer interface groove) formed at the dimer interface by inward-facing sidechains of the bottom sheet and the two small “top” sheets (Fig. 1c).
- Two identical “edge grooves” (one on each subunit) created by strands β4 and β8 (often referred to as the β4/β8 groove; Fig. 1d).
Fig. 1.
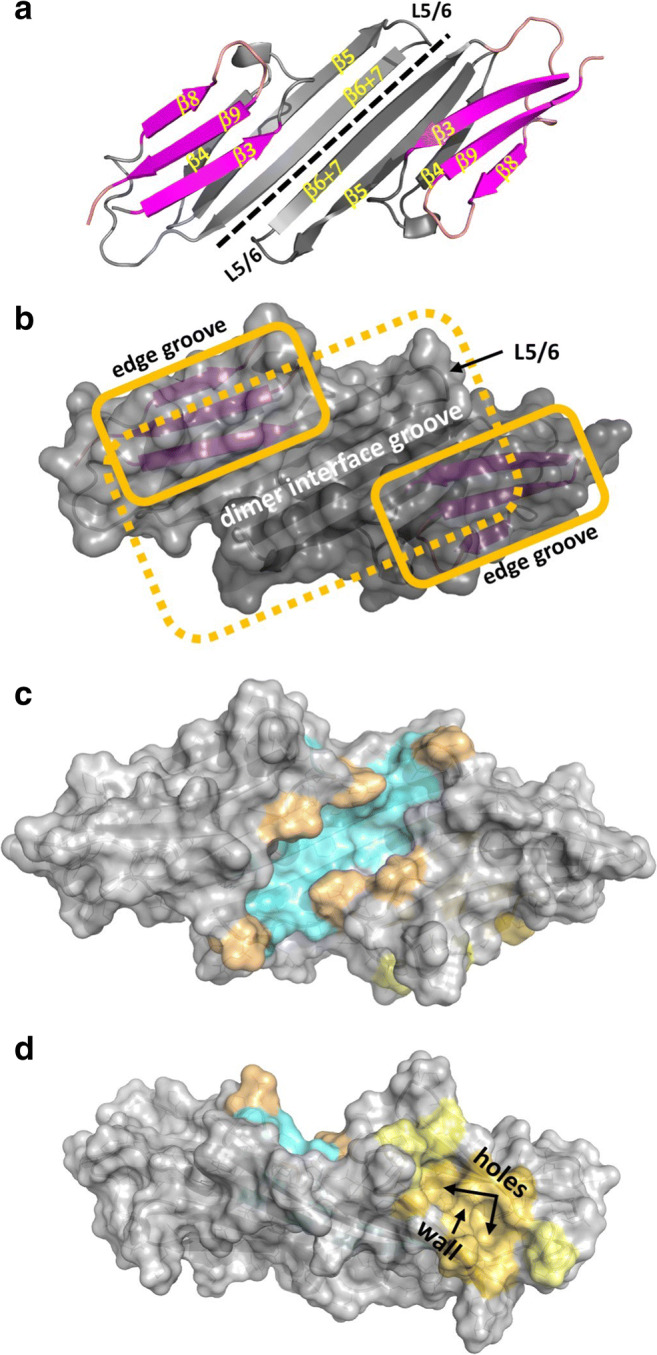
The architecture, nomenclature, and grooves of human sHSP α-crystallin domains (ACD). The ACD structure of HSPB1 (PDB 4MJH) is shown as exemplary in all panels. a Cartoon representation of the ACD dimer, with β-strand and loop nomenclature illustrated. The dimer interface is highlighted by a dashed line. The large “bottom” β-sheet is in gray; the two smaller “top” sheets are in magenta. b Surface rendition of the same view as in panel a reveals the dimer interface groove and its boundary as formed by loop L5/6 in the “loop-up” position. Dashed yellow line shows outline of bottom sheet; solid yellow lines outline the top sheets. c Same view as in panels a and b, with sidechains that create the dimer interface groove floor colored cyan and sidechains that create the edges of the dimer interface groove colored buff. d Rotated view reveals an ACD edge groove. The two holes and the wall between them are marked for easy identification
In essence, the ACD dimer can be thought of as a template that presents three grooves into which other proteins or other segments of sHSPs can bind. I believe the grooves may hold a key to understanding sHSP function. For example, binding grooves might help determine the specificity profile for a given sHSP and its binding partners. That said, the amino acid sidechains that create the two types of grooves are almost identical among HSPB1–HSPB6 and HSPB8 (Fig. 2). But each sHSP has at least one groove-forming sidechain that is different and may impart slightly different binding preference or affinity. In most cases, the differences are subtle: for example, the amino acid sidechains that create the dimer interface grooves of HSPB1 and HSPB5 are identical, and those that define their edge grooves differ only at one position.
