

Abstract
Coronavirus disease 2019 (COVID-19) is a viral respiratory disease which caused global health emergency and announced as pandemic disease by World Health Organization. Lack of specific drug molecules or treatment strategy against this disease makes it more devastating. Thus, there is an urgent need of effective drug molecules to fight against COVID-19. The main protease (Mpro) of SARS CoV-2, a key component of this viral replication, is considered as a prime target for anti-COVID-19 drug development. In order to find potent Mpro inhibitors, we have selected eight polyphenols from green tea, as these are already known to exert antiviral activity against many RNA viruses. We have elucidated the binding affinities and binding modes between these polyphenols including a well-known Mpro inhibitor N3 (having binding affinity −7.0 kcal/mol) and Mpro using molecular docking studies. All eight polyphenols exhibit good binding affinity toward Mpro (−7.1 to −9.0 kcal/mol). However, only three polyphenols (epigallocatechin gallate, epicatechingallate and gallocatechin-3-gallate) interact strongly with one or both catalytic residues (His41 and Cys145) of Mpro. Molecular dynamics simulations (100 ns) on these three Mpro–polyphenol systems further reveal that these complexes are highly stable, experience less conformational fluctuations and share similar degree of compactness. Estimation of total number of intermolecular H-bond and MM-GBSA analysis affirm the stability of these three Mpro–polyphenol complexes. Pharmacokinetic analysis additionally suggested that these polyphenols possess favorable drug-likeness characteristics. Altogether, our study shows that these three polyphenols can be used as potential inhibitors against SARS CoV-2 Mpro and are promising drug candidates for COVID-19 treatment.
Communicated by Ramaswamy H. Sarma
Keywords: COVID-19, SARS CoV-2 main protease, docking, molecular dynamics simulation, green tea polyphenols/catechins
Graphical Abstract

1. Introduction
The novel corona virus disease (COVID-19) is spreading in the whole world causing deaths of 308,962 people till the 16th of May 2020 (https://www.worldometers.info/coronavirus/). The World Health Organization (WHO) has already declared COVID-19 as a pandemic disease (Cucinotta & Vanelli, 2020). Symptoms of this disease mainly include fever, cough, sore throat, runny nose and difficulty in breathing (Chen et al., 2020; Ren et al., 2020; Zhu et al., 2020). The disease was initially originated from Wuhan, China and expanded to nearly 215 countries (Zhu et al., 2020). USA, Spain and UK are among the most affected countries due to COVID-19 disease. In the USA alone, a total number of 1,484,287 people are infected and 88,507 people died due to COVID-19. In Spain and UK, 274,367 and 236,711 people are infected and 27,459 and 33,998 people have died there, respectively. This viral disease has not only challenged human health but also enormously affected the global economy.
The etiological agent of COVID-19 is severe acute respiratory syndrome corona virus-2 (SARS CoV-2) which belongs to the genus β-coronavirus (Zheng, 2020). Before SARS CoV-2, two more epidemic diseases were caused by corona viruses namely severe acute respiratory syndrome (SARS) and middle east respiratory syndrome (MERS) (Drosten et al., 2003; Zaki et al., 2012). In 2003, SARS had killed ∼750 people, whereas MERS was the reason behind the death of ∼860 people in 2012 (Mahase, 2020). On the contrary, the mortality rate due to SARS CoV-2 is exceptionally high (Mahase, 2020). SARS CoV-2 is a non-segmented enveloped positive-sense single-stranded RNA virus with ∼29.9 kb genome size (Wu et al., 2020; Zhu et al., 2020). The virus is thought to be originated from the bat and transmitted to humans by other sources (Zheng, 2020). Later on, human to human transmission of this disease is confirmed (Chan et al., 2020). Till date, no drug molecules or specific therapies have been developed to combat COVID-19. Considering the risk factors associated with this viral infection, an effective drug molecule is urgently required for effective treatment and to limit the transmission of this disease.
Many efforts involve the targeting of nonstructural protein, spike protein, the RNA-dependent RNA polymerase (RdRp) and the angiotensin-converting enzyme II (ACE2) entry receptor for the anti-COVID-19 drug development (Abdelli et al., 2020; Babadaei et al., 2020; Basit et al., 2020; Elfiky, 2020a, 2020b; Hasan et al., 2020; Nejadi Babadaei et al., 2020; Sinha et al., 2020; Wahedi et al., 2020). Targeting the components responsible for replication of SARS CoV-2 may be also a good strategy to identify effective drugs for the treatment of COVID-19. Like SARS and MERS corona viruses, the SARS CoV-2 genome also contains two open reading frames namely ORF1a and ORF1ab (Boopathi et al., 2020). These two ORFs help in translating two overlapping viral polyproteins pp1a and pp1ab required for viral replication and transcription (Grum-Tokars et al., 2008; Marra et al., 2003; Thiel et al., 2003). The functional polypeptides are released from the pp1a and pp1ab polyproteins by proteolytic processing by the papain-like proteinase (PLpro) and the 3C-like protease (3CLpro) (Grum-Tokars et al., 2008; Marra et al., 2003; Thiel et al., 2003). PLpro and 3CLpro are responsible for the cleaving of three sites and 11 sites, respectively within the viral genome (Harcourt et al., 2004; Thiel et al., 2003). As 3CLpro cleaves most of the sites of the polypeptide, it is also known as the main protease or Mpro. Different studies have identified Mpro as a cysteine protease with a Cys-His catalytic dyad (His41 and Cys145) in the active site of the protease (Blanchard et al., 2004; Dai et al., 2020; Jin et al., 2020; Osman et al., 2020). As Mpro plays a vital role in polyprotein processing and virus maturation, it is considered to be an important target for designing antiviral drugs against SARS CoV-2 (Anand et al., 2003; Yan et al., 2003). In addition, there is no human homolog of Mpro which makes it an ideal antiviral target (Kim et al., 2016). Apart from this, the high-resolution crystal structure of Mpro protease along with its inhibitor is recently made available, which has eased the way in designing structure-based Mpro-specific inhibitors to combat this viral disease (Jin et al., 2020; Yang et al., 2003). Utilizing computational approach, investigators have identified many small molecules including HIV and malaria drugs as SARS CoV-2 protease inhibitor (Adeoye et al., 2020; Khan et al., 2020; Muralidharan et al., 2020). Interestingly, many antiviral phytochemicals, bioactive compounds from Moroccan medicinal plants (Crocin, Digitoxigenin and β-Eudesmol) and chemical compounds from Indian spices (Carnosol, Rosmanol and Arjunglucoside-I) are proposed to be effective SARS CoV-2 Mpro inhibitors (Aanouz et al., 2020; Das et al., 2020; Enmozhi et al., 2020; Gyebi et al., 2020; Islam et al., 2020; Joshi et al., 2020; Umesh et al., 2020).
