

Abstract
CARD14 gain‐of‐function mutations cause psoriasis in humans and mice. Together with BCL10 and the protease MALT1, mutant CARD14 forms a signaling node that mediates increased NF‐κB signaling and proinflammatory gene expression in keratinocytes. However, it remains unclear whether psoriasis in response to CARD14 hyperactivation is keratinocyte‐intrinsic or requires CARD14 signaling in other cells. Moreover, the in vivo effect of MALT1 targeting on mutant CARD14‐induced psoriasis has not yet been documented. Here, we show that inducible keratinocyte‐specific expression of CARD14E138A in mice rapidly induces epidermal thickening and inflammation as well as increased expression of several genes associated with psoriasis in humans. Keratinocyte‐specific MALT1 deletion as well as oral treatment of mice with a specific MALT1 protease inhibitor strongly reduces psoriatic skin disease in CARD14E138A mice. Together, these data illustrate a keratinocyte‐intrinsic causal role of enhanced CARD14/MALT1 signaling in the pathogenesis of psoriasis and show the potential of MALT1 inhibition for the treatment of psoriasis.
Keywords: cytokines, inflammation, MALT1, psoriasis, therapeutic
Subject Categories: Immunology, Molecular Biology of Disease, Skin
Expression of a pathogenic human CARD14 mutant transgene in keratinocytes is sufficient to drive psoriasis‐like dermatitis in mice, which can be inhibited by treatment with a MALT1 protease inhibitor.
Introduction
Psoriasis is a skin disease with both autoinflammatory and autoimmune components that is characterized by red, scaly, and sharply demarcated plaques which appear in a relapsing, remitting pattern. It is a common disease which affects 2–3% of the world population. Furthermore, psoriasis is often linked with comorbidities such as psoriatic arthritis and imposes a heavy psychological burden 1, 2. Current therapies for psoriasis mainly consist of immunosuppressive small molecules such as cyclosporine and methotrexate, or biologics that target key cytokines such as IL‐17a, IL‐23, and TNF. Even though the treatment options have increased over the past few years, they are still not effective for all patients, can be associated with unwanted side effects, and have a high production cost, demonstrating the need for alternative treatments 3.
According to the current view on the pathogenesis of psoriasis, the disease is mainly maintained by a pathogenic interplay between keratinocytes and immune cells such as dendritic cells, T cells, and neutrophils. Activation of dendritic cells in the skin results in IL‐12 and IL‐23 production, leading to recruitment and activation of Th17 cells, which produce inflammatory mediators such as IL‐17a and TNF. These stimulate keratinocyte hyperproliferation and secretion of cytokines and chemokines, leading to further recruitment of immune cells and thus resulting in a chronic inflammatory loop 2. However, the factors that kindle these processes are still not entirely clear.
Genetic effects play a substantial part in the etiology of psoriasis. This is illustrated by the strong association of several single nucleotide polymorphisms and psoriasis susceptibility loci (PSORS) with psoriasis 4. Caspase recruitment domain‐containing protein 14 (CARD14) is a gene that is located in the PSORS2 locus and for which several gain‐of‐function mutations have been associated with psoriasis and pityriasis rubra pilaris in humans 5, 6, 7, 8. Transgenic expression of psoriasis‐associated mutants of CARD14 leads to excessive proinflammatory signaling in keratinocytes in vitro 9, 10, and heterozygous mice harboring mutant CARD14ΔE138, CARD14E138A, or CARD14ΔQ136 were recently shown to spontaneously develop a psoriatic phenotype 11, 12. CARD14, also known as CARMA2, is closely related to CARD9, CARD10 (CARMA3), and CARD11 (CARMA1) 13, which upon activation, each form an oligomeric CBM signaling complex with B‐cell lymphoma 10 (BCL10) and mucosa‐associated lymphoid tissue lymphoma translocation protein 1 (MALT1). In this CBM complex, MALT1 acts as a scaffold for other proteins that mediate NF‐κB and JNK/p38 MAP kinase signaling 9, 10. CBM complex formation also activates MALT1 protease activity, resulting in the cleavage of a limited number of substrates whose cleavage is believed to further fine‐tune inflammatory signaling (reviewed in refs. 14 and 15). The biological relevance of MALT1 protease activity is highlighted by the autoimmune phenotype of mice expressing catalytically inactive MALT1 (reviewed in ref. 16). On the other hand, inhibition of MALT1 suppresses T‐ and B‐cell activation and prevents disease development in preclinical models of autoimmune disease and oncogenic MALT1‐driven lymphoma, illustrating the therapeutic potential of MALT1 inhibition 17, 18, 19, 20, 21, 22.
We have previously demonstrated that inhibition of MALT1 proteolytic activity reduces CARD14E138A‐induced secretion of inflammatory cytokines in primary human keratinocytes 9. To further investigate whether targeting MALT1 might be a valid strategy in the treatment of psoriasis, we have created transgenic mice that inducibly express a human psoriasis‐associated hyperactive CARD14E138A mutant specifically in keratinocytes 7 and tested the effect of genetic or pharmacological inhibition of MALT1. We show here that inducible expression of CARD14E138A in keratinocytes is sufficient to cause psoriasis‐like skin inflammation in mice, illustrating a keratinocyte‐intrinsic effect of CARD14E138A. Using this elegant novel mouse model, we demonstrate that genetic deletion of MALT1 as well as oral treatment with a small compound MALT1 inhibitor suppresses CARD14E138A‐induced psoriasiform inflammation, highlighting the therapeutic potential of MALT1 targeting for the treatment of psoriasis.
Results and Discussion
Keratinocyte‐specific deletion of MALT1 reduces CARD14E138A‐induced psoriasiform inflammation
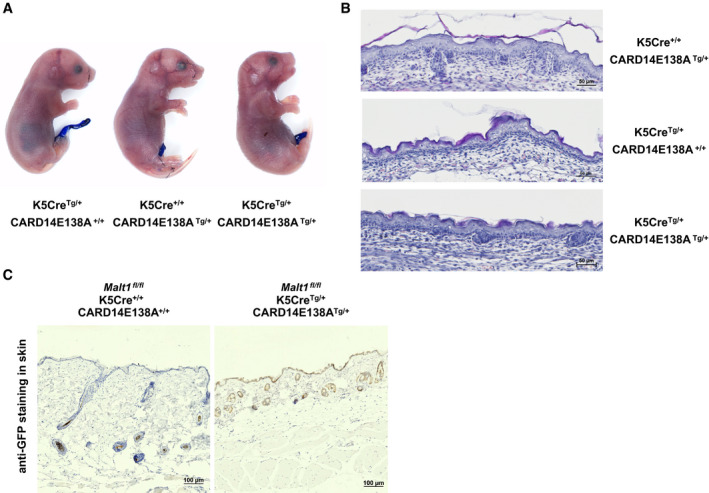
Several CARD14 gain‐of‐function mutations have been identified in psoriasis patients, and mice with heterozygous expression of some of these mutants (CARD14ΔE138, CARD14E138A, or CARD14ΔQ136) were recently shown to spontaneously develop a psoriatic phenotype 11, 12. To further investigate the physiological role of MALT1 in CARD14‐induced psoriatic disease, we analyzed the effect of MALT1 deficiency on the development of disease symptoms induced by keratinocyte‐specific expression of the human CARD14 E138A transgene in mice. The CARD14 E138A mutation was chosen because it was identified in a patient with pustular psoriasis and displays potent activation of NF‐κB 7, 9. It was previously reported that mice carrying a similar mutation in the endogenous Card14 gene show increased mortality 11, 12. Although the reasons for this are still unclear, it is not unlikely that mutant CARD14 expression in different cell types or tissues besides skin keratinocytes, such as endothelial cells, γδ T cells, Langerhans cells, esophagus, and colon, contributes to the observed lethality 5, 23, 24. We therefore decided to generate conditional CARD14E138A transgenic mice by knocking in the human CARD14 E138A cDNA preceded by a loxP‐flanked stop cassette under control of the ROSA26 promoter using RMCE‐mediated recombination 25 (Fig 1A), which were subsequently crossed with male mice expressing Cre recombinase under the keratin 5 (K5) promoter in order to obtain epidermal CARD14E138A expression in the skin 26. Further crossing of these mice into mice containing a floxed (fl) Malt1 allele was done to obtain MALT1 deficiency in epidermal cells. We did not observe a normal mendelian segregation pattern in MALT1 sufficient conditions because no mice expressing both the K5cre transgene and CARD14 E138A transgene were obtained (Fig 1B) (P < 0.0001, chi‐square test). However, upon caesarian section, pups at E18.5 that express the CARD14E138A transgene in skin tissue could be recovered (Fig 1C). Even though these pups did not show any overt phenotype and were indistinguishable compared to wild‐type littermates (Fig 1D), they failed to survive longer than 24 h after birth. Toluidine blue staining showed that the epidermal skin barrier of CARD14E138A transgenic mice is intact and histological sections of the skin did not show clear abnormalities (Fig EV1A and B). Importantly, mice expressing epidermal CARD14E138A in the absence of MALT1 were born at expected mendelian segregation of the genotypes (P = 0.9824, chi‐square test; Fig 1E) and did not show macroscopically visible signs of disease. Body weight and ear thickness of these mice were comparable to littermate controls, and histological analysis of ear and skin sections did not indicate epidermal thickening or inflammation in mice between 4 and 7 months old (Fig 1F–H). Immunohistochemistry staining for GFP confirmed expression of the bicistronic CARD14E138A/GFP transgene in the epidermis of the ears (Fig EV1C). Together, these data show that constitutive K5cre‐driven CARD14E138A expression results in perinatal lethality and highlight an important in vivo role for MALT1 in CARD14 signaling. The quick onset of death of K5cre‐CARD14E138A expressing pups is quite surprising and is unlikely to be attributed to CARD14E138 expression in the skin only (see also below).
