

Abstract
The emergence of the coronavirus disease-2019 pandemic has led to an outbreak in the world. The SARS-CoV-2 is seventh and latest in coronavirus family with unique exonucleases for repairing any mismatches in newly transcribed genetic material. Therefore, drugs with novel additional mechanisms are required to simultaneously target and eliminate the virus. Thus, a newly deciphered N protein is taken as a target that belongs to SARS-CoV-2. They play a vital role in RNA transcription, viral replication and new virion formation. This study used virtual screening, molecular modeling and docking of the 8987 ligands from Asinex and PubChem databases against this novel target protein. Three hotspot sites having DScore ≥1 (Site 1, Site 2 and Site 3) for ligand binding were selected. Subsequently, high throughput screening, standard precision and extra precision docking process and molecular dynamics concluded three best drugs from two libraries. Two antiviral moieties from Asinex databases (5817 and 6799) have docking scores of −10.29 and −10.156; along with their respective free binding energies (ΔG bind) of −51.96 and −64.36 on Site 3. The third drug, Zidovudine, is from PubChem database with docking scores of −9.75 with its binding free energies (ΔG bind) of −59.43 on Site 3. The RMSD and RMSF were calculated for all the three drugs through molecular dynamics simulation studies for 50 ns. Zidovudine shows a very stable interaction with fluctuation starting at 2.4 Å on 2 ns and remained stable at 3 Å from 13 to 50 ns. Thus, paving the way for further biological validation as a potential treatment.
Communicated by Ramaswamy H. Sarma
Keywords: COVID-19, docking, molecular dynamics, nucleocapsid phosphoprotein, SARS-CoV-2
1. Introduction
Coronaviruses (CoV) are the positively single stranded RNA enveloped viruses having spherical or pleomorphic shape with club shaped glycoproteins on their outer surface (Fehr & Perlman, 2015). Thus, deriving its name from a Latin word meaning ‘crown or halo’ (Paules et al., 2020). There are seven recognized strains of these viruses such as 229E, NL63, OC43, HKU1, MERS-CoV, SARS-CoV-1, and the latest and novel SARS-CoV-2 which is responsible for the current outbreak (Zhu et al., 2020). The last two epidemics due to SARS-CoV-1 and MERS-CoV were associated with 10% and 37% mortality, respectively (Huang et al., 2020). The SARS-CoV-2 is causing the COVID-19 disease with variable mortality in different countries of almost all continents. It has been declared as the pandemic by World Health Organization (WHO) (Eurosurveillance, 2020). It directly affects the respiratory tract leading to the Acute Respiratory Distress Syndrome (ARDS) and death in the want of oxygen (Chen et al., 2020).
There are four structural proteins in CoV, including trimeric spike (S glycoprotein) responsible for attachment, fusion and entry with the help of angiotensin converting enzyme-2 (ACE2) on the human cell (Hasan et al., 2020). A pentameric small envelope protein (E protein) regulates ion channel activity (Gupta et al., 2020). The others are matrix protein (M protein) and nucleocapsid protein (N protein) for virion formation and replication (Brian & Baric, 2005). Along with these some main proteases (Mpro) are also present (Boopathi et al., 2020). In addition to that, unlike other RNA viruses, it has exonucleases to repair mismatches in newly transcribed double stranded RNA and provide resistance to many of the therapies (Shannon et al., 2020). So drugs with additional mechanisms are required to target the several viral proteins simultaneously. The nucleocapsid N proteins are present in large amount as structural protein and they are the least variable and highly conserved structure of CoV (Lin et al., 2014). The N-terminal Domain (NTD), the C-Terminal Domains (CTD) and a central Ser/Arg (SR) rich linker are three main domains of N proteins which are capable of binding to the RNA, oligomerization and phosphorylation, respectively (Chang et al., 2013; Lo et al., 2013). These bind with the RNA genome to form ribonucleoprotein complex necessary to produce virion core and RNA synthesis for viral replication (Cong et al., 2019; McBride et al., 2014). Furthermore, studies have suggested their role in regulating host–pathogen interactions such as reorganizations of actin filaments, progression of the host cell cycle, and finally, apoptosis of their cells (Du et al., 2008; Surjit et al., 2006).
The structure of nucleocapsid phosphoproteins is just like an overall right hand like fold with an extended central loop divided into an area called palm and fingers (e.g. 6M3M and 6VYO) (Kang et al., 2020). One of the fingers is having more basic amino acid residues and therefore called as basic finger. So instead of fewer location based on NTD and CTD, the protein binds with the basic amino acid residue moieties significantly with (A50, T57, H59, R92, I94, S105, R107, R149, Y172) which are located in the basic finger and its junction with the palm. The studies suggest that the suitable target is the hot spot located at the interface of basic finger and the palm (Dinesh et al., 2020).
All the SARS-CoV-2 proteins are potential drug targets thus inhibiting N-proteins will impair the viral replication machinery (Gordon et al., 2020). Many studies have been conducted with different targets of this virus such as RNA-dependent RNA polymerase (Elfiky, 2020b), Spike protein/ACE2 (Hasan et al., 2020) and main proteases (Elmezayen et al., 2020; Pant et al., 2020). Others have been conducted to find the potential treatments like phytochemicals (Aanouz et al., 2020; Elfiky, 2020a; Enmozhi et al., 2020; Islam et al., 2020). However, because of the lack of safety and toxicity studies, they will take time for further validation and clinical trials. Therefore, designing and repurposing the drugs from FDA approved drug for inhibiting N protein binding with RNA domains will lead to the inhibition of viral replication and subsequent virion formation of SARS-CoV-2.