Plant-derived natural polyphenols are well known in preventing a wide range of diseases which include viral diseases as well. Recently, Purohit and coworkers have revealed that many bioactive molecules including a polymerized polyphenol (Oolonghomobisflavan-A) from tea plant (Camellia sinensis L.) act as effective SARS CoV-2 Mpro inhibitors (Bhardwaj et al., 2020). Green tea (C. sinensis) also contains eight native monomeric catechins or polyphenolic compounds and those are epigallocatechin gallate (EGCG), epigallocatechin (EGC), epicatechin gallate (ECG), epicatechin (EC), gallocatechin-3-gallate (GCG), gallocatechin (GC), catechin gallate (CG) and catechin (C) (Ai et al., 2019). These green tea polyphenols/catechins are promising compounds in exhibiting antiviral activities. They show antiviral activity against a wide range of human viruses including influenza, hepatitis B, hepatitis C, herpes simplex virus and HIV (Calland et al., 2012; Fassina et al., 2002; Ide et al., 2016; Lyu et al., 2005; Xu et al., 2008). These polyphenols are even active against dengue virus (DENV), Chikungunya virus (CHIKV) and Zika virus (ZIKV) (Carneiro et al., 2016; Ismail & Jusoh, 2017; Mahajan et al., 2020; Weber et al., 2015). But whether these polyphenols show any antiviral activity against SARS CoV-2 by inhibiting the enzymatic activity of its Mpro is far from clear. In this current study, we have undertaken a thorough attempt to find out the potentiality of these eight green tea polyphenols/catechins to inhibit the Mpro using in silico docking studies, molecular dynamics (MD) simulations and binding free energy calculations. This study provides three green tea polyphenols (EGCG, ECG and GCG) which may be served as inhibitors against SARS CoV-2 Mpro.
2. Materials and methods
2.1. Preparation of the ligands
The structures of eight green tea catechins or polyphenols [EGCG, EGC, ECG, EC, GCG, GC, CG and C] were downloaded from PubChem crystal database server in SDF format (https://pubchem.ncbi.nlm.nih.gov). Then all the sdf files were converted to pdb files using pymol software and each of the polyphenol structures were optimized with B3LYP/6-31G* type of basis set by using Gaussian09 software (Frisch & Clemente). Following optimization, each of these was loaded to AutoDock Tools for the final preparation of ligands. Each of the optimized polyphenol structure was then inserted to AutoDock Tools and standard processes were used to obtain the pdbqt files.
2.2. Preparation of Mpro
The crystal structure of the SARS CoV-2 Mpro was taken from the RCSB Protein Data Bank (http://www.rcsb.org) (PDB ID: 6LU7) (Jin et al., 2020). Then we checked whether there were any improper bonds, missing hydrogens, side chain anomalies present or not. After correcting all those aspects the structure file was inserted into AutoDock Tools and standard procedures were followed to get pdbqt file format of Mpro (Morris et al., 2008, 2009).
2.3. Molecular docking
The docking of Mpro with the eight polyphenols of green tea was performed with the aid of AutoDock Vina, which is now widely used as a successor of AutoDock Tools (Morris et al., 2008, 2009). The binding affinities of polyphenols–Mpro were determined and analyzed using the same software. By seeing the position of active site region, the center of the grid box was chosen to be at X: −11.75, Y: 15.135, Z: 68.856 with a suitable gridbox volume where the ligands can easily be fitted and which covers the entire active site pocket. The available crystal structure (PDB: 6LU7) with a bound N3 molecule was used as a reference to calibrate and optimize the docking procedures. Following calibration and optimization, the same grid box size and other parameters were used for docking studies of all the eight polyphenols and the entire set up was run to obtain different docked conformations. Amongst them, the best suited conformations with lowest root mean square deviation (RMSD) values were selected to calculate the binding energetic between Mpro and green tea polyphenols. The output from AutoDock Vina was rendered with PyMOL (DeLano, 2002).
2.4. MD simulation
All the MD simulations were carried out by the GROningen MAchine for Chemical Simulations (GROMACS) version 5.1.2 (Abraham et al., 2015). The OPLS-AA/L force field and TIP3P water model embedded in GROMACS were used for all the MD simulations (Kaminski et al., 2001). The topology file of Mpro protein was prepared by the GROMACS, while the ligand topologies were obtained from the LigParGen (a server from the Jorgensen group to produce OPLS topologies). In order to satisfy minimum image conventions the system was initially accommodated in a cubic box with a distance of 1.4 nm between protein complex and the box. All bond lengths of protein and polyphenols were constrained using the LINCS algorithm while water molecules were restrained by SETTLE algorithm (Hess et al., 1997; Miyamoto & Kollman, 1992). A total of 30105, 30065, 30073 and 30073 water molecules were added to a cubic simulation box containing the unligated Mpro, Mpro–ECG, Mpro–EGCG, Mpro–GCG complexes, respectively. Each system was energy-minimized using steepest descent algorithm and equilibrated to achieve the appropriate volume. Short- and long-range non-bonded interactions were calculated by applying twin-range cutoffs of 0.9 and 1.4 nm, respectively. The leap frog algorithm with time step 2 fs was used for integrating the equation of motion and the neighbor list was updated at every five steps. Long-range electrostatics is treated using the Particle Mesh Ewald method, with a Fourier grid spacing of 0.16 (Darden et al., 1993). Periodic boundary conditions were applied in all three directions. Equilibration of the systems was carried out in two main stages. First, the system was allowed to heat gradually to 300 K in NVT ensemble using v-rescale algorithm for 10 ns. Then, NPT ensemble was used for 5 ns by restraining the complexes (unligated Mpro, Mpro–ECG, Mpro–EGCG, Mpro–GCG) while slowly allowing the solvent molecules to relax around it and finally another 10 ns NPT equilibration was carried out by gradually removing the restraints on the complexes. The pressure was maintained using Berendsen barostat. For every system, the average temperature and pressure values remained close to the desired values. The equilibrated systems were then subjected to unrestrained production MD simulations of 100 ns each, maintaining target pressure (1 bar) and temperature (300 K). Reproducibility of the results was checked by five different repeating simulations with different initial velocities and equilibration times. The overall trends in the RMSD, root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA) and hydrogen bond numbers remained the same. RMSD, RMSF, Rg, SASA and total number of H-bond count were calculated from the MD simulations (Purohit, 2014; Rajendran, 2016; Rajendran et al., 2018; Sharma et al., 2020; Singh et al., 2020).
2.5. MM–GBSA
To predict the theoretical free energies of binding of ligands to receptor, generally two most commonly used methods are (a) the molecular mechanics generalized Born surface area (MM-GBSA) and (b) molecular mechanics Poisson–Boltzmann surface area (MM-PBSA). Both methods are equally efficient to predict the correct binding affinities (Chen, 2016; Chen et al., 2015; Hou et al., 2011; Venugopal et al., 2020). Here, we used the MM-GBSA method to calculate the relative binding free energies of green tea polyphenols to Mpro protein. The free energy of binding can be calculated as ΔGbind = ΔH − TΔS.
ΔH = ΔEelec + ΔEvdW + ΔGpolar + ΔGnon-polar, where Eelec and EvdW are the electrostatic and van der Waal’s contributions, and Gploar and Gnon-polar are the polar and non-polar solvation terms, respectively. The non-polar energy is calculated from the SASA while the polar contribution of the free energy can be estimated using generalized Born model with an external dielectric constant of 80 and internal dielectric constant of 1. The entropic contribution is neglected here due to similar type of ligands bind to the receptor. Therefore, our calculated values are referred to as relative binding free energies. The theoretical binding free energy of the potent inhibitors of Mpro was identified using the prime module of Schrodinger suit (Schrödinger Release 2020-1: Prime, Schrödinger, LLC, New York, NY, 2020). MM-GBSA is a popular method to calculate binding energy, which uses energy properties of free ligand, free receptor and receptor–ligand complex for binding affinity calculation. Binding energies were estimated for the three polyphenols selected based on the binding affinity of AutoDock Vina docking and three complex structures were selected. Then with these docking structures MM-GBSA were calculated.
