

Abstract
MiR-142-3p has been reported to act as a tumor suppressor in breast cancer. However, the regulatory effect of miR-142-3p on drug resistance of breast cancer cells and its underlying mechanism remain unknown. Here, we found that miR-142-3p was significantly downregulated in the doxorubicin (DOX)-resistant MCF-7 cell line (MCF-7/DOX). MiR-142-3p overexpression increased DOX sensitivity and enhanced DOX-induced apoptosis in breast cancer cells. High-mobility group box 1 (HMGB1) is a direct functional target of miR-142-3p in breast cancer cells and miR-142-3p negatively regulated HMGB1 expression. Moreover, overexpression of HMGB1 dramatically reversed the promotion of apoptosis and inhibition of autophagy mediated by miR-142-3p up-regulation. In conclusion, miR-142-3p overexpression may inhibit autophagy and promote the drug sensitivity of breast cancer cells to DOX by targeting HMGB1. The miR-142-3p/HMGB1 axis might be a novel target to regulate the drug resistance of breast cancer patients.
Keywords: Breast cancer, MCF-7 cell line, HMGB1, MiR-142-3p, Drug resistance, Chemosensitivity
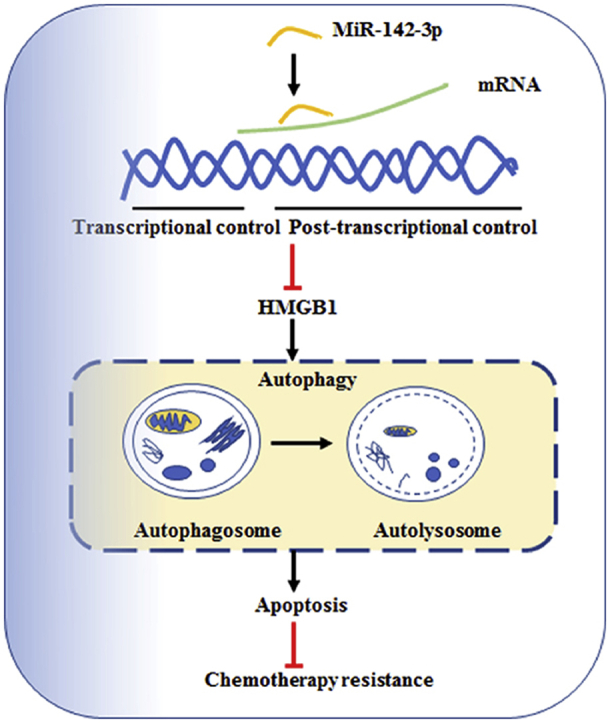
Graphical abstract
High-mobility group box 1 (HMGB1) is a direct functional target of miR-142-3p in breast cancer cells. MiR-142-3p overexpression may inhibit autophagy and promote the drug sensitivity of doxorubicin (DOX) by targeting HMGB1. The miR-142-3p/HMGB1 axis might be an important pathway regulating the sensitivity of breast cancer cells.
1. Introduction
Breast carcinoma is the most common malignancy and the second leading cause of cancer-related death among women1,2. The standard treatment for breast cancer patients is surgical resection, radiotherapy and neoadjuvant chemotherapy3. Doxorubicin (DOX) is considered as a first-line chemotherapeutic agent in the treatment of breast cancer4. Despite the development of potent chemotherapeutic agents with higher efficacy and lesser toxicity, the resistance to chemotherapy is still the main reason of the poor prognosis5. Therefore, understanding the possible molecular mechanisms of DOX resistance may lead to the improvement of clinical outcomes. Among the many factors, therapy-induced autophagy represents a novel mechanism of resistance to anticancer therapy6.
Autophagy is an evolutionarily conserved process, which is characterized as new cell formation through degrading and recycling of cellular components7. There is mounting evidence that autophagy contributes to tumor chemotherapy resistance and cancer cell survival under various types of stresses8, 9, 10. Recently, autophagy inhibitors have been identified and used to enhance the sensitivity of various cancers toward chemotherapy11,12. Therefore, combination treatment with autophagy inhibitors may be useful in increasing the sensitivity of breast cancer cells for chemotherapeutic treatment. It has been reported that microRNAs (miRNAs) can modulate chemotherapy and radiotherapy via autophagy13,14.
MiRNAs represent a new class of small, noncoding endogenous RNAs with 19–25 nucleotides in length15. MiRNAs can negatively regulate target gene expression in a posttranscriptional manner through cleaving, destabilizing the targeted mRNAs or preventing their translation15. Emerging evidence has reported that miRNAs can mediate a series of important biological processes and diseases, such as cell proliferation, differentiation and apoptosis, as well as fibrosis16. Dysregulation of miRNAs expression has been reported to enhance the sensitivity of anticancer agents in various types of cancers17, 18, 19. MiR-142-3p was initially identified in hematopoietic cells as an oncogenic biomarker for T cell acute lymphoblastic leukemia20. Additionally, miR-142-3p is widely reported to act as a tumor suppressor, involved in tumorigenesis, tumor growth, and invasiveness21, 22, 23, 24, 25.
High-mobility group box 1 (HMGB1) is a non-histone, nuclear DNA binding protein that belongs to the HMGB superfamily. HMGB1 has been found to participate in the organization of DNA and gene transcription and play a role in several cellular processes, including inflammation, cell differentiation, and tumor cell migration26,27. Meanwhile, inhibiting HMGB1 expression can break telomere homeostasis, suppress the repair of DNA damage, and thus increase the sensitivity of human breast cancer cells28,29. Therefore, HMGB1 might be a critical gene promoting chemoresistance of breast cancer cells. To explore the regulatory role of miR-142-3p in autophagy and chemoresistance, we investigated the function of miR-142-3p in chemoresistance of MCF-7 cells and the relationship between miR-142-3p and HMGB1 to rule out the potential mechanism of multidrug resistance.
