

Abstract
Coronavirus disease-2019 (COVID-19) is a global health emergency and the matter of serious concern, which has been declared a pandemic by WHO. Till date, no potential medicine/ drug is available to cure the infected persons from SARS-CoV-2. This deadly virus is named as novel 2019-nCoV coronavirus and caused coronavirus disease, that is, COVID-19. The first case of SARS-CoV-2 infection in human was confirmed in the Wuhan city of the China. COVID-19 is an infectious disease and spread from man to man as well as surface to man . In the present work, in silico approach was followed to find potential molecule to control this infection. Authors have screened more than one million molecules available in the ZINC database and taken the best two compounds based on binding energy score. These lead molecules were further studied through docking against the main protease of SARS-CoV-2. Then, molecular dynamics simulations of the main protease with and without screened compounds were performed at room temperature to determine the thermodynamic parameters to understand the inhibition. Further, molecular dynamics simulations at different temperatures were performed to understand the efficiency of the inhibition of the main protease in the presence of the screened compounds. Change in energy for the formation of the complexes between the main protease of novel coronavirus and ZINC20601870 as well ZINC00793735 at room temperature was determined on applying MM-GBSA calculations. Docking and molecular dynamics simulations showed their antiviral potential and may inhibit viral replication experimentally.
Communicated by Ramaswamy H. Sarma
Keywords: SARS-CoV-2, ZINC database, inhibitors, docking, MD simulations, MM-GBSA

Introduction
Coronavirus disease-2019 (COVID-19) is a global health emergency and the matter of serious concern, which has been declared as a pandemic by WHO. In the present time, the virus engulfing the entire world is known as new coronavirus or severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) or coronavirus-19 as in Figure 1 (Arshad Ali et al., 2020). This virus results in respiratory illness in humans, that is, severe acute respiratory system (SARS). It may vary from mild to severe in humans (Duan & Zhu, 2020; Mitra et al., 2019). It is believed that the actual reservoir of this virus were bats and then infected another animal and then to humans. The spread of infection in humans during SARS, MERS and COVID-19 is explained in Figure 2. The virus has got its name from Latin word corona which means crown (Agarwal et al., 2020; Ai et al., 2020). First case of coronavirus infection in human was detected in the Wuhan city of China. Those people were working in the sea food market (Bhardwaj et al., 2020).
Figure 1.

(a) General structure of coronavirus and (b) prefusion CoVs spikes.
Figure 2.

Spread of the infection from BAT to humans in SARS, MERS and COVID-19.
This outbreak is causing more than 3,70,657 death and more than 60,40,609 million infections as on 1 June 2020. Further, USA is the most affected nation in the world with 17,34,040 cases and 1,02,640 deaths. Despite the huge population of India (∼130 crores), 1,90,535 are the infected cases and 5394 deaths are reported as on 1 June 2020. The infected persons have respiratory problems and the problem is due to the binding of the virus on angiotensin-converting enzyme-2 (ACE-2) in alveoli of the lungs and control the cell (Ji et al., 2020; Thomas-Ruddel et al., 2020). It released the RNA into the cell and it controls the cell. Then, there is translation for the synthesis of polyproteins and then proteolysis occurs to give the constituents. There is a RNA-dependent RNA polymerase used to give the copies of single-stranded RNA and it binds with constituents of polyproteins to give virus. Then, viruses come out from the cell and start infecting other cells (Babadaei et al., 2020; Borkotoky & Banerjee, 2020; Enmozhi et al., 2020; Muralidharan et al., 2020). With the increase in infection, the liquid in alveoli increases and creates problem in respiration of human as in Figure 3.
Figure 3.