2.6. Pharmacokinetic properties analysis
Various pharmacokinetic properties of these eight polyphenols and N3 (chosen as model inhibitor) were predicted using pkCSM-pharmacokinetics and Swiss ADME online softwares (Daina et al., 2017; Pires et al., 2015). The drug-likeness properties such as absorption, distribution, metabolism and excretion parameters of these polyphenols along with the toxicity were mainly analyzed.
3. Result and discussion
The theoretical study of SARS CoV-2 found a new horizon when Yang and coworkers solved the crystal structure of Mpro with N3 complex (Jin et al., 2020). The crystal structure revealed that Mpro is a cysteine protease which is a homodimer consisting of two identical protomers. Each protomer has three distinct domains (domain I, II and III) along with a specific substrate-binding site as shown in Figure 1. This substrate-binding site mainly consists of a cysteine–histidine dyad (His41 and Cys145) which controls the catalytic activity of SARS CoV-2 Mpro. The substrate-binding pocket is located between domains I (magenta) and domain II (cyan) of each protomer (Figure 1). The compound N3 is reported to be a potential inhibitor of Mpro which mainly binds to the active/catalytic site of the protease Mpro. Docking of N3 compound with Mpro protease revealed that the structural orientation of N3 helps its binding to the catalytic cleft or substrate-binding region of Mpro (Figure 1 and Table 1).
Figure 1.

Surface representation of SARS CoV-2 Mpro with N3 inhibitor. The protomer of Mpro from SARS CoV-2 has been shown with its domains – Domain I (colored with magenta), Domain II (colored with cyan) and Domain III (colored with yellow) while blue color represents the linker. The inhibitor N3 (represented by the red stick) is attached to the substrate-binding pocket of Mpro.
Table 1.
Structure and binding energy of green tea polyphenols with Mpro along with N3 inhibitor as standard.
| Complex | Structure | Binding energy (kcal/mol) |
|---|---|---|
| N3 | 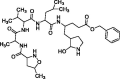 |
−7.0 |
| Epigallocatechin (EGC) | 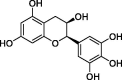 |
−7.0 |
| Gallocatechin (GC) | 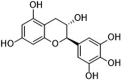 |
−7.1 |
| Catechin (C) | 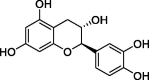 |
−7.1 |
| Epicatechin (EC) | 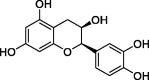 |
−7.2 |
| Catechin gallate (CG) | 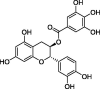 |
−7.2 |
| Epigallocatechin gallate (EGCG) | 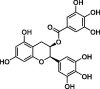 |
−7.6 |
| Epicatechingallate (ECG) | 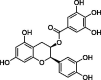 |
−8.2 |
| Gallocatechin-3-gallate (GCG) | 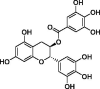 |
−9.0 |
3.1. Molecular docking studies
Once the crystal structure of N3 inhibitor bound to Mpro was elucidated, many such theoretical studies commenced in order to find other suitable inhibitors for Mpro to control COVID-19 (Elmezayen et al., 2020; Huynh et al., 2020; Islam et al., 2020; Mahanta et al., 2020; Mittal et al., 2020). In the current study, we used this N3 complex as a standard inhibitor of Mpro and docked this complex with the protease. The binding energy of this particular binding was found to be −7.0 kcal/mol which is identical to the previously published estimation by other investigators (Huynh et al., 2020). Analysis of N3 docked complex suggested that this complex is stabilized by multiple hydrophobic and hydrogen bond interactions (particularly with His41 and Cys145) (Tables 2 and 3). Thus, we can also conclude like others that binding of N3 to both the catalytic residues (His41 and Cys145) of Mpro may be an important factor behind the inhibition of its protease activity. But the pharmacokinetics analysis revealed that N3 shows hepatotoxicity which makes it carcinogenic for humans (Table 4). Data presented in this table also reflected a negative tolerance dose of this compound for human. All these adverse pharmacokinetic behaviors make N3 an unsafe drug for COVID-19 treatment.
Table 2.
Hydrogen bond interactions of N3 and different green tea polyphenols with the SARS CoV-2 Mpro.
| Complex | Number of H-bonds | Amino acids of Mpro i nvolved in H-bonding | Hydrogen bond distance (Å) |
|---|---|---|---|
| N3 | 8 | His41 | 2.3 |
| Glu166 | 2.8 | ||
| Cys145 | 2.1 | ||
| Phe140 | 3.2 | ||
| Thr190 | 2.8 | ||
| His164 | 2.0 | ||
| Gly143 | 2.9 | ||
| Gln189 | 2.9 | ||
| Epigallocatechin (EGC) | 4 | Ser144 | 2.5 |
| His163 | 2.9, 3.3 | ||
| Gln192 | 2.6 | ||
| Gallocatechin (GC) | 4 | Phe140 | 2.4 |
| Glu166 | 2.3 | ||
| Arg 188 | 2.0 | ||
| Gln192 | 2.7 | ||
| Catechin (C) | 5 | Leu141 | 2. |
| Ser144 | 2.5 | ||
| His163 | 3.0, 3.3 | ||
| Gln192 | 2.6 | ||
| Epicatechin (EC) | 5 | Ser 144 | 2.4 |
| His163 | 2.9, 3.3 | ||
| Thr190 | 2.2 | ||
| Gln192 | 2.6 | ||
| Catechin gallate (CG) | 5 | Ser144 | 2.4 |
| His163 | 3.1 | ||
| Arg188 | 2.8 | ||
| Thr190 | 2.5, 1.9 | ||
| Epigallocatechin gallate (EGCG) | 9 | Thr26 | 2.2, 1.9 |
| His41 | 2.8 | ||
| Cys145 | 2.6 | ||
| Ser144 | 2.3, 2.7 | ||
| Glu166 | 2.9 | ||
| Gln189 | 2.1 | ||
| Gly143 | 2.7 | ||
| Epicatechingallate (ECG) | 8 | Ser144 | 2.4, 2.7 |
| Gly143 | 2.1 | ||
| Cys145 | 2.1 | ||
| Thr26 | 2.3, 1.9 | ||
| His41 | 2.8 | ||
| Glu166 | 2.9 | ||
| Gallocatechin-3-gallate (GCG) | 9 | Phe140 | 2.1, 2.1 |
| His163 | 2.9 | ||
| Ser144 | 2.2, 2.5, 2.7 | ||
| Cys145 | 2.7 | ||
| Gly143 | 2.3, 2.7 |
Table 3.