2. Materials and methods
2.1. Materials
Five-week-old female BALB/c nude mice were obtained from the Vital River Laboratories (Beijing, China; the license number: SCXK 2016-0002). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were bought from Gibco (Grand Island, NY, USA). Penicillin, streptomycin, annexin V, propidium iodide (PI), DOX, 3-(4,5-dimethylthia-zol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), and 12-hydroxylauric acid were products of Sigma–Aldrich (St. Louis, MO, USA). TRIzol reagent and Lipofectamine 2000 were from Invitrogen (Carlsbad, CA, USA). MiR-142-3p mimics, miR-142-3p inhibitor, corresponding negative control (miRNA control), siRNA specially targeting HMGB1 (si-HMGB1), pcDNA-HMGB1, and matched negative control (si-NC, pcDNA) were purchased from Genepharma (Shanghai, China). The bicinchoninic acid assay (BCA) kit was from Thermo (San Diego, CA, USA). QuantiTect® reverse transcription kit (Qiagen, Hilden, Germany), pGL3 luciferase vector, and luciferase reporter assay kit were from Promega Corporation (Madison, WI, USA). SuperSignal™ reagent was from Thermo Fisher (Rockford, IL, USA). The primary antibodies used were anti-HMGB1 (Abcam, Cambridge, MA, USA), anti-autophagy-related protein 5 (ATG5) (Abcam), anti-LC3B (Sigma–Aldrich), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology, Santa Cruz, CA, USA). The HRP-conjugated secondary antibodies were from Proteintech Group (Chicago, IL, USA).
2.2. Cell lines
Human breast cancer cell line MCF-7 was purchased from Cell Bank of the Chinese Academy of Sciences (Shanghai, China). DOX-resistant MCF cell line (MCF/DOX) was established from parental cells MCF-7 by exposure to gradually increasing concentrations of DOX. All cells were incubated in DMEM containing 10% FBS and 1% penicillin–streptomycin in a humidified incubator at 37 °C with 5% CO2. MCF/DOX cells were cultured in culture medium with 3 μmol/L DOX to maintain drug resistance phenotype.
2.3. MTT assay
Cell viability and survival rates were estimated using MTT assay. For each cell line, the cells were seeded at 2500 cells per well in 96-well plates and incubated for 24 h. Then cells were treated with horizontal dilutions of anticancer agents DOX for 72 h. Subsequently, 20 μL MTT solution (5.0 mg/mL in phosphate-buffered saline, PBS) was added and the plates were incubated for another 4 h at 37 °C. Then the cultured medium was discarded, and 100 μL DMSO was used to dissolve the purple formazan crystals for 10 min. Absorbance at 490 nm was measured by the Mithras2 LB 943 multimode reader (Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany). The untreated cells were used as control. The cell viability ratio was calculated by Eq. (1):
| (1) |
Cytotoxicity was expressed as concentration of DOX inhibiting cell growth by 50% (IC50).
2.4. Apoptosis assay
Apoptotic cells were detected by initially staining with annexin V and PI solution followed by flow cytometry analysis. Following treatment, the cells were harvested and washed twice with PBS. The cell pellet was resuspended with 100 μL annexin V-binding buffer and 5 μL annexin V-FITC/PI for 30 min at 4 °C in the dark. Then, labeled cells were analyzed by a flow cytometer (Partec PAS, Partec GmbH, Gorlitz, Germany).
2.5. Bioinformatics prediction and dual-luciferase reporter assay
For the luciferase reporter experiments, the wild-type (WT) of the 3′ UTR segment of the HMGB1 gene containing the miR-142-3p binding sequences was amplified by PCR from human genomic DNA. To examine whether miR-142-3p regulates the expression of HMGB1, the wild type of HMGB1 containing the putative miR-142-3p target binding sites and its mutant were synthesized and inserted into 3′ UTR of firefly luciferase gene in the pGL3 luciferase vector. The cells were then co-transfected with the WT or mutant type (MT) pGL3-HMGB1-3′ UTR combined with miR-NC, miR-142-3p, anti-miR-NC or anti-miR-142-3p by Lipofectamine 2000. The cells were harvested 24 h after transfection, then luciferase activity was measured using a dual luciferase reporter assay kit and normalized against Renilla luciferase activity.
2.6. Cell transfection
Cells were seeded into 12-well plates and incubated at 37 °C for 24 h to reach approximately 70% confluence. Subsequently, the cells were transfected using Lipofectamine 2000 agent according to the instructions. The cells were then incubated at 37 °C with 5% CO2 for 6 h. Subsequently, the medium in each well was replaced by DMEM with 10% FBS and incubated for 48 h. Transfection efficiencies were determined by real time quantitative reverse transcription-PCR and Western blotting.
2.7. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from adherent cells using TRIzol reagent. A total of 500 ng of total RNA was reverse transcribed to cDNA using QuantiTect® reverse transcription kit according to the manufacturer's instructions. In brief, 1 μg template RNA, 2 μL 7×DNA Wipeout buffer and diethyl pyrocarbonate (DEPC) treated water in a final volume of 14 μL were mixed. After incubation at 42 °C for 2 min, they were chilled on ice. Then 1 μL of Quantiscript reverse transcriptase, 4 μL of 5×Quantiscript RT buffer, and 1 μL of 1×RT primer mix were added, and the tube was incubated at 42 °C for 15 min. Finally, the tube was incubated for 3 min at 95 °C to inactivate Quantiscript reverse transcriptase. The cDNA samples were stored at −20 °C until use.