Fig. 2.

Comparison of human sHSP ACD sequences. All sequences shown except HSPB8 have experimentally determined atomic-level ACD structures (Janowska et al. 2019). The sequences that compose each of the six β-strands are highlighted in dashed boxes, with the strands that create the bottom sheet in gray and the top sheet in magenta. Residues whose sidechains create the ACD grooves are denoted in blue symbols for dimer interface (#, end of groove; %, floor of groove) and orange symbols for the edge groove (&, end of groove; +, floor of groove). CTR I/V-X/I/V motifs are denoted with underlining
The remarkable conservation of groove structure and composition raises the question as to how the grooves may be altered by conditions known to modulate chaperone activity. There are several notable variations observed in available ACD structures that may provide clues. First, in a small number of structures, the alignment of the two anti-parallel β6 + 7 strands that form the dimer interface is shifted from the predominantly observed register. The floor of the dimer interface groove is created by six residues from each subunit: four groove-facing sidechains from the β6 + 7 strands and two conserved loop residues (Figs. 1c and 2). Of the twelve sidechains that line the groove, six are hydrophobic and four are positively charged. A shift of the two β6 + 7 strands relative to one another will change both the length and pattern of the groove. In principle, different groove configurations could either enhance or inhibit certain groove binding interactions. Specific conditions that lead to register shifts between two sHSP subunits remain a mystery. Intriguingly, redox-sensitive formation of a disulfide bond between a Cys residue in the center of the β6 + 7 strand of HSPB1 serves to lock the “canonical” alignment, but functional consequences for this reversible modification remain somewhat unclear (Pasupuleti et al. 2010; PDB 2N3J).
A second variation observed among ACD dimer structures that could affect the dimer interface groove is in the loop that links strands β5 and β6 + 7 (L5/6; Fig. 1a). ACD structures in the PDB fall into two classes with L5/6 either in a “loop-up” or a “loop-down” position (Fig. 1 shows an example of the loop-up conformation). Again, we have no clear idea of what favors one loop position over the other, although an inter-subunit interaction between a conserved Asp residue in L5/6 (Asp109 in HSPB5) and a conserved Arg residue in β6 + 7 (Arg120 in HSPB5) is observed in many “loop-up” structures. NMR studies indicate that the loops are dynamic in solution, indicating that the conformations captured in crystals, and their implied stabilizing interactions are transient. The L5/6 loops define the ends of the dimer interface groove, so a loop’s position and sequence will alter the groove (Fig 1a, b). Reported patient missense mutations in either the conserved Asp (D109H-HSPB5) or conserved Arg (R120G in HSPB5 and analogous positions in others) imply functional importance of the loop position, but structural studies to date have failed to provide clarity regarding the structural consequences of loss of the interaction. For example, a recent NMR structure of the ACD dimer of HSPB5 mutant, R120G, has a shifted dimer register with its L5/6 loop up (PDB ID 6BP9), but a crystal structure of the R120G ACD dimer is in the canonical register with L5/6 down (PDB 2Y1Z), indicative of the inherent plasticity of this structural feature. No structure has been reported for the D109H variant of HSPB5.
Another intriguing way that the dimer interface groove might be altered is via metal binding. Histidine residues in loops L3/4 and 5/6 and in β6 + 7 of HSPB5 have been implicated in binding divalent metal ions such as zinc and copper (Mainz et al. 2012). Notably, these histidine residues are well conserved among the sHSPs considered here. The sole structure available for a metal-bound ACD species has a Zn2+ ion bound between three copies of the ACD in the crystal lattice (PDB 3L1E). Although this arrangement may be an artifact of crystal packing, it identifies similar amino acid positions in HSB4 which are capable of binding a metal ion. In this structure, loop L5/6 is in a “loop-down” conformation, and the dimer interface groove is highly elongated.