Binding interactions of N3 and different polyphenols of green tea with the active site of SARS CoV-2 Mpro.
| Complex | Non-covalent interactions (other than H-bonding) |
|
|---|---|---|
| Polar | Hydrophobic | |
| N3 | Ser144, Asn142, | Phe140, Leu141, Met165, Glu166, Leu167, Pro168, His172, Ala191, Gln192 |
| Epigallocatechin (EGC) | Ser46, Asn142 | Leu141, Met165, Glu166, Ala191 |
| Gallocatechin (GC) | Ser46, Asn142 | Met49, Leu141, Met165, Gln189 |
| Catechin (C) | Thr24, Thr25, Thr45, Ser46 | Leu27, Met49 |
| Epicatechin (EC) | Thr45, Ser46, Gln189 | Met49, Leu141, Met165 |
| Catechin gallate (CG) | Gln189 | Phe140, Leu141, Met165, Glu166, His172 |
| Epigallocatechin gallate (EGCG) | Thr24, Thr25, Ser46, Asn142, Gln192 | Met49, Leu141, His163, Met165 |
| Epicatechingallate (ECG) | Thr24, Thr25, Thr45, Ser46, Asn142, Gln189 | Met49, Leu27, Leu141, Met165 |
| Gallocatechin-3-gallate (GCG) | Asn142, Gln189, Thr190, Gln192 | Met49, Leu141, Met165, Glu166, Arg188 |
Table 4.
Pharmacokinetic properties of N3 and green tea polyphenols.
| Compound | MW | H-Ac | H-Do | Nrot | TPSA | LogP | IA | TC | LD50 | HT | AT | MTD | NLV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N3 | 684.82 | 10 | 5 | 23 | 196.88 | 2.1352 | 62.162 | 0.843 | 4.355 | Yes | No | −0.341 | 2 |
| Epigallocatechin (EGC) | 306.27 | 7 | 6 | 1 | 130.61 | 1.2517 | 54.128 | 0.328 | 2.492 | No | No | 0.506 | 1 |
| Gallocatechin (GC) | 306.27 | 7 | 6 | 1 | 130.61 | 1.2517 | 54.128 | 0.328 | 2.492 | No | No | 0.506 | 1 |
| Catechin (C) | 290.27 | 6 | 5 | 1 | 110.38 | 1.5461 | 68.829 | 0.183 | 2.428 | No | No | 0.438 | 0 |
| Epicatechin (EC) | 290.27 | 6 | 5 | 1 | 110.38 | 1.5461 | 68.829 | 0.183 | 2.428 | No | No | 0.438 | 0 |
| Catechin gallate (CG) | 442.37 | 10 | 7 | 4 | 177.14 | 2.5276 | 62.096 | −0.169 | 2.558 | No | No | 0.449 | 1 |
| Epigallocatechin gallate (EGCG) | 458.37 | 11 | 8 | 4 | 197.37 | 2.2332 | 47.395 | 0.292 | 2.522 | No | No | 0.441 | 2 |
| Epicatechin gallate (ECG) | 442.37 | 10 | 7 | 4 | 177.14 | 2.5276 | 62.096 | −0.169 | 2.558 | No | No | 0.449 | 1 |
| Gallocatechin-3-gallate (GCG) | 458.37 | 11 | 8 | 4 | 197.37 | 2.2332 | 47.395 | 0.292 | 2.522 | No | No | 0.441 | 2 |
MW, molecular weight (g/mol); H-Ac, no. of hydrogen bond acceptor; H-Do, no. of hydrogen bond donors; Nrot, no. of rotatable bonds; TPSA, topological polar surface area (Å2); LogP, predicted octanol/water partition coefficient; IA, intestinal absorption (% absorbed); TC, total clearance (log ml/min/kg); LD50, oral rat acute toxicity; HT, hepatotoxicity; AT, AMES toxicity; MTD, maximum tolerated dose for human (log mg/kg/day); NLV, no. of Lipinski rule violation.
In search of effective drugs for COVID-19 treatment, we hypothesized that green tea catechins or polyphenols (structure given in Table 1) may be suitable drug candidates by exerting inhibitory effects like N3. Their antiviral activity against diverse viruses including RNA viruses further strengthens our hypothesis (Calland et al., 2012; Carneiro et al., 2016; Ide et al., 2016; Ismail & Jusoh, 2017; Weber et al., 2015). But, before testing our hypothesis, first, we analyzed the pharmacokinetic and the drug-likeness properties of these eight green tea polyphenols. All the polyphenols exhibited negative AMES toxicity and hepatotoxicity which suggested that these compounds are not carcinogenic in nature (Table 4). Besides these, the oral rat acute toxicity (LD50) also supports this phenomenon (Table 4). All the polyphenols show good absorption in the human intestine as well as good excretion out (except catechin gallate and epicatechin gallate) from the body. Also, these polyphenolic compounds have a maximum tolerance dose (expressed in terms of log mg/kg/day) ranging from 0.438 to 0.506 which makes them suitable for human use. Altogether this valuable information indicated that green tea polyphenols/catechins can be safely used as drugs.
Once we understood that these polyphenols are safe, we studied their inhibition potency against SARS CoV-2 Mpro using molecular docking study. Amongst them, five polyphenols (EGC, GC, C, EC and CG) bound to Mpro having binding energy comparable to that of ‘Mpro–N3 binding’ (Table 1). However, these four polyphenols fitted within the substrate-binding pocket of Mpro having slightly different binding mode(s). For EGC–Mpro complex, two polar interactions (with Ser46 and Asn142 of Mpro), four hydrophobic interactions (with Leu141, Met165, Glu166 and Ala191 of Mpro) and four hydrogen bonds [with Ser144 (2.5 Å), His163 (2.9 Å & 3.3 Å) and Gln192 (2.6 Å) of Mpro] were observed (Supplemental Figure 1, Tables 2 and 3). When GC was docked into the active site of Mpro, we observed a similar number of polar, hydrophobic and hydrogen bond interactions, that is, two polar interactions (with Ser46, and Asn142 of Mpro), four hydrophobic interactions (with Met49, Leu141, Met165 and Gln189 of Mpro) and four hydrogen bonds [with Phe140 (2.4 Å), Glu166 (2.3 Å), Arg188 (2.0 Å) and Gln192 (2.7 Å) of Mpro] (Supplemental Figure 2, Tables 2 and 3). When the other three polyphenols (C, EC and CG) were docked individually to Mpro, these complexes were stabilized by more number of hydrogen bond interactions (5 nos.) and many non-covalent (polar and hydrophobic) interactions (Supplemental Figures 3–5, Tables 2 and 3). Leu141, Ser144, His163 and Gln192 are the four amino acid residues of Mpro involved in hydrogen bonding with C (Supplemental Figure 3, Table 2). However, EC formed hydrogen bonds with Ser 144, His163, Thr190 and Gln192 of Mpro and the amino acid residues of Mpro engaged in the hydrogen bonding with CG are Ser144, His163, Arg188 and Thr190, respectively (Supplemental Figures 4 and 5, Table 2). Critical analysis of docking results revealed that none of these five polyphenols have any interaction with the two key catalytic residues of Mpro (His41 and Cys145). Therefore, it can be concluded that these five polyphenols (EGC, GC, C, EC and CG) may not inhibit the catalytic/proteolytic activity of Mpro and thus may be ineffective towards the treatment of COVID-19 disease.
