

Abstract
RNA modifications have recently emerged as an important layer of gene regulation. N6‐methyladenosine (m6A) is the most prominent modification on eukaryotic messenger RNA and has also been found on noncoding RNA, including ribosomal and small nuclear RNA. Recently, several m6A methyltransferases were identified, uncovering the specificity of m6A deposition by structurally distinct enzymes. In order to discover additional m6A enzymes, we performed an RNAi screen to deplete annotated orthologs of human methyltransferase‐like proteins (METTLs) in Drosophila cells and identified CG9666, the ortholog of human METTL5. We show that CG9666 is required for specific deposition of m6A on 18S ribosomal RNA via direct interaction with the Drosophila ortholog of human TRMT112, CG12975. Depletion of CG9666 yields a subsequent loss of the 18S rRNA m6A modification, which lies in the vicinity of the ribosome decoding center; however, this does not compromise rRNA maturation. Instead, a loss of CG9666‐mediated m6A impacts fly behavior, providing an underlying molecular mechanism for the reported human phenotype in intellectual disability. Thus, our work expands the repertoire of m6A methyltransferases, demonstrates the specialization of these enzymes, and further addresses the significance of ribosomal RNA modifications in gene expression and animal behavior.
Keywords: behavior, Drosophila, m6A, Mettl5, ribosome, RNA methyltransferase
Subject Categories: Neuroscience; Post-translational Modifications, Proteolysis & Proteomics; RNA Biology
This study identifies the Mettl5 Drosophila homolog as an m6A 18S rRNA methyltransferase required for normal walking behavior.
Introduction
N6‐methyladenosine (m6A) was discovered on mammalian mRNA in the last seventies 1, 2. The recent development of transcriptome‐wide modification mapping approaches and identification of the m6A mRNA machinery sparked new interest in the field. Mapping approaches revealed the prevalence of m6A within thousands of mRNAs and long noncoding RNAs (lncRNAs) 3, 4, while genetic manipulation of m6A players revealed its diverse roles in development and diseases through regulation of mRNA fate 5.
Deposition of m6A is catalyzed by a subfamily of methyltransferases characterized by the conserved catalytic motif [D/N/S/H]PP[Y/F/W] 6. In mRNA, the methylation is installed on adenosine within a conserved consensus sequence context, DRACH (where D = A/G/U, R = A/G and H = A/C/U), by a multi‐subunit methyltransferase complex. In mammals, methyltransferase‐like 3 (METTL3) is the catalytic subunit and forms a stable heterodimer with METTL14, which facilitates binding to mRNA substrates 7, 8, 9. The role of other subunits, which include Wilms tumor 1‐associated protein (WTAP), Vir‐like m6A methyltransferase associated (VIRMA) 10, RNA binding motif 15 (RBM15) 11, Zinc‐finger CCH domain‐containing protein (ZC3H13) 12, 13, 14, and HAKAI 10, 15, is less understood. This complex is highly conserved from insects to mammals but is only partially present in the yeast Saccharomyces cerevisiae and absent in the worm Caenorhabditis elegans 16, 17, 18.
In addition to its well‐characterized occurrence on mRNA, m6A is also known to occur on circular RNAs, small nuclear RNAs (snRNAs), microRNAs (miRNAs), long noncoding RNAs (lncRNAs), and ribosomal RNAs (rRNAs), whereas its presence on transfer RNAs (tRNAs) was only reported in bacteria so far 18. Recent reports found that METTL16 binds a subset of mRNAs and adds m6A on MAT2A transcripts, as well as on U6 snRNA 19, 20, 21, 22. In contrast, m6A deposition on rRNA is less understood. rRNAs are the second most heavily modified class of RNAs, after tRNAs, with ~ 2% of rRNA nucleotides bearing post‐transcriptional modifications 23. 2′‐O‐methylation of the sugar backbone and pseudouridylation are the most abundant modifications, while base modifications represent only about 5% of total rRNA modifications in yeast and humans 24. Among these, m6A is reported to be present on human 18S and 28S rRNA at positions 1832 and 4220, respectively 25. In a recent report, ZCCHC4 was shown to generate m6A on the 28S rRNA; impacting ribosome subunit distribution, global translation, and cancer cell proliferation 26. This study, along with others 27, 28, 29, 30, demonstrated that base modifications on rRNA may play important roles in gene expression.
In order to identify additional m6A methyltransferases, we carried out a screen in Drosophila S2R+ cells, in which we knocked down all annotated orthologs of human methyltransferase‐like proteins (METTLs) and assessed the effect on global m6A levels on total and messenger RNAs. Our screen identified the previously uncharacterized Drosophila gene: CG9666, the ortholog of METTL5. We show that CG9666 contains the conserved catalytic “asparagine‐proline‐proline‐phenylalanine” (“NPPF”) motif found in most m6A methyltransferase enzymes and that it is required for m6A deposition on 18S rRNAs. In addition, a proteomic screen identified the ortholog of human TRMT112, CG12975, as a co‐factor of CG9666. In yeast and archaea, TRMT112 homologs were shown to bind and activate several methyltransferases (Bud23, Trm9, Trm11, and Mtq2) 31. Here, we found that CG12975 forms a stable and conserved complex with CG9666. This is consistent with a recent report in human cells showing the importance of the METTL5‐TRMT112 interaction for m6A deposition on 18S RNA 23. The lack of m6A modification on 18S rRNA does not affect rRNA processing, yet flies lacking this modification display impaired orientation in walking behavioral assays. Interestingly, recent exome sequencing in Pakistani and Yemenite families identified METTL5 as a novel gene associated with recessive intellectual disability 30. Altogether, these findings demonstrate the importance of m6A modification on 18S rRNA for normal behavior and suggest that this function is conserved from flies to human.
Results and Discussion
Mettl5 controls m6A levels in total RNA
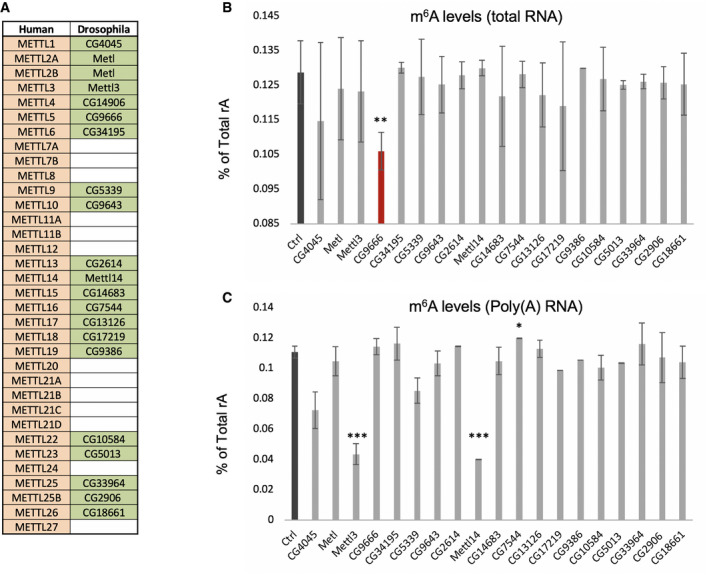
In order to identify novel enzymes required for m6A deposition on RNA, we conducted a targeted RNAi screen in Drosophila S2R+ cells followed by m6A quantification using mass spectrometry. Candidates were selected based on their sequence homology with annotated human METTLs (Fig 1A). Out of the 32 annotated human METTL enzymes, 20 have a Drosophila ortholog. Most of these enzymes are not related to each other by sequence homology. Few candidates were predicted to catalyze m6A modification based on characteristics of their catalytic domain. These include the already known mRNA m6A methyltransferase, Mettl3, and uncharacterized homologs of METTL4 (CG14906), METTL5 (CG9666), and METTL16 (CG7544). We generated double‐stranded RNA for the 20 Drosophila genes and incubated S2R+ cells for a total period of 6 days, to ensure sufficient depletion (Appendix Fig S1A and B). Total and poly(A)+ RNA were then purified and subjected to mass spectrometry analysis. As expected, the knock down (KD) of Mettl3 and Mettl14 resulted in a substantially decreased m6A level within the mRNA fraction, but had no effect on total RNA (Fig 1B and C and Appendix Fig S1C). In contrast, the KD of CG9666 led to a reduction of m6A on total RNA, but did not alter the abundance of m6A on mRNA. KD of other predicted methyltransferases did not significantly affect m6A levels, suggesting that they are involved in different types of enzymatic reactions or that they regulate only a subset of m6A sites. The latter case is likely true for CG7544, as the vertebrate ortholog METTL16 has only few confirmed m6A targets. Similarly, human METTL4 was recently shown to specifically catalyze m6A on U2 snRNA 24. Thus, our screen identified CG9666, hereafter named Mettl5, as a potential new m6A methyltransferase in Drosophila.