A fourth variation observed among ACD structures is the presence or absence of an additional β-strand (β2) aligned next to β3. When present, the strand fills part of the dimer interface groove, thereby changing its size and shape. I will return to this aspect when discussing the NTR.
The edge grooves of ACDs are created predominantly by hydrophobic sidechains that emanate from strands β4 and β8 and neighboring strands (β5 and β9; see Figs. 1c and 2). Packing of these sidechains creates two well-defined holes into which other hydrophobic moieties can bind. A small sidechain in the middle position of β8 creates a wall between the two holes (Figs. 1d and 2). The nature of the wall-forming sidechain appears to be less important than its size (Ser, Ala, and Cys are present in the sHSPs under consideration here). Substitution of a native wall sidechain with a larger one, such as Gln, occludes the edge groove, creating a sHSP with impaired binding activity (Delbecq et al. 2012). This “groove bump” strategy has now been validated in several human sHSPs and could prove useful for parsing out the role of edge grooves and their binding partners in cellular functions of sHSPs (Baughman et al. 2020).
Are there mechanisms by which edge grooves are altered naturally? Unlike the dimer interface groove, there are no obvious structural variations of edge grooves among available ACD structures. One intriguing possibility is presented by the eye lens-specific sHSP, HSPB4 (aka αA-crystallin). HSPB4 contains a Cys residue as its β8 strand wall-forming position. This Cys can form an intramolecular disulfide with a Cys on the neighboring β9 strand in HSPB4 (Cherian-Shaw et al. 1999). For such a bond to form, the sidechain of the β8 Cys131 must leave its wall-creating position to swing towards Cys142, potentially distorting the edge groove. Consistent with this notion, in a comparison of the reduced and oxidized forms of HSPB4 by hydrogen-deuterium exchange (HDX/MS), the edge groove as well as much of the rest of the ACD is deprotected in the oxidized species, indicating a weakening or alteration in structure (Kaiser et al. 2019). How this might affect HSPB4 function in the eye lens where it resides is yet to be determined. On a more speculative level, a conserved His residue in loop L3/4 of HSPB5 that helps create one end of the edge groove (His83 in HSPB5) is implicated as a metal-binding residue, suggesting that metal binding might also disrupt or distort the groove.
The ACD grooves help to recruit subunits into oligomers
If ACD structures have evolved as presentation templates for the binding grooves described above, what do we know about what binds in them? The best-known interaction involves a tripeptide motif found in the C-terminal region of many sHSPs called the I/V-X-I/V (Ile or Val residues separated by any other residue). The Ile/Val sidechains insert into the two holes in the ACD edge groove creating a knob-into-hole interaction. The interaction has been observed in multiple ACD crystal structures that include some or all of a CTR sequence, either in trans or in cis. Importantly, the I/V-X-I/V motif from one subunit can bind into the edge groove from another dimer, and this intermolecular interaction has been shown to play a key role in the recruitment of sHSP subunits into oligomers (Delbecq et al. 2015). Consistent with the model, neither HSPB6 nor HSPB8 contains a CTR I/V-X-I/V motif (Fig. 2), and both exist predominantly as homodimers. In both cases, they can be recruited into hetero-oligomers with sHSPs that do contain a CTR I/V-X-I/V, implying that the CTR motif of one type of dimer can bind to and recruit another via its edge groove.
Although the role for CTR/ACD groove interactions in sHSP oligomerization is well established, whether the interaction directly imparts function is unknown. Even when the CTR I/V-X-I/V is bound to the edge groove, the rest of the CTR remains highly flexible and is solvent-exposed. Current thinking is that CTRs, which tend to contain high hydrophilicity, serve as solubility aids for sHSPs and may contribute to their ability to be present at remarkably high cellular concentrations. Paradoxically, ablation of CTR I/V-X-I/V motifs by substitution of the Ile residues with Gly tends to yield larger oligomers and to enhance chaperone activity in vitro. But whether the enhanced activity arises from larger oligomers is a thorny question. What is only now becoming clear is that one cannot consider a given groove interaction in isolation, as each groove is multi-functional and disruption of one interaction will have an impact on others.