A brief understanding of the attack of the virus to make the person sick.
Further, coronavirus affect the respiratory, gastrointestinal, central nervous system in human being. This virus belongs to the family Coronaviridae in the order Nidovirales have positive-stranded RNA (Lai et al., 2020). It has large genome among the RNA containing viruses (Almazan et al., 2004; Berkhout & van Hemert, 2015; Boursnell et al., 1987). Heterocyclic molecules are popular in biological sciences and are being used as antibacterial, antidiabetic, antifungal, anticancerous, antiviral agents (Kumari et al., 2017; Singh et al., 2010, 2019).
Recently, many research groups are working for the development of promising medicines or drugs for the treatment of infection or symptoms. The 3D crystal structure of main protease of SARS-CoV-2 in complex with an inhibitor N3 (PDB ID: 6LU7) is available on RCSB protein databank and used for the study (Ahmed et al., 2020; Arshad Ali et al., 2020; Boldog et al., 2020; Burley et al., 2018). Computational tools play an important role in understanding the interaction between small molecules and receptor, that is, biomolecule (Rajendran et al., 2018; Sarma et al., 2020; Sharma et al., 2020). In this context, molecular docking and molecular dynamics (MD) simulations play a very crucial role to finding the binding affinity of drug-like molecules with the receptor. It is very much like lock–key model. This approach is used to find the new drug against a target of the research interest. In the molecular docking, the molecule binds to the different sites of the receptor and explains the binding affinity in the form of energy (Purohit, 2014; Singh et al., 2020). Based on it, the best receptor site is screened. It is also used to screen the molecules against a receptor based on the binding affinity. It generates possible conformation or the orientation or pose in which a molecule or compound binds to the receptor (Kumar et al., 2019; Kumari et al., 2014; Singh et al., 2019; Vishvakarma et al., 2013, 2015, 2019). MD simulations is used for simulation of the protein with or without small molecules to understand the dynamic behavior of molecular systems. It is explained in terms of time and taking all the entities in a box for the said simulation times at fixed temperature. It can also be called as isothermal MD simulations. Replica exchange molecular dynamics (REMD) is an interesting approach to investigate the inhibition of the protease in the presence/absence of small molecules on varying the temperature. It is used to study the stability/inhibition on increasing temperature. It is also known as non-isothermal MD simulations (Sarma et al., 2020; Sharma et al., 2020; Singh et al., 2020; Sinha et al., 2020).
This work aims to contribute to the development of a promising inhibitor for main protease of SARS-CoV-2. Authors have screened more than one million molecules available in the ZINC database. Screening of the molecules was done through RASPD, a web server and then, bioactive score and ‘Lipinski’s Rule of Five’ using SwissADME and Molinspiration web server were calculated for the best screened two compounds (Kumar et al., 2019; Lipinski et al., 2012; Mukherjee & Jayaram, 2013). The best two molecules were further studied using molecular docking and MD simulations. MD simulations were performed to determine the binding affinity of small molecules with the main protease of SARS-CoV-2 at room temperature. MD simulations at different temperature were performed to study the impact of temperature for the the formation of the complex. In other words, it can be said to understand the inhibition of the main protease of SARS-CoV-2 using the best two molecules at high temperatures. For a better understanding of the inhibition, different trajectories were used to understand the binding or inhibition of the main protease of the SARS-CoV-2 causing COVID-19.
Experimental details
Three-dimensional structure of the main protease of SARS-CoV-2 (PDB ID: 6LU7) was taken from the Protein Data Bank file (Burley et al., 2018; Lai et al., 2020). Missing atoms were added and water molecules were removed from the main protease using Chimera and Notepad++ (Pettersen et al., 2004). Promising or screened molecules from the ZINC database were saved in .pdb format using Marvin Sketch (Akhmadiev et al., 2019). However, the top two hit molecules were further studied based on bioactive score (Kumar et al., 2019; Lohidakshan et al., 2018; Naz et al., 2020).
Two compounds were used for docking with the main protease of novel coronavirus to create a complex system using ParDOCK web server (Gupta et al., 2007). ParDOCK is a useful online server for understanding the binding affinity of ligand with the receptor and discovering hit molecules against the target protein. The binding affinity of screened molecules against the main protease of coronavirus was obtained in terms of kcal/mol. ParDOCK is a web server to calculate the energy using the docking at the active site as in Equation (1) (Du et al., 2011)
| (1) |
where E is the total non-bonded energy while Eel, EvdW and Ehbb are the energy due to electrostatic interaction, van der Waals interactions and hydrophobic interaction, respectively.
Molecular Dynamics (MD) simulations of the main protease of SARS-CoV-2 with and without screened molecules were performed using pmemd modules of AMBER18 suite by utilizing the Amber ff14SB force field (Jagannadh et al., 1996; Song et al., 2019). Three-dimensional structures of the best two screened molecules from the ZINC database were drawn using Marvin sketch and then they were optimized using Gaussian 09 with the B3LYP/6-31G*. The parameters of the screened compounds for MD simulations were generated using antechamber module of AMBER (Kirby et al., 2018; Zhang et al., 2009). The complex system was solvated using TIP3P 10.0 water box model as an explicit solvent and simulated in truncated octahedron box at 10 Å with periodic boundary conditions (Cai et al., 2018; Cummins et al., 2019; Genheden et al., 2012). Trajectories analyses were performed using the CPPTRAJ modules (Roe & Cheatham, 2013, 2018). Authors analyzed different trajectories like root mean square deviation (RMSD), a root mean square fluctuation (RMSF), hydrogen bonds (HBs) obtained from the MD simulation along with the change in binding free energy calculation using the AMBER18 (Agarwal et al., 2019; Al-Refaei et al., 2020; Cob-Calan et al., 2019; Nazarian et al., 2015; Pola et al., 2018).
RMSD trajectory is used to study structural stability of the protease with and without the compound as in Equation (2), while RMSF trajectory helps to investigate the flexibility in the presence or absence of compound (Cob-Calan et al., 2019). These values were calculated by using the following equations:
| (2) |
where N is the number of atoms, Xm, Ym, Zm are the Cartesian coordinates of the initial structure and X1, Y1, Z1 are the Cartesian coordinates of trajectory at frame t.
These value are calculated as given in Equation (3)
| (3) |
where T is the number of trajectory frames and x̅ is the time-averaged position.
Relative change in binding free energy calculations
Molecular mechanics - Generalized Born solvent accessibility (MM-GBSA) method was used to calculate enthalpy for the formation of complex, number of HBs and relative change in binding free energy are conducted to deeply explore the molecular basis for the binding of complex system with screened molecules. The MM-GBSA method is based on end point method and it is efficient, reliable and broadly used to calculate accurate relative change in binding free energies (Al-Anazi et al., 2018; Balaji & Ramanathan, 2012). This method combines the Generalized Born (GB) electrostatics with molecular mechanics (MM) and solvent accessibility (SA) models or continuum solvent approaches, to estimate binding energies. From trajectories of MD productions, the relative change in enthalpy for the formation of the complex for the MD simulation time of 100 and 10 ns were calculated using Equations (4)–(7) (Bai et al., 2020; Balasubramanian et al., 2019; Bea et al., 2006; Du et al., 2011; Greenidge et al., 2014; Greenidge et al., 2016)
| (4) |
| (5) |
| (6) |
| (7) |
ΔEMM is the change in MM energy; ΔGsolv the solvation free energy; ΔGGB the polar contribution (Madhavaram et al., 2019; Morris et al., 2020).
Results and discussion
RASPD is a web server to screen to get the potential molecules against a receptor. Authors have taken the top 50 molecules as in Table 1 and then the best two molecules were chosen for further study with the main protease of the SARS-CoV-2 as in Table 2. However, scoring of one million small molecule database by RASPD method to identify hits molecules against the main protease of novel coronavirus. The authors used the best two molecules for further studies.
Table 1.
Binding energy of top 50 molecules against the main protease of new coronavirus.
| S. no. | ZINC ID | RASPD (kcal/mol) | S. no. | ZINC ID | RASPD (kcal/mol) |
|---|---|---|---|---|---|
| 1 | ZINC20601870 | −13.9 | 26 | ZINC11785819 | −12.3 |
| 2 | ZINC00793735 | −13.4 | 27 | ZINC11912657 | −12.3 |
| 3 | ZINC02803090 | −13.1 | 28 | ZINC11997969 | −12.3 |
| 4 | ZINC12576410 | −13.1 | 29 | ZINC12023122 | −12.3 |
| 5 | ZINC02060288 | −13 | 30 | ZINC12182905 | −12.3 |
| 6 | ZINC12845408 | −12.9 | 31 | ZINC12191737 | −12.3 |
| 7 | ZINC08680620 | −12.6 | 32 | ZINC12218089 | −12.3 |
| 8 | ZINC02992412 | −12.5 | 33 | ZINC12247286 | −12.3 |
| 9 | ZINC12127094 | −12.5 | 34 | ZINC12283408 | −12.3 |
| 10 | ZINC13475661 | −12.5 | 35 | ZINC14741222 | −12.3 |
| 11 | ZINC16676053 | −12.5 | 36 | ZINC14953594 | −12.3 |
| 12 | ZINC04557820 | −12.4 | 37 | ZINC15003313 | −12.3 |
| 13 | ZINC12152045 | −12.4 | 38 | ZINC19113739 | −12.3 |
| 14 | ZINC12510698 | −12.4 | 39 | ZINC19147957 | −12.3 |
| 15 | ZINC14880667 | −12.4 | 40 | ZINC19731562 | −12.3 |
| 16 | ZINC14987207 | −12.4 | 41 | ZINC20775277 | −12.3 |
| 17 | ZINC14998924 | −12.4 | 42 | ZINC20912417 | −12.3 |
| 18 | ZINC16392797 | −12.4 | 43 | ZINC22033904 | −12.3 |
| 19 | ZINC20138061 | −12.4 | 44 | ZINC02504256 | −12.2 |
| 20 | ZINC20599396 | −12.4 | 45 | ZINC06142347 | −12.2 |
| 21 | ZINC02954911 | −12.3 | 46 | ZINC08869898 | −12.2 |
| 22 | ZINC09777726 | −12.3 | 47 | ZINC09008208 | −12.2 |
| 23 | ZINC09892479 | −12.3 | 48 | ZINC09065379 | −12.2 |
| 24 | ZINC11783287 | −12.3 | 49 | ZINC09601672 | −12.2 |
| 25 | ZINC11785459 | −12.3 | 50 | ZINC09833600 | −12.2 |
Table 2.
Best two hit molecules from screening results of RASPD based on ranking score.
| S. no. | ZINC ID | RASPD | 2D structures |
|---|---|---|---|
| 1. | ZINC20601870 | −13.9 | 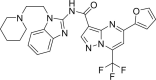 |
| 2. | ZINC00793735 | −13.4 | 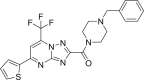 |
These top two screened molecules showed good bioactive score or binding affinity as in Table 1. The binding energy is represented in kcal/mol and with changes from oxygen to sulfur as well in other parts, effective binding is observed with ZINC00793735 than ZINC20601870. There are various rules to understand the bioavailability of the molecules but Lipinski’s rule of five is consider to be best and the screened compound follow it. (1) According to Lipinski filter, a compound to have good bioavailability should follow: the molecular weight (MW) ≤500, value of partition coefficient in octanol and water (mlog P) ≤4.15, heavy atoms N or O (Hacc) ≤10 and number of NH or OH (Hdon) ≤5. (2) According to Ghose filter, a compound to have good bioavailability should follow: 160 ≤ MW ≤ 480, value of wlog P − 0.4 ≤ wlog p ≤ 5.6, value of molar refractivity (MR) 40 ≤ MR ≤ 130, number of atoms 20 ≤ atom ≤ 70. (3) According to Veber filter, a compound to have good bioavailability should follow: the number of rotatable bond ≤10 and the value of topological polar surface area (TPSA) ≤140. Egan rule comprises wlog p ≤ 5.88 and TPSA ≤ 131.6. (4) According to Muegge filter, a compound to have good bioavailability should follow: 200 ≤ 600, −2 ≤ xlog p ≤ 5, TPSA ≤ 150, number of rings ≤7, number of carbon >4, number of heteroatoms >1 and number of rotatable bonds ≤15. These hits do not show any violations for the Lipinski Rule of Five as in Table 3.
Table 3.
Physiochemical properties, lipophilicity, water-solubility, pharmacokinetics, drug-likeness, and bioactivity score of the top two hit drug molecules from designed library against main protease of novel coronavirus calculated using SwissADME and Molinspiration.
| Properties | Screened best two molecules from designed compounds against main protease of novel coronavirus |
|
|---|---|---|
| ZINC20601870 | ZINC00793735 | |
| Log S | −5.22 | −5.02 |
| Heavy atoms | 38 | 33 |
| MW (g/mol) | 523.51 | 472.49 |
| No. of rotational bonds | 8 | 6 |
| No. H-bond acceptors | 9 | 8 |
| Num. H-bond donors | 1 | 0 |
| Log Po/w(iLOGP) | 3.31 | 3.34 |
| GPCR ligand | −0.07 | −0.26 |
| Ion channel modulator | −0.53 | −0.47 |
| Kinase inhibitor | 0.09 | −0.40 |
| Nuclear receptor ligand | −0.85 | −0.82 |
| Protease inhibitor | −0.50 | −0.42 |
| Enzyme inhibitor | −0.40 | −0.41 |
| Lipinski | Yes; 1 violation: MW >500 | Yes; 0 violation |
| Log Kp in cm/s | −7.00 | −6.70 |
| TPSA(Å2) | 93.49 | 94.87 |
| Bioavailability score | 0.55 | 0.55 |
| Synthetic accessibility | 3.90 | 3.50 |
| Physiochemical space for oral bioavailability | 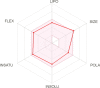 |
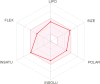 |
Docking studies of the screened molecules against the main protease of the SARS-CoV-2 were performed using ParDOCK, a web server. First of all water, metals and ligand molecules were removed from the target proteins and it was loaded as an input file for docking (Basit et al., 2020; Beura & Prabhakar, 2020; Bhardwaj et al., 2020; Boopathi et al., 2020; Borkotoky & Banerjee, 2020). The drug site is observed using the previous knowledge of the original ligand interaction site. Docking method is used to find the interaction between the small molecule and the receptor and form a stable complex with minimum binding affinity. Binding energy for the formation of the complex of main protease of n-coronavirus with ZINC20601870, ZINC00793735 and N3 are −3.96, −6.20 and −6.43 kcal/mol, respectively. ZINC20601870 shows hydrogen bond interaction with the main protease of the new coronavirus. From this, the residues of CYS-145, HIE-163, HIE-41, MET-49, HIE-164, GLU-166, MET-165, THR-26, GLY-143 and ASN-142 interacted with ZINC20601870. Another screened molecule, ZINC00793735 interacts with MET-165, HIE-41, LEU-141 and GLN-189. Further, inhibitor N3 interacts with amino acids of the main protease of new coronavirus. It shows pi-interactions with MET-165, ARG-188, HIE-41, CYS-145, MET-49 and hydrogen bonding with GLN-189, HIE-163, GLY-143, HIE-164, CYS-145, HIE-41 as in Table 4. Docked posed and interaction view of the screened molecules with the main protease of SARS-CoV-2 is given in Figure 4. Binding energy of ZINC00793735 is comparable to the N3 ligand and is better than the ZINC20601870.
Table 4.
Interaction of ZINC20601870, ZINC00793735 and N3 with the main protease of new coronavirus-2.
| CMPD | Π Interactions |
H. Bond interactions |
||
|---|---|---|---|---|
| Interacted residue | Distance (Å) | Interacted residue | Distance (Å) | |
| ZINC20601870 | CYS-145, HIE-41, MET-49 | 5.69/5.47, 5.05/6.24, 6.17 | ASN-142, HIE-41, HIE-163, HIE-164, GLU-166, MET-165, THR-26, GLY-143 | 4.50, 5.23, 5.64, 5.74, 4.82, 5.94, 4.72, 4.66 |
| ZINC00793735 | MET-165, HIE-41, LEU-141 | 6.91/4.46, 5.24, 6.53 | GLN-189 | 3.36/3.89 |
| N3 | MET-165, ARG-188, HIE-41, CYS-145, MET-49 | 5.60, 7.59, 4.91, 4.93/5.50/4.56, 4.43 | GLN-189, HIE-163, GLY-143, HIE-164, CYS-145, HIE-41 | 4.28/4.75, 4.67, 3.0, 6.33, 3.83, 5.98 |
Figure 4.