Real-time quantitative PCR was performed on the LightCycler 480 real-time PCR system (Roche Applied Science, Mannheim, Baden-Wuerttemberg, Germany). The primer sequences used in the PCR were as Table 1. The reaction mixture of a 10 μL final volume contained 1 μmol/L primer each, 5 μL of 2×SYBR Green Master, and 4 μL of cDNA. The conditions of PCR protocols were as follows: the initial template was activated at 95 °C for 5 min, followed by 50 cycles at various temperatures/times (95 °C for 15 s, 65 °C for 30 s, and 72 °C for 1 min). The gene expression level was normalized the Ct values to the internal control GAPDH and was calculated in percentage relative to the untreated cells.
Table 1.
Primers of RT-PCR.
| Name | Sequence (5′–3′) |
|---|---|
| HMGB1 Forward | GCTGACAAGGCTCGTTATGAA |
| HMGB1 Reverse | CCTTTGATTTTGGGG CGGTA |
| miR-142-3p Forward | TGCGGTGTAGTGTTTCCTACTT |
| miR-142-3p Reverse | CCAGTGCAGGGTCCGAGGT |
| GAPDH Forward | CCCTCAACGACCACTTTGTC |
| GAPDH Reverse | AGGGGAGATTCAGTGTGGTG |
2.8. Western blotting assay
For isolation of total protein fractions, cells were collected, washed with ice-cold PBS, and lysed using RIPA lysis buffer. Protein concentration was determined using BCA protein quantitation kit. Total protein samples were loaded into the wells of 12.5% SDS-polyacrylamide gel for electrophoresis gel, along with molecular weight marker and transferred onto nitrocellulose membrane. Membranes were blocked at room temperature for 30 min with TBST containing 5% dry milk. Then membranes were incubated with specific primary antibodies: anti-HMGB1 (1:500, 25 kDa), anti-ATG5 (1:500, 32 kDa), anti-LC3B (1:1000, 16 kDa LC3-II and 18 kDa LC3-I), and anti-GAPDH (1:10,000, 36 kDa). The SuperSignal™ reagent was used to visualize the signals after the HRP-conjugated secondary antibodies were bound to primary antibodies.
2.9. Tumor xenografts
Six-week-old female nude athymic BALB/c nu/nu mice were used for xenograft studies. The animal experiment was performed in accordance with Institutional Guidelines and the Guide for the Care and Use of Laboratory Animals (NIH publication No. 85-23, revised 1996). A total of 100 μL miR-NC or stably overexpressing miR-142-3p MCF-7 cells (6 × 106) were implanted subcutaneously into the right flank regions of the mice. Seven days later, the mice were divided into miR-NC + PBS (n = 6); miR-142-3p + PBS (n = 6); miR-NC + DOX (n = 6); miR-142-3p + DOX (n = 6). The treatment of mice was carried out intravenously (through tail vein) every 3 days for 2 weeks (total of 4 applications) using 5 μg/g of body weight of DOX. The mice were killed after 30 days later and the tumors were dissected out. Tumor volume was calculated using the following standard formula Eq. (2):
| (2) |
2.10. Statistical analysis
The data were expressed as means ± standard deviations (SD). An analysis of ANOVA variance with a Tukey post hoc test was used for multiple comparisons. Correlation was calculated using the function ReglinP function and inverted Student's t-test. All statistics were calculated using the STATISTICA program (StatSoft Inc., Tulsa, Oklahoma, USA). A P value < 0.05 was considered as significant.
3. Results
3.1. Characteristics of MCF-7 and MCF-7/DOX cells
To further confirm the DOX resistance towards MCF-7 and MCF-7/DOX cells, the MCF-7 and MCF-7/DOX cells were treated with different concentrations of DOX (0.1625, 0.3125, 0.625, 1.25, 2.5, and 5 μmol/L) for 48 h, respectively. MCF-7 cells showed higher sensitivity to DOX compared with MCF-7/DOX cells (Fig. 1A). Additionally, HMGB1 had higher expression in MCF-7/DOX cells compared with MCF-7 cells (Fig. 1B and C). Furthermore, the levels of ATG5 and the accumulation of LC3-II from LC3-I conversion were substantially improved in MCF-7/DOX cells compared with MCF-7 cells (Fig. 1D). Taken together, these results indicated that HMGB1, and autophagy may be implicated in the development of drug resistance.
Figure 1.

The levels of HMGB1 and autophagy-related proteins in human drug-resistant breast cancer cell line MCF-7/DOX and their parental cell line MCF-7. (A) Cell survival rates were detected by MTT assay in MCF-7 and MCF-7/DOX treated with various concentrations (0.1625, 0.3125, 0.625, 1.25, 2.5, and 5 μmol/L) of DOX for 48 h. The levels of HMGB1 (B and C) and autophagy-related proteins (ATG5, LC3-I, and LC3-II, D) in MCF-7 and MCF-7/DOX cells were detected by Western blot and qRT-PCR, respectively. Columns show the mean values of three experiments (±SD). *P < 0.05 and #P < 0.05.
3.2. The effect of miR-142-3p on MCF-7 and MCF-7/DOX cells
To investigate the role of miR-142-3p in chemoresistance of breast cancer cells, the expression level of miR-142-3p was measured in parental MCF-7 and DOX-resistant MCF-7/DOX cells. The results demonstrated that miR-142-3p expression significantly decreased in MCF-7/DOX cells compared with parental MCF-7 cells (Fig. 2A). Then, we enforced miR-142-3p expression in MCF-7/DOX cells and knocked down its expression in MCF-7 cells (Fig. 2B and C).