Figure 1. Mettl5 regulates m6A level in total RNA .
-
AList of human methyltransferases and their orthologs in Drosophila melanogaster. Empty cells indicate that no ortholog could be identified (see Material and Methods for details).
-
B, CLC‐MS/MS measurements of m6A levels in total RNA (B) or in poly(A) + RNA (C) upon KD of predicted methyltransferases in Drosophila S2R+ cells. m6A abundance in total RNA is significantly reduced when Mettl5 is depleted, while its depletion has no effect on m6A level in mRNA. As expected, the KD of Mettl3 and Mettl14 reduce m6A levels in mRNA. Bar chart represents the mean ± standard deviation of three technical measurements from three biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 (two‐tailed t‐test).
Mettl5 is predominantly expressed in the cytoplasm and is enriched in the brain
Sequence analysis shows that Mettl5 is probably an ancient protein with full‐length orthologs detected in all eukaryotic genomes (except fungi), but also in all completely sequenced archaeal species examined. The presence of orthologs in a few species of bacteria and phylogenetic analysis (Fig 2A and Appendix Fig S2) suggests horizontal transfer from archaea to bacteria. Conservation is high across eukarya (e.g., 53% identity between human, plant Arabidopsis thaliana, and fly sequences). The protein harbors N‐terminal signatures characteristics of methyltransferase enzymes (amino acid co‐ordinates in the fly sequence 39–146) with homology to many families (PFAM domain PF05175; MTS; methyltransferase small domain) and a C‐terminal part (aa 147–213), which is unique to this family. Of note, all orthologs contain the characteristic NPPF motif, indicating that the catalytic activity is likely conserved throughout evolution (Fig 2B).
Figure 2. Phylogenetic and expression analyses of Mettl5.
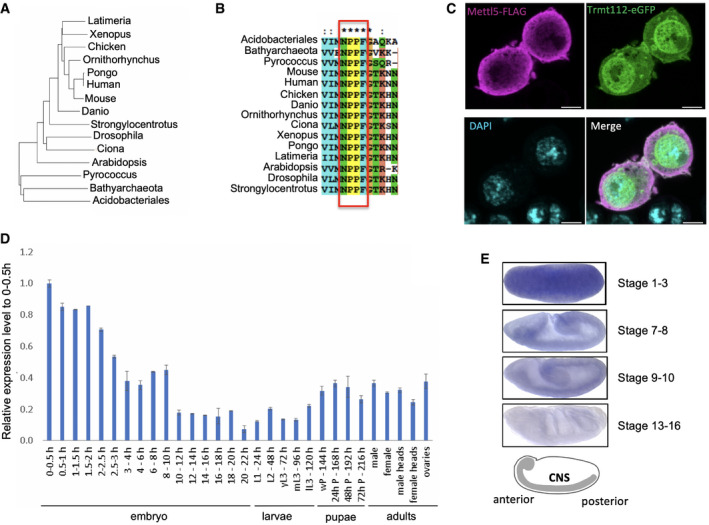
- Phylogenetic tree of the alignment of representative Mettl5 orthologs from selected species (see Materials and Methods for details). Prokaryotic sequences from archaea (Pyrococcus, Bathyarchaeota) and bacteria (Acidobacteriales) are included as outliers.
- Multiple sequence alignment of Mettl5 orthologs showing conservation of the NPPF motif. Asterisks indicate perfect conservation.
- Subcellular localization of Mettl5‐FLAG (purple) and Trmt112‐eGFP (green) expressed under the control of their own promoter in S2R+ cells. 63× magnified merge of immunofluorescently labeled S2R+ cells. Scale bar: 4.47 μm.
- Developmental expression of Mettl5 transcript assayed by RT–qPCR analysis. The figure shows mean ± standard deviation of three technical measurements from three biological replicates.
- In situ hybridization of Mettl5 transcript at different embryonic stages. The central nervous system (CNS) is highlighted in the schematics. Data retrieved from FlyExpress 7 (http://www.flyexpress.net/search.php?type=image&search=FBgn0036856).
To get more insight into Mettl5 function, we examined its subcellular distribution and its expression during fly development. We found that Mettl5‐FLAG expressed under its own regulatory promoter accumulates predominantly in the cytoplasm (Fig 2C). This is in sharp contrast with the localization of Mettl5 in human cells where it is predominantly localized to the nucleolus 23. Mettl5 is expressed at high level in early embryo, and its expression gradually decreases and remains low in the larval stages (Fig 2D). A mild increase is observed at metamorphosis, and this level remains constant in the adult phase. According to fly express (http://www.flyexpress.net/search.php?type=image&search=FBgn0036856), Mettl5 is broadly expressed during embryogenesis and displays some enrichment in the nervous system (Fig 2E).
Mettl5 promotes m6A deposition on Drosophila 18S rRNA
In order to get deeper insights into the molecular and functional role of Mettl5 in vivo, we generated loss‐of‐function alleles using the CRISPR/Cas9 methodology. Two guide RNAs encompassing the methyltransferase domain were designed. Using this approach, we obtained two distinct mutations (Fig 3A). The first allele (Mettl5 ∆2AA) lacks six nucleotides, resulting in a two amino acid deletion just upstream the predicted methyltransferase signatures (at positions 36 and 37; the AA in PHIAACMAH). These two amino acids are conserved from flies to human (Appendix Fig S2). The second allele (Mettl5 fs) is an insertion–deletion (indel) mutation consisting of a deletion of thirteen nucleotides combined with an insertion of three nucleotides (Appendix Fig S3). This indel mutation results in a frameshift at the amino acid position 36 and in a premature stop codon after amino acid 107, indicating that if the resulting mRNA is not eliminated by the nonsense‐mediated mRNA decay pathway, only a truncated protein lacking the full methyltransferase domain is produced.
Figure 3. Drosophila Mettl5 is required for m6A deposition on 18S rRNA .