Interactions between the main protease of novel coronavirus with ZINC20601870, ZINC00793735 and N3.
The RMSD values of the ZINC20601870, ZINC00793735 and N3 in interactions with the main protease of the novel coronavirus are given in Table 5. It can be seen that they bind at the same site and with acceptable RMSD values.
Table 5.
RMSD values of the ZINC20601870, ZINC00793735 and N3 on interaction with the main protease of the novel coronavirus.
| Complex with main protease of the novel coronavirus | RMSD value (Å) |
|---|---|
| ZINC20601870 | 0.00 |
| ZINC00793735 | 0.040 |
| N3 | 0.162 |
One thing common in both the screened compounds is 3,5-dimethyl-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidyl part ring as given below: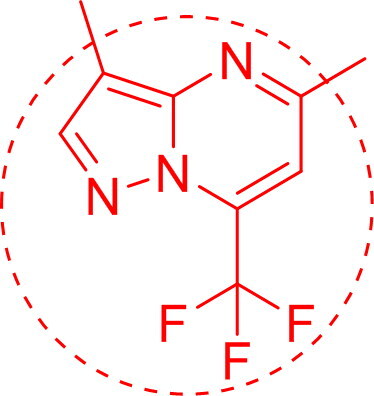
The superposition analysis is the best method for the comparison of more than one drug molecules and also allows the predictions of similarities between binding sites or entire proteins. In this comparison, the screened molecules were docked into the cavity of the reported binding site for the analysis of structure–function relationships between two drug molecules. In Figure 5, ZINC20601870, ZINC00793735 and N3 are superimposed on interaction with the main protease of the novel coronavirus.
Figure 5.