The ACD grooves organize and/or sequester NTRs
There is a growing body of evidence that knob-into-hole binding is not limited to the CTR I/V-X-I/V motif. Two different bits of sequence from the NTR of HSPB6 have been observed inserted in an ACD edge groove: (1) 3IPVPV at the extreme distal end of the NTR (PDB 5LTW) and (2) 61ALPVAQ near the C-terminal end of the NTR just before the structured ACD (PDB 4JUS). In the structure of a hetero-tetramer of HSPB2 and HSPB3 (PDB 6F2R), the distal region of HSPB3, containing the sequence 4IILRH, is also inserted into an edge groove. While appropriately wary of putative interactions that may arise from crystal contacts, I believe that they are indicative of what can occur. In support of this, recent results from my lab indicate that the most distal region of HSPB1’s NTR interacts with the edge groove in solution, consistent with observations from the protein crystals just mentioned. But HSPB1 does not contain a canonical I/V-X-I/V motif, and we have proposed that the alternating hydrophobic sequence 6VPFSL is responsible for binding (Clouser et al. 2019). Altogether, the currently available structural observations indicate that (1) the very distal-most part of the NTR can insert into ACD edge grooves and (2) edge-groove binding motifs are not limited to the previously defined I/V-X-I/V sequences.
Extrapolating from the emerging observations, it appears that the NTRs of other human sHSPs (HSPB1, B3-B6, and B8) could have similar distal NTR-to-edge groove binding capacity (Table 1). This is in addition to the CTR I/V-X-I/V motifs, leading to a situation where, at least in HSPB1, B4, and B5, there are more edge-groove binding sequences within an sHSP than there are edge grooves. This is true regardless of the oligomeric state, as each sHSP polypeptide creates one ACD edge groove and may have edge-groove binding sequences in both its NTR and CTR. A survey of NTR sequences reveals additional potential edge-groove binding tripeptides in other regions of human sHSP NTRs, so the numbers of edge-groove binders could be even higher (Table 1). In solution, the interactions are fairly weak and transient, and there is a dynamic equilibrium with some fraction of each edge-groove binding sequence untethered at a given moment and grooves existing in empty and bound states (Fig. 3a).
Table 1.
Observed and potential ACD groove binding sequences in human sHSPs
| sHSP | Distal NTR edge-binding sequence | Other alternating hydrophobic sequences (NTR) | I/V-X-I/V CTR sequence |
Conserved NTR motif |
|---|---|---|---|---|
| HSPB1 | 6VPFSLa | – | 179ITIPVTFf | 26SRLFDQa |
| HSPB2 | – |
16YEF 47YYV |
160VYISLLb | 22SRLGEQ |
| HSPB3 | 4IILb |
35YAL 43VDL |
– | 29CRL-DH |
| HSPB4 | 3VTI |
32FEY 36LLPFL |
159IPVg | 20SRLFDQ |
| HSPB5 | 3IAIc |
15FPF 47FYL |
159IPIh | 21SRLFDQ |
| HSPB6 | 3IPVPVd |
53YYL 60VALPVe |
– | 26GRLFDQd |
| HSPB7 | – |
45LSM 63LAF |
168IKI | – |
| HSPB8 | 6MPF |
51WAL 79FGV |
– | 28SRLLDD |
Analysis performed by Janowska et al. (2019)
Experimentally observed binding sequences are highlighted in bold font
aClouser et al., 2019
bPDB 6F2R
cJanowska M and Klevit RE, unpublished
dPDB 5LTW
ePDB 4JUS
fPDB 4MJH
gPDB 3N3E
hPDB 4M5S, 4M5T, 2Y1Y, 2Y1Z, 2YGD
Fig. 3.