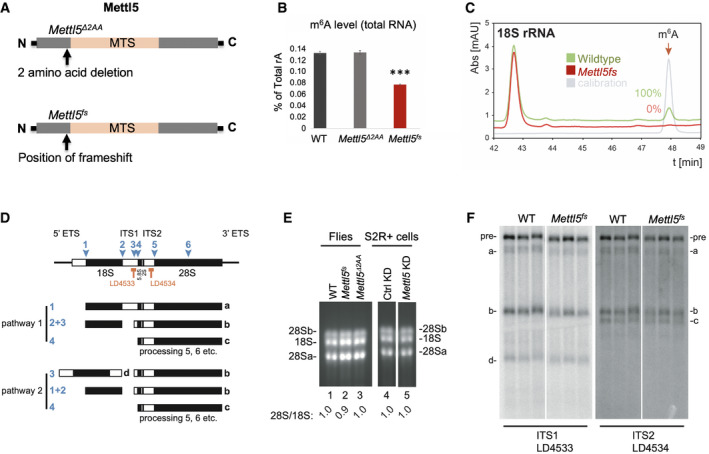
- Representation of the two Mettl5 mutant alleles generated in this work, consisting either of a two amino acid deletion upstream of the methyltransferase domain (top, Mettl5 ∆2AA) or of a frameshift mutation leading to a premature stop codon (bottom, Mettl5 fs).
- LC‐MS/MS measurements of m6A levels in total RNA of WT and Mettl5 mutant flies. Bars represent mean ± standard deviation of measurements of three biological replicates. ***P < 0.001 (two‐tailed t‐test).
- Purified 18S rRNA analyzed for its m6A content by quantitative HPLC. The 18S rRNA was extracted from 40S subunits isolated on sucrose gradients. The calibration control is a commercial source of m6A (in gray). m6A elutes at 48 min.
- Pre‐rRNA processing in Drosophila: four mature rRNAs (the small ribosomal subunit 18S, and the large ribosomal subunit 5.8S, 2S, and 28Sa and 28Sb) are produced by sequential RNA cleavage following two alternative pathways, as depicted. Processing sites are indicated (1–6). The major pre‐rRNA intermediates (a, b, c, and d) are highlighted.
- Mature rRNA analysis on ethidium‐stained denaturing agarose gels. The same amounts of total RNA extracted from the indicated flies and from S2R+ cells depleted or not of Mettl5 were loaded. The 28S/18S ratio was established by densitometry.
- Analysis of 18S rRNA maturation in WT and mutant flies (Mettl5 fs). Total RNA extracted from the indicated animals was resolved on denaturing agarose gels and processed for Northern blotting with specific probes (complementary to ITS1 or ITS2 sequences). The pre‐rRNAs detected accumulate to normal levels indicating that processing is unaffected.
We found that both mutations give rise to viable and fertile flies with no obvious defect at the morphological level. In order to address their impact on m6A levels, RNA was isolated from whole flies and analyzed by mass spectrometry. We found that the level of m6A from Mettl5 ∆2AA flies is similar to wild type (WT) on both total and poly(A) RNA. However, a 45% decrease in total RNA of Mettl5 fs flies was observed, consistent with a reduction of rRNA methylation, while the level on mRNA remained unchanged (Fig 3B and Appendix Fig S4). This is in agreement with our experiments in S2R+ cells indicating that Mettl5 is required for proper m6A levels. Furthermore, this suggests that Mettl5 fs is a strong loss‐of‐function allele, while Mettl5 ∆2AA has little to no impact on Mettl5 methyltransferase activity.
Recently, METTL5 was identified as a m6A methyltransferase for 18S human rRNA 23. To address whether this function is conserved in flies, the 18S and 28S rRNAs were individually isolated from mutant and isogenic control flies by velocity centrifugation, digested to nucleosides, and analyzed by HPLC (Fig 3C and Appendix Fig S5). Using commercial m6A as calibration control, we established that this modified nucleoside elutes at 48 min (Fig 3C and Appendix Fig S5, gray). Analysis of purified 18S rRNA from flies confirmed the presence of the modification in wild‐type animals, as expected, and revealed its absence in the Mettl5 fs mutant (Fig 3C). In contrast, the level of m6A on 28S rRNA was not substantially affected (Appendix Fig S5). These results indicate that Mettl5 is required for m6A deposition on 18S rRNA in Drosophila, which is consistent with the activity of human METTL5 23.
Since another ribosomal RNA modification, 2′‐O‐methylation, was recently shown to occur substoichiometrically at specific positions 32, 33, 34, we wondered whether m6A on 18S rRNA might also be partially modified. To address this question, we used the standard molar response factor on HPLC profiles (28). We made use of the UV254 molar response factors (Mr) of unmodified nucleosides (A, C, G, and U) and of selected modified (ac4C and m6A) nucleosides. The Mr values of A, C, G, U, ac4C, and m6A are, respectively, of: 431, 215, 463, 290, 172, and 340 (ref. 35). The number of A, C, G, U, ac4C, and m6A nucleosides on fly 18S rRNA is, respectively, of: 564, 376, 473, 583, 2, and 1. For each nucleoside, the peak area was established on four independent HPLC profiles using four independent RNA preparations. Each peak area was divided by its respective standard molar response factor. The levels of m6A were estimated by comparing the value obtained for m6A with that of ac4C or of each of the unmodified nucleosides. In each case, the level of m6A modification was estimated to be of 100%. This indicates that m6A on fly 18S rRNA is fully methylated and does not appear to be regulated, at least in the conditions tested.
To test whether Mettl5‐mediated 18S rRNA m6A modification is required for ribosome biogenesis, we analyzed mature rRNA steady‐state levels and pre‐rRNA processing in the Mettl5 fs and control flies. In Drosophila, five mature rRNAs (the 18S, 5.8S, 2S, 28Sa, and 28Sb—the 28S rRNA is fragmented in fly following cleavage at site 6) are produced from a long polycistronic transcript synthesized by RNA polymerase I (see Fig 3D). Total RNA was extracted from Mettl5 fs and from control wild‐type and Mettl5 ∆2AA flies, resolved on denaturing agarose gels, and processed for Northern blotting with probes specific to major pre‐rRNA intermediates. First, we analyzed the steady‐state accumulation of the large mature rRNAs (18S, 28Sa, and 28Sb) by ethidium bromide staining (Fig 3E). The ratio of 28S/18S was unaffected in the Mettl5 fs mutant flies. This was also the case in S2R+ cells depleted for Mettl5 (Fig 3E). Next, we analyzed the levels of individual pre‐rRNA intermediates (Fig 3F). We found that Mettl5 fs mutant flies do not display any qualitative difference in pre‐rRNA processing. Altogether, we conclude that the Mettl5‐mediated m6A site is not required for pre‐rRNA processing or 18S rRNA production (Fig 3E and F).
Trmt112 is a conserved Mettl5 co‐factor
In human cells, METTL5 has recently been shown to act as an m6A 18S rRNA methyltransferase in concert with the noncatalytic co‐activator, TRMT112 36. To test whether this mode of action is conserved in fly, we immunoprecipitated FLAG‐Myc‐tagged Mettl5 from S2R+ cells and submitted the co‐precipitated proteins to mass spectrometry analysis. We found that seven proteins were significantly enriched in the pull‐down fraction (omitting the ribosomal proteins; Fig 4A and Table EV1). Among them, we found the homolog of TRMT112, the previously uncharacterized CG12975. To validate this interaction, we cloned CG12975 (hereafter called Trmt112) along with the HA epitope in the N‐terminal region. Immunoprecipitation of HA‐Trmt112 pulled down FLAG‐Myc‐Mettl5, revealing that both proteins are in the same complex, independently of RNA (the interaction pertains in the presence of RNase T1; Fig 4B). Reciprocally, this was confirmed by immunoprecipitating FLAG‐Myc‐Mettl5 and blotting the eluates with anti‐GFP that revealed Trmt112‐eGFP (Fig 4B). Consistent with a role of Mettl5 in regulating m6A on ribosomal RNA, we found that Trmt112 KD in S2R+ cells also reduced m6A level on total RNA (Fig 4C).