On the contrary, three other polyphenols (EGCG, ECG and GCG) interacted with one or both of the catalytic residues (His41 and Cys145) of Mpro by hydrogen bonding (Figures 2–4, Table 2). Both EGCG and ECG formed hydrogen bond with His41 and Cys145 amino acid residues of Mpro (Figures 2 and 3, Table 2). Besides these two key residues, several other amino acid residues from the active site of Mpro were involved in hydrogen bonding as well as different non-covalent interactions (Figures 2 and 3, Tables 2 and 3). Additionally, we noticed that ‘Mpro–GCG docked complex’ was stabilized by nine hydrogen bond interactions [with Phe140 (2.1 Å & 2.1 Å), His163 (2.9 Å), Ser144 (2.2 Å, 2.5 Å & 2.7 Å), Cys145 (2.7 Å) and Gly143 (2.3 Å & 2.7 Å) of Mpro] and multiple non-covalent interactions (polar, hydrophobic, etc.) (Figure 4, Tables 2 and 3). The binding energy of these three polyphenols are higher than that of ‘Mpro–N3 docked complex’ (−7.6 to −9.0 kcal/mol) (Table 1). Since, these three polyphenols (EGCG, ECG and GCG) interacted with the catalytic residue(s) of the Mpro protease and exhibited higher binding affinity than a well-known Mpro protease inhibitor (N3); we can say that they may possibly inhibit the catalytic activity of Mpro protease and maybe the good drug candidates for the treatment of COVID-19 disease.
Figure 2.

Molecular docking of EGCG with Mpro. Stereoview of the docked conformation of the Mpro–EGCG complex showing the possibility of hydrogen bonding interactions with the amino acid residues of Mpro (panel A). Surface representation showing the interaction of EGCG (green stick) at the substrate-binding region of Mpro (panel B). EGCG forms hydrogen bonding with many amino acid residues including His41 and Cys145 of Mpro.
Figure 3.

Molecular docking of ECG with Mpro. The docked conformation of the Mpro–ECG complex depicting the possible hydrogen bonding interactions with various amino acids of Mpro (panel A). Surface representation showing the binding of ECG (red) with Mpro (panel B). ECG forms hydrogen bonding with many amino acid residues including His41 and Cys141 of Mpro.
Figure 4.

Molecular docking of GCG with Mpro. Various hydrogen bonds with different amino acid residues of Mpro with GCG are shown in panel A as docked stereoview conformation. Binding of GCG (orange) at the active site of Mpro is illustrated in panel B as surface representation. GCG interacts with nine amino acid residues including Cys145 of Mpro via H-bonds.
In order to consolidate this conclusion, we selected Mpro–EGCG, Mpro–ECG and Mpro–GCG complex for MD study.
3.2. MD simulation
The MD simulations for the Mpro–EGCG, Mpro–ECG and Mpro–GCG complexes along with unligated Mpro/Mpro (unligated) and Mpro–N3 complex were performed for 100 ns. MD trajectories were initially analyzed with the aid of RMSD and RMSF so as to understand the stability and the fluctuations of these Mpro–polyphenol complex structures. The RMSD values of Mpro (unligated) and Mpro–ECG complex from 2 to 30 ns remained almost constant (∼1.3 Å) (Figure 5). After that RMSD values of these two systems increased gradually till 60 ns and thereafter it converged to a constant value (Figure 5). On the other hand, the RMSD values of Mpro–N3, Mpro–EGCG and Mpro–GCG complexes at 2 ns were 1.50, 1.55 and 1.45, respectively and these values remained almost the same till 30 ns. Afterward, RMSD values for these three complexes experienced a slight deviation (decrease for two Mpro–polyphenol complexes and increase for Mpro–N3 complex) up to 45 ns and beyond this time period, it converged to a constant value (Figure 5). The average RMSD values of Mpro (unligated), Mpro–N3, Mpro–ECG, Mpro–EGCG, Mpro–GCG complexes are 1.68, 1.56, 1.61, 1.53, 1.43 Å, respectively. Such low RMSD value (<2 Å) clearly suggested that all these three Mpro–polyphenol complexes are stable. From these data, it can be further inferred that the Mpro–GCG complex is more stable than the other two Mpro–polyphenol complexes. These findings additionally indicated that the stability of these three Mpro–polyphenol complexes are comparable or relatively more than that of Mpro–N3 complex.
Figure 5.

Determination of RMSD of unligated Mpro and Mpro–N3/ECG/EGCG/GCG complex. The MD simulations for each system were performed for 100 ns. These MD trajectories were analyzed with the aid of RMSD.
The RMSF of alpha-carbon atoms of all system were analyzed and represented in Figure 6. All the five systems [Mpro (unligated), Mpro–N3 and three Mpro–polyphenol complexes] exhibited a similar kind of fluctuation pattern across all three domains (Figure 6, Supplemental Figures 6–8). In all these systems, the fluctuations for the amino acid residues corresponding to domain I was lowest (Figure 6, Supplemental Figure 6). Whereas, the same for the amino acid residues corresponding to domain III was highest (Figure 6, Supplemental Figure 8). The average RMSF values of Mpro (unligated), Mpro–N3, Mpro–ECG, Mpro–EGCG, Mpro–GCG complexes are 1.28, 1.15, 1.22, 1.15, 1.17 Å, respectively. These values indicated that all the Mpro–polyphenol complexes as well as Mpro–N3 complex experienced relatively less conformational fluctuation than Mpro (unligated) system. The fluctuation of various specific amino acid residues (Thr25, Thr26, Leu27, His41 in domain I and Phe140, Leu141, Gly143, Ser144, Cys145, His163, His164, Glu166 of domain II) in these three Mpro–polyphenol systems were less than that in Mpro (unligated) system. Such less fluctuations of these residues in these three Mpro–polyphenol complexes further indicated that these residues within the active site of Mpro interact with ECG/EGCG/GCG. Moreover, these findings suggested that the two Mpro–polyphenol complexes (Mpro–EGCG and Mpro–GCG) and Mpro–N3 complex experienced comparable conformational fluctuation.
Figure 6.

Determination of RMSF of unligated Mpro and Mpro–N3/ECG/EGCG/GCG complex. The RMSF values for Mpro (unligated) and Mpro–N3/ECG/EGCG/GCG complex were estimated from the respective 100 ns MD trajectories. The values were then plotted separately for the domain I (amino acid residues 8–101) as shown in panel A, domain II (amino acid residues 102–184) as shown in panel B and domain III (amino acid residues 201–303) as shown in panel C.
MD trajectories corresponding to three Mpro–polyphenol and Mpro (unligated) systems were further analyzed with the aid of Rg, SASA and the existence of total number of intermolecular hydrogen bonds. Rg, which indicates the compactness of the system, was determined (Supplemental Figure 9(A)) and the calculated average Rg values are listed in Table 5. The obtained average Rg value for Mpro (unligated) was 2.5 Å. Interestingly, the Rg value for all these three Mpro–polyphenol complexes were slightly lower (2.20–2.24 Å) than that of Mpro (unligated) system. Therefore, it can be suggested that these Mpro–polyphenol complexes are relatively more rigid than the Mpro (unligated) system. Subsequently, the degree of expansion of protein volume in each system was assessed by estimating the SASA from individual MD trajectories (Supplemental Figure 9(B)) and the determined average SASA values are listed in Table 5. The SASA value corresponding to the ‘Mpro-ECG complex’ (14208.34 Å2) was slightly higher than that of Mpro (unligated) system (14124.45 Å2), suggesting a slight expansion of Mpro during the interaction with ECG. Whereas, the obtained SASA value for the ‘Mpro–EGCG complex’ and ‘Mpro–GCG complex’ 14113.53 and 13954.64 Å2, respectively, were a little lower compared to that of Mpro (unligated) system (14124.45 Å2) which indicated that Mpro suffers less expansion upon binding with EGCG and GCG.
Table 5.