Intrinsic interactions between NTR sub-regions and ACD grooves limit the conformational states of an NTR. a A simplified view of how the small number of intrinsic NTR-to-ACD groove interactions defines possible states. The accounting shown is for a dimeric sHSP unit composed of subunits A and B. Additional states will arise in oligomers if an interaction can take place between dimeric units, as in the case of the CTR I/V-X-I/V motif binding into the edge groove of a neighboring dimer. b Cartoons illustrate how binding of the distal region (pink) to an edge groove or the conserved motif (yellow) to a dimer interface creates loops that could serve to limit or alter the conformational states available to the NTR by constraining the polypeptide chain
As if multiple edge-groove binding sequences within an sHSP were not enough, other proteins can also bind in the groove. Exemplary are co-chaperone, Bag3, and aggregation-prone clients, such as aβ and tau. Similar to the intrinsic ones, these interactions are weak and transient, predicting an even more complicated dynamic equilibrium in the presence of co-chaperones and/or clients. Presumably, the more potential binders, both intrinsic and extrinsic, that are present, the lower the population of empty grooves. Altogether, the emerging picture is that ACD edge grooves serve as hubs for what could be a myriad of interacting partners under cellular conditions. It is possible that small changes in conditions or in the available cadre of binders might tip the balance in favor of one interaction over another. But whether edge groove binding is directly responsible for chaperone activity is an open question whose answer is like to be client dependent. We currently favor a model in which the edge grooves are like the palm of a highly skilled juggler who is able to keep many balls in the air and in a limited region of space simultaneously, with each ball contacting the palm for a split second (Baughman 2019).
As its name indicates, the dimer interface groove is only present when two ACDs come together to form a dimer. Stably folded ACD monomers are difficult to detect, and the dimer is considered to be the predominant species and the main building block for oligomers. Two types of intrinsic (sHSP-derived) binding are observed in deposited structures: (1) NTR sequences that are proximal to the ACD align anti-parallel to the β3 strand, often referred to as the β2 strand, and (2) the sole conserved sequence within NTRs, “RLFDQ,” binds in the middle of the groove (Table 1). At a given dimer interface, there are two docking sites for the β2 type of binding and a single site for the conserved motif. The sites are non-overlapping, so it is possible for both types of binding to occur simultaneously in a single groove, although to date no experimentally determined structure has been deposited that contains two β2 strands and a bound conserved sequence. Model building based on existing structures reveals that two β2 strands and a conserved motif can fit into a single dimer interface groove without steric clashes (Clouser et al. 2019). Nevertheless, there is one more conserved motif than there is binding groove, so similar to the edge groove situation, at least some binders must be untethered at any time (Fig. 3a). Recent studies on HSPB1 revealed an interplay between the two types of dimer interface binding: disease-associated mutations in either the conserved region or the boundary region led not only to dissociation of the mutated sequence but also to the other binding region, as evidenced from HDX/MS data (Clouser et al. 2019). To date, no non-sHSP sequences have been observed bound to a dimer interface groove, so whether a situation similar to the edge grooves occurs remains to be seen. Here, it is worth noting that the only small (non-peptide) molecule reported to bind to a human sHSP to date binds in the dimer interface groove of HSPB5 (Makley et al. 2015).
All this groove binding begs the question, “What is its function?”. Although client binding to grooves may delay aggregation in some cases, this cannot be the whole story as the NTR is known to be required for chaperone activity in many cases. An alternate hypothesis is that groove binding of intrinsic NTR sequences helps to sequester them and keep these highly hydrophobic regions from aggregating themselves. Anecdotally, there have been no successful examples in which a sHSP NTR has been expressed and purified on its own, suggesting that the NTR needs its ACD and grooves. In HDX/MS studies of an HSPB1 dimer, we observed several sub-regions of the NTR to be protected from solvent exchange, notably those discussed above as groove binders. These three sub-regions, called the distal, conserved motif, and boundary, are at the beginning, middle, and end of the NTR. Tethering these regions, even transiently, will limit the physical and conformational space available to an NTR. Let us consider the case of HSPB1 and its approximately 90-residue NTR (Fig. 3b). A distal-to-edge groove binding creates a ~ 80-residue loop rather than a 90-residue free polypeptide chain. Binding of the conserved motif (residues 26–31) to the dimer interface would create an even shorter loop, further constraining the NTR. Again, the result will be a highly dynamic equilibrium of states, but these will have substantially less disorder than an untethered NTR would possess. At a coarse-grained level, one can “count” the number of states (Fig. 3b): (1) a distal region can be untethered, bound to its own edge groove, bound to the other edge groove within a dimer, or bound to a neighboring edge groove within an oligomer; 2) a conserved region can be untethered, bound to its dimer interface groove or (possibly?) bound to a neighboring groove; and 3) a boundary region can be untethered or bound at the dimer interface groove. In light of this relatively small number of defined interactions/states, we have proposed that the NTRs possess “quasi-order” (Clouser et al. 2019). The interactions that give rise to quasi-order are conserved on both sides, i.e., the grooves and the NTR sub-regions, strongly suggesting that this feature is shared among the human sHSPs. Other intrinsic interactions (NTR-to-ACD and/or NTR-to-NTR) may also occur, but these are likely to be idiosyncratic to a particular sHSP. I am optimistic that the repertoire of interactions can be defined using approaches similar to those used to discover the intrinsic HSPB1 NTR interactions (Clouser et al. 2019; Collier et al. 2019; PDB 6GJH).