Figure 2.

The effect of miR-142-3p on MCF-7 and MCF-7/DOX cells. (A) qRT-PCR analysis of the relative miR-142-3p expression in parental MCF-7 and DOX-resistant MCF-7/DOX cells. qRT-PCR analysis of relative miR-142-3p expression in MCF-7 cells transfected with anti-miR-142-3p (B) or MCF-7/DOX cells transfected with miR-142-3p mimic (C). Columns show the mean values of three experiments (±SD). *P < 0.05.
To identify the chemosensitivity of MCF-7/DOX and their parental MCF-7 cells to DOX, MTT assay and flow cytometry were employed to determine the cell viability and apoptosis in anti-miR-142-3p-transfected MCF-7 cells and miR-142-3p-transfected MCF-7/DOX cells after treatment with different doses of DOX for 48 h. As demonstrated by MTT assay, depletion of miR-142-3p dramatically improved cell viability and DOX resistance in MCF-7 cells compared with anti-miR-NC group (Fig. 3A). Moreover, miR-142-3p overexpression led to a marked decrease of cell viability and DOX resistance in MCF-7/DOX cells when compared with miR-NC-transfected cells (Fig. 3B). Additionally, inhibition of miR-142-3p remarkably abated DOX-induced apoptosis in MCF-7 cells and overexpression of miR-142-3p strengthened DOX-induced apoptosis in MCF-7/DOX cells (Fig. 3C and D). Therefore, above results indicated that miR-142-3p overexpression improved drug sensitivity in MCF-7/DOX cells by inhibiting cell viability and promoting apoptosis.
Figure 3.

MiR-142-3p up-regulation sensitized breast cancer cells to DOX. Transfected MCF-7 (A) and MCF-7/DOX cells (B) were treated with different doses of DOX (0.1625, 0.3125, 0.625, 1.25, 2.5 and 5 μmol/L) for 48 h, respectively, then MTT assay was used to examine the cell viability and IC50 value of DOX. Transfected MCF-7 (C) and MCF-7/DOX cells (D) were treated with or without DOX for 48 h, followed by the assessment of apoptotic rate by flow cytometry, respectively. Columns show the mean values of three experiments (±SD). *P < 0.05 and #P < 0.05.
3.3. MiR-142-3p targets HMGB1 in breast cancer cells
Bioinformatics analysis was performed to predict the potential target genes for miR-142-3p. HMGB1 was identified to be a candidate target of miR-142-3p and include the binding sites for the seed region of miR-142-3p (Fig. 4A). To confirm whether miR-142-3p could directly target HMGB1, the vectors containing the WT or MT of HMGB1 3′ UTR binding sites for miR-142-3p were constructed with in MCF-7 and MCF-7/DOX cells. As shown in Fig. 4B and C, down-regulation of miR-142-3p prominently promoted the luciferase activity of HMGB1-WT reporter in MCF-7 cells, while enforced expression of miR-142-3p substantially attenuated the luciferase activity of HMGB1-WT reporter in MCF-7/DOX cells. However, these effects were not observed in the mutated HMGB1-MT groups in both MCF-7 and MCF-7/DOX cells, suggesting that HMGB1 is the target gene of miR-142-3p.
Figure 4.

MiR-142-3p inhibited HMGB1 expression by binding to its 3′ UTR. (A) Diagram of putative miR-142-3p binding sequence in HMGB1 3′ UTR and its mutant in luciferase reporter assay. Luciferase reporter assay was performed to measure luciferase activity in MCF-7 (B) or MCF-7/DOX cells (C), respectively. Western blot analysis of HMGB1 protein levels in MCF-7 (D) and MCF-7/DOX cells (E). Columns show the mean values of three experiments (±SD). *P < 0.05.
Subsequently, Western blot analysis was performed to examine the protein level of HMGB1. As shown in Fig. 4D and E, transfection with miR-142-3p inhibitor upregulated HMGB1 expression in MCF-7 cells, whereas transfection with miR-142-3p mimic decreased HMGB1 expression. Above all, miR-142-3p suppressed HMGB1 expression in breast cancer cells by binding to its 3′ UTR.
3.4. HMGB1 overexpression induces DOX resistance in breast cancer cells
Furthermore, we investigated the underlying mechanism of HMGB1 by up-regulating HMGB1 in MCF-7 cells (Fig. 5A) and silencing HMGB1 in MCF-7/DOX cells (Fig. 5B). As shown in Fig. 5C, overexpression of HMGB1 improved cell viability and DOX resistance in MCF-7 cells. Moreover, down-regulating HMGB1 lowered cell viability and DOX resistance in MCF-7/DOX cells compared with control group (Fig. 5E). Simultaneously, the overexpression of HMGB1 inhibited the DOX-induced apoptosis in MCF-7 cells (Fig. 5D), while following the knockdown of HMGB1 enhanced apoptosis in MCF-7/DOX cells (Fig. 5F). Collectively, HMGB1 overexpression facilitated the development of DOX resistance in breast cancer cells.
Figure 5.

HMGB1 overexpression induced DOX resistance in breast cancer cells. Western blot analysis of HMGB1 protein levels in transfected MCF-7 (A) and MCF-7/DOX cells (B). (C) MTT assay was used to assess the cell viability and DOX IC50 value in pcDNA- or pcDNA-HMGB1-transfected MCF-7 cells after treatment with different doses of DOX (0.1625, 0.3125, 0.625, 1.25, 2.5 and 5 μmol/L) for 48 h. (D) Flow cytometry analysis of apoptosis in pcDNA- or pcDNA-HMGB1-transfected MCF-7 cells with or without DOX treatment. (E) MTT assay was applied to determine the cell viability and DOX IC50 value in si-NC- or siHMGB1-transfected MCF-7/DOX cells after treatment with different doses of DOX (0.1625, 0.3125, 0.625, 1.25, 2.5 and 5 μmol/L) for 48 h. (F) Flow cytometry analysis of apoptosis in si-NC or si-HMGB1-transfected MCF-7/DOX cells with or without DOX treatment. Columns show the mean values of three experiments (±SD). *P < 0.05 and #P < 0.05.