Figure 1. Perinatal lethality of CARD14E138A transgenic mice is rescued by MALT1 deficiency.

- Schematic representation of the Rosa26LSL‐CARD14‐E138A transgene construct (LSL = LoxP‐stop‐LoxP).
- Ratio of genotypes obtained of 1‐week‐old pups after crossing K5creTg/+ CARD14E138A+/+ male mice with K5cre+/+ CARD14E138ATg/+ female mice; CARD14E138A+/+ = Rosa26+/+, CARD14E138ATg/+ = Rosa26LSL‐CARD14‐E138A/+
- Representative Western blot showing CARD14 protein levels in skin and liver lysates of E18.5 pups. Actin is shown as a loading control.
- Representative images of K5creTg/+ CARD14E138A+/+ and K5creTg/+ CARD14E138ATg/+ pups at E18.5.
- Ratio of genotypes obtained of 1‐week‐old pups from crossing Malt1 fl/fl K5creTg/+ CARD1414E138A+/+ male mice with Malt1 fl/fl K5cre+/+ CARD14E138ATg/+ female mice.
- Representative image of 6‐month‐old Malt1 fl/fl K5creTg/+ CARD14E138A+/+ and Malt1 fl/fl K5creTg/+ CARD14E138ATg/+ mice.
- Body weight and ear thickness of untreated WT and Malt1 fl/fl K5creTg/+ CARD14E138ATg/+ male and female mice (between 4 and 7 months old). Each symbol represents one mouse; the line represents the mean value (n ≥ 4 mice per group). WT = Malt1 fl/fl K5cre+/+ CARD14E138A+/+, Malt1 fl/fl K5cre+/+ CARD14E138ATg/+, and Malt1 fl/fl K5creTg/+ CARD14E138A+/+ mice. Statistical difference between two groups was determined using a Mann–Whitney U‐test (ns: P > 0.05)
- Representative H&E‐stained histological sections of ear and skin tissue of Malt1 fl/fl K5cre+/+ CARD14E138A+/+ and Malt1 fl/fl K5creTg/+ CARD14E138ATg/+ mice (scale bar represents 100 μm).
Source data are available online for this figure.
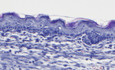
Figure EV1. Effect of constitutive skin epidermal CARD14E138A expression on skin barrier function and structure.
- Toluidine blue staining of K5creTg/+ CARD14E138A+/+, K5cre+/+ CARD14E138ATg/+, and K5creTg/+ CARD14E138ATg/+ E18.5 pups.
- Histological sections of skin tissue of K5cre+/+ CARD14E138ATg/+, K5creTg/+ CARD14E138A+/+, and K5creTg/+ CARD14E138ATg/+ pups at E18.5 stained with hematoxylin and eosin (scale bar represents 50 μm). CARD14E138A+/+ = Rosa26+/+, CARD14E138ATg/+ = Rosa26LSL‐CARD14‐E138A/+.
- Immunohistochemical staining of GFP in sections of skin tissue from Malt1 fl/fl K5cre+/+ CARD14E138A+/+ and Malt1 fl/fl K5creTg/+ CARD14E138ATg/+ mice of 5 months old (scale bar represents 100 μm).
Because the lethality induced by constitutive epidermal expression of CARD14E138A prevented us from analyzing the effect of MALT1 targeting on mutant CARD14‐induced psoriasis, we crossed CARD14 E138A transgenic mice with mice containing a K14creER transgene, allowing the tamoxifen‐inducible expression of CARD14E138A in stratified epithelia as found in skin and certain other tissues such as tongue and esophagus 27, 28. To account for possibly unanticipated effects of Cre expression, mice that contain the K14creER transgene but not the CARD14E138A transgene were used as wild‐type controls (Fig 2A ). Mice with both transgenes (hereafter referred to as inducible epidermal CARD14E138A (ieCARD14E138A)) were born at normal mendelian ratios and did not show any sign of disease up to 6 weeks of age. From 6 weeks on, some mild but significant swelling of the ears was observed in ieCARD14E138A mice in the absence of tamoxifen, which might be caused by leaky expression of the CARD14E138A transgene due to marginal spontaneous K14creER activity. Otherwise, the mice were healthy and did not show differences in body weight (Fig EV2A). To investigate the effect of MALT1 deficiency, we also generated ieCARD14E138A mice with floxed Malt1 alleles, which allow CARD14E138A induction together with MALT1 deletion in epidermal cells upon tamoxifen treatment (hereafter referred to as Malt1 EKO ieCARD14E138A mice). Similar to Malt1 +/+ ieCARD14E138A mice, Malt1 EKO ieCARD14E138A mice showed mild ear thickening before treatment with tamoxifen (Fig EV2B). It should be mentioned that complete Cre‐mediated MALT1 deletion requires two recombination events, while only a single recombination is needed for CARD14E138A transgene expression. Therefore, the minor leaky K14creER activity may not be sufficient to induce complete MALT1 deletion in the absence of tamoxifen. In addition, due to MALT1 protein stability, MALT1 protein expression levels do not diminish as fast as CARD14E138A protein expression levels rise. Tamoxifen treatment of Malt1 +/+ ieCARD14E138A mice increased ear swelling progressively (twofold), and the ears became red and scaly (Figs 2B and EV2C). Furthermore, ieCARD14E138A mice showed a rapid decrease in body weight, up to 18 percent, 4 days after tamoxifen administration, at which point mice were euthanized to minimize suffering (Fig 2B). It should be mentioned that no clear macroscopical signs of psoriatic disease could be observed at other skin regions 4 days after tamoxifen treatment.
Figure 2. Keratinocyte‐specific deletion of MALT1 reduces CARD14E138A‐induced psoriasiform inflammation.

- Schematic representation of the experimental induction of CARD14E138A transgene using tamoxifen. WT = K14creERTg/+ Rosa26+/+, ieCARD14E138A = K14creERTg/+ Rosa26LSL‐CARD14‐E138A/+
- Changes in relative body weight and ear thickness upon CARD14E138A induction with tamoxifen. Malt1 EKO = Malt1 fl/fl. The combined results of five independent experiments are shown (Malt1 +/+ WT n = 17, Malt1 fl/fl WT n = 8, Malt1 +/+ ieCARD14E138A n = 18, Malt1 fl/fl ieCARD14E138A n = 12).
- Representative H&E‐stained histological sections of ear tissue of tamoxifen‐treated mice (scale bar represents 200 μm). Arrowheads indicate infiltrates of immune cells.
- Epidermal thickness measured on ear sections. Each symbol represents the mean of at least ten measurements for each ear; the line represents the mean value. The combined results of three independent experiments are shown.