Peeking from behind the veil of enigma: how quasi-order might shine a light on a mechanistic understanding of sHSPs
The ability of an NTR to be in dynamic equilibrium between tethered/sequestered and untethered states suggests several mechanistic alternatives that need not be mutually exclusive. First, these NTR sub-regions could be directly involved in client binding, in which case their untethered states (and conditions that favor these) would enhance chaperone function. Alternatively, NTR tethering could limit the accessibility and/or conformational landscape of intervening NTR sequences within the NTR, and these may be responsible for client binding and chaperone function. Either way, events or conditions that alter the ACD grooves in ways that alter binding could modulate chaperone activity. This prediction is borne out in recent results showing that a groove bump mutation in HSPB1 enhances its chaperone activity towards tau even though it disrupts tau binding to the edge groove (Baughman et al. 2020). Intriguingly, the oxidized form of HSPB4 that presumably has a distorted/disrupted edge groove due to its edge-groove Cys being involved in a disulfide bond exhibits increased protection of its most distal NTR sub-region and altered chaperone activity against model clients (Kaiser et al. 2019).
In this paper, I have carefully avoided consideration of oligomeric structure and dynamics. Despite remarkable technological developments in cryo-electron microscopy and its ability to deal with heterogeneous systems, the infamous heterogeneity and plasticity of sHSPs renders them still among the most challenging of structural targets (Kaiser et al. 2019). And the heterogeneity itself raises fundamental questions: Are some oligomeric species more functionally relevant than others? How will we know? If we cannot find ways to grapple with these questions, does it make sense to expend enormous amounts of human and scientific resources to attempt to define the structures? It is a thorny chicken-and-egg situation. Perhaps if we had sufficiently well-defined oligomeric structures, they would suggest testable hypotheses. Only time will tell.
Regardless of whether emerging structural biology approaches can ultimately surmount the sHSP oligomer challenge, I believe the notion of NTRs as being composed of multiple sub-regions which will be fruitful. While no one (to my knowledge) has succeeded in preparing biochemically tractable samples of full-length NTRs, peptides that represent individual sub-region sequences are often soluble and can be useful reagents. As an example, a distal sub-region peptide binds to the ACD edge groove in a manner that recapitulates that observed by NMR for full-length HSPB1. Furthermore, a peptide that contains a stress-dependent phosphorylation site of HSPB1 displays phosphorylation-dependent binding to the ACD (Clouser et al. 2019). Although still early days, I am optimistic that similar strategies will allow for many of the intrinsic sHSP interactions to be parsed out, providing a list of the ways in which a given NTR can be tethered and sequestered by its ACD. Such parsing of NTRs that have historically been viewed as virtually intractable can move the field towards deeper mechanistic understanding and help in the design of sensible cellular investigations.
Conceptually, there is reason to believe that the interactions discussed here occur within the context of oligomers. The situation that leads to quasi-order, that is, the presence of more binding sequences than binding grooves, may provide at least a partial explanation for the infamous polydispersity, i.e., non-discrete numbers of subunits, in oligomers. So, we can think of oligomers as assemblies that contain both tethered and untethered NTR and CTR sequences. In addition, the high local concentration of NTRs may promote inter-NTR interactions that could sequester untethered NTRs to inhibit sHSP aggregation.
In conclusion, it was not that long ago that I thought that the structural biology of sHSPs might be terminally stuck at atomic-level ACD structure and coarse-grained and/or pseudo-atomic models of oligomers and that these would not provide sufficient insight into sHSP function. With new ways of thinking about (and approaching) the intrinsic properties of sHSPs, I am optimistic about the prospects for achieving sub-molecular, if not atomic-level, information that will finally allow the veil of enigma to be removed from the sHSPs.