Figure 4. Mettl5 interacts with Trmt112 to install m6A on 18S rRNA .
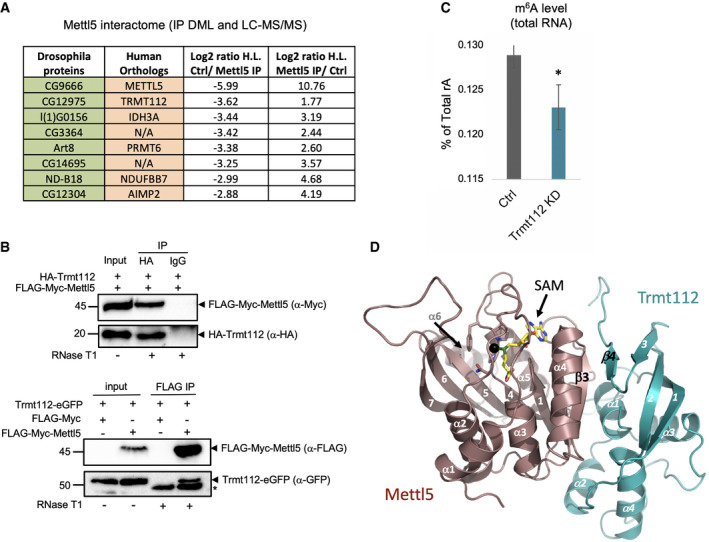
- Mettl5 partners identified as significantly enriched candidates in mass spectrometric analysis of Mettl5‐FLAG‐Myc affinity purifications (ribosomal proteins not listed for simplicity). Measurements of two biological replicates.
- Western blot validation of the co‐immunoprecipitated proteins Mettl5‐FLAG‐Myc and Trmt112, bearing either a N‐terminal HA tag (top) or a C‐terminal GFP tag (bottom), in S2R+ cells. The arrows point at the indicated proteins. HA‐Trmt12 migrates below the antibody light chains (top). Trmt112‐eGFP migrates above the heavy chains indicated by an asterisk (bottom).
- Trmt112 depletion reduces m6A levels in total RNA from Drosophila S2R+ cells. Mean ± standard deviation of three technical measurements from three biological replicates. *P < 0.05 (two‐tailed t‐test).
- The sequences of D. melanogaster Mettl5 and Trmt112 were modeled in the experimentally determined atomic resolution structure of the human METTL5‐TRM112 complex (based on PDB model 6H2V), revealing high conservation and formation of a parallel β‐zipper involving main chain atoms at the complex interface. Mettl5 and Trmt112 are colored light brown and blue, respectively. The NPPF signature, known to co‐ordinate planar nitrogen groups to be methylated, is shown as sticks. The S‐adenosyl‐methionine (SAM) is shown in stick representation (yellow) with the methyl group to be transferred to the 18S rRNA represented as a black sphere. Important secondary structure elements, including β3 on Mettl5 and β4 on Trmt112, are indicated.
We next addressed the expression pattern of Trmt112. Like Mettl5, Trmt112 transcript displays strong expression in early embryos (Appendix Fig S6). In subsequent developmental stages, its expression follows a “wavy” pattern with a specific enrichment in adult ovaries. In contrast to Mettl5, Trmt122 localizes predominantly in the nucleus but is also found in the cytoplasm (Fig 2C). This difference in subcellular distribution is not surprising considering that Trmt112 is well‐known to interact with additional methyltransferases.
Recently, the structure of human METTL5‐TRMT112 was solved at atomic resolution by X‐ray crystallography 36. This revealed that the heterodimeric complex assembles through formation of a parallel ß‐zipper involving main chain atoms between ß‐strand 3 of METTL5 and ß‐strand 4 of TRMT112, resulting in a remarkable continuous eleven‐stranded ß‐sheet in the complex. To evaluate whether formation of a Mettl5‐Trmt112 complex is conserved in Drosophila, we modeled the fly Mettl5 and Trmt112 sequences using the 3‐D structure of the human complex as template (Fig 4D). This illustrates a near‐perfect structural conservation, with most of the major residues involved at the interface between the two subunits of the complex being conserved between human and fly (Fig 4D and Appendix Fig S7). Upon binding, Trmt112 masks a large hydrophobic area on Mettl5, a region which would otherwise be unfavorable in the water‐based environment. Our modeling that predicts the structural conservation of the Mettl5‐Trmt112 heterodimer indicates that like in human Trmt112 might be required to stabilize Mettl5 in Drosophila.
Mettl5 is required for locomotion and orientation
METTL5 deficiency has recently been associated with neurological symptoms in autosomal recessive intellectual disability (ARID), including microcephaly and intellectual disability with altered behavioral and social skills 38. Patients with this syndrome carry frameshift mutations in METTL5, resulting in truncated products, which, incidentally, is highly reminiscent of our fly mutation Mettl5 fs. These truncated products are predicted to lack important secondary structure elements of Mettl5, which is expected to alter protein function and is consistent with the destabilization of the corresponding proteins in cell culture experiments 30.
To address whether Mettl5 mutant flies also display neurological defects, we performed the Buridan's paradigm assay 37. In this method, individual flies are set onto a lit‐up platform surrounded by water, in which two inaccessible black stripes serve as visible landmarks for the flies. Flies move from one stripe to another in a robust manner, enabling the measurement of their activity, orientation, and walking speed in 15‐min intervals (Fig 5A). Wild‐type flies were used as control and were compared to homozygous mutant animals. We also used trans‐heterozygous flies to exclude off‐target effects (see Material and Methods).
Figure 5. Mettl5 is required for fly orientation.
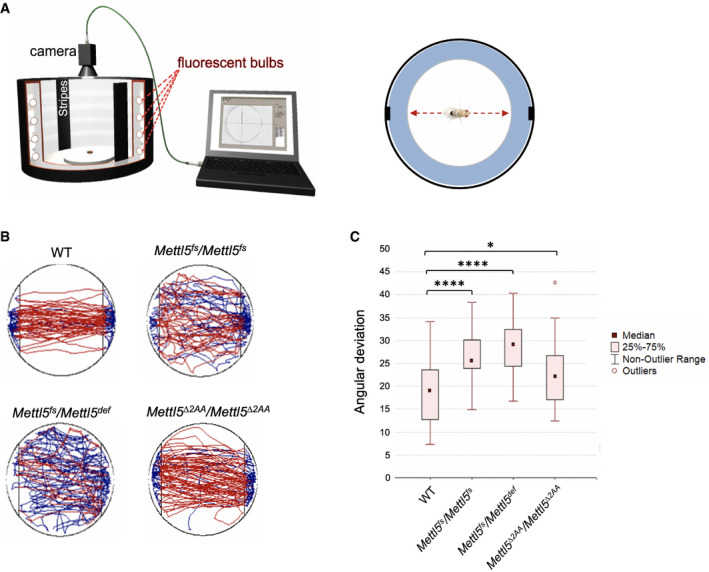
- Cartoon depicting the experiment with the main components of the setup showing the arena in white, the visual landmarks as black stripes and the camera to record the fly (left, from Ref. 37), and cartoon showing the normal behavior of a WT fly in the arena. The black rectangles represent the visual landmarks and the red arrow the main trajectory undertaken by WT flies relative to the position of the landmarks (right).
- Representative trajectories of WT and Mettl5 mutant flies analyzed by Buridan's paradigm. The blue lines indicate when the fly stops and changes direction.