3.5. MiR-142-3p overexpression enhanced drug sensitivity of breast cancer cells through inhibiting autophagy related proteins expression by targeting HMGB1
To investigate the mechanism of interaction between miR-142-3p and HMGB1 regulating drug sensitivity, MCF-7 cells were transfected with anti-miR-NC, anti-miR-142-3p, or anti-miR-142-3p + si-HMGB1, and MCF-7/DOX cells were treated with miR-NC, miR-142-3p, or miR-142-3p + pcDNA-HMGB1, respectively. Our results demonstrated that silencing HMGB1 remarkably reversed anti-miR-142-3p-induced DOX resistance in MCF-7 cells (Fig. 6A), while HMGB1 overexpression greatly weakened miR-142-3p-elicted DOX sensitivity in MCF-7/DOX cells (Fig. 6B). Additionally, knockdown of HMGB1 weakened the anti-apoptotic effect mediated by anti-miR-142-3p in MCF-7 cells under DOX treatment (Fig. 6C). Furthermore, HMGB1 overexpression reversed the promotion of apoptosis by miR-142-3p in MCF-7/DOX cells with DOX treatment (Fig. 6D). Collectively, these results indicated that miR-142-3p increased the sensibility of breast cancer cells to DOX by targeting HMGB1.
Figure 6.

MiR-142-3p upregulation enhanced drug sensitivity of breast cancer cells through suppressing autophagy by targeting HMGB1. Transfected MCF-7 (A) and MCF-7/DOX cells (B) were treated with different doses of DOX (0.1625, 0.3125, 0.625, 1.25, 2.5 and 5 μmol/L) for 48 h, respectively, then MTT assay was used to determine the cell viability and IC50 value. Transfected MCF-7 (C) and MCF-7/DOX cells (D) were treated with 0.15 or 3 μmol/L DOX for 48 h, respectively, followed by the detection of apoptotic rate by flow cytometry. (E) The levels of ATG5, LC3-I and LC3-II in MCF-7 cells transfected with anti-miR-NC, anti-miR-142-3p, or anti-miR-142-3p + si-HMGB1 were detected by Western blot. (F) The levels of ATG5, LC3-I, and LC3-II in MCF-7/DOX cells transfected with miR-NC, miR-142-3p, or miR-142-3p + pcDNA-HMGB1 were detected by Western blot. Columns show the mean values of three experiments (±SD). *P < 0.05.
Then, the levels of autophagy related proteins in MCF-7/DOX cells were detected by Western blotting. As presented in Fig. 6E, the levels of ATG5 and LC3-II/LC3-I increases significantly after MCF-7 cells were transfected with miR-142-3p inhibitor, while this effect was counteracted by HMGB1 down-regulation. MiR-142-3p overexpression strikingly reduced the levels of autophagy-related proteins ATG5 and LC3-II/LC3-I in MCF-7/DOX cells, while HMGB1 overexpression effectively recuperated miR-142-3p-induced decrease of ATG5 and LC3-II/LC3-I levels (Fig. 6F). Taken together, these results indicated that miR-142-3p upregulation enhanced drug sensitivity of breast cancer cells through suppressing autophagy by targeting HMGB1.
3.6. MiR-142-3p inhibited autophagy and sensitized MCF-7 cells to DOX in vivo
The animals were implanted with MCF-7 overexpression miR-142-3p stable cells or control cells and injected DOX or PBS. Importantly, compared with control group, the combination of miR-142-3p and DOX can significantly decrease the tumor volume and weight (Fig. 7A–C). Meanwhile, real time PCR results indicated DOX treatment decreased the level of miR-42-3p in the tumor, while miR-142-3p overexpression abolished the inhibitory effect of DOX (Fig. 7D). Western blotting results demonstrated that treatment with DOX significantly increased ATG5 and HMGB1 protein expression and induced higher transfer of LC3-I to LC3-II in tumors whereas the combination of miR-142-3p and DOX group exhibited lower ATG5 and HMGB1 expression and decreased transfer of LC3-I to LC3-II than only treatment with DOX group (Fig. 7E). In total, miR-142-3p overexpression improved the therapeutic effect of DOX by regulating HMGB1.
Figure 7.

Overexpression of miR-142-3p enhanced the therapeutic effect of DOX by inhibiting autophagy in vivo. (A) Tumor volumes were detected in different treatment groups. The excised tumor masses were photographed (B) and weighed (C). (D) The levels of miR-142-3p levels were detected in tumors. (E) The levels of autophagy related proteins were measured in tumors. Columns show the mean values of three experiments (±SD). *P < 0.05 and #P < 0.05.