Pictorial view for the superimposition of (a) ZINC20601870 with ZINC00793735; (b) ZINC20601870, ZINC00793735 with N3 on interaction with the main protease of the novel coronavirus.
Based on the position of the proton and its charge, an amino acid, histidine exists in three forms: HIP (+1 charged, both δ- and ε-nitrogens protonated), HID (neutral, δ-nitrogen protonated) and HIE (neutral, ε-nitrogen protonated). In this study, authors found that HIE residues of main protease of coronavirus-2 showed π–π T-shaped stacking, π–alkyl interactions with ZINC20601870 and ZINC00793735 screened drug molecules while HIE residue do not shows any type of π interactions with N3 (reported drug molecule).
AMBER18 suite was used to perform MD simulations to study the structural stability and flexibility of the newly formed drug–target complex with their molecular interactions. To assess overall stability, the RMSD curve for target and its complex with ZINC20601870 and ZINC00793735 with respect to apo protein after the complex system reached equilibrium within 2 ns and continue over 100 ns as in Figure 6. The RMSD plot showed that most of the complex system of ZINC20601870 was relatively more stable than the complex of ZINC00793735. The RMSD value of complex with ZINC20601870 is found to be 0.89–2.95 Å, while another complex with ZINC00793735 is found to be 0.96–3.21 for 100 ns simulation time.
Figure 6.

RMSD plots of main protease of novel coronavirus-2 with and without ZINC20601870 and ZINC00793735 at 300 K for simulations time of 100 ns.
RMSD trajectory can be used to validate the results obtained from the docking results. RMSD trajectories using MD simulations shows the interaction of the small molecule with the receptor. If the RMSD value is less than 1.5 Å, it is considered to be good and acceptable. But, with value of more than 3 for RMSD, it is clearly rejected. RMSD values are used to find the stability of the receptor or main protease of SARS-CoV-2 with and without screened compounds and also to study the conformational changes of the receptor. RMSD value of the trajectory depends on the binding affinity and binding energy of the main protease of new coronavirus in the presence of the screened compounds. The RMSD of the main protease is represented in Angstrom (Å) and it is expected that lesser the values means more reliability. Further, binding of the screened compounds with the main protease can be explained by another trajectory obtained from the MD simulation, that is, RMSF. It provides information in the form of fluctuation of the main protease with and without screened compounds due to the fluctuation in energy. It provides useful information for the structural stability on protein. It is used to measure the differences in dynamics in reference to the average position of residues, that is, Cα-atoms (Elfiky, 2020a, 2020b; Elfiky & Azzam, 2020; Elmezayen et al., 2020).
From MD simulations results, CPPTRAJ module was used to analyze RMSF of main protease of new coronavirus with best two compounds at 100 ns time period. The amino acids contribute for the formation of the complex between main protease with the ZINC20601870 and ZINC00793735 and showed structural fluctuations that need to be studied by RMSF. RMSF plots determine the flexibility of each amino acid or the residue in the main protease of the complex. The active residue of main protease of new or novel-coronavirus are CYS-145, HIE-163, HIE-41, MET-49, HIE-164, GLU-166, MET-165, THR-26, GLY-143 and ASN-142, which are surrounded to the ZINC20601870 molecule while other residues, MET-165, HIE-41, LEU-141 and GLN-189 which surrounds to the ZINC00793735, showed significant fluctuations compared to other residues of apo protein and less fluctuations of complex is observed as in Figure 7. The RMSF value for C-α atoms for complex of main protease of novel coronavirus with ZINC20601870 was found to be 1.3–11.80 at 300 K, while RMSF value of complex with ZINC00793735 was found to be 1.38-12.10 at 300 K. The structural fluctuations formed are minimum. It is observed that these inhibitors could have the interaction with residue of target proteins.
Figure 7.

RMSF plots for residue number of main protease of novel coronavirus-2 with ZINC20601870 and ZINC00793735 at 300 K for simulations time of 100 ns.
Relative change in enthalpy for the formation of the complex between the filtered compounds and the main protease of the SARS-CoV-2 is determined on using MM-GBSA method by using Equations (5)–(8). From this method, the change in enthalpy (ΔH) versus simulations time was plotted for the complex of ZINC20601870 and ZINC00793735 as in Figure 8.
Figure 8.

Relative changes in enthalpy for the formation of complexes of main protease of SARS-CoV-2 with ZINC20601870 and ZINC00793735 for simulation time of 100 ns.
Relative change in enthalpy (ΔH) was calculated for the formation of complexes of main protease of SARS-CoV-2 with the screened compounds, ZINC20601870 and ZINC00793735 using MM-GBSA methods as in Table 6 using Equations (5)–(8). Change in enthalpy (ΔH) for the formation of complex with ZINC20601870 and ZINC00793735 with the main protease of novel coronavirus is found to be −24.21 and −26.53 kcal/mol, respectively, while the change in free energy (ΔG) for the formation of complex with ZINC20601870 and ZINC00793735 with the main protease of novel coronavirus is found to be −2.66 and −4.55 kcal/mol.
Table 6.
Calculated the relative terms for change in enthalpy, entropy and binding free energy of difference in drug, target protein and drug-target complex of both systems using MM-GBSA method at 100 ns simulations time.
| Energy component | Complex of ZINC20601870 with main protease of SARS-CoV-2 | Complex of ZINC00793735 with main protease of SARS-CoV-2 |
|---|---|---|
| Average (kcal/mol) | Average (kcal/mol) | |
| VDWAALS | −38.88 | −40.14 |
| EEL | −9.12 | −9.85 |
| EGB | 28.40 | 28.21 |
| ESURF | −4.61 | −4.74 |
| ΔGgas | −48.0 | −50.0 |
| ΔGsolv | 23.79 | 23.47 |
| ΔHtotal | −24.21 | −26.53 |
| ΔS | −21.54 | −21.97 |
| ΔG | −2.66 | −4.55 |
Trajectory of the hydrogen bond play very important and play important role for binding affinity or binding energy and structural stability of the complex formed between the screened compounds and the main protease of novel coronavirus. It is used to find % occupancy of time of HBs formed and angle at which frame where the maximum number of HBs formed during simulation times. It gives the information about the number of hydrogen bonds formed in the complex. Number of hydrogen bonds formed for the complex for the main protease of SARS-CoV-2 with the hit compounds, ZINC20601870 and ZINC00793735 are three and two, respectively, as in Figures 9 and 10. Average number of HBs of drug molecule as per residue during 100 ns for different donor–acceptor average distance cutoffs is 2.86 (strong bonding) with larger average angle. It was found that HBs between N1 of ZINC20601870 molecules (acceptor N1) and residues are GLN_189@HE22 (donor H) while in ZINC00793735 (acceptor F1) and residues GLN_192@HE22 (donor N & H) at 100 ns as in Table 7. It was assumed that the formed HBs have distance between donor residue H & N in the backbone with accepter N1 atom in the ZINC20601870 showed shorter distance (2.92 Å) and the angle of H–N–H is 155.55° with 14.02% occupancy. In another complex, the formed HBs have distance between donor residue H & N in the backbone with accepter F1 atom in the ZINC00793735 showed shorter distance (2.89 Å) less than ZINC20601870 and the angle of H–N–F is 154.10° with 02.92% occupancy.
Figure 9.

Hydrogen bonding (HB) plot for interacted residue of main protease of novel coronavirus with ZINC20601870 at 300 K for simulations time of 100 ns.
Figure 10.