- Orientation evaluated with the help of Buridan's paradigm for Mettl5 mutant flies. Orientation was measured as the angular deviation from the straight path needed to move from one landmark to another in the arena. Number of flies tested per genotype: 30. Mean ± standard deviation. Shapiro–Wilk test was used to test for normal distribution in each group. Normally distributed groups were tested by t‐test. Due to multiple comparison, Bonferroni correction was applied (*P < 0.05; ****P < 0.0001).
Although Mettl5 fs flies displayed normal activity, we found they were severely disoriented as they changed their walking directions more often compared to wild‐type flies (Fig 5B and C; Appendix Fig S8A). Both homozygous and trans‐heterozygous animals show similar behavior, confirming the specificity of the phenotypes toward Mettl5 loss of function. In contrast, Mettl5 ∆2AA flies behaved more similar to WT, although a mild angular deviation from the walking tracks could be observed. We conclude that Mettl5 is required for normal walking behavior in Drosophila and that its methyltransferase activity plays an important role in this process.
To exclude the possibility that Mettl5 fs flies are disorientated due to blindness, we re‐analyzed in depth our data from the Buridan's assay. If the flies were blind, they would be expected to have the same probability to stay in the area around the black stripes as to stay in the area 90° to the black stripes. Therefore, we split this circled platform into 48 imaginary areas and calculated the frequency with which the flies were in the eight areas around the black stripes compared to the frequency they were in the eight areas 90° to the black stripe. We observed that for all genotypes, the probability that the flies are around the black stripes was highly significantly increased (Appendix Fig S8B). Therefore, this higher propensity to be close to the black stripes demonstrates that the Mettl5 mutant flies indeed are not blind.
In human cells, METTL5 is mostly found in the nucleolus 33. In fly, Mettl5 is detected mostly in the cytoplasm, suggesting the existence of ribosome biogenesis specificities. It is possible that in flies, pre‐ribosome‐bound‐Mettl5 follows pre‐40S ribosomes to the cytoplasm, while in human cells METTL5 dissociates from pre‐40S early on in the nucleus. Small ribosomal RNA base modifications are typically generated late in rRNA processing, but cross‐species differences have been reported. For example, the dimethylation installed at the 3′‐end of 18S rRNA by DIM1 occurs in the cytoplasm in budding yeast, while it is deposited in the nucleus in human cells 39.
A brief comparison of the absence of phenotype of the METTL5 knock out in human cells and the behavioral phenotype reported here in fly illustrates the benefits of using complex models (animal) and sophisticated assays (behavioral) to approach the role of conserved rRNA modifications present at essential functional sites on the ribosome. Considering that the m6A modification deposited by Mettl5 on the 18S rRNA is right at the decoding site, in direct vicinity to the P‐site, makes it a likely possibility that the phenotypes observed in mutant flies are explained by differential translation; however, this will require additional work to be tested directly. In conclusion, we propose that the underlying mechanism for the METTL5‐associated human neurological disorder is loss of 18S rRNA m6A modification.
Materials and Methods
Drosophila stocks and genetics
Drosophila melanogaster Canton‐S with mutant alleles for CG9666 was generated using the CRISPR/Cas9 system, as described previously 36. Guide RNA sequences used were CTTCGCCGCACATAGCCGCGTGCA and AAACTGCACGCGGCTATGTGCGGC as well as CTTCTTCGGCACGAAACACAATGC and AAACGCATTGTGTTTCGTGCCGAAT. For the first allele (Mettl5 ∆2AA), a deletion of six base pairs (bp) from base pair 104 to 109 in the genome region chr3L (genome assembly BDGP release 6) containing CG9666 was produced. For the second allele (Mettl5 fs), a deletion of 13 bp from base pair 104 to 118 in the genome region chr3L and an insertion of 3 bp were generated. Trans‐heterozygous CG9666 fly mutants were produced by crossing Mettl5 fs flies with a deficiency fly line for CG9666 (BL8083‐CG9666def, Bloomington Drosophila Stock Center) to rule out possible off‐target effects by the CRISPR/Cas9 system.
Drosophila cell line
Drosophila S2R+ are embryonically derived cells obtained from the Drosophila Genomics Resource Center (DGRC; Flybase accession FBtc0000150). Mycoplasma contamination was not detected (verified by analyzing RNA sequencing data).
Cloning
The plasmids used for immunohistochemistry and immunoprecipitation assays in S2R+ cells were constructed by cloning the corresponding cDNA or gene region of Mettl5 and Trmt112 in the Gateway‐based vectors with N‐terminal 3XFLAG–6XMyc tag (pPFMW) for Mettl5 and N‐terminal 3XHA tag (pAHW) or C‐terminal eGFP tag (pAc5.1b) for Trmt112. Furthermore, for the immunofluorescence assay, the UAS promoter in pPFMW and the Actin promoter in pAc5.1b were replaced with the endogenous promoters of Mettl5 and Trmt112, respectively.
RNA isolation and mRNA purification
Total RNA from flies as well as S2R+ cells was isolated using TRIzol reagent (Invitrogen), and DNA was removed with DNase‐I treatment (NEB). Subsequently, poly(A)+ RNA was isolated by three rounds of purification with Dynabeads Oligo (dT)25 (Invitrogen). Following this, all samples were tested for their quality by capillary electrophoresis using the Bioanalyzer (Agilent).
RT–PCR
qRT–PCR was performed to gain insight into expression levels of CG9666 and CG12975 during development and to assess KD efficiency of fly putative methyltransferase genes (primer list in Table EV2). Staging experiment was performed as previously reported 40, and the RNA was isolated as described above. The RNA was subjected to reverse transcription using the M‐MLV Reverse Transcriptase Kit (Promega). Following cDNA synthesis, transcript levels were quantified in technical triplicates for each gene in each developmental stage via qPCR using SYBR Green PCR Master Mix (Thermo Fisher Scientific) and the ViiA 7 Real‐Time PCR System (Thermo Fisher Scientific). The qPCR primers TCGAAGGATATTGAGGTGGA and TGTCCAAAACTAAATGCAACTGA were used to measure CG9666 expression levels and CTCAGCACATACAACTTCTTGACC and CGCTCTCCACCACTTCCTTT to measure CG12975 expression levels.
Immunostaining
Transfection of the tagged constructs in S2R+ cells was performed using Effectene reagent (Qiagen), as described in Ref. 36, co‐transfecting both Mettl5 and Trmt112 constructs under the control of their endogenous promoters. Immunostaining of the cells was performed 72 h after transfection, as previously described 36. To this end, the cells were incubated with primary antibody (mouse anti‐Myc, Enzo 9E10, 1:1,000) in 0.2% Triton X‐100 PBS supplemented with 10% donkey serum) at 4°C overnight and secondary antibody (anti‐mouse AlexaFluor 568, 1:1,000 in 10% donkey serum in PBST) as well as DAPI (1:1,000) for 2 h at RT. Images were taken with Leica SP5 confocal microscope using 63× oil immersion objective.
Cell culture, RNAi and transfection
Drosophila S2R+ cells were grown in Schneider's medium (Gibco) supplemented with 10% FBS (Sigma) and 1% penicillin–streptomycin (Sigma). For RNA interference (RNAi) as well as transfection experiments, S2R+ cells were seeded at a density of 106 cells/ml in serum‐free and serum‐supplemented medium, respectively. For RNAi experiments, PCR templates for the dsRNA were prepared using the T7 megascript Kit (NEB; primer list in Table EV2). dsRNA against bacterial β‐galactosidase gene (lacZ) was used as a control for all RNA interference (RNAi) experiments. A 7.5 μg of dsRNA was added to 106 cells. After 6 h of cell starvation, serum‐supplemented medium was added to the cells. dsRNA treatment was repeated after 48 and 96 h, and cells were collected 24 h after the last treatment. The Effectene Transfection Reagent Kit (Qiagen) was used to transfect vector constructs together with the Actin‐Gal4 driver construct in all overexpression experiments following the manufacturer's protocol.