4. Discussion
DOX is the most commonly used anti-cancer drug in the treatment of a variety of cancers. However, the development of resistance and cardiotoxicity of doxorubicin is a common event in clinic use. Previous studies indicate that the development of drug resistance is closely associated with altered expression of miRNAs in cancer cells30. MiRNAs are widely considered as crucial regulators in apoptosis, cell cycle progression, growth, proliferation and drug resistance in human cancers31,32. For breast cancer therapy, a series of miRNAs has been reported to be involved in the modulation of chemosensitivity, such as miR-216b33, microRNA-708-3p34, and miR-129-5p35. MiR-142-3p has been found to participate in malignant cell proliferation and metastasis in many cancer types. For example, miR-142-3p overexpression inhibits cell proliferation in pancreatic cancer23, osteosarcoma cells36, cervical cancer38and colon cancer39. However, the association between miR-142-3p and the DOX resistance remains unclear in breast cancer cells. In the present study, we found that miR-142-3p overexpression could sensitize MCF-7 cells to DOX, while knockdown of miR-142-3p could reduce sensitivity of MCF-7 cells. Further mechanistic study identified that miR-142-3p sensitized MCF-7 cells to DOX through inhibiting autophagy related proteins expression by targeting HMGB1 in vitro and in vivo.
HMGB1 acts as a typical regulator of autophagy and apoptosis, which can regulate inflammation, cell differentiation, cell migration, and tumor progression23,36. HMGB1 has been found to be a direct target of miR-142-3p in non-small-cell lung carcinoma37, cardiomyocytes38, and osteoarthritis39. Subsequently, we further revealed the potential mechanism of miR-142-3p in enhancing the breast cancer cells sensitivity to DOX. By bioinformatics analysis and luciferase reporter experiment, we found that HMGB1 was identified to act as a direct target of miR-142-3p. HMGB1 is overexpressed in many types of cancer40,41 including breast cancer42. In the current study, HMGB1 was highly expressed at both mRNA and protein levels in MCF-7/DOX cells compared with their parental MCF-7 cells. Recent reports have revealed that HMGB1 contributed to drug resistance of tumors during autophagy43. Furthermore, we found that HMGB1 overexpression lowered DOX sensitivity and weakened DOX-induced apoptosis in MCF-7 cells, while silencing HMGB1 enhanced DOX sensitivity and promoted DOX-induced apoptosis in MCF-7/DOX cells. Further studies revealed that anti-miR-142-3p-induced DOX resistance was reversed by silencing HMGB1 in MCF-7 cells, and miR-142-3p-elicted DOX responsiveness was impaired following HMGB1 overexpression in MCF-7/DOX cells. All these results indicated that miR-142-3p enforced the re-sensitization of breast cancer cells by targeting HMGB1.
Autophagy is a process of cell self-digestion, which maintains cellular homeostasis through degrading and reusing those unwanted or dysfunctional components in cells44. Autophagy plays dual roles in tumor development. Autophagy acts as a suppressor by clearing mutant or misfolded proteins and alleviating cellular stress at the beginning of tumor development, on the other hand, autophagy enables tumor cells to form a barrier to treatment with chemotherapeutic drugs under stressful conditions6. Chaachouay et al.45 reported that autophagy promotes the resistance of breast cancer cells to ionizing radiation. Thus, we speculated inhibition of autophagy might overcome drug resistance and be beneficial to cancer therapy. As expected, our results showed that treatment with DOX resulted in the increase of autophagy activity. MiR-142-3p overexpression dramatically sensitized the ability of DOX to induced cell apoptosis by inhibiting autophagy. Increasing evidence demonstrates that miR-142-3p was able to regulate autophagy and drug resistance by directly binding to HMGB1 target46. Furthermore, restoration of miR-142-3p increased the DOX sensitivity and significantly reduced the growth of doxorubicin-resistant human breast cancer xenografts through targeting HMGB1 in vitro and in vivo.
5. Conclusions
These results show that miR-142-3p can suppress autophagy and enhance sensitivity of breast cancer cells to DOX by targeting HMGB1. Meanwhile, we have already explored the functions of miR-142-3p on the cellular and animal models, but further studies are still required to focus on the clinical samples. Moreover, the intensive mechanistic study of miR-142-3p regulating autophagy through targeting HMGB1 will be implemented in the following experiments. Additionally, the attendant limitations of the present study will be overcome in the future. In conclusion, the miR-142-3p/HMGB1 axis can induce cell apoptosis by regulating the autophagy and might be an important pathway regulating the sensitivity of breast cancer cells.
Acknowledgments
The authors gratefully acknowledge the financial support by National Natural Science Foundation of China (Nos. 81330007 and U1601227), the Science and Technology Programs of Guangdong Province (Nos. 2014A050503047 and 2015B020225006, China), National Natural Science Foundation of China (81700382).
Author contributions
Lu Liang, Jijun Fu and Siran Wang participated in the most of the experiments. Huiyu Cen, Lingmin Zhang and Safur Rehman Mandukhail performed cell proliferation assay, cell transfection, RT-PCR and Western blotting assay. Lingran Du and Qianni Wu performed the animal studies. Xiyong Yu and Peiquan Zhang conceived and supervised the project. Lu Liang wrote the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Contributor Information
Peiquan Zhang, Email: pqzhang@gzhmu.edu.cn.
Xiyong Yu, Email: yuxycn@aliyun.com.