Average values of the Rg, SASA and the total number of intermolecular hydrogen bond formed for the simulated systems.
| System | Rg (Å) | SASA (Å2) | Total no. of intermolecular hydrogen bonds formed |
|---|---|---|---|
| Mpro (unligated) | 22.52 | 14124.45 | 518 |
| Mpro–ECG | 22.22 | 14208.34 | 520 |
| Mpro–EGCG | 22.45 | 14113.53 | 528 |
| Mpro–GCG | 22.07 | 13954.64 | 540 |
The conformational stability of these three Mpro–polyphenol complexes was analyzed by estimating the total number of intermolecular hydrogen bonds formed during the entire simulation time span (Table 5). The average number of intermolecular hydrogen bonds in Mpro (unligated) and Mpro–ECG complex was almost equal suggesting similar conformational stability in these two systems (Table 5). However, more number of intermolecular hydrogen bonds was evidenced in other two Mpro–polyphenol complexes. Between these two complexes, the lowest number of intermolecular hydrogen bonds (528) was observed when EGCG was complexed with Mpro. Whereas, in the Mpro–GCG complex, the average number of hydrogen bonds was found to be 540 suggesting the highest conformational stability of this complex. These findings are in agreement with the binding energy data obtained from molecular docking studies.
3.3. MM-GBSA
Drugs that showed a high binding score in AutoDock Vina are further selected to calculate binding energy using the MM-GBSA method. The MM-GBSA free energy values of ECG–Mpro, EGCG–Mpro and GCG–Mpro are calculated from the MD trajectories. The MM-GBSA free energy values of GCG–Mpro, EGCG–Mpro and ECG–Mpro complexes are −53.5, −48.9 and −43.56 kcal/mol, respectively. From the MM-GBSA values (Table 6) it is clear that GCG–Mpro complex shows higher free binding energy than other complexes which is also supported by our AutoDock Vina binding energy values. Higher number of hydrogen bonds and hydrophobic interactions may play the key component in MM-GBSA value for GCG–Mpro complex system. Even lower SASA value in case of GCG–Mpro complex also indicated that the non-polar residues are buried in the solvent which may favor the stability of the complex through the synergistic effects of hydrogen bonding and hydrophobic interactions.
Table 6.
MM-GBSA values of different Mpro–polyphenol complexes.
| System | Binding free energy (kcal/mol) |
|---|---|
| Mpro–GCG | −53.54 |
| Mpro–EGCG | −48.92 |
| Mpro–ECG | −43.56 |
4. Conclusion
In summary, our molecular docking study revealed that five polyphenols [EGC, GC, C, EC and CG] which do not interact with the His41 and Cys145 from catalytic dyad of Mpro, has binding energy more than −7.0 kcal/mol. Other three polyphenols [EGCG, ECG and GCG] has the interaction with one or both of these residues. The binding of these three polyphenols ranges between −7.6 and −9.0 kcal/mol with the lowest affinity for GCG and the highest affinity for EGCG. RMSD, RMSF, Rg and SASA investigations strongly support these findings. Even, binding free energy estimations using the MM-GBSA method also reveal that Mpro–GCG complex (−53.54 kcal/mol) is relatively more stable than Mpro–ECG (−48.92 kcal/mol) and Mpro–EGCG complex (−43.56 kcal/mol). Altogether, our findings reveal that green tea catechins/polyphenols (especially EGCG, ECG and GCG) can be potent anti-COVID-19 drug candidates. Additionally, this study opens up futuristic testing (in vitro and in vivo) possibilities of these three green tea polyphenols against COVID-19.
Supplementary Material
Glossary
Abbreviations
- COVID-19
corona virus disease 2019
- SARS CoV-2
severe acute respiratory syndrome corona virus-2
- Mpro
main protease
- MD
molecular dynamics
- RMSD
root mean square deviation
- RMSF
root mean square fluctuation
- Rg
radius of gyration
- SASA
solvent accessible surface area
Funding Statement
RG acknowledges IIT Bhubaneswar for providing fellowship.
Acknowledgements
The authors thank IIT Delhi HPC facility for computational resources.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Aanouz I., Belhassan A., El-Khatabi K., Lakhlifi T., El-Ldrissi M., & Bouachrine M. (2020). Moroccan Medicinal plants as inhibitors against SARS-CoV-2 main protease: Computational investigations. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1758790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelli I., Hassani F., Bekkel Brikci S., & Ghalem S. (2020). In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1763199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., & Lindahl E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1–2, 19–25. 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- Adeoye A. O., Oso B. J., Olaoye I. F., Tijjani H., & Adebayo A. I. (2020). Repurposing of chloroquine and some clinically approved antiviral drugs as effective therapeutics to prevent cellular entry and replication of coronavirus. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1765876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai Z., Liu S., Qu F., Zhang H., Chen Y., & Ni D. (2019). Effect of stereochemical configuration on the transport and metabolism of catechins from green tea across Caco-2 monolayers. Molecules, 24(6), 1185 10.3390/molecules24061185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J. R., & Hilgenfeld R. (2003). Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science, 300(5626), 1763–1767. 10.1126/science.1085658 [DOI] [PubMed] [Google Scholar]
- Babadaei M. M. N., Hasan A., Vahdani Y., Bloukh S. H., Sharifi M., Kachooei E., Haghighat S., & Falahati M. (2020). Development of remdesivir repositioning as a nucleotide analog against COVID-19 RNA dependent RNA polymerase. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1767210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basit A., Ali T., & Rehman S. U. (2020). Truncated human angiotensin converting enzyme 2; A potential inhibitor of SARS-CoV-2 spike glycoprotein and potent COVID-19 therapeutic agent. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1768150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj V. K., Singh R., Sharma J., Rajendran V., Purohit R., & Kumar S. (2020). Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1766572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard J. E., Elowe N. H., Huitema C., Fortin P. D., Cechetto J. D., Eltis L. D., & Brown E. D. (2004). High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chemistry & Biology, 11(10), 1445–1453. 10.1016/j.chembiol.2004.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi S., Poma A. B., & Kolandaivel P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calland N., Albecka A., Belouzard S., Wychowski C., Duverlie G., Descamps V., Hober D., Dubuisson J., Rouille Y., & Seron K. (2012). (−)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology, 55(3), 720–729. 10.1002/hep.24803 [DOI] [PubMed] [Google Scholar]
- Carneiro B. M., Batista M. N., Braga A. C. S., Nogueira M. L., & Rahal P. (2016). The green tea molecule EGCG inhibits Zika virus entry. Virology, 496, 215–218. 10.1016/j.virol.2016.06.012 [DOI] [PubMed] [Google Scholar]
- Chan J. F., Yuan S., Kok K. H., To K. K., Chu H., Yang J., Xing F., Liu J., Yip C. C., Poon R. W., Tsoi H. W., Lo S. K., Chan K. H., Poon V. K., Chan W. M., Ip J. D., Cai J. P., Cheng V. C., Chen H., Hui C. K., & Yuen K. Y. (2020). A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. The Lancet, 395(10223), 514–523. 10.1016/S0140-6736(20)30154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. (2016). Drug resistance mechanisms of three mutations V32I, I47V and V82I in HIV-1 protease toward inhibitors probed by molecular dynamics simulations and binding free energy predictions. RSC Advances, 6(63), 58573–58585. 10.1039/C6RA09201B [DOI] [Google Scholar]