- Analysis of infiltrating immune cells in the ears of tamoxifen‐treated mice using flow cytometry. Cell count of neutrophils (CD45+ CD3/CD19− CD64− CD11b+ Ly6G+), eosinophils (CD45+ CD3/CD19− CD64− Ly6G− SiglecF+), T cells (CD45+ CD3/CD19+, MHCII−), and DCs (CD45+ CD3/CD19− CD64− MHCII+CD11c+) in single cell suspensions of the ear (n ≥ 4).
- mRNA expression levels of the indicated genes in ears relative to reference genes (Hprt1, Rpl13a, and Tbp1). Each symbol represents one mouse; the line represents the mean value (n ≥ 3).
Figure EV2. Psoriasis‐like symptoms upon leaky and tamoxifen‐induced CARD14E138A expression in the ears of ieCARD14E138A mice.

- Body weight and ear thickness of untreated 8‐week‐old WT and ieCARD14E138A males and females. Each symbol represents one mouse; the line represents the mean value (n ≥ 9 mice per group). WT = K14creERTg/+ Rosa26+/+, ieCARD14E138A = K14creERTg/+ Rosa26LSL‐CARD14‐E138A/+. Statistical difference between WT and ieCARD14E138A groups was determined using an unpaired t‐test (normality of data was tested with a D'Agostino‐Pearson normality test) (***P < 0.001, ****P < 0.0001).
- Ear thickness of untreated 8‐week‐old Malt1 +/+ WT, Malt1 +/+ ieCARD14E138A, Malt1 fl/fl WT, and Malt1 fl/fl ieCARD14E138A mice. Each symbol represents one mouse; the line represents the mean value (n ≥ 7). Statistical difference between two groups was determined using a Mann–Whitney U‐test (****P < 0.0001).
- Representative images from Malt1 +/+ WT, Malt1 +/+ ieCARD14E138A, Malt1 fl/fl WT, and Malt1 fl/fl ieCARD14E138A mice 4 days after tamoxifen treatment.
- Immunohistochemical staining of the proliferation marker Ki67 in sections of ear tissue from WT and ieCARD14E138A mice 4 days after tamoxifen treatment (scale bare represents 100 μm).
In contrast to the clear psoriatic phenotype of Malt1 +/+ ieCARD14E138A mice in the ears, increased ear swelling and loss of body weight were much less pronounced in tamoxifen‐treated Malt1 EKO ieCARD14E138A mice (Fig 2B), indicating an important role for MALT1. Hematoxylin and eosin staining of ear sections showed acanthosis, hyperkeratosis, and parakeratosis in Malt1 +/+ ieCARD14E138A mice, which are histological features of human psoriasis (Fig 2C) 2, 29. In line with this, blinded measurements showed that the thickness of the epidermis was significantly increased and also immunostaining for Ki67, a marker for proliferation, showed an increased amount of proliferating cells in the basal layer of ear skin of Malt1 +/+ ieCARD14E138A mice (Figs 2D and EV2D). Furthermore, several large infiltrates of immune cells, reminiscent of Munro's abscesses, were visible in the stratum corneum (Fig 2C, arrowheads). Thickening of the epidermis and inflammatory cell infiltration in the stratum corneum were reduced in Malt1 EKO ieCARD14E138A mice compared to Malt1 +/+ ieCARD14E138A mice (Fig 2C and D). Flow cytometry analysis of the ears showed that neutrophils are one of the most abundant infiltrating immune cell types, which is consistent with the large pustules observed in ear sections and the acute nature of our model (Fig 2E). Also T cells and dendritic cells were increased, while the amount of eosinophils, which are associated with Th2 immunity, remained very low. Again, the number of infiltrating neutrophils and dendritic cells was significantly reduced in the ears of Malt1 EKO ieCARD14E138A mice (Fig 2E).
mRNA analysis of the ears of Malt1 +/+ ieCARD14E138A mice showed a strong induction of proinflammatory cytokines and chemokines that are strongly linked to and expressed during human psoriasis, such as Il17a, Il23, Il36γ, Cxcl1, Cxcl2, and Tnf (Fig 2F) 30. Furthermore, expression of antimicrobial peptides such as S100a8 and Lcn2, which are also highly increased in human psoriatic lesions, was upregulated (Fig 2F) 31. As expected, human transgene CARD14 E138A mRNA levels were high in ieCARD14E138A mice and absent in wild‐type mice (Fig 2F). Most importantly, upregulation of most of the analyzed cytokines, chemokines, and antimicrobial peptides was much less pronounced in ears of MALT1‐deficient ieCARD14E138A mice (Fig 2F). Of note, Il17a mRNA levels were similar in Malt1 EKO ieCARD14E138A and Malt1 +/+ ieCARD14E138A mice, which might reflect equal T‐cell counts in the ears of both mouse lines (Fig 2E and F). Both the chemokines CXCL1 and CXCL2 and the antimicrobial peptide LCN2 have been shown to recruit and modulate neutrophil activation and in this way contribute to psoriatic inflammation 32, 33. Therefore, the reduced Cxcl1/2 and Lcn2 levels are in line with the reduced neutrophil influx in ears of Malt1 EKO ieCARD14E138A mice and might explain the protective effect of MALT1 deficiency. Taken together, our data demonstrate that human CARD14E138A expression in keratinocytes is sufficient to drive a pathogenic inflammatory cascade mimicking several pathological features of human psoriasis. Most importantly, our data are the first to demonstrate a keratinocyte‐intrinsic in vivo role of MALT1 in skin inflammation. However, it should be mentioned that the above described protective effect of MALT1 deletion on CARD14E138A‐induced skin inflammation does not allow us to distinguish between the role of MALT1 scaffold versus MALT1 proteolytic activities.
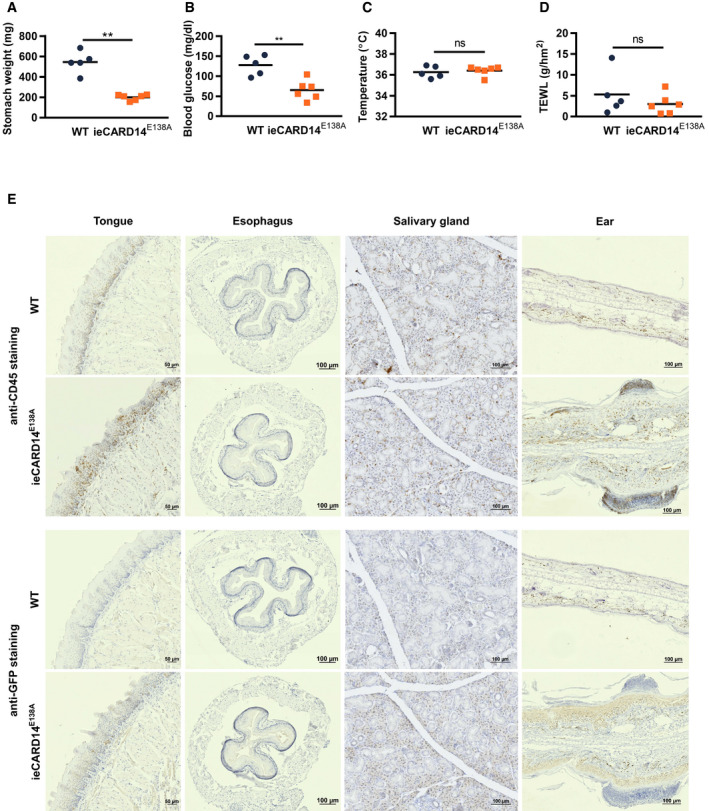
The quick onset of death of CARD14E138A expressing pups as well as the rapid weight loss of up to 18% over 4 days of tamoxifen‐induced transgene induction in ieCARD14E138A mice can hardly be attributed to health problems in the skin only. Closer examination showed that the stomach of ieCARD14E138A mice was less filled and that blood glucose levels were lower 4 days after tamoxifen treatment (Fig EV3A and B), suggesting a defect in eating behavior. We could not detect a significant difference in body temperature (Fig EV3C) or transepidermal water loss through the skin (Fig EV3D). It should be mentioned that K14creER has been shown to not only target keratinocytes of the skin, but also keratinocytes of the oral epithelia, tongue, and esophagus, as well as mammary epithelium, vaginal epithelium, salivary glands, and cornea 27, 28. As high‐quality antibodies against CARD14 that can be used for immunohistochemistry are not available, we performed immunohistochemistry for GFP, which is expressed with CARD14E138A as a bicistronic transgene (Fig 1A). This revealed that the transgene is not only expressed in the ears, but also in the stratified epithelium of the tongue and the epithelium of salivary glands, and very faintly in the esophagus (Fig EV3E). In addition, we could detect clear infiltration of CD45+ inflammatory cells in the tongue (Fig EV3E). Together these results let us speculate that epithelial expression of the CARD14E138A transgene in the oral epithelium induces inflammatory reactions that lead to irritation and affect eating behavior and weight loss of ieCARD14E138A mice. Similar effects may also explain the observed mortality in pups in which CARD14E138A expression is under control of K5cre.