References
- 1.Siegel R., Ma J., Zou Z., Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Shioi Y., Kashiwaba M., Inaba T., Komatsu H., Sugai T., Wakabayashi G. Long-term complete remission of metastatic breast cancer, induced by a steroidal aromatase inhibitor after failure of a non-steroidal aromatase inhibitor. Am J Case Rep. 2014;15:85–89. doi: 10.12659/AJCR.890023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rapoport B.L., Demetriou G.S., Moodley S.D., Benn C.A. When and how do I use neoadjuvant chemotherapy for breast cancer? Curr Treat Options Oncol. 2014;15:86–98. doi: 10.1007/s11864-013-0266-0. [DOI] [PubMed] [Google Scholar]
- 4.Sharifi S., Barar J., Hejazi M.S., Samadi N. Doxorubicin changes Bax/Bcl-xL ratio, Caspase-8 and 9 in breast cancer cells. Adv Pharmaceut Bull. 2015;5:351–359. doi: 10.15171/apb.2015.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gottesman M.M. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 6.Yang Z.J., Chee C.E., Huang S., Sinicrope F.A. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buyuklu M., Kandemir F.M., Ozkaraca M., Set T., Bakirci E.M., Topal E. Protective effect of curcumin against contrast induced nephropathy in rat kidney: what is happening to oxidative stress, inflammation, autophagy and apoptosis?. Eur Rev Med Pharmacol Sci. 2014;18:461–470. [PubMed] [Google Scholar]
- 8.Chang Y., Yan W., He X., Zhang L., Li C., Huang H. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology. 2012;143:177–187. doi: 10.1053/j.gastro.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 9.McAlpine F., Williamson L.E., Tooze S.A., Chan E.Y. Regulation of nutrient-sensitive autophagy by uncoordinated 51-like kinases 1 and 2. Autophagy. 2013;9:361–373. doi: 10.4161/auto.23066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang P., Zhang J., Zhang L., Zhu Z., Fan J., Chen L. MicroRNA 23b regulates autophagy associated with radioresistance of pancreatic cancer cells. Gastroenterology. 2013;145:1133–1143. doi: 10.1053/j.gastro.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 11.Maycotte P., Aryal S., Cummings C.T., Thorburn J., Morgan M.J., Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–212. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McAfee Q., Zhang Z., Samanta A., Levi S.M., Ma X.H., Piao S. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109:8253–8258. doi: 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z., Wang N., Liu P., Chen Q., Situ H., Xie T. MicroRNA-25 regulates chemoresistance-associated autophagy in breast cancer cells, a process modulated by the natural autophagy inducer isoliquiritigenin. Oncotarget. 2014;5:7013–7026. doi: 10.18632/oncotarget.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Q., Liu T., Yuan Y., Guo Z., Xie G., Du S. MiR-200c inhibits autophagy and enhances radiosensitivity in breast cancer cells by targeting UBQLN1. Int J Cancer. 2015;136:1003–1012. doi: 10.1002/ijc.29065. [DOI] [PubMed] [Google Scholar]
- 15.Flynt A.S., Lai E.C. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet. 2008;9:831–842. doi: 10.1038/nrg2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Croce C.M., Calin G.A. miRNAs, cancer, and stem cell division. Cell. 2005;122:6–7. doi: 10.1016/j.cell.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 17.Chen M.J., Wu D.W., Wang G.C., Wang Y.C., Chen C.Y., Lee H. MicroRNA-630 may confer favorable cisplatin-based chemotherapy and clinical outcomes in non-small cell lung cancer by targeting Bcl-2. Oncotarget. 2018;9:13758–13767. doi: 10.18632/oncotarget.24474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun F.D., Wang P.C., Luan R.L., Zou S.H., Du X. MicroRNA-574 enhances doxorubicin resistance through down-regulating SMAD4 in breast cancer cells. Eur Rev Med Pharmacol Sci. 2018;22:1342–1350. doi: 10.26355/eurrev_201803_14476. [DOI] [PubMed] [Google Scholar]
- 19.Min A., Zhu C., Peng S., Rajthala S., Costea D.E., Sapkota D. MicroRNAs as important players and biomarkers in oral carcinogenesis. BioMed Res Int. 2015;2015:186904. doi: 10.1155/2015/186904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lv M., Zhang X., Jia H., Li D., Zhang B., Zhang H. An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-alpha and cAMP/PKA pathways. Leukemia. 2012;26:769–777. doi: 10.1038/leu.2011.273. [DOI] [PubMed] [Google Scholar]
- 21.Chen H.H., Huang W.T., Yang L.W., Lin C.W. The PTEN-AKT-mTOR/RICTOR pathway in nasal natural killer cell lymphoma is activated by miR-494-3p via PTEN but inhibited by miR-142-3p via RICTOR. Am J Pathol. 2015;185:1487–1499. doi: 10.1016/j.ajpath.2015.01.025. [DOI] [PubMed] [Google Scholar]
- 22.Wu L., Cai C., Wang X., Liu M., Li X., Tang H. MicroRNA-142-3p, a new regulator of RAC1, suppresses the migration and invasion of hepatocellular carcinoma cells. FEBS Lett. 2011;585:1322–1330. doi: 10.1016/j.febslet.2011.03.067. [DOI] [PubMed] [Google Scholar]
- 23.MacKenzie T.N., Mujumdar N., Banerjee S., Sangwan V., Sarver A., Vickers S. Triptolide induces the expression of miR-142-3p: a negative regulator of heat shock protein 70 and pancreatic cancer cell proliferation. Mol Cancer Ther. 2013;12:1266–1275. doi: 10.1158/1535-7163.MCT-12-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiou G.Y., Chien C.S., Wang M.L., Chen M.T., Yang Y.P., Yu Y.L. Epigenetic regulation of the miR142-3p/interleukin-6 circuit in glioblastoma. Mol Cell. 2013;52:693–706. doi: 10.1016/j.molcel.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 25.Wang X.S., Gong J.N., Yu J., Wang F., Zhang X.H., Yin X.L. MicroRNA-29a and microRNA-142-3p are regulators of myeloid differentiation and acute myeloid leukemia. Blood. 2012;119:4992–5004. doi: 10.1182/blood-2011-10-385716. [DOI] [PubMed] [Google Scholar]