- Chen J., Wang X., Zhu T., Zhang Q., & Zhang J. Z. (2015). A comparative insight into amprenavir resistance of mutations V32I, G48V, I50V, I54V, and I84V in HIV-1 protease based on thermodynamic integration and MM-PBSA methods. Journal of Chemical Information and Modeling, 55(9), 1903–1913. 10.1021/acs.jcim.5b00173 [DOI] [PubMed] [Google Scholar]
- Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y., Xia J., Yu T., Zhang X., & Zhang L. (2020). Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. The Lancet, 395(10223), 507–513. 10.1016/S0140-6736(20)30211-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucinotta D., & Vanelli M. (2020). WHO declares COVID-19 a pandemic. Acta Bio-Medica : Atenei Parmensis, 91(1), 157–160. 10.23750/abm.v91i1.9397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W., Zhang B., Su H., Li J., Zhao Y., Xie X., Jin Z., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., … Liu H. (2020). Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 10.1126/science.abb4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Michielin O., & Zoete V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7, 42717 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T., York D., & Pedersen L. (1993). Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 98(12), 10089–10092. https://ui.adsabs.harvard.edu/abs/1993JChPh.9810089D 10.1063/1.464397 [DOI] [Google Scholar]
- Das S., Sarmah S., Lyndem S., & Singha Roy A. (2020). An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1763201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. (2002). The PyMOL Molecular Graphics System (2002). DeLano Scientific; http://www.pymol.org [Google Scholar]
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H. R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R. A., Berger A., Burguiere A. M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J. C., Muller S., … Doerr H. W. (2003). Identification of a novel coronavirus in patients with severe acute respiratory syndrome. The New England Journal of Medicine, 348(20), 1967–1976. 10.1056/NEJMoa030747 [DOI] [PubMed] [Google Scholar]
- Elfiky A. A. (2020. a). Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1761881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. (2020. b). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1761882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen A. D., Al-Obaidi A., Sahin A. T., & Yelekci K. (2020). Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1758791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi S. K., Raja K., Sebastine I., & Joseph J. (2020). Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. Journal of Biomolecular Structure and Dynamics, 1–7. 10.1080/07391102.2020.1760136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassina G., Buffa A., Benelli R., Varnier O. E., Noonan D. M., & Albini A. (2002). Polyphenolic antioxidant (−)-epigallocatechin-3-gallate from green tea as a candidate anti-HIV agent. AIDS, 16(6), 939–941. 10.1097/00002030-200204120-00020 [DOI] [PubMed] [Google Scholar]
- Frisch M., Clemente F.. Gaussian 09, Revision A. 01, MJ Frisch, GW Trucks, HB Schlegel, GE Scuseria, MA Robb, JR Cheeseman, G. Scalmani, V. Barone, B. Mennucci, GA Petersson, H. Nakatsuji, M. Caricato, X. Li, HP Hratchian, AF Izmaylov, J. Bloino, G. Zhe.
- Grum-Tokars V., Ratia K., Begaye A., Baker S. C., & Mesecar A. D. (2008). Evaluating the 3C-like protease activity of SARS-coronavirus: Recommendations for standardized assays for drug discovery. Virus Research, 133(1), 63–73. 10.1016/j.virusres.2007.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi G. A., Ogunro O. B., Adegunloye A. P., Ogunyemi O. M., & Afolabi S. O. (2020). Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CL(pro)): An in silico screening of alkaloids and terpenoids from African medicinal plants. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1764868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt B. H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K. M., Smith C. M., Rota P. A., & Baker S. C. (2004). Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. Journal of Virology, 78(24), 13600–13612. 10.1128/JVI.78.24.13600-13612.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan A., Paray B. A., Hussain A., Qadir F. A., Attar F., Aziz F. M., Sharifi M., Derakhshankhah H., Rasti B., Mehrabi M., Shahpasand K., Saboury A. A., & Falahati M. (2020). A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1754293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B., Bekker H., Berendsen H. J. C., & Fraaije J. G. E. M. (1997). LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry, 18(12), 1463–1472. [DOI] [Google Scholar]
- Hou T., Wang J., Li Y., & Wang W. (2011). Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. Journal of Chemical Information and Modeling, 51(1), 69–82. 10.1021/ci100275a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh T., Wang H., & Luan B. (2020). In silico exploration of molecular mechanism of clinically oriented drugs for possibly inhibiting SARS-CoV-2’s main protease. Journal of Physical Chemistry Letters., 11(11), 4413–4420. 10.1021/acs.jpclett.0c00994 [DOI] [PubMed] [Google Scholar]
- Ide K., Kawasaki Y., Kawakami K., & Yamada H. (2016). Anti-influenza virus effects of catechins: A molecular and clinical review. Current Medicinal Chemistry, 23(42), 4773–4783. 10.2174/0929867324666161123091010 [DOI] [PubMed] [Google Scholar]
- Islam R., Parves M. R., Paul A. S., Uddin N., Rahman M. S., Mamun A. A., Hossain M. N., Ali M. A., & Halim M. A. (2020). A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1761883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail N. A., & Jusoh S. A. (2017). Molecular docking and molecular dynamics simulation studies to predict flavonoid binding on the surface of DENV2 E protein. Interdisciplinary Sciences, Computational Life Sciences, 9(4), 499–511. 10.1007/s12539-016-0157-8 [DOI] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., … Yang H. (2020). Structure of M(pro) from COVID-19 virus and discovery of its inhibitors. Nature. 582, 289–293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Joshi R. S., Jagdale S. S., Bansode S. B., Shankar S. S., Tellis M. B., Pandya V. K., Chugh A., Giri A. P., & Kulkarni M. J. (2020). Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. Journal of Biomolecular Structure and Dynamics, 1–16. 10.1080/07391102.2020.1760137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski G. A., Friesner R. A., Tirado-Rives J., & Jorgensen W. L. (2001). Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. The Journal of Physical Chemistry B, 105(28), 6474–6487. 10.1021/jp003919d [DOI] [Google Scholar]
- Khan S. A., Zia K., Ashraf S., Uddin R., & Ul-Haq Z. (2020). Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1751298 [DOI] [PubMed] [Google Scholar]
- Kim Y., Liu H., Galasiti Kankanamalage A. C., Weerasekara S., Hua D. H., Groutas W. C., Chang K. O., & Pedersen N. C. (2016). Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathogens, 12(3), e1005531 10.1371/journal.ppat.1005531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu S. Y., Rhim J. Y., & Park W. B. (2005). Antiherpetic activities of flavonoids against herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2) in vitro. Archives of Pharmacal Research, 28(11), 1293–1301. 10.1007/BF02978215 [DOI] [PubMed] [Google Scholar]
- Mahajan P., Tomar S., Kumar A., Yadav N., Arya A., & Dwivedi V. D. (2020). A multi-target approach for discovery of antiviral compounds against dengue virus from green tea. Network Modeling Analysis in Health Informatics and Bioinformatics, 9(1), 20 10.1007/s13721-020-0222-4 [DOI] [Google Scholar]