Co‐immunoprecipitation and Western blot analysis
For the co‐immunoprecipitation assay, HA‐tagged or eGFP‐tagged Trmt112 and FLAG‐Myc‐tagged Mettl5 constructs were co‐transfected in S2R+ cells, as described above. As controls for GFP co‐IP, empty vectors were used. Seventy‐two hours after transfection, cells were harvested, washed in DPBS, and pelleted by centrifugation at 500 g for 5 min. The pellets were lysed in 1 ml of lysis buffer (50 mM Tris–HCl at pH 7.4, 150 mM NaCl, 0.05% NP‐40) supplemented with protease inhibitors and rotated head over tail for 30 min at 4°C. Following five cycles of sonication of 30 s on and 30 s off at the low power setting, the lysates were centrifuged at 20,000 g for 10 min at 4°C to remove the remaining cell debris. Protein concentrations were determined using Bradford reagent (Bio‐Rad). For the immunoprecipitation, 2 mg of proteins was incubated with 15 μl of pre‐washed Protein G beads (Thermo Fischer Scientific) for 1 h at 4°C as a pre‐clearing step, to deplete for proteins enrichment based on nonspecific binding to the beads. The lysate with the unbound protein fraction was then added to 15 μl of Protein G beads, which were previously incubated at room temperature for 30 min with 8 μg of either of the following antibodies: mouse anti‐HA (clone 12CA5, produced in‐house), normal mouse IgG (Santa Cruz Biotechnology, sc‐2025), and mouse anti‐FLAG (Sigma‐Aldrich, M2‐F1804) as a pre‐coating step to saturate the beads with the antibody. Lysates and pre‐coated beads were incubated with head over tail rotation for 2 h at 4°C. The beads were then washed three times with washing buffer (50 mM Tris–HCl at pH 7.4, 150 mM NaCl) and incubated at 70°C for 10 min in 1× NuPAGE LDS buffer (Thermo Fisher) supplemented with 100 mM DTT for denaturation and elution of the immunoprecipitated proteins. Inputs were subjected to the same treatment to denature the proteins prior to Western blot analysis. For Western blot analysis, proteins were separated on a 12% SDS–PAGE gel and transferred to a PVDF membrane (Bio‐Rad). After blocking with 5% milk in PBS with 0.05% Tween (PBST) for 1 h at room temperature, the membrane was incubated with primary antibody in blocking solution overnight at 4°C. The primary antibodies used were mouse anti‐Myc (1:2,000; Enzo, 9E10), mouse anti‐FLAG (1:2,000, Sigma, M2), mouse anti‐HA (1:2,000, in‐house, 12CA5), and mouse anti‐GFP (1:200, Santacruz, B‐2). The membrane was washed three times in PBST for 15 min prior incubation for 1 h at room temperature with anti‐mouse secondary antibody (Jackson ImmunoResearch) in 5% milk. Protein bands were detected using SuperSignalWest Pico chemiluminescent substrate (Thermo Scientific).
IP, Dimethyl labeling of the samples and proteomic analysis
For proteomic analysis of Mettl5 interactors, the immunoprecipitation was performed using two biological replicates transfected with FLAG‐Myc empty plasmid as control and two biological replicates transfected with Mettl5 FLAG‐Myc construct, for forward and reverse experiment. Seventy‐two hours after transfection, cells were harvested, washed in DPBS, and pelleted by centrifugation at 500 g for 5 min. The pellets were lysed in 1 ml of lysis buffer (50 mM Tris–HCl at pH 7.4, 150 mM NaCl, 0.05% NP‐40) supplemented with protease inhibitors and rotated head over tail for 30 min at 4°C. Following five cycles of sonication of 30 s on and 30 s off at the low power setting, the lysates were centrifuged at 20,000 g for 10 min at 4°C to remove the remaining cell debris. Protein concentrations were determined using Bradford reagent (Bio‐Rad). The antibody used for IP was anti‐Myc coupled with magnetic beads (Pierce, 9E10). A 2 mg of proteins was incubated with 15 μl of pre‐washed beads for 2 h at 4°C. The beads were washed three times for 10 min with lysis buffer, and immunoprecipitated proteins were eluted at 70°C for 10 min in 1× NuPAGE LDS buffer (Thermo Fisher) supplemented with 100 mM DTT.
Protein lysates were firstly subjected to tryptic digestion, as previously described 41. Subsequently, the peptides from the four samples were mixed with either formaldehyde‐H2 (4% in water, 1 μl) or formaldehyde‐D2 (4% in water, 1 μl) and vortexed. Freshly prepared sodium cyanoborohydride (260 mM, 1 μl) was added. The mixture was vortexed again and then let stand for 5 min, for the dimethyl labeling reaction to occur, as described earlier (Hsu JL et al, 2003). For the forward experiment, the control sample was labeled with formaldehyde‐H2 and the lysate from Mettl5 immunoprecipitation with formaldehyde‐D2. The reverse experiment was performed vice versa. The samples were then subjected to mass spectrometry measurements as described previously 42. Raw files were processed with MaxQuant (version 1.5.2.8, Cox and Mann, 2008) and searched against the UniProt database of annotated Drosophila proteins (D. melanogaster: 41,850 entries, downloaded January 8, 2015).
LC‐MS/MS analysis of m6A levels
Ribonucleoside (rA, N6 mA) standards, ammonium acetate, and LC/MS grade acetonitrile were purchased from Sigma‐Aldrich. 13C9‐A was purchased from Silantes, GmbH (Munich, Germany). 2H3‐N6 mA was obtained from TRC, Inc (Toronto, Canada). All solutions were prepared using Millipore quality water (Barnstead GenPure xCAD Plus, Thermo Scientific). A 0.1–1 μg of RNA was degraded to nucleosides with 0.003 U nuclease P1 (Roche), 0.01 U snake venom phosphodiesterase (Worthington), and 0.1 U alkaline phosphatase (Fermentas). Separation of the nucleosides from the digested RNA samples was performed with an Agilent 1290 UHPLC system equipped with RRHD Eclipse Plus C18 (95Å, 2.1 × 50 mm, 1.8 μm, Zorbax, USA) with a gradient of 5 mM ammonium acetate (pH 7, solvent A) and acetonitrile (solvent B). Separations started at a flow rate of 0.4 ml/min and linearly increased to 0.5 ml/ml during first 7 min. Then, washing and re‐conditioning was done at 0.5 ml/min for an additional 3 min and linearly decrease to 0.4 ml/min during the last minute. The gradients were as follows: solvent B linear increase from 0 to 7% for first 3 min, followed by isocratic elution at 7% solvent B for another 4 min; then switching to 0% solvent B for last 4 min, to recondition the column. Quantitative MS/MS analysis was performed with an Agilent 6490 triple quadrupole mass spectrometer in positive ion mode. The details of the method and instrument settings are described elsewhere 43. MRM transitions used in this study were 269.2→137.2 (rA), 278.2→171.2 (13C9‐rA), 282.1→150.1 (N6mrA), and 285.1→153.1 (2H3‐N6mrA). The quantification of all samples utilized biological triplicates and the average values with one s.d. are shown.