Figure EV3. Tamoxifen‐induced K14Cre‐driven CARD14E138A expression in the tongue and salivary glands induces oral inflammation and decreased food uptake.
-
A–DStomach weight (A), blood glucose levels (B), body temperature (C), and transepidermal water loss (TEWL) (D) of WT and ieCARD14E138A mice three (C) or four (A, B and D) days after tamoxifen treatment. WT = K14creERTg/+ Rosa26+/+, ieCARD14E138A = K14creERTg/+ Rosa26LSL‐CARD14‐E138A/+. Each symbol represents one mouse; the line represents the mean value (n ≥ 4 mice per group). Statistical difference between WT and ieCARD14E138A groups was determined using a Mann–Whitney U‐test (ns: P > 0.05, **P < 0.01).
-
ERepresentative images of immunohistochemical staining of CD45 and GFP in sections of tongue, esophagus, salivary gland, and ear tissue from WT and ieCARD14E138A mice 4 days after tamoxifen treatment (CD45‐ and GFP‐stained sections of each organ are derived from the same WT and ieCARD14E138A mouse; scale bar represents 50 μm in sections of tongue tissue and 100 μm in sections of esophagus, salivary gland, and ear tissue).
Inhibition of MALT1 proteolytic activity attenuates CARD14E138A‐induced psoriasis
Therapeutic targeting of MALT1 proteolytic activity by small compound inhibitors has shown promising effects in preclinical models of inflammatory disease and cancer 17, 18, 19, 20, 21, 22. However, therapeutic targeting of MALT1 in the context of psoriasis has not yet been reported. The inducible and rapidly developing skin phenotype of ieCARD14E138A mice as well as the causal link with a mutation present in humans makes these mice particularly interesting to test the effect of MALT1 small compound inhibitors in psoriatic dermatitis. We therefore investigated the effect of systemic treatment with MLT‐827, a potent small compound inhibitor of MALT1 proteolytic activity that has been originally developed by Novartis 34, on the development of psoriasiform dermatitis in ieCARD14E138A mice. MALT1 is ubiquitously expressed, and systemic treatment with a small compound MALT1 inhibitor is expected to affect MALT1 activity in different cell types that may contribute to disease development, including keratinocytes and T cells. MLT‐827 was administered two times daily by oral gavage, starting at the same time as tamoxifen treatment (Fig 3A). It should be mentioned that due to some leaky CARD14E138A expression, mice already exhibit increased ear thickness before tamoxifen and MALT1 inhibitor treatment. Using immunoblot analysis of ear tissue extracts, we could show that CARD14E138A expression induces cleavage of CYLD and BCL10, two known substrates of MALT1 35, 36. Cleavage of both substrates was completely prevented in mice treated with the MALT1 inhibitor MLT‐827 (Fig 3B), illustrating in vivo MALT1 target engagement of the compound. Compared to vehicle‐treated mice, MALT1 inhibitor treatment almost completely blocked ear swelling induced by CARD14E138A and reduced the accompanying loss in body weight (Fig 3C). In line with this, hematoxylin and eosin staining of ear sections showed that the epidermis is less thickened in MALT1 inhibitor‐treated mice (Fig 3D and E). However, the reduction in epidermal thickening was rather limited compared to the strong reduction in total ear thickness, suggesting that reduced ear swelling mainly reflects reduced edema and inflammatory cell infiltration in the skin. Next, we examined inflammatory cytokine secretion by lymphocytes of the ear‐draining lymph nodes of mice treated with MLT‐827. CD3/CD28‐induced IL‐6 and IL‐17a production was significantly lower in the lymphocytes isolated from MALT1 inhibitor‐treated mice when compared to vehicle‐treated mice (Fig 3F), suggesting that MALT1 inhibition can impair the development of a Th17‐response in CARD14E138A‐induced psoriasis. Consistent with our earlier results, mRNA levels encoding several proinflammatory mediators were strongly upregulated in ears of vehicle‐treated ieCARD14E138A mice (Figs 3G and EV4), which was significantly diminished in MLT‐827 treated mice. In particular, the strong inhibition of Il17a, Il23, Cxcl1, and Tnf expression by MLT‐827 demonstrates that MALT1 inhibition can tackle psoriasis‐like inflammation induced by CARD14E138A. Furthermore, also the expression of other cytokines such as Il36γ, Il1β, and Il6 and antimicrobial peptides such as S100a8 and Lcn2 was significantly reduced upon MALT1 inhibitor treatment in CARD14E138A mice (Figs 3F and EV4). Together, these data show that MALT1 proteolytic activity plays a key role in CARD14E138A‐induced psoriasiform dermatitis and illustrate that MALT1 inhibitor treatment may be an interesting novel therapeutic approach in psoriasis. It should be mentioned that in our mouse model MLT‐827 treatment is not only targeting MALT1 in keratinocytes, but also other cell types that play a role in skin inflammation. For example, T‐cell receptor‐induced MALT1 activation mediates T‐cell proliferation and Th17 differentiation 19, 22, 37, while MALT1 proteolytic activity has also been shown to regulate endothelial permeability and activation 38, 39. Therefore, MALT1 inhibition might attenuate CARD14E138A‐induced skin inflammation at multiple levels. Interestingly, a few early reports describe amelioration of psoriasis features in psychiatric patients treated with the antipsychotic chlorpromazine, which was recently shown to also inhibit MALT1 40, 41, 42, further supporting the use of MALT1 inhibitors for the management of psoriasis. Furthermore, accumulating evidence suggests a more ubiquitous role for CARD14‐MALT1 signaling axis in inflammatory skin conditions. For instance, CARD14 mutations have not only been identified in psoriasis patients but also in patients suffering from pityriasis rubra pilaris, a rare inflammatory skin disease phenotypically related to psoriasis 5, indicating that MALT1 targeting may also be of interest in psoriasis‐related skin diseases. Noteworthy, while dominant gain‐of‐function mutations in CARD14 are associated with psoriasis and related diseases, loss‐of‐function mutations in CARD14 were recently reported to be associated with a severe variant of atopic dermatitis 43. It will be interesting to investigate the effect of MALT1 targeting in the context of atopic dermatitis to obtain a complete view on the potential of MALT1 targeting in inflammatory skin disease. Of note, several groups have shown that mice expressing a catalytically inactive MALT1 mutant suffer from multiorgan inflammation and autoimmunity, likely caused by a reduced frequency of regulatory T cells 19, 37, 44, 45, 46. In contrast, pharmacological inhibition of MALT1 in preclinical mouse models did not lead to obvious side effects, suggesting that MALT1 inhibitor treatment may be safe 18, 20. Genetic inactivation of MALT1 may indeed not be representative for pharmacological inhibition of MALT1 as the latter may affect less the development of regulatory T cells in the thymus and never lead to a complete MALT1 inhibition. Nevertheless, it will be important to closely monitor possible side effects upon long‐term systemic treatment with MALT1 inhibitors. Alternatively, in the case of psoriasis, it might also be possible to deliver MALT1 inhibitors topically, with less risk of systemic side effects.
Figure 3. Inhibition of MALT1 proteolytic activity attenuates CARD14E138A‐induced psoriasis.

-
ASchematic representation of the experimental set‐up of CARD14E138A induction and MALT1 inhibitor treatment.
-
BCleavage of MALT1 substrates CYLD and BCL10 as analyzed by Western blotting of lysates of ear tissue of mice 4 days after treatment with tamoxifen and MALT1 inhibitor or vehicle. Actin is shown as a loading control. Each lane represents one mouse.