Hydrogen bonding (HB) plot for interacted residue of main protease of novel coronavirus with ZINC00793735 at 300 K for simulations time of 100 ns.
Table 7.
Hydrogen bond analysis for the complexes of main protease of novel coronavirus with ZINC20601870 and ZINC00793735 at 300 K for simulations time of 100 ns.
| Temperature | Acceptor | Donor | Donor | Occupancy | Distance | Angle |
|---|---|---|---|---|---|---|
| ZINC20601870 | DRG_307@N1 | GLN_189@HE22 | GLN_189@NE2 | 14.02% | 2.92 | 155.55 |
| ZINC00793735 | DRG_307@F1 | GLN_192@HE22 | GLN_192@NE2 | 02.92% | 2.89 | 154.10 |
Non-isothermally molecular dynamics (MD) simulations for the main protease of SARS-CoV-2 with screened molecules
Non-isothermally MD simulations were performed for 10 ns at different temperature, that is, 325, 350, 375 and 400 K. Huge deviation is found at 350, 375 and 400 K and least deviation at 325 K for the complex between the main protease of novel corona virus with ZINC20601870 (Figure 11) and ZINC00793735 (Figure 12) is observed. Other trajectories like RMSF and the number of HBs at particular temperature were analyzed. RMSD value confirmed the structural stability of drug–target complex at variable temperature. If the temperature of complex system of main protease of SARS-CoV-2 with ZINC20601870 and ZINC00793735 increases then the deviations increases gradually from 325 to 400 K on average at the end of the simulations. For MD simulations for the complex with ZINC20601870 at 10 ns, the RMSD values at 350, 375 and 400 K go above 3 is not acceptable and therefore, inhibition cannot be studied. Further, the MD simulations at 325 K need to be studied and need a comparison with the MD simulations at 300 K as in Figure 10. For MD simulations for the complex with ZINC00793735 at 10 ns, the RMSD values at 375 and 400 K go above 3 is not acceptable and therefore, inhibition cannot be studied. Further, the MD simulations at 325 and 350 K need to be studied and need a comparison with the MD simulations at 300 K as in Figure 11.
Figure 11.

RMSD plot of main protease of new-coronavirus with ZINC20601870 at 325, 350, 375 and 400 K for simulations time of 10 ns.
Figure 12.

RMSD plot of main protease of new-coronavirus with ZINC00793735 at 325, 350, 375 and 400 K for simulations time of 10 ns.
Non-isothermally, the active residue of main protease of coronavirus CYS-145, HIE-163, HIE-41, MET-49, HIE-164, GLU-166, MET-165, THR-26, GLY-143 and ASN-142 which are surrounded to the ZINC20601870 molecule while another residues of MET-165, HIE-41, LEU-141 and GLN-189 which are surrounded to the ZINC00793735, showed significant fluctuations compared to other residues of apo protein and less fluctuations of complex is observed. The analysis of RMSF versus the residue number for the drug–target complex is illustrated as in Figures 13 and 14 was used to calculate fluctuation of drug–target complex of ZINC20601870 and ZINC00793735 after binding of drug into the cavity of target protein at non-isothermally. The RMSF value for C-α atoms for complex of main protease of novel coronavirus with ZINC20601870 was found to be 1.2–10.31 at 325 K, 0.94–10.79 at 350 K, 1.5–23.52 at 375 K & 1.29–43.20 at 400 K while RMSF value of complex with ZINC00793735 was found to be 1.01–9.28 at 325 K, 0.74–8.4 at 350 K, 1.33–13.51 at 375 K & 1.26–45.86 at 400 K observed. The structural fluctuations formed are minimum. It is observed that these inhibitors could have the interaction with residue of target proteins. On increasing the temperature of the system, the fluctuation is formed more for 375 K to 400 K for both the system and less fluctuations are observed at 350 K.
Figure 13.

RMSF plots for residue number of main protease of new-coronavirus with ZINC20601870 at 325, 350, 375 and 400 K for 10 ns simulations time.
Figure 14.

RMSF plot for residue number of main protease of new-coronavirus with ZINC00793735 at 325, 350, 375 and 400 K for 10 ns simulations time.
The total numbers of average HBs formed during MD simulations in each time frame were predicted non-isothermally. In this complex, the number of hydrogen bonds analysis suggested the presence of two and three intermolecular hydrogen bonds for the complex of main protease of SARS-CoV-2 with the ZINC20601870 and ZINC00793735 throughout the MD simulations time for 10 ns as in Figures 15 and 16.
Figure 15.

HBs plot for interacted residue of main protease of new-coronavirus with ZINC20601870 at 325, 350, 375 and 400 K for simulations time of 10 ns.
Figure 16.