Phylogenetic analysis
Orthologs of CG9666 were searched for representative species at increasing taxonomic distances from Homo sapiens to eukarya with the assistance of the Protein Path Tracker online tool (PPT; 44). Identifiers and species names are the following: Q8K1A0, Mus musculus; Q9NRN9, H. sapiens; F1NZ60, Gallus gallus; F1QVR8, Danio rerio; F6R9A4, Ornithorhynchus anatinus; F6WB92, Ciona intestinalis; F7AIW5, Xenopus tropicalis; H2P7S0, Pongo abelli; H3B2F1, Latimeria chalumnae; Q84TF1, A. thaliana; Q8MSW4, D. melanogaster; and W4YQG0, Strongylocentrotus purpuratus. Multiple sequence alignment of the sequences collected was obtained with MUSCLE as implemented at EBI 45. ClustalW 46 was used to represent the alignment and to produce a phylogenetic tree.
Modeling the Drosophila melanogaster Mettl5 and Trmt112 sequences into the human 3‐D structure of the METTL5‐TRMT112 complex
The METTL5 and TRMT112 proteins from human and D. melanogaster share 59 and 50% sequence identity, respectively. To generate a model of the D. melanogaster Mettl5‐Trmt112 complex, we processed the co‐ordinates of the crystal structure of the human METTL5‐TRMT112 complex 36 together with sequence alignments between human and fruit fly proteins using the ROBETTA server 47. The resulting model exhibits a RMSD value of 0.4 Å over 250 Cα atoms compared to the structure of the human complex. This is typically what is seen when comparing the crystal structures of two proteins sharing between 50 to 60% sequence identity.
Buridan's behavioral paradigm analysis in Drosophila
Behavioral tests were performed on 5‐day‐old flies using the Canton‐S strain as wild‐type control. Wings were cut under cold anesthesia to one‐third of their length on the evening before the experiment. Activity and orientation behavior were analyzed using Buridan's paradigm as described before 48. All statistical groups were tested for normal distribution with the Shapiro–Wilk test. t‐Test analysis of variance with Bonferroni correction was used to compare different conditions. N = 30 for all genotypes. The sample size was chosen based on a previous study 49, and its power was validated with result analysis. Blinding was applied during the experiment.
Pre‐rRNA processing analysis
A 5 μg total RNA extracted from 10 flies was separated on 1.2% denaturing agarose gels and processed for Northern blotting analysis with specific probes, as described in Ref. 50. The probes used are as follows: LD4533 (ITS1) and LD4534 (ITS2). Mature rRNAs were visualized by ethidium bromide staining of the gels. The ratio of mature rRNAs was established by densitometry on a ChemiDoc MP (Bio‐Rad).
Ribosomal RNA modification analysis
18S rRNA was purified on 10–30% sucrose gradient (NaCl 300 mM, Tris–HCl pH8.0 50 mM, MgCl2 2 mM, EGTA 1 mM, Triton X‐100 1%, sodium deoxycholate 0.1%), digested to nucleosides with 2 U P1 nuclease (Sigma N8630) and 10 μl alkaline phosphatase (Sigma P4252‐100U), and analyzed by HPLC as described in 51.
Statistics
In Buridan's paradigm, Shapiro–Wilk's test was used to test for normal distribution in each group. Homogeneity of variances was tested using Levene's test. Normally distributed groups with homogeneous variances were tested by Student's t‐test. Due to multiple comparison, Bonferroni correction was applied.
m6A measurements were taken from three biological replicates, and the variance between the groups that are being statistically compared is similar; therefore, the two‐tailed unpaired Student's t‐test was applied.
Author contributions
JL, MS, DLJL, and J‐YR conceived the study. JL, MS, LW, MP, MAV, MM, MG, CN, DLJL, and J‐YR performed the methodology. PM and MAA‐N performed the phylogenetic analysis. JL and MS wrote the draft of the manuscript. All authors reviewed and edited the manuscript. DLJL and J‐YR supervised the study.
Conflict of interest
The authors declare that they have conflict of interest.
Supporting information
Appendix
Table EV1
Table EV2
Review Process file
Acknowledgements
We thank the Bloomington Drosophila Stock Center and the Zurich ORFeome Project (FlyORF) for fly reagents; the Drosophila Genomics Resource Center at Indiana University for plasmids and cell line; Roland Strauss and his group for the help with Buridan's paradigm; members of the Roignant and Lafontaine laboratories for helpful discussion; the Genomics and Bioinformatics IMB core facilities for great support. Support by IMB Proteomics core facility is gratefully acknowledged (instrument is funded by DFG INST 247/766‐1 FUGG). In particular, we wish to thank Anja Freiwald from IMB Proteomics core facility for sample preparation and Dr. Mario Dejung from Proteomics core facility for data processing. Research in the laboratory of J.‐Y.R. is supported by the Deutsch‐Israelische Projektkooperation (DIP) RO 4681/6‐1 and the Epitran COST action (CA16120). Research in the laboratory of D.L.J.L. is supported by the Belgian Fonds de la Recherche Scientifique (F.R.S./FNRS), the Université Libre de Bruxelles (ULB), the Région Wallonne (DGO6) [grant RIBOcancer no. 1810070], the Fonds Jean Brachet, and the International Brachet Stiftung. Research in the laboratory of M.G. is supported by the Agence Nationale pour la Recherche [grant number ANR‐16‐CE11‐0003], the CNRS, Ecole Polytechnique, and the CNRS PICS program [grand number PICS07484].
EMBO Reports (2020) 21: e49443
Contributor Information
Denis LJ Lafontaine, Email: denis.lafontaine@ulb.ac.be.
Jean‐Yves Roignant, Email: jean-yves.roignant@unil.ch.