-
CChanges in relative ear thickness and body weight in vehicle‐ and MALT1 inhibitor‐treated mice upon CARD14E138A induction. M1 inh = MALT1 inhibitor, WT = K14creERTg/+ Rosa26+/+, ieCARD14E138A = K14creERTg/+ Rosa26LSL‐CARD14‐E138A/+. Combined results of two independent experiments are shown (WT + vehicle: n = 9, WT + M1 inh: n = 8, ieCARD14E138A + vehicle: n = 9, ieCARD14E138A + M1 inh: n = 10).
-
D, E(D) Representative histological sections of ear tissue stained with hematoxylin and eosin and (E) measurements of epidermal thickness of tamoxifen‐treated mice. Combined results of two independent experiments are shown. Each symbol represents the mean of at least ten epidermal thickness measurements for each ear; the line represents the mean value.
-
FIL‐6 and IL‐17a production by ear‐draining lymphocytes upon ex vivo restimulation with anti‐CD3/anti‐CD28 for 3 days (n ≥ 3 biological replicates).
-
GHeatmap showing relative mRNA expression levels of the indicated genes in ear tissue normalized to reference genes. Values represent median of each group (n ≥ 3).
Figure EV4. MALT1 inhibition reduces proinflammatory gene expression in ears of ieCARD14E138A mice.
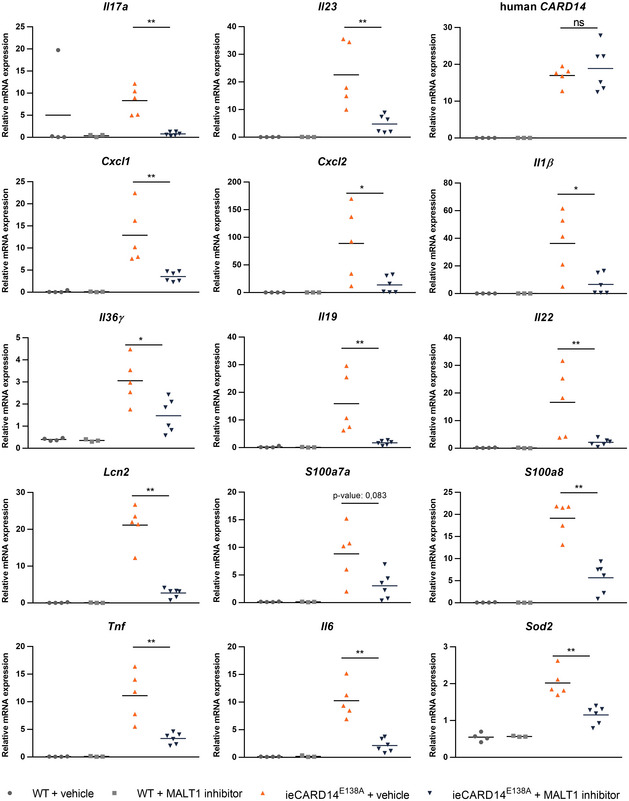
mRNA expression levels of the indicated genes in ears relative to reference genes (Hprt1 and Tbp). Each symbol represents one mouse; the line represents the mean value (n ≥ 3). Statistics: Differences between ieCARD14E138A + vehicle and ieCARD14E138A + MALT1 inhibitor groups were determined using a Mann–Whitney U‐test (*P < 0.05, **P < 0.01). WT = K14creERTg/+ Rosa26+/+, ieCARD14E138A = K14creERTg/+ Rosa26LSL‐CARD14‐E138A/+.
Materials and Methods
Rosa26LSL‐CARD14‐E138A transgenesis
Cloning of RMCE targeting constructs
Plasmids of the cloned genes were deposited in the BCCM/GeneCorner plasmid collection along with detailed descriptions of cloning strategy and plasmid sequence (http://bccm.belspo.be/about-us/bccm-genecorner). Human CARD14E138A (LMBP : 9623) was cloned into pENTR3C. The ENTR clone was integrated in the pDV1_RMCE (LMBP: 8870) destination vector using Gateway LR reactions to generate a RMCE targeting vector for CARD14E138A (LMBP: 09578).
RMCE targeting
RMCE compatible mouse ES cells (G4 ROSALUC 25) were cultured in standard ES‐cell medium containing 500 ml Knockout™‐DMEM (Thermo Fisher Scientific) supplemented with 15% FBS (Hyclone), 100 μM non‐essential amino acids (Thermo Fisher Scientific), 1× GlutaMAX (Thermo Fisher Scientific), 100 μM β‐mercaptoethanol (Thermo Fisher Scientific), and 2,000 U/ml leukemia inhibitory factor (Protein Service Facility, VIB). ES cells were co‐transfected with 2.5 μg of a FlpE expressing plasmid and 2.5 μg of targeting vector using lipofectamin 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions 47. G418 selection (200 μg/ml) was applied 48 h after transfection. After 7–10 days, individual G418‐resistant, RMCE‐targeted ES‐cell clones were picked and further expanded. Correct targeting events were confirmed by PCR; the targeted allele generates a band of 560 bp (primers: FW 5′ AAA GTC GCT CTG AGT TGT TAT 3′ and REV 5′ GCG GCC TCG ACT CTA CGA TA 3′), as previously described 25. Correctly targeted ES cells were aggregated with outbred Swiss morula, which were then implanted into pseudopregnant Swiss mice to generate chimeric mice that transmitted the transgene to their offspring.
Mice
Mice were bred and maintained under specific pathogen‐free conditions and housed in individually ventilated cages in accordance with the institutional and national guidelines regarding the care and use of laboratory animals. Animal protocols were approved by the ethics committee of Ghent University (EC2017_013, EC2017_102, and EC2018_056). To induce Cre excision of the Lox‐STOP‐Lox (LSL) cassette, hemizygous Rosa26LSL‐CARD14‐E138A transgenic mice were bred with hemizygous K5cre and K14creER mouse strains. This generated double transgenic mice that constitutively express CARD14E138A transgene or express CARD14E138A transgene inducibly upon tamoxifen treatment in epidermal cells 26, 27. As controls, littermates that only express the K5cre or K14creER transgene were used.
Malt1 tm1a(EUCOMM)Hmgu/+ (C57BL6/N background) mice were derived from ES cells purchased from EUCOMM. Malt1 tm1a mice were bred with mice of the FLP deleter strain (C57BL6/J background) to remove the FRT‐flanked LacZ and neomycin selection cassette and to generate the Malt1 tm1c floxed allele 48. Malt1 tm1c mice were bred with Rosa26LSL‐CARD14‐E138A transgenic and K5Cre or K14creER transgenic mouse strains to obtain mice with CARD14E138A‐transgene‐expressing, MALT1‐deficient keratinocytes. To induce expression of the CARD14E138A transgene, both male and female mice of 8–9 weeks old were injected intraperitoneally with 2.5 mg tamoxifen (T‐5648, Sigma‐Aldrich) dissolved in corn oil (C8267, Sigma‐Aldrich). Mice were weighed, and ear thickness was measured every other day using a G‐1A dial thickness gauge (Peacock, Japan) by an observer blinded to the treatments and genotypes. For MALT1 inhibitor treatment, mice were treated two times per day using oral gavage with 30 mg/kg MALT1 inhibitor (MLT‐827; generously provided by Galapagos n.v., Mechelen) dissolved in Kolliphor® HS 15 (42966, Sigma)/methylcellulose 0.5% (AX021233, VWR chemicals) (2/98% ratio) under continuous stirring and protected from light.
Genotyping
The Malt1 floxed allele or knockout allele was genotyped with the primers MALTcKO‐F (GTTTCTCAGGTCTTTAGTTCATGTC), CoMLT‐3‐R (TATACTCTACATCTCCATGGT), and MALTcKO‐R (TTGTTTTGCAGATCTCTGCC) resulting in 280 bp (WT), 448 bp (FL), and 345 (KO) products. Rosa26LSL‐CARD14E138A transgenes were genotyped using the following primers: Fwd 5′ AACCCTGACGTCTACACCCT 3′, Rev 5′ CACTCGGTCAGCTTGGATGT 3′ resulting in a 95 bp PCR product.