- 26.Gruber H.E., Hoelscher G.L., Bethea S., Ingram J., Cox M., Hanley E.N., Jr. High-mobility group box-1 gene, a potent proin-lammatory mediators, is upregulated in more degenerated human discs in vivo and its receptor upregulated by TNF-α exposure in vitro. Exp Mol Pathol. 2015;98:427–430. doi: 10.1016/j.yexmp.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Chen M., Liu Y., Varley P., Chang Y., He X.X., Huang H. High-mobility group box-1 promotes hepatocellular carcinoma progression through miR-21-mediated matrix metalloproteinase activity. Cancer Res. 2015;75:1645–1656. doi: 10.1158/0008-5472.CAN-14-2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amornsupak K., Insawang T., Thuwajit P., O-Charoenrat P., Eccles S.A., Thuwajit C. Cancer-associated fibroblasts induce high mobility group box 1 and contribute to resistance to doxorubicin in breast cancer cells. BMC Canc. 2014;14:955. doi: 10.1186/1471-2407-14-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ke S., Zhou F., Yang H., Wei Y., Gong J., Mei Z. Downregulation of high mobility group box 1 modulates telomere homeostasis and increases the radiosensitivity of human breast cancer cells. Int J Oncol. 2015;46:1051–1058. doi: 10.3892/ijo.2014.2793. [DOI] [PubMed] [Google Scholar]
- 30.Zheng T., Wang J., Chen X., Liu L. Role of microRNA in anticancer drug resistance. Int J Cancer. 2010;126:2–10. doi: 10.1002/ijc.24782. [DOI] [PubMed] [Google Scholar]
- 31.Giovannetti E., Erozenci A., Smit J., Danesi R., Peters G.J. Molecular mechanisms underlying the role of microRNAs (miRNAs) in anticancer drug resistance and implications for clinical practice. Crit Rev Oncol Hematol. 2012;81:103–122. doi: 10.1016/j.critrevonc.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 32.O'Bryan S., Dong S., Mathis J.M., Alahari S.K. The roles of oncogenic miRNAs and their therapeutic importance in breast cancer. Eur J Cancer. 2017;72:1–11. doi: 10.1016/j.ejca.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Zou J., Kuang W., Hu J., Rao H. miR-216b promotes cell growth and enhances chemosensitivity of colorectal cancer by suppressing PDZ-binding kinase. Biochem Biophys Res Commun. 2017;488:247–252. doi: 10.1016/j.bbrc.2017.03.162. [DOI] [PubMed] [Google Scholar]
- 34.Lee J.W., Guan W., Han S., Hong D.K., Kim L.S., Kim H. MiR-708-3p mediates metastasis and chemoresistance through inhibition of epithelial-to-mesenchymal transition in breast cancer. Cancer Sci. 2018;109:1404–1413. doi: 10.1111/cas.13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu X., Ma J., Chu J., Shao Q., Zhang Y., Lu G. MiR-129-5p sensitizes the response of Her-2 positive breast cancer to trastuzumab by reducing Rps6. Cell Physiol Biochem. 2017;44:2346–2356. doi: 10.1159/000486122. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y.Q., Qi J., Xu J.Q., Hao P. MicroRNA-142-3p, a novel target of tumor suppressor menin, inhibits osteosarcoma cell proliferation by down-regulation of FASN. Tumour Biol. 2014;35:10287–10293. doi: 10.1007/s13277-014-2316-z. [DOI] [PubMed] [Google Scholar]
- 37.Xiao P., Liu W.L. MiR-142-3p functions as a potential tumor suppressor directly targeting HMGB1 in non-small-cell lung carcinoma. Int J Clin Exp Pathol. 2015;8:10800–10807. [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y., Ouyang M., Wang Q., Jian Z. MicroRNA-142-3p inhibits hypoxia/reoxygenation-induced apoptosis and fibrosis of cardiomyocytes by targeting high mobility group box 1. Int J Mol Med. 2016;38:1377–1386. doi: 10.3892/ijmm.2016.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X., Guo Y., Wang C., Yu H., Yu X., Yu H. MicroRNA-142-3p inhibits chondrocyte apoptosis and inflammation in osteoarthritis by targeting HMGB1. Inflammation. 2016;39:1718–1728. doi: 10.1007/s10753-016-0406-3. [DOI] [PubMed] [Google Scholar]
- 40.Wild C.A., Brandau S., Lotfi R., Mattheis S., Gu X., Lang S. HMGB1 is overexpressed in tumor cells and promotes activity of regulatory T cells in patients with head and neck cancer. Oral Oncol. 2012;48:409–416. doi: 10.1016/j.oraloncology.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 41.Wu D., Ding Y., Wang S., Zhang Q., Liu L. Increased expression of high mobility group box 1 (HMGB1) is associated with progression and poor prognosis in human nasopharyngeal carcinoma. J Pathol. 2008;216:167–175. doi: 10.1002/path.2391. [DOI] [PubMed] [Google Scholar]
- 42.Flohr A.M., Rogalla P., Meiboom M., Borrmann L., Krohn M., Thode-Halle B. Variation of HMGB1 expression in breast cancer. Anticancer Res. 2001;21:3881–3885. [PubMed] [Google Scholar]
- 43.Liu L., Yang M., Kang R., Wang Z., Zhao Y., Yu Y. DAMP-mediated autophagy contributes to drug resistance. Autophagy. 2011;7:112–114. doi: 10.4161/auto.7.1.14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 45.Chaachouay H., Ohneseit P., Toulany M., Kehlbach R., Multhoff G., Rodemann H.P. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol. 2011;99:287–292. doi: 10.1016/j.radonc.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y., Liu Y., Xu X. Upregulation of miR-142-3p improves drug sensitivity of acute myelogenous leukemia through reducing P-glycoprotein and repressing autophagy by targeting HMGB1. Transl Oncol. 2017;10:410–418. doi: 10.1016/j.tranon.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]