HBs plot for interacted residue of main protease of new-coronavirus with ZINC00793735 at 325, 350, 375 and 400 K for simulations time of 10 ns.
The % occupancy, average distance and angle of HBs formed between ZINC20601870 and ZINC00793735 with target protein were analyzed through MD trajectories as in Table 8. In this, the MD simulations were performed at 325, 350, 375 and 400 K. At temperature of 350 K, the % occupancy for ZINC20601870 is found maximum. It indicates that binding affinity of drug molecules into the cavity of protein is high. The HBs have distance between atom of donor residue N in the backbone with accepter O atom in the molecules showed shorter distance in case of ZINC20601870 while in case of ZINC00793735, it does not affect the distance effectively.
Table 8.
Hydrogen bond analysis for main protease of SARS-coronavirus-2 with complex of ZINC20601870 and ZINC00793735 at 325, 350, 375 and 400 K for simulations time of 10 ns.
| Temperature (K) | Acceptor | Donor | Donor | Occupancy (%) | Distance | Angle |
|---|---|---|---|---|---|---|
| ZINC20601870 | ||||||
| 325 | DRG_307@O | GLU_166@H | GLU_166@N | 01.64 | 2.90 | 159.52 |
| 350 | DRG_307@F | ALA_194@H | ALA_194@N | 0.75 | 2.89 | 154.48 |
| 375 | DRG_307@O | THR_190@HG1 | THR_190@OG1 | 02.17 | 2.76 | 155.85 |
| 400 | DRG_307@N1 | ARG_76@HH21 | ARG_76@NH2 | 0.55 | 2.88 | 149.99 |
| ZINC00793735 | ||||||
| 325 | DRG_307@F1 | GLN_192@HE22 | GLN_192@NE2 | 03.94 | 2.89 | 152.21 |
| 350 | DRG_307@O | GLN_189@H | GLN_189@N | 22.27 | 2.84 | 157.23 |
| 375 | DRG_307@O | GLN_189@H | GLN_189@N | 18.39 | 2.85 | 157.20 |
| 400 | DRG_307@O | ASN_72@H | ASN_72@N | 0.54 | 2.88 | 160.69 |
Conclusions
In the present work, authors have screened the compounds from ZINC database and best two compounds are taken for further study. Based on docking, the binding energy for the formation of the complex of main protease of n-coronavirus with ZINC20601870, ZINC00793735 and N3 is −3.96, −6.20 and −6.43 kcal/mol, respectively. Binding energy of ZINC00793735 is comparable to the N3 ligand and better that the ZINC20601870. Authors found that HIE residues of main protease of coronavirus-2 showed π–π T-shaped stacking, π–alkyl interactions with ZINC20601870 and ZINC00793735 screened drug molecules while HIE residue do not shows any type of π-interactions with N3 (reported drug molecule). The best two compounds were docked against the main protease of the nCoV-2 and then they were studied through MD simulations. The binding of the screened compounds with the receptor was studied with the help of different trajectories of MD simulations to know about the stability, fluctuation, hydrogen bonding, etc. Binding energy for the formation of the complexes for ZINC20601870 and ZINC00793735 with the main protease of SARS-CoV-2 at room temperature was calculated using MM-GBSA. Change in free energy for the formation of each complex at room temperature was calculated to know the effective binding as well as the spontaneity of the formation of the complex. Complex of ZINC00793735 showed better binding affinity with the main protease of SARS-CoV-2. At room temperature, change in enthalpy (ΔH) for the formation of complex with ZINC20601870 and ZINC00793735 with the main protease of novel coronavirus is found to be −24.21 and −26.53 kcal/mol, respectively, while the change in free energy (ΔG) for the formation of complex with ZINC20601870 and ZINC00793735 with the main protease of novel coronavirus is found to be −2.66 and −4.55 kcal/mol. At non-isothermally or REMD means at different temperature, prediction of structural stability for a short time period was studied. On increasing the temperature of complexes, the deviations, fluctuations increase and also less number of hydrogen bonds are found during simulations time. It is suggested the ZINC00793735 can be used as potential antiviral agent against the main protease of SARS-CoV-2.
Funding Statement
Durgesh Kumar (DK) thankfully acknowledges to Council of Scientific and Industrial Research (CSIR, Reference no. 08/593/0002/2015/EMR I) for the fellowship. Another author, Prof. Ramesh Chandra (RC) thankfully acknowledges the financial assistance provided by the University of Delhi under DST-PURSE grant, Council of Scientific and Industrial Research (CSIR, Reference no. 02/265/16/EMR II), SERB-DST (Reference no. EEQ/2016/00489), Government of India.
Acknowledgements
One of the authors, Durgesh Kumar (DK) thankfully acknowledges the guidance provided by Prof. B. Jayaram, Incharge, SCFBio, Indian Institute of Technology, New Delhi, India and also for providing facilities and training; and also thankful to the Department of Chemistry, University of Delhi, Delhi, India for providing facilities to purse his research work.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Agarwal A., Nagi N., Chatterjee P., Sarkar S., Mourya D., Sahay R., & Bhatia R. (2020). Guidance for building a dedicated health facility to contain the spread of the 2019 novel coronavirus outbreak. Indian Journal of Medical Research. 10.4103/ijmr.IJMR_518_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal G., Gupta S., Gabrani R., Gupta A., Chaudhary V. K., & Gupta V. (2019). Virtual screening of inhibitors against envelope glycoprotein of Chikungunya Virus: A drug repositioning approach. Bioinformation, 15(6), 439–447. 10.6026/97320630015439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S. F., Quadeer A. A., & McKay M. R. (2020). Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses, 12(3), 254 10.3390/v12030254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai T., Yang Z., Hou H., Zhan C., Chen C., & Lv W. (2020). Correlation of chest CT and RT-PCR testing in coronavirus disease 2019 (COVID-19) in China: A report of 1014 cases. Radiology, 1–23. 10.1148/radiol.2020200642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmadiev N. S., Galimova A. M., Akhmetova V. R., Khairullina V. R., Galimova R. A., Agletdinov E. F., Ibragimov A. G., & Kataev V. A. (2019). Molecular docking and preclinical study of five-membered S,S-palladaheterocycle as hepatoprotective agent. Advanced Pharmaceutical Bulletin, 9(4), 674–684. 10.15171/apb.2019.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Anazi M., Al-Najjar B. O., & Khairuddean M. (2018). Structure-based drug design studies toward the discovery of novel chalcone derivatives as potential epidermal growth factor receptor (EGFR) inhibitors. Molecules, 23(12), 3203 10.3390/molecules23123203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Refaei M. A., Makki R. M., & Ali H. M. (2020). Structure prediction of transferrin receptor protein 1 (TfR1) by homology modelling, docking, and molecular dynamics simulation studies. Heliyon, 6(1), e03221 10.1016/j.heliyon.2020.e03221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazan F., Galan C., & Enjuanes L. (2004). The nucleoprotein is required for efficient coronavirus genome replication. Journal of Virology, 78(22), 12683–12688. 10.1128/JVI.78.22.12683-12688.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arshad Ali S., Baloch M., Ahmed N., Arshad Ali A., & Iqbal A. (2020). The outbreak of coronavirus disease 2019 (COVID-19) – An emerging global health threat. Journal of Infection and Public Health, 13(4), 644–646. 10.1016/j.jiph.2020.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babadaei M. M. N., Hasan A., Vahdani Y., Bloukh S. H., Sharifi M., & Kachooei E. (2020). Development of remdesivir repositioning as a nucleotide analog against COVID-19 RNA dependent RNA polymerase. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1767210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J., Ma X., & Sun X. (2020). Investigation on the interaction of food colorant Sudan III with bovine serum albumin using spectroscopic and molecular docking methods. The Journal of Environmental Science and Health, Part A, Toxic/Hazardous Substances and Environmental Engineering, 55, 1–8. 10.1080/10934529.2020.1729616 [DOI] [PubMed] [Google Scholar]