Histology
Ears, skin, tongue, salivary gland, and esophagus were dissected and incubated in 4% PFA in PBS at 4°C overnight, followed by dehydration and embedding in paraffin. Sections of 5 μm were deparaffinized followed by staining with hematoxylin and eosin. For immunohistochemistry staining, antigen retrieval was performed by heating the slides in a citrate‐based antigen unmasking solution (H‐3300, Vector Laboratories) and endogenous peroxidase activity was blocked by immersing slides in 3% H2O2. Nonspecific binding was blocked by incubating sections in 5% normal goat serum and 1% BSA. The following primary antibodies were used: anti‐GFP (2956, Cell Signaling Technology), anti‐CD45 (ab10558, Abcam), and anti‐Ki67 (12202, Cell Signaling Technology). After overnight incubation with the primary antibody at 4°C, the tissue sections were sequentially incubated with a biotinylated anti‐rabbit secondary Ab (E0432, Dako), followed by incubation with Vectastain Elite ABC kit (PK–6100, Vector Laboratories), followed by detection with diaminobenzidine chromogen (ImmPACT® DAB, SK‐4105, Vector Laboratories) and counterstaining with hematoxylin. Sections were mounted by use of Entellan mounting medium (Merck Millipore). Images were acquired with Axio Scan.Z1 slide scanner (Zeiss), and image analysis was done using Zen lite (Zeiss) software. Average epidermal thickness was determined as the mean of at least 10 measurements for each sample.
Cytokine production by lymphocytes
For CD3/CD28 stimulation, a 96‐well plate was coated overnight at 4°C with 200 μl PBS containing 1 μg/ml anti‐CD3 (553057, BD Biosciences) and 1 μg/ml anti‐CD28 (553294, BD Biosciences). Single cell suspensions from ear‐draining lymph nodes were obtained by homogenizing the lymph node through a 70 μm cell sieve. Isolated lymphocytes were counted, and 200,000 lymphocytes were seeded in an anti‐CD3/CD28‐coated plate for in vitro restimulation. After 72 h, the medium was collected and assayed for IL‐6 (171‐G5007M) and IL‐17a (171‐G5013M) production by Bio‐Plex Pro (Bio‐Rad) according to manufacturer's instructions on a Bio‐Plex 200 system (Bio‐Rad).
Western blotting
After sacrifice, ears or back skin were harvested and immediately frozen and stored at −70°C. To make protein lysates, tissues were homogenized and lyzed using Precellys 24 (Bertin technologies with CK26 beads) in Laemmli buffer (50 mM Tris–HCl pH 8, 2% SDS, 10% glycerol). Debris was removed by two centrifugation steps at 16,000 g for 10 min. Pierce™ BCA Protein Assay Kit (23225, Thermo Fisher Scientific) was used to determine protein concentration. 0.005% bromophenol blue and 5% β‐mercaptoethanol were added to the samples, and equal amounts of proteins were loaded and separated by 10% SDS–PAGE. Proteins were then transferred to nitrocellulose membrane with 0.45 μm pores (Protran, Perkin Elmer). The membranes were blocked in 5% milk powder in TBS/0.2% Tween 20 (TBST) for 1 h at room temperature (RT) and probed with specific primary antibodies in 5% milk powder in TBST. The following antibodies were used: anti‐CARD14 (10400‐1‐AP, Proteintech), anti‐CYLD (sc‐74435, Santa Cruz), anti‐BCL10 cleavage‐specific (gift from Thijs Baens, Cistim Leuven vzw, Leuven, Belgium), anti‐BCL10 (sc‐5273, Santa Cruz), anti‐MALT1 (32494, Cell Signaling Technology), and anti‐β‐actin‐HRP (sc‐47778, Santa Cruz). Secondary HRP‐conjugated anti‐mouse or anti‐rabbit IgG antibody was purchased from Thermo Fisher Scientific (31432 and 31464). Proteins were detected using the Western Lightning ECL detection system (Perkin Elmer) according to the manufacturer's instructions.
RNA extraction, cDNA synthesis, and quantitative real‐time PCR
After sacrifice, ears were harvested at the base and incubated overnight in RNA later at 4°C before long‐term storage at −70°C. For RNA extraction, ears were homogenized and lysed using Precellys 24 (Bertin technologies with CK26 beads) in TRIzol reagent (Invitrogen). After phenol–chloroform phase separation, RNA was isolated using RNeasy mini kit (Qiagen). Synthesis of cDNA was performed using an iScript Advanced cDNA synthesis kit (Bio‐Rad), according to manufacturer's instructions. Quantitative PCR was performed with a LightCycler 480 (Roche) using SensiFAST™ SYBR ® No‐ROX kit (Bioline) with a total of 10 ng of cDNA and 300 nM of specific primers in a total volume of 10 μl. Real‐time PCR reactions were performed in triplicates. The following specific primers were used (5′–3′): Hprt1 forward, AGTGTTGGATACAGGCCAGAC; Hprt1 reverse, CGTGATTCAAATCCCTGAAGT; Tbp forward, TCTACCGTGAATCTTGGCTGTAAA; Tbp reverse, TTCTCATGATGACTGCAGCAAA; Rpl13a forward, CCTGCTGCTCTCAAGGTT; Rpl13a reverse, TGGTTGTCACTGCCTGGTACTT; Il17a forward, GGACTCTCCACCGCAATGA; Il17a reverse, TCAGGCTCCCTCTTCAGGAC; Il23 forward, CCCGTATCCAGTGTGAAGATG; Il23 reverse, GGGCTATCAGGGAGTAGAGCA; Il1f9 (Il36γ) forward, TCCTGACTTTGGGGAGGTTTT; Il1f9 (Il36γ) reverse, TCACGCTGACTGGGGTTACT; hCARD14 forward, GTCAACACGGACGGTTATAAGA; hCARD14 reverse, GTTGACCCGGATGTAGAATGAG; S100a7a forward, TGCTCTTGGATAGTGTGCCTC; S100a7a reverse, GCTCTGTGATGTAGTATGGCTG; Ccl20 forward, GTACTGCTGGCTCACCTCTG; Ccl20 reverse, CTTCATCGGCCATCTGTCTTGTG; Cxcl1 forward, GAGCCTCTAACCAGTTCCAG; Cxcl1 reverse, TGAGTGTGGCTATGACTTCG; Cxcl2 forward, ACAGAAGTCATAGCCACTCTC; Cxcl2 reverse, TTAGCCTTGCCTTTGTTCAG; Tnf forward, ACCCTGGTATGAGCCCATATAC; Tnf reverse, ACACCCATTCCCTTCACAGAG; Il1β forward, CACCTCACAAGCAGAGCACAAG; Il1β reverse, GCATTAGAAACAGTCCAGCCCATAC; Il19 forward, CTCCTGGGCATGACGTTGATT; Il19 reverse, GCATGGCTCTCTTGATCTCGT; Il22 forward, CAGCTCCTGTCACATCAGCGGT; Il22 reverse, AGGTCCAGTTCCCCAATCGCCT; Il6 forward, GAGGATACCACTCCCAACAGACC; Il6 reverse, AAGTGCATCATCGTTGTTCATACA; S100a8 forward, AAATCACCATGCCCTCTACAAG; S100a8 reverse, CCCACTTTTATCACCATCGCAA; S100a9 forward, ATACTCTAGGAAGGAAGGACACC; S100a9 reverse, TCCATGATGTCATTTATGAGGGC; Lcn2 forward, TGGCCCTGAGTGTCATGTG; Lcn2 reverse CTCTTGTAGCTCATAGATGGTGC; and Sod2 forward, CAGACCTGCCTTACGACTATGG; Sod2 reverse CTCGGTGGCGTTGAGATTGTT. Analysis was done using qBase+ software (Biogazelle, Gent, Belgium). Values were normalized to the appropriate amount of reference genes, as determined by geNorm analysis in the qBase software.
Flow cytometry
Ear samples were incubated overnight at 4°C with 200 μg/ml Dispase II (from Bacillus polymerase grade 2; Roche) with the dermal side down to facilitate isolation of cells. Next, the ear skin was manually minced into small pieces and was further digested with 1.5 mg/ml collagenase type 4 (Worthington Biochemical, Lakewood, NJ) and 10 U of DNase (Roche) in RPMI medium buffered with HEPES and supplemented with 2% FCS. The suspension was resuspended for 30 min and provided with fresh digestion buffer for a total of 90 min at 37°C. After digestion, the cell suspension was passed through 100 and 40 μm cell strainers to remove debris and clots.