- Mahanta S., Chowdhury P., Gogoi N., Goswami N., Borah D., Kumar R., Chetia D., Borah P., Buragohain A. K., & Gogoi B. (2020). Potential anti-viral activity of approved repurposed drug against main protease of SARS-CoV-2: An in silico based approach. Journal of Biomolecular Structure and Dynamics, 1–15. 10.1080/07391102.2020.1768902 [DOI] [PubMed] [Google Scholar]
- Mahase E. (2020). Coronavirus covid-19 has killed more people than SARS and MERS combined, despite lower case fatality rate. BMJ, 368, m641 10.1136/bmj.m641 [DOI] [PubMed] [Google Scholar]
- Marra M. A., Jones S. J., Astell C. R., Holt R. A., Brooks-Wilson A., Butterfield Y. S., Khattra J., Asano J. K., Barber S. A., Chan S. Y., Cloutier A., Coughlin S. M., Freeman D., Girn N., Griffith O. L., Leach S. R., Mayo M., McDonald H., Montgomery S. B., … Roper R. L. (2003). The genome sequence of the SARS-associated coronavirus. Science, 300(5624), 1399–1404. 10.1126/science.1085953 [DOI] [PubMed] [Google Scholar]
- Mittal L., Kumari A., Srivastava M., Singh M., & Asthana S. (2020). Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. Journal of Biomolecular Structure and Dynamics, 1–26. 10.1080/07391102.2020.1768151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S., & Kollman P. A. (1992). Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. Journal of Computational Chemistry, 13(8), 952–962. 10.1002/jcc.540130805 [DOI] [Google Scholar]
- Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., & Olson A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M., Huey R., & Olson A. J. (2008). Using AutoDock for ligand–receptor docking. Current Protocols in Bioinformatics, 24, 8.14.1–8.14.40. 10.1002/0471250953.bi0814s24 [DOI] [PubMed] [Google Scholar]
- Muralidharan N., Sakthivel R., Velmurugan D., & Gromiha M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics, 1–6. 10.1080/07391102.2020.1752802 [DOI] [PubMed] [Google Scholar]
- Nejadi Babadaei M. M., Hasan A., Haj Bloukh S., Edis Z., Sharifi M., Kachooei E., & Falahati M. (2020). The expression level of angiotensin-converting enzyme 2 determine the severity of COVID-19: Lung and heart tissue as targets. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1767211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman E. E. A., Toogood P. L., & Neamati N. (2020). COVID-19: Living through another pandemic. ACS Infectious Diseases. 10.1021/acsinfecdis.0c00224 [DOI] [PubMed] [Google Scholar]
- Pires D. E., Blundell T. L., & Ascher D. B. (2015). pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. Journal of Medicinal Chemistry, 58(9), 4066–4072. 10.1021/acs.jmedchem.5b00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit R. (2014). Role of ELA region in auto-activation of mutant KIT receptor: A molecular dynamics simulation insight. Journal of Biomolecular Structure & Dynamics, 32(7), 1033–1046. 10.1080/07391102.2013.803264 [DOI] [PubMed] [Google Scholar]
- Rajendran V. (2016). Structural analysis of oncogenic mutation of isocitrate dehydrogenase 1. Molecular Biosystems, 12(7), 2276–2287. 10.1039/c6mb00182c [DOI] [PubMed] [Google Scholar]
- Rajendran V., Gopalakrishnan C., & Sethumadhavan R. (2018). Pathological role of a point mutation (T315I) in BCR-ABL1 protein-A computational insight. Journal of Cellular Biochemistry, 119(1), 918–925. 10.1002/jcb.26257 [DOI] [PubMed] [Google Scholar]
- Ren L. L., Wang Y. M., Wu Z. Q., Xiang Z. C., Guo L., Xu T., Jiang Y. Z., Xiong Y., Li Y. J., Li X. W., Li H., Fan G. H., Gu X. Y., Xiao Y., Gao H., Xu J. Y., Yang F., Wang X. M., Wu C., … Wang J. W. (2020). Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chinese Medical Journal, 133(9), 1015–1024. 10.1097/CM9.0000000000000722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma J., Bhardwaj V. K., Das P., & Purohit R. (2020). Identification of naturally originated molecules as gamma-aminobutyric acid receptor antagonist. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1720818 [DOI] [PubMed] [Google Scholar]
- Singh R., Bhardwaj V., & Purohit R. (2020). Identification of a novel binding mechanism of Quinoline based molecules with lactate dehydrogenase of Plasmodium falciparum. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1711809 [DOI] [PubMed] [Google Scholar]
- Sinha S. K., Shakya A., Prasad S. K., Singh S., Gurav N. S., Prasad R. S., & Gurav S. S. (2020). An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1762741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Ivanov K. A., Putics Á., Hertzig T., Schelle B., Bayer S., Weißbrich B., Snijder E. J., Rabenau H., Doerr H. W., Gorbalenya A. E., & Ziebuhr J. (2003). Mechanisms and enzymes involved in SARS coronavirus genome expression. The Journal of General Virology, 84(Pt 9), 2305–2315. 10.1099/vir.0.19424-0 [DOI] [PubMed] [Google Scholar]
- Umesh K. D., Selvaraj C., Singh S. K., & Dubey V. K. (2020). Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1763202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal P. P., Das B. K., Soorya E., & Chakraborty D. (2020). Effect of hydrophobic and hydrogen bonding interactions on the potency of ss-alanine analogs of G-protein coupled glucagon receptor inhibitors. Proteins: Structure, Function, and Bioinformatics, 88(2), 327–344. 10.1002/prot.25807 [DOI] [PubMed] [Google Scholar]
- Wahedi H. M., Ahmad S., & Abbasi S. W. (2020). Stilbene-based natural compounds as promising drug candidates against COVID-19. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1762743 [DOI] [PubMed] [Google Scholar]
- Weber C., Sliva K., von Rhein C., Kummerer B. M., & Schnierle B. S. (2015). The green tea catechin, epigallocatechin gallate inhibits chikungunya virus infection. Antiviral Research, 113, 1–3. 10.1016/j.antiviral.2014.11.001 [DOI] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y. M., Wang W., Song Z. G., Hu Y., Tao Z. W., Tian J. H., Pei Y. Y., Yuan M. L., Zhang Y. L., Dai F. H., Liu Y., Wang Q. M., Zheng J. J., Xu L., Holmes E. C., & Zhang Y. Z. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579(7798), 265–269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Wang J., Deng F., Hu Z., & Wang H. (2008). Green tea extract and its major component epigallocatechin gallate inhibits hepatitis B virus in vitro. Antiviral Research, 78(3), 242–249. 10.1016/j.antiviral.2007.11.011 [DOI] [PubMed] [Google Scholar]
- Yan L., Velikanov M., Flook P., Zheng W., Szalma S., & Kahn S. (2003). Assessment of putative protein targets derived from the SARS genome. FEBS Letters, 554(3), 257–263. 10.1016/S0014-5793(03)01115-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G. F., Anand K., Bartlam M., Hilgenfeld R., & Rao Z. (2003). The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proceedings of the National Academy of Sciences of the United States of America, 100(23), 13190–13195. 10.1073/pnas.1835675100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki A. M., van Boheemen S., Bestebroer T. M., Osterhaus A. D., & Fouchier R. A. (2012). Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. The New England Journal of Medicine, 367(19), 1814–1820. 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- Zheng J. (2020). SARS-CoV-2: An emerging coronavirus that causes a global threat. International Journal of Biological Sciences, 16(10), 1678–1685. 10.7150/ijbs.45053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., & Tan W. (2020). A novel coronavirus from patients with pneumonia in China, 2019. The New England Journal of Medicine, 382(8), 727–733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.