- Balaji B., & Ramanathan M. (2012). Prediction of estrogen receptor β ligands potency and selectivity by docking and MM-GBSA scoring methods using three different scaffolds . Journal of Enzyme Inhibition and Medicinal Chemistry, 27(6), 832–844. 10.3109/14756366.2011.618990 [DOI] [PubMed] [Google Scholar]
- Balasubramanian P. K., Balupuri A., Bhujbal S. P., & Cho S. J. (2019). 3D-QSAR-aided design of potent c-Met inhibitors using molecular dynamics simulation and binding free energy calculation. Journal of Biomolecular Structure & Dynamics, 37(8), 2165–2178. 10.1080/07391102.2018.1479309 [DOI] [PubMed] [Google Scholar]
- Basit A., Ali T., & Rehman S. U. (2020). Truncated human angiotensin converting enzyme 2; A potential inhibitor of SARS-CoV-2 spike glycoprotein and potent COVID-19 therapeutic agent. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1768150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bea I., Gotsev M. G., Ivanov P. M., Jaime C., & Kollman P. A. (2006). Chelate effect in cyclodextrin dimers: A computational (MD, MM/PBSA, and MM/GBSA) study. The Journal of Organic Chemistry, 71(5), 2056–2063. 10.1021/jo052469o [DOI] [PubMed] [Google Scholar]
- Berkhout B., & van Hemert F. (2015). On the biased nucleotide composition of the human coronavirus RNA genome. Virus Research, 202, 41–47. 10.1016/j.virusres.2014.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura S., & Prabhakar C. (2020). In-silico strategies for probing chloroquine based inhibitors against SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 1–25. 10.1080/07391102.2020.1772111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj V. K., Singh R., Sharma J., Rajendran V., Purohit R., & Kumar S. (2020). Identification of bioactive molecules from Tea plant as SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1766572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldog P., Tekeli T., Vizi Z., Denes A., Bartha F. A., & Rost G. (2020). Risk assessment of novel coronavirus COVID-19 outbreaks outside China. Journal of Clinical Medicine, 9(2), 1–12. 10.3390/jcm9020571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi S., Poma A. B., & Kolandaivel P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkotoky S., & Banerjee M. (2020). A computational prediction of SARS-CoV-2 structural protein inhibitors from Azadirachta indica (Neem). Journal of Biomolecular Structure and Dynamics, 1–17. 10.1080/07391102.2020.1774419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursnell M. E., Brown T. D., Foulds I. J., Green P. F., Tomley F. M., & Binns M. M. (1987). Completion of the sequence of the genome of the coronavirus avian infectious bronchitis virus. Journal of General Virology., 68(1), 57–77. 10.1099/0022-1317-68-1-57 [DOI] [PubMed] [Google Scholar]
- Burley S. K., Berman H. M., Christie C., Duarte J. M., Feng Z., Westbrook J., Young J., & Zardecki C. (2018). RCSB protein data bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Science: A Publication of the Protein Society, 27(1), 316–330. 10.1002/pro.3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X., Gao C., Su B., Tan F., Yang N., & Wang G. (2018). Expression profiling and microbial ligand binding analysis of high-mobility group box-1 (HMGB1) in turbot (Scophthalmus maximus L.). Fish & Shellfish Immunology, 78, 100–108. 10.1016/j.fsi.2018.04.025 [DOI] [PubMed] [Google Scholar]
- Cob-Calan N. N., Chi-Uluac L. A., Ortiz-Chi F., Cerqueda-García D., Navarrete-Vázquez G., Ruiz-Sánchez E., & Hernández-Núñez E. (2019). Molecular docking and dynamics simulation of protein beta-tubulin and antifungal cyclic lipopeptides. Molecules, 24(18), 3387 10.3390/molecules24183387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins P. L., Kannappan B., & Gready J. E. (2019). Ab initio molecular dynamics simulation and energetics of the ribulose-1,5-biphosphate carboxylation reaction catalyzed by Rubisco: Toward elucidating the stereospecific protonation mechanism. The Journal of Physical Chemistry. B, 123(12), 2679–2686. 10.1021/acs.jpcb.8b12088 [DOI] [PubMed] [Google Scholar]
- Du J., Sun H., Xi L., Li J., Yang Y., Liu H., & Yao X. (2011). Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM-GBSA calculation. Journal of Computational Chemistry, 32(13), 2800–2809. 10.1002/jcc.21859 [DOI] [PubMed] [Google Scholar]
- Duan L., & Zhu G. (2020). Psychological interventions for people affected by the COVID-19 epidemic. The Lancet Psychiatry, 7(4), 300–302. 10.1016/S2215-0366(20)30073-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. (2020. a). Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics, 1–16. 10.21203/rs.3.rs-22458/v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. (2020. b). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 1–15. 10.1080/07391102.2020.1761882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A., & Azzam E. B. (2020). Novel guanosine derivatives against MERS CoV polymerase: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1758789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen A. D., Al-Obaidi A., Şahin A. T., & Yelekçi K. (2020). Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1758791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi S. K., Raja K., Sebastine I., & Joseph J. (2020). Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1760136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genheden S., Kuhn O., Mikulskis P., Hoffmann D., & Ryde U. (2012). The normal-mode entropy in the MM/GBSA method: Effect of system truncation, buffer region, and dielectric constant. Journal of Chemical Information and Modeling, 52(8), 2079–2088. 10.1021/ci3001919 [DOI] [PubMed] [Google Scholar]
- Greenidge P. A., Kramer C., Mozziconacci J. C., & Sherman W. (2014). Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. Journal of Chemical Information and Modeling, 54(10), 2697–2717. 10.1021/ci5003735 [DOI] [PubMed] [Google Scholar]
- Greenidge P. A., Lewis R. A., & Ertl P. (2016). Boosting pose ranking performance via rescoring with MM-GBSA. Chemical Biology & Drug Design, 88(3), 317–328. 10.1111/cbdd.12763 [DOI] [PubMed] [Google Scholar]
- Gupta A., Gandhimathi A., Sharma P., & Jayaram B. (2007). ParDOCK: An all atom energy based Monte Carlo docking protocol for protein–ligand complexes. Protein and Peptide Letters, 14(7), 632–646. 10.2174/092986607781483831 [DOI] [PubMed] [Google Scholar]
- Jagannadh B., Kunwar A. C., Thangavelu R. P., & Osawa E. (1996). New technique for conformational sampling of cyclic molecules using the AMBER force field: Application to 18-crown-6. The Journal of Physical Chemistry, 100(34), 14339–14342. 10.1021/jp960929z [DOI] [Google Scholar]
- Ji T., Chen H. L., Xu J., Wu L. N., Li J. J., & Chen K. (2020). Lockdown contained the spread of 2019 novel coronavirus disease in Huangshi city, China: Early epidemiological findings. Clinical Infectious Diseases, 1–17. 10.1093/cid/ciaa390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby M. E., Simperler A., Krevor S., Weiss D. J., & Sonnenberg J. L. (2018). Computational tools for calculating log β values of geochemically relevant uranium organometallic complexes. The Journal of Physical Chemistry. A, 122(40), 8007–8019. 10.1021/acs.jpca.8b06863 [DOI] [PubMed] [Google Scholar]
- Kumar D., Kumari K., Jayaraj A., & Singh P. (2019). Development of a theoretical model for the inhibition of nsP3 protease of Chikungunya virus using pyranooxazoles. Journal of Biomolecular Structure and Dynamics, 1–17. 10.1080/07391102.2019.1650830 [DOI] [PubMed] [Google Scholar]
- Kumar D., Singh P., Jayaraj A., Kumar V., Kumari K., & Patel R. (2019). A theoretical model to study the interaction of Erythro-Noscapines with nsP3 protease of Chikungunya Virus. ChemistrySelect, 4(17), 4892–4900. 10.1002/slct.201803360 [DOI] [Google Scholar]
- Kumari K., Vishvakarma V. K., Singh P., Patel R., & Chandra R. (2017). Microwave: An important and efficient tool for the synthesis of biological potent organic compounds. Current Medicinal Chemistry, 24(41), 4579–4595. 10.2174/0929867324666170529100929 [DOI] [PubMed] [Google Scholar]