For phenotyping of immune cells in cell suspensions of ear skin, cells were stained with CD16/CD32 (553142, BD), MHCII‐eFluor 450 (# 48‐5321‐80, eBioscience), CD64‐BV711 (Biolegend, 139311), Siglec F‐PE (BD, 552126), CD45‐APC‐eFluor780 (47‐0451‐82, eBioscience), CD8‐PerCP‐Cy5.5 (45‐0081, eBioscience), CD3‐PE‐Cy5 (55‐0031, Tonbo Biosciences), CD19‐PE‐Cy5 (15‐0193, eBioscience), CD11c‐PE‐Cy7 (117317, Biolegend), CD11b‐BV605 (563015, BD), γδ‐TCR‐APC (17‐5711, eBioscience), Ly6G‐AF700 (561236, BD), and CD4‐APC‐eFluor780 (47‐0042, eBioscience). Dead cells were excluded from the analysis by using fixable viability dye eFluor506 (eBioscience), and 123count eBeads (01‐1234‐42, Invitrogen) were used for quantification of cells. Acquisition of multi‐color samples was done on an LSRFortessa flow cytometer (BD Bioscience). Final analysis and graphical output were performed using FlowJo software (Tree Star Inc.).
Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis between two groups was done with GraphPad Prism 7 using a Mann–Whitney U‐test for unpaired data. Analysis of data from Bio‐Plex analysis was performed with GraphPad Prism 7 using two‐way ANOVA to determine significant differences. Relative body weight and ear thickness measured at consecutive equally spaced time points were analyzed as repeated measurements data (also called longitudinal data) using the residual maximum likelihood (REML) as implemented in Genstat v19 49. Briefly, a linear mixed model (random terms underlined) of the form y = μ + genotype + time + genotype.time + replicate + mouse.time was fitted to the longitudinal data. The term mouse.time represents the residual error term with dependent errors because the repeated measurements are taken in the same individual, causing correlations among observations. Times of measurement were set as equally spaced, and the autoregressive model of order 1 was selected as the best correlation model based on the Aikake information coefficient. Significances of genotype effects across time (i.e., genotype.time) and of pairwise differences between genotype effects across time were assessed by an approximate F‐test, of which the denominator degrees of freedom were calculated using algebraic derivatives as implemented in Genstat v19.
Data availability
Plasmids were deposited in the BCCM/GeneCorner plasmid collection along with detailed descriptions of cloning strategy and plasmid sequence (http://bccm.belspo.be/about-us/bccm-genecorner). All other data are available from the authors upon reasonable requests.
Author contributions
EVN, JS, ISA, and RB contributed to the conception and design of the study. EVN, JS, TH, and ISA acquired the data. MH, GB, and YD provided technical assistance. EVN, JS, ISA, and RB analyzed and interpreted the data. EVN, JS, ISA, and RB drafted the article or revised it critically for important intellectual content. All authors approved the final version of the manuscript.
Conflict of interest
R.B. is inventor on a patent application (Inhibitors of MALT1 proteolytic activity and uses thereof; WO09065897; applicants: VIB and UGent) and was involved in a research collaboration with Galapagos n.v. that is no longer running. The authors have no additional financial interests.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Acknowledgments
We thank Galapagos n.v. for providing MLT‐827. M. Baens is acknowledged for providing cleaved BCL10 specific antibody. This work was supported by grants from the VIB, the Research Foundation—Flanders (FWO) (G035517N and G090914N), the Foundation Against Cancer (FAF‐F/2016/812), and the Ghent University Concerted Research Actions (GOA). I.S.A. is supported by a postdoctoral fellowship and research grants (1503418N and 1503815N) of the FWO. E.V.N. is supported by a predoctoral fellowship of the FWO. Marnik Vuylsteke is acknowledged for help and advice with the statistical analysis. We also thank prof. Wim Declercq for kindly providing K5cre and K14creER transgenic mice. Julie Deckers is acknowledged for advice on flow cytometry analysis and we are grateful to Kelly Lemeire for performing Ki67 immunohistochemistry staining on tissue sections.
EMBO Reports (2020) 21: e49237
References
- 1. Connor CJ, Liu V, Fiedorowicz JG (2015) Exploring the physiological link between psoriasis and mood disorders. Dermatol Res Pract 2015: 409637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lowes MA, Suarez‐Farinas M, Krueger JG (2014) Immunology of psoriasis. Annu Rev Immunol 32: 227–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hawkes JE, Chan TC, Krueger JG (2017) Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol 140: 645–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Capon F (2017) The genetic basis of psoriasis. Int J Mol Sci 18: 2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fuchs‐Telem D, Sarig O, van Steensel MA, Isakov O, Israeli S, Nousbeck J, Richard K, Winnepenninckx V, Vernooij M, Shomron N et al (2012) Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am J Hum Genet 91: 163–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jordan CT, Cao L, Roberson ED, Duan S, Helms CA, Nair RP, Duffin KC, Stuart PE, Goldgar D, Hayashi G et al (2012) Rare and common variants in CARD14, encoding an epidermal regulator of NF‐kappaB, in psoriasis. Am J Hum Genet 90: 796–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jordan CT, Cao L, Roberson ED, Pierson KC, Yang CF, Joyce CE, Ryan C, Duan S, Helms CA, Liu Y et al (2012) PSORS2 is due to mutations in CARD14. Am J Hum Genet 90: 784–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Nuffel E, Schmitt A, Afonina IS, Schulze‐Osthoff K, Beyaert R, Hailfinger S (2017) CARD14‐mediated activation of paracaspase MALT1 in keratinocytes: implications for psoriasis. J Invest Dermatol 137: 569–575 [DOI] [PubMed] [Google Scholar]
- 9. Afonina IS, Van Nuffel E, Baudelet G, Driege Y, Kreike M, Staal J, Beyaert R (2016) The paracaspase MALT1 mediates CARD14‐induced signaling in keratinocytes. EMBO Rep 17: 914–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Howes A, O'Sullivan PA, Breyer F, Ghose A, Cao L, Krappmann D, Bowcock AM, Ley SC (2016) Psoriasis mutations disrupt CARD14 autoinhibition promoting BCL10‐MALT1‐dependent NF‐kappaB activation. Biochem J 473: 1759–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mellett M, Meier B, Mohanan D, Schairer R, Cheng P, Satoh TK, Kiefer B, Ospelt C, Nobbe S, Thome M et al (2018) CARD14 gain‐of‐function mutation alone is sufficient to drive IL‐23/IL‐17‐mediated psoriasiform skin inflammation in vivo . J Invest Dermatol 138: 2010–2023 [DOI] [PubMed] [Google Scholar]
- 12. Wang M, Zhang S, Zheng G, Huang J, Songyang Z, Zhao X, Lin X (2018) Gain‐of‐function mutation of Card14 leads to spontaneous psoriasis‐like skin inflammation through enhanced keratinocyte response to IL‐17A. Immunity 49: 66–79.e65 [DOI] [PubMed] [Google Scholar]
- 13. Staal J, Driege Y, Haegman M, Borghi A, Hulpiau P, Lievens L, Gul IS, Sundararaman S, Goncalves A, Dhondt I et al (2018) Ancient origin of the CARD‐Coiled Coil/Bcl10/MALT1‐like paracaspase signaling complex indicates unknown critical functions. Front Immunol 9: 1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Juilland M, Thome M (2018) Holding all the CARDs: how MALT1 controls CARMA/CARD‐dependent signaling. Front Immunol 9: 1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruland J, Hartjes L (2019) CARD‐BCL‐10‐MALT1 signalling in protective and pathological immunity. Nat Rev Immunol 19: 118–134 [DOI] [PubMed] [Google Scholar]
- 16. Demeyer A, Staal J, Beyaert R (2016) Targeting MALT1 proteolytic activity in immunity, inflammation and disease: good or bad? Trends Mol Med 22: 135–150 [DOI] [PubMed] [Google Scholar]