References
- 1. Desrosiers R, Friderici K, Rottman F (1974) Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA 71: 3971–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perry R, Kelley D (1974) Existence of methylated messenger RNA in mouse L cells. Cell 1: 37–42 [Google Scholar]
- 3. Dominissini D, Moshitch‐Moshkovitz S, Schwartz S, Salmon‐Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob‐Hirsch J, Amariglio N, Kupiec M et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A‐seq. Nature 485: 201–206 [DOI] [PubMed] [Google Scholar]
- 4. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149: 1635–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi H, Wei J, He C (2019) Where, when, and how: context‐dependent functions of RNA methylation writers, readers, and erasers. Mol Cell 74: 640–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bujnicki JM, Feder M, Radlinska M, Blumenthal RM (2002) Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT‐A70 subunit of the human mRNA:m(6)A methyltransferase. J Mol Evol 55: 431–444 [DOI] [PubMed] [Google Scholar]
- 7. Sledz P, Jinek M (2016) Structural insights into the molecular mechanism of the m(6)A writer complex. eLife 5: e18434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang P, Doxtader KA, Nam Y (2016) Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell 63: 306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C et al (2016) Structural basis of N(6)‐adenosine methylation by the METTL3‐METTL14 complex. Nature 534: 575–578 [DOI] [PubMed] [Google Scholar]
- 10. Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H et al (2018) VIRMA mediates preferential m(6)A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov 4: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR (2016) m6A RNA methylation promotes XIST‐mediated transcriptional repression. Nature 537: 369–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo J, Tang HW, Li J, Perrimon N, Yan D (2018) Xio is a component of the Drosophila sex determination pathway and RNA N(6)‐methyladenosine methyltransferase complex. Proc Natl Acad Sci USA 115: 3674–3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, Masiello I, Hares T, Villasenor R, Hess D et al (2018) Zc3 h13/Flacc is required for adenosine methylation by bridging the mRNA‐binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev 32: 415–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wen J, Lv R, Ma H, Shen H, He C, Wang J, Jiao F, Liu H, Yang P, Tan L et al (2018) Zc3 h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self‐renewal. Mol Cell 69: 1028–1038.e1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruzicka K, Zhang M, Campilho A, Bodi Z, Kashif M, Saleh M, Eeckhout D, El‐Showk S, Li H, Zhong S et al (2017) Identification of factors required for m(6) A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. N Phytol 215: 157–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Balacco DL, Soller M (2019) The m(6)A writer: rise of a machine for growing tasks. Biochemistry 58: 363–378 [DOI] [PubMed] [Google Scholar]
- 17. Lence T, Soller M, Roignant JY (2017) A fly view on the roles and mechanisms of the m6A mRNA modification and its players. RNA Biol 14: 1232–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lence T, Paolantoni C, Worpenberg L, Roignant JY (2019) Mechanistic insights into m(6)A RNA enzymes. Biochim Biophys Acta Gene Regul Mech 1862: 222–229 [DOI] [PubMed] [Google Scholar]
- 19. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, Conrad NK (2017) The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 169: 824–835.e814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, Kumagai S, Ochiai K, Suzuki T, Igarashi K (2017) S‐adenosylmethionine synthesis is regulated by selective N(6)‐adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep 21: 3354–3363 [DOI] [PubMed] [Google Scholar]
- 21. Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Hobartner C, Sloan KE, Bohnsack MT (2017) Human METTL16 is a N(6)‐methyladenosine (m(6)A) methyltransferase that targets pre‐mRNAs and various non‐coding RNAs. EMBO Rep 18: 2004–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mendel M, Chen KM, Homolka D, Gos P, Pandey RR, McCarthy AA, Pillai RS (2018) Methylation of structured RNA by the m(6)A writer METTL16 is essential for mouse embryonic development. Mol Cell 71: 986–1000.e1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sloan KE, Warda AS, Sharma S, Entian KD, Lafontaine DLJ, Bohnsack MT (2017) Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol 14: 1138–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharma S, Lafontaine DLJ (2015) ‘View from a bridge’: a new perspective on eukaryotic rRNA base modification. Trends Biochem Sci 40: 560–575 [DOI] [PubMed] [Google Scholar]
- 25. Piekna‐Przybylska D, Decatur WA, Fournier MJ (2008) The 3D rRNA modification maps database: with interactive tools for ribosome analysis. Nucleic Acids Res 36: D178–D183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, Chen K, Lu Z, Chen H, Shi YG et al (2019) N(6‐)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol 15: 88–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lafontaine DL, Preiss T, Tollervey D (1998) Yeast 18S rRNA dimethylase Dim1p: a quality control mechanism in ribosome synthesis? Mol Cell Biol 18: 2360–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu PC, Thiele DJ (2001) Novel stress‐responsive genes EMG1 and NOP14 encode conserved, interacting proteins required for 40S ribosome biogenesis. Mol Biol Cell 12: 3644–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eschrich D, Buchhaupt M, Kotter P, Entian KD (2002) Nep1p (Emg1p), a novel protein conserved in eukaryotes and archaea, is involved in ribosome biogenesis. Curr Genet 40: 326–338 [DOI] [PubMed] [Google Scholar]
- 30. Schosserer M, Minois N, Angerer TB, Amring M, Dellago H, Harreither E, Calle‐Perez A, Pircher A, Gerstl MP, Pfeifenberger S et al (2015) Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat Commun 6: 6158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bourgeois G, Letoquart J, van Tran N, Graille M (2017) Trm112, a protein activator of methyltransferases modifying actors of the eukaryotic translational apparatus. Biomolecules 7: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sharm S, Marchand V, Motorin Y, Lafontaine DLJ (2017) Identification of sites of 2′‐O‐methylation vulnerability in human ribosomal RNAs by systematic mapping. Sci Rep 7: 11490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Erales J, Marchand V, Panthu B, Gillot S, Belin S, Ghayad SE, Garcia M, Laforets F, Marcel V, Baudin‐Baillieu A et al (2017) Evidence for rRNA 2′‐O‐methylation plasticity: Control of intrinsic translational capabilities of human ribosomes. Proc Natl Acad Sci USA 114: 12934–12939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krogh N, Jansson MD, Hafner SJ, Tehler D, Birkedal U, Christensen‐Dalsgaard M, Lund AH, Nielsen H (2016) Profiling of 2′‐O‐Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res 44: 7884–7895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gehrke CW, Kuo KC (1990) Ribonucleoside analysis by reversed‐phase high performance liquid chromatography. J Chromatogr 471: 3–36 [DOI] [PubMed] [Google Scholar]
- 36. van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, Bohnsack KE, Bohnsack MT, Jaffrey SR, Graille M et al (2019) The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res 47: 7719–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Colomb J, Reiter L, Blaszkiewicz J, Wessnitzer J, Brembs B (2012) Open source tracking and analysis of adult Drosophila locomotion in Buridan's paradigm with and without visual targets. PLoS ONE 7: e42247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Richard EM, Polla DL, Assir MZ, Contreras M, Shahzad M, Khan AA, Razzaq A, Akram J, Tarar MN, Blanpied TA et al (2019) Bi‐allelic variants in METTL5 cause autosomal‐recessive intellectual disability and microcephaly. Am J Hum Genet 105: 869–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zorbas C, Nicolas E, Wacheul L, Huvelle E, Heurgue‐Hamard V, Lafontaine DL (2015) The human 18S rRNA base methyltransferases DIMT1L and WBSCR22‐TRMT112 but not rRNA modification are required for ribosome biogenesis. Mol Biol Cell 26: 2080–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, Kreim N, Andrade‐Navarro MA, Poeck B, Helm M et al (2016) m6A modulates neuronal functions and sex determination in Drosophila . Nature 540: 242–247 [DOI] [PubMed] [Google Scholar]
- 41. Hsu JL, Huang SY, Chow NH, Chen SH (2003) Stable‐isotope dimethyl labeling for quantitative proteomics. Anal Chem 75: 6843–6852 [DOI] [PubMed] [Google Scholar]
- 42. Bluhm A, Casas‐Vila N, Scheibe M, Butter F (2016) Reader interactome of epigenetic histone marks in birds. Proteomics 16: 427–436 [DOI] [PubMed] [Google Scholar]
- 43. Schomacher L, Han D, Musheev MU, Arab K, Kienhofer S, von Seggern A, Niehrs C (2016) Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat Struct Mol Biol 23: 116–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mier P, Perez‐Pulido AJ, Andrade‐Navarro MA (2018) Automated selection of homologs to track the evolutionary history of proteins. BMC Bioinformatics 19: 431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R et al (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- 47. Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, Baker D (2013) High‐resolution comparative modeling with RosettaCM. Structure 21: 1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Strauss R, Hanesch U, Kinkelin M, Wolf R, Heisenberg M (1992) No‐bridge of Drosophila melanogaster: portrait of a structural brain mutant of the central complex. J Neurogenet 8: 125–155 [DOI] [PubMed] [Google Scholar]
- 49. Poeck B, Triphan T, Neuser K, Strauss R (2008) Locomotor control by the central complex in Drosophila‐an analysis of the tay bridge mutant. Dev Neurobiol 68: 1046–1058 [DOI] [PubMed] [Google Scholar]
- 50. Tafforeau L, Zorbas C, Langhendries JL, Mullineux ST, Stamatopoulou V, Mullier R, Wacheul L, Lafontaine DL (2013) The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre‐rRNA processing factors. Mol Cell 51: 539–551 [DOI] [PubMed] [Google Scholar]
- 51. Sharma S, Langhendries JL, Watzinger P, Kotter P, Entian KD, Lafontaine DL (2015) Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res 43: 2242–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Table EV2
Review Process file