- Kumari M., Maurya J. K., Singh U. K., Khan A. B., Ali M., Singh P., & Patel R. (2014). Spectroscopic and docking studies on the interaction between pyrrolidinium based ionic liquid and bovine serum albumin. Spectrochimica Acta. Part A, Molecular and Biomolecular Spectroscopy, 124, 349–356. 10.1016/j.saa.2014.01.012 [DOI] [PubMed] [Google Scholar]
- Lai C. C., Shih T. P., Ko W. C., Tang H. J., & Hsueh P. R. (2020). Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. International Journal of Antimicrobial Agents, 55(3), 105924 10.1016/j.ijantimicag.2020.105924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A., Lombardo F., Dominy B. W., & Feeney P. J. (2012). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 64, 4–17. 10.1016/S0169-409X(96)00423-1 [DOI] [PubMed] [Google Scholar]
- Lohidakshan K., Rajan M., Ganesh A., Paul M., & Jerin J. (2018). Pass and Swiss ADME collaborated in silico docking approach to the synthesis of certain pyrazoline spacer compounds for dihydrofolate reductase inhibition and antimalarial activity. Bangladesh Journal of Pharmacology, 13(1), 23–29. 10.3329/bjp.v13i1.33625 [DOI] [Google Scholar]
- Madhavaram M., Nampally V., Gangadhari S., Palnati M. K., & Tigulla P. (2019). High-throughput virtual screening, ADME analysis, and estimation of MM/GBSA binding-free energies of azoles as potential inhibitors of Mycobacterium tuberculosis H37Rv. Journal of Receptor and Signal Transduction Research, 39(4), 312–320. 10.1080/10799893.2019.1660895 [DOI] [PubMed] [Google Scholar]
- Mitra S., Das A., Ghosh D., & Sengupta A. (2019). Postoperative systemic acyclovir in Juvenile-onset recurrent respiratory papillomatosis: The outcome. Ear, Nose, & Throat Journal, 98(1), 28–31. 10.1177/0145561318823311 [DOI] [PubMed] [Google Scholar]
- Morris K. F., Geoghegan R. M., Palmer E. E., George M. Jr., & Fang Y. (2020). Molecular dynamics simulation study of AG10 and tafamidis binding to the Val122Ile transthyretin variant. Biochemistry and Biophysics Reports, 21, 100721 10.1016/j.bbrep.2019.100721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee G., & Jayaram B. (2013). A rapid identification of hit molecules for target proteins via physico-chemical descriptors. Physical Chemistry Chemical Physics: PCCP, 15(23), 9107–9116. 10.1039/c3cp44697b [DOI] [PubMed] [Google Scholar]
- Muralidharan N., Sakthivel R., Velmurugan D., & Gromiha M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics, 1–6. 10.1080/07391102.2020.1752802 [DOI] [PubMed] [Google Scholar]
- Naz S., Farooq U., Khan S., Sarwar R., Mabkhot Y. N., & Saeed M. (2020). Pharmacophore model-based virtual screening, docking, biological evaluation and molecular dynamics simulations for inhibitors discovery against alpha-tryptophan synthase from Mycobacterium tuberculosis. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1715259 [DOI] [PubMed] [Google Scholar]
- Nazarian S. M., Shaon K. Y., Schwankhaus J. D., Chacko J. G., Hudgins P. A., & Brat D. J. (2015). Bilateral optic neuropathy after erythematous rash. Bilateral anterior optic neuropathy due to RMSF. Journal of Neuro-Ophthalmology: The Official Journal of the North American Neuro-Ophthalmology Society, 35(2), 201–204. https://collections.lib.utah.edu/ark:/87278/s66b08p3 10.1097/WNO.0000000000000261 [DOI] [PubMed] [Google Scholar]
- Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., & Ferrin T. E. (2004). UCSF Chimera – a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pola M., Rajulapati S. B., Potla Durthi C., Erva R. R., & Bhatia M. (2018). In silico modelling and molecular dynamics simulation studies on L-Asparaginase isolated from bacterial endophyte of Ocimum tenuiflorum. Enzyme and Microbial Technology, 117, 32–40. 10.1016/j.enzmictec.2018.06.005 [DOI] [PubMed] [Google Scholar]
- Purohit R. (2014). Role of ELA region in auto-activation of mutant KIT receptor: A molecular dynamics simulation insight. Journal of Biomolecular Structure & Dynamics, 32(7), 1033–1046. 10.1080/07391102.2013.803264 [DOI] [PubMed] [Google Scholar]
- Rajendran V., Gopalakrishnan C., & Sethumadhavan R. (2018). Pathological role of a point mutation (T315I) in BCR-ABL1 protein –A computational insight. Journal of Cellular Biochemistry, 119(1), 918–925. 10.1002/jcb.26257 [DOI] [PubMed] [Google Scholar]
- Roe D. R., & Cheatham T. E. 3rd. (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9(7), 3084–3095. 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
- Roe D. R., & Cheatham T. E. 3rd. (2018). Parallelization of CPPTRAJ enables large scale analysis of molecular dynamics trajectory data. Journal of Computational Chemistry, 39(25), 2110–2117. 10.1002/jcc.25382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma P., Shekhar N., Prajapat M., Avti P., Kaur H., & Kumar S. (2020). In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1753580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma J., Bhardwaj V. K., Das P., & Purohit R. (2020). Identification of naturally originated molecules as γ-aminobutyric acid receptor antagonist. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1720818 [DOI] [PubMed] [Google Scholar]
- Singh P., Kumar D., Vishvakarma V. K., Yadav P., Jayaraj A., & Kumari K. (2019). Computational approach to study the synthesis of noscapine and potential of stereoisomers against nsP3 protease of CHIKV. Heliyon, 5(12), e02795 10.1016/j.heliyon.2019.e02795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P., Kumar P., Katyal A., Kalra R., Dass S. K., Prakash S., & Chandra R. (2010). Synthesis and electrochemical studies of charge-transfer complexes of thiazolidine-2,4-dione with sigma and pi acceptors. Spectrochimica Acta. Part A, Molecular and Biomolecular Spectroscopy, 75(3), 983–991. 10.1016/j.saa.2009.12.019 [DOI] [PubMed] [Google Scholar]
- Singh R., Bhardwaj V., & Purohit R. (2020). Identification of a novel binding mechanism of Quinoline based molecules with lactate dehydrogenase of Plasmodium falciparum. Journal of Biomolecular Structure and Dynamics, 1–15. 10.1080/07391102.2020.1711809 [DOI] [PubMed] [Google Scholar]
- Sinha S. K., Shakya A., Prasad S. K., Singh S., Gurav N. S., & Prasad R. S. (2020). An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1762741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L. F., Lee T. S., Zhu C., York D. M., & Merz K. M. Jr.(2019). Using AMBER18 for relative free energy calculations. Journal of Chemical Information and Modeling, 59(7), 3128–3135. 10.1021/acs.jcim.9b00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Ruddel D., Winning J., Dickmann P., Ouart D., Kortgen A., & Janssens U. (2020). Coronavirus disease 2019 (COVID-19): update for anesthesiologists and intensivists March 2020. Anaesthesist, 1–10. 10.1007/s00101-020-00760-3 [DOI] [PubMed] [Google Scholar]
- Vishvakarma V. K., Kumari K., Patel R., Dixit V. S., Singh P., Mehrotra G. K., Chandra R., & Chakrawarty A. K. (2015). Theoretical model to investigate the alkyl chain and anion dependent interactions of Gemini surfactant with bovine serum albumin. Spectrochimica Acta. Part A, Molecular and Biomolecular Spectroscopy, 143, 319–323. 10.1016/j.saa.2015.01.068 [DOI] [PubMed] [Google Scholar]
- Vishvakarma V. K., Shukla N., Reetu Kumari K., Patel R., & Singh P. (2019). A model to study the inhibition of nsP2B–nsP3 protease of dengue virus with imidazole, oxazole, triazole thiadiazole, and thiazolidine based scaffolds. Heliyon, 5(8), e02124 10.1016/j.heliyon.2019.e02124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishvakarma V. K., Singh P., Dubey M., Kumari K., Chandra R., & Pandey N. D. (2013). Quantitative structure–activity relationship analysis of thiazolidineones: Potent antidiabetic compounds. Drug Metabolism and Drug Interactions, 28(1), 31–47. 10.1515/dmdi-2012-0036 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Zeng X. H., Xia X. M., & Lingle C. J. (2009). N-terminal inactivation domains of beta subunits are protected from trypsin digestion by binding within the antechamber of BK channels. The Journal of General Physiology, 133(3), 263–282. 10.1085/jgp.200810079 [DOI] [PMC free article] [PubMed] [Google Scholar]