Acknowledgements
I thank all members of my research team, past and present, who have had the fortitude to journey with me on a quest to understand how small heat shock proteins work. I especially acknowledge recent members H. Baughman, A. Clouser, M. Janowska, N. Stone, and C. Woods for their exciting breakthroughs. I gratefully acknowledge the continuous support of our research by the National Eye Institute (2 R01 EY017370).
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Baughman HER (2019) Chaperone effects on tau amyloid formation. Ph D Thesis Univ of Washington
- Baughman HER, Pham T-HT, Adams CS, Nath A, Klevit RE. Release of a disordered domain enhances HspB1 chaperone activity toward tau. Proc Natl Acad Sci U S A. 2020;117(6):2923–2929. doi: 10.1073/pnas.1915099117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian-Shaw M, Smith JB, Jiang XY, Abraham EC. Intrapolypeptide disulfides in human alphaA-crystallin and their effect on chaperone-like function. Mol Cell Biochem. 1999;199(1–2):163–167. doi: 10.1023/A:1006906615469. [DOI] [PubMed] [Google Scholar]
- Clouser AF, Baughman HER, Basanta B, Guttman M, Nath A, Klevit RE (2019) Interplay of disordered and ordered regions of a human small heat shock protein yields an ensemble of “quasi-ordered” states. bioRxiv. 10.1101/750646 [DOI] [PMC free article] [PubMed]
- Collier MP, Alderson TR, de Villiers CP, Nicholls D, Gastall HY, Allison TM, Degiacomi MT, Jiang H, Mlynek G, Fürst DO, der Ven PFM v, Djinovic-Carugo K, Baldwin AJ, Watkins H, Gehmlich K, Benesch JLP. HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C. Sci Adv. 2019;5(5):eaav8421. doi: 10.1126/sciadv.aav8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbecq SP, Jehle S, Klevit R. Binding determinants of the small heat shock protein, αB-crystallin: recognition of the 'IxI' motif. EMBO J. 2012;31(24):4587–4594. doi: 10.1038/emboj.2012.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbecq SP, Rosenbaum JC, Klevit RE. A mechanism of subunit recruitment in human small heat shock protein oligomers. Biochemistry. 2015;54(28):4276–4284. doi: 10.1021/acs.biochem.5b00490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck M, Weinkauf S, Buchner J. Small heat shock proteins: simplicity meets complexity. J Biol Chem. 2019;294(6):2121–2132. doi: 10.1074/jbc.REV118.002809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowska MK, Baughman HER, Woods CN, Klevit RE (2019) Mechanisms of Small Heat Shock Proteins. Cold Spring Harb Perspect Biol. 10.1101/cshperspect.a034025 [DOI] [PMC free article] [PubMed]
- Kaiser CJO, Peters C, Schmid PWN, Stavropoulou M, Zou J, Dahiya V, Mymrikov EV, Rockel B, Asami S, Haslbeck M, Rappsilber J, Reif B, Zacharias M, Buchner J, Weinkauf S. The structure and oxidation of the eye lens chaperone αA-crystallin. Nat Struct Mol Biol. 2019;26(12):1141–1150. doi: 10.1038/s41594-019-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainz A, Bardiaux B, Kuppler F, Multhaup G, Felli IC, Pierattelli R, Reif B. Structural and mechanistic implications of metal binding in the small heat-shock protein αB-crystallin. J Biol Chem. 2012;287(2):1128–1138. doi: 10.1074/jbc.M111.309047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makley LN, McMenimen KA, DeVree BT, Goldman JW, McGlasson BN, Rajagopal P, Dunyak BM, McQuade TJ, Thompson AD, Sunahara R, Klevit RE, Andley UP, Gestwicki JE. Pharmacological chaperone for α-crystallin partially restores transparency in cataract models. Science. 2015;350(6261):674–677. doi: 10.1126/science.aac9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasupuleti N, Gangadhariah M, Padmanabha S, Santhoshkumar P, Nagaraj RH. The role of the cysteine residue in the chaperone and anti-apoptotic functions of human Hsp27. J Cell Biochem. 2010;110(2):408–419. doi: 10.1002/jcb.22552. [DOI] [PMC free article] [PubMed] [Google Scholar]