- 17. Fontan L, Qiao Q, Hatcher JM, Casalena G, Us I, Teater M, Durant M, Du G, Xia M, Bilchuk N et al (2018) Specific covalent inhibition of MALT1 paracaspase suppresses B cell lymphoma growth. J Clin Invest 128: 4397–4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fontan L, Yang C, Kabaleeswaran V, Volpon L, Osborne MJ, Beltran E, Garcia M, Cerchietti L, Shaknovich R, Yang SN et al (2012) MALT1 small molecule inhibitors specifically suppress ABC‐DLBCL in vitro and in vivo . Cancer Cell 22: 812–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jaworski M, Marsland BJ, Gehrig J, Held W, Favre S, Luther SA, Perroud M, Golshayan D, Gaide O, Thome M (2014) Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J 33: 2765–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mc Guire C, Elton L, Wieghofer P, Staal J, Voet S, Demeyer A, Nagel D, Krappmann D, Prinz M, Beyaert R et al (2014) Pharmacological inhibition of MALT1 protease activity protects mice in a mouse model of multiple sclerosis. J Neuroinflammation 11: 124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagel D, Spranger S, Vincendeau M, Grau M, Raffegerst S, Kloo B, Hlahla D, Neuenschwander M, Peter von Kries J, Hadian K et al (2012) Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC‐DLBCL. Cancer Cell 22: 825–837 [DOI] [PubMed] [Google Scholar]
- 22. Nakamura Y, Igaki K, Komoike Y, Yokoyama K, Tsuchimori N (2019) Malt1 inactivation attenuates experimental colitis through the regulation of Th17 and Th1/17 cells. Inflamm Res 68: 223–230 [DOI] [PubMed] [Google Scholar]
- 23. Harden JL, Lewis SM, Pierson KC, Suarez‐Farinas M, Lentini T, Ortenzio FS, Zaba LC, Goldbach‐Mansky R, Bowcock AM, Lowes MA (2014) CARD14 expression in dermal endothelial cells in psoriasis. PLoS One 9: e111255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanaka M, Kobiyama K, Honda T, Uchio‐Yamada K, Natsume‐Kitatani Y, Mizuguchi K, Kabashima K, Ishii KJ (2018) Essential role of CARD14 in murine experimental psoriasis. J Immunol 200: 71–81 [DOI] [PubMed] [Google Scholar]
- 25. Haenebalcke L, Goossens S, Naessens M, Kruse N, Farhang Ghahremani M, Bartunkova S, Haigh K, Pieters T, Dierickx P, Drogat B et al (2013) Efficient ROSA26‐based conditional and/or inducible transgenesis using RMCE‐compatible F1 hybrid mouse embryonic stem cells. Stem Cell Rev 9: 774–785 [DOI] [PubMed] [Google Scholar]
- 26. Ramirez A, Page A, Gandarillas A, Zanet J, Pibre S, Vidal M, Tusell L, Genesca A, Whitaker DA, Melton DW et al (2004) A keratin K5Cre transgenic line appropriate for tissue‐specific or generalized Cre‐mediated recombination. Genesis 39: 52–57 [DOI] [PubMed] [Google Scholar]
- 27. Vasioukhin V, Degenstein L, Wise B, Fuchs E (1999) The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci USA 96: 8551–8556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang X, Zinkel S, Polonsky K, Fuchs E (1997) Transgenic studies with a keratin promoter‐driven growth hormone transgene: prospects for gene therapy. Proc Natl Acad Sci USA 94: 219–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tirumalae R (2013) Psoriasiform dermatoses: microscopic approach. Indian J Dermatol 58: 290–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chiricozzi A, Romanelli P, Volpe E, Borsellino G, Romanelli M (2018) Scanning the immunopathogenesis of psoriasis. Int J Mol Sci 19: E179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morizane S, Gallo RL (2012) Antimicrobial peptides in the pathogenesis of psoriasis. J Dermatol 39: 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, Hogg N (2013) Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 121: 4930–4937 [DOI] [PubMed] [Google Scholar]
- 33. Shao S, Cao T, Jin L, Li B, Fang H, Zhang J, Zhang Y, Hu J, Wang G (2016) Increased Lipocalin‐2 contributes to the pathogenesis of psoriasis by modulating neutrophil chemotaxis and cytokine secretion. J Invest Dermatol 136: 1418–1428 [DOI] [PubMed] [Google Scholar]
- 34. Bardet M, Unterreiner A, Malinverni C, Lafossas F, Vedrine C, Boesch D, Kolb Y, Kaiser D, Gluck A, Schneider MA et al (2018) The T‐cell fingerprint of MALT1 paracaspase revealed by selective inhibition. Immunol Cell Biol 96: 81–99 [DOI] [PubMed] [Google Scholar]
- 35. Rebeaud F, Hailfinger S, Posevitz‐Fejfar A, Tapernoux M, Moser R, Rueda D, Gaide O, Guzzardi M, Iancu EM, Rufer N et al (2008) The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol 9: 272–281 [DOI] [PubMed] [Google Scholar]
- 36. Staal J, Driege Y, Bekaert T, Demeyer A, Muyllaert D, Van Damme P, Gevaert K, Beyaert R (2011) T‐cell receptor‐induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J 30: 1742–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu JW, Hoffman S, Beal AM, Dykon A, Ringenberg MA, Hughes AC, Dare L, Anderson AD, Finger J, Kasparcova V et al (2015) MALT1 protease activity is required for innate and adaptive immune responses. PLoS One 10: e0127083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klei LR, Hu D, Panek R, Alfano DN, Bridwell RE, Bailey KM, Oravecz‐Wilson KI, Concel VJ, Hess EM, Van Beek M et al (2016) MALT1 protease activation triggers acute disruption of endothelial barrier integrity via CYLD cleavage. Cell Rep 17: 221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Y, Huang S, Huang X, Li X, Falcon A, Soutar A, Bornancin F, Jiang Z, Xin HB, Fu M (2018) Pharmacological inhibition of MALT1 protease activity suppresses endothelial activation via enhancing MCPIP1 expression. Cell Signal 50: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. D'Silva JL, Fisher RA (1956) Chlorpromazine in the management of psoriasis. Ill Med J 110: 135–136 [PubMed] [Google Scholar]
- 41. Jacobs KA, Andre‐Gregoire G, Maghe C, Thys A, Li Y, Harford‐Wright E, Trillet K, Douanne T, Alves Nicolau C, Frenel JS et al (2020) Paracaspase MALT1 regulates glioma cell survival by controlling endo‐lysosome homeostasis. EMBO J 39: e102030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shimamoto Y, Shimamoto H (1990) Annular pustular psoriasis associated with affective psychosis. Cutis 45: 439–442 [PubMed] [Google Scholar]
- 43. Peled A, Sarig O, Sun G, Samuelov L, Ma CA, Zhang Y, Dimaggio T, Nelson CG, Stone KD, Freeman AF et al (2019) Loss‐of‐function mutations in caspase recruitment domain‐containing protein 14 (CARD14) are associated with a severe variant of atopic dermatitis. J Allergy Clin Immunol 143: 173–181.e110 [DOI] [PubMed] [Google Scholar]
- 44. Bornancin F, Renner F, Touil R, Sic H, Kolb Y, Touil‐Allaoui I, Rush JS, Smith PA, Bigaud M, Junker‐Walker U et al (2015) Deficiency of MALT1 paracaspase activity results in unbalanced regulatory and effector T and B cell responses leading to multiorgan inflammation. J Immunol 194: 3723–3734 [DOI] [PubMed] [Google Scholar]
- 45. Gewies A, Gorka O, Bergmann H, Pechloff K, Petermann F, Jeltsch KM, Rudelius M, Kriegsmann M, Weichert W, Horsch M et al (2014) Uncoupling Malt1 threshold function from paracaspase activity results in destructive autoimmune inflammation. Cell Rep 9: 1292–1305 [DOI] [PubMed] [Google Scholar]
- 46. Demeyer A, Skordos I, Driege Y, Kreike M, Hochepied T, Baens M, Staal J, Beyaert R (2019) MALT1 proteolytic activity suppresses autoimmunity in a T cell intrinsic manner. Front Immunol 10: 1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schaft J, Ashery‐Padan R, van der Hoeven F, Gruss P, Stewart AF (2001) Efficient FLP recombination in mouse ES cells and oocytes. Genesis 31: 6–10 [DOI] [PubMed] [Google Scholar]
- 48. Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM (2000) High‐efficiency deleter mice show that FLPe is an alternative to Cre‐loxP. Nat Genet 25: 139–140 [DOI] [PubMed] [Google Scholar]
- 49. VSN International (2017) Genstat for Windows, 19th edn Hemel Hempstead, UK: VSN International. Genstat.co.uk; [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Data Availability Statement
Plasmids were deposited in the BCCM/GeneCorner plasmid collection along with detailed descriptions of cloning strategy and plasmid sequence (http://bccm.belspo.be/about-us/bccm-genecorner). All other data are available from the authors upon reasonable requests.