2. Material and methods
2.1. Target selection and molecular library preparation
Our objective was to design an inhibitor against RNA binding domain (RBD) of structural N protein of SARS-CoV-2. The three dimensional structure of N protein 6VYO of SARS-CoV-2 was retrieved from the Protein Data Bank (PDB) (https://www.rcsb.org/) for RBD. The resolution of the selected target protein structure is 1.70 Å and the structure was identified by the X-ray diffraction method. It was selected because of its high resolution value and the latest structure of nucleocapsid phosphoprotein of SARS-CoV-2 during the study.
The five active sites were obtained which were designated as Site 1, Site 2, Site 3, Site 4 and Site 5. Out of these, three sites were selected for further analysis based on the DScore of more than one (DScore of Sites 1, 2 and 3 were1.039, 0.99 and 1.045, respectively). The 2D structure of ligands were downloaded from Asinex and PubChem open chemistry databases. Thus, a total of 8987 (eight thousands nine hundred and eighty-seven) ligands were grouped into two libraries (Lib A and Lib B). The Lib A included 8722 antiviral compounds downloaded from Asinex Elite molecular database (http://www.asinex.com/antiviral/), whereas, Lib-B had 265 FDA approved moieties categorized as drug for infectious disease that were downloaded from NCBI PubChem (https://pubchem.ncbi.nlm.nih.gov/) (Figure 1).
Figure 1.

Virtual screening workflow targeting N protein of SARS CoV-2 (PDB ID: 6VYO) with Asinex and PubChem database moieties.
2.2. Protein preparation
The Schrodinger’s Protein Preparation Wizard was used for the preparation of three-dimensional structure of target protein (PDB ID: 6VYO). It was done by correcting bond orders, the addition of hydrogen bonds (HBs), creating zero-order bonds to metal atoms, creating disulphide bonds, conversion of selenomethionine to methionine, filling in missing side chains prime, deleting waters from het groups and generating het states using EpikPh 7 to ±2. The optimization of protein HBs was done through renovation by the overlying and minimizing hydrogen using OPLS3e force field (Madhavi Sastry et al., 2013).
2.3. Receptor grid generation and active site prediction
The recognition of the active site or the binding site of the target structure is a key step in drug designing through computational docking of the novel chemical moieties. The active site of 6VYO target was found to be a pocket like region containing a mix of hydrophilic and hydrophobic regions with a variety of hydrogen donors and acceptors. The active sites for receptor–ligand binding interaction of 6VYO proteins were predicted using sitemap modules in Maestro 12.0 Schrodinger suite (Halgren, 2009). The receptor grid was generated using the generation module in Maestro 12.0 Schrodinger suite by preferring active site residues after their prediction. The size of grid box was centroid of selected residues of the protein receptor. The atoms of protein were fixed within the default parameters of the radii of Vander Waal’s scaling factor of 1 Å with partial charge cut-off of 0.25 Å using OPLS3e force field and 20 Å docked ligand length. The dimensions of the grid box and receptor setup were x = 20 Å, y = 20 Å, z = 20 Å and x = 10 Å, y = 10 Å, z = 10 Å during docking study, respectively, with a grid space of 1 Å.
2.4. Ligands preparation
The 8722 antiviral drugs and 265 FDA approved drugs used for infectious diseases were selected for virtual screening and molecular docking study against SARS-CoV-2 target proteins (6VYO) from Asinex Elite molecular database (http://www.asinex.com/antiviral/) and NCBI PubChem database (https://pubchem.ncbi.nlm.nih.gov/), respectively. The ligands were prepared by using the ligand preparation module of molecular modeling package with suitable parameters like optimization, ring conformation, 2D to 3D conversion, determination of promoters, tautomers and ionization states at pH 7.0 along with partial atomic charges using OPLS3e force field A.
2.5. Virtual screening and molecular docking
Docking study of selected ligands (8722 antiviral drugs, 265 FDA approved drugs for infectious diseases) for high throughput virtual screening (HTVS) against a target protein (PDB ID: 6YVO) was done. The HTVS with flexible docking was performed on Schrodinger’s ligand docking module between ligands and active Site 3 of target protein (6YVO). After HTVS, standard precision (SP) using ligand-docking process filtered out 500 ligands from Asinex database and 200 ligands from PubChem database. Out of those, 100 ligands from each library were selected which had best configuration with highest docking score of virtual screening following extra precision (XP) with flexible docking (Friesner et al., 2006; Halgren et al., 2004). These 100 ligands from each library were docked with Site 1 and Site 2 as well. Finally, the top 10 ligands from both libraries (Lib A and Lib B) were selected which were common against all the three sites for further analysis (Figure 1).
2.6. Calculation of binding free energies by prime MM-GBSA approach
The ligands and receptor complexes were analyzed for their binding free energies by prime MM-GBSA (Molecular Mechanics Generalized Born Surface Area) module of Schrodinger suite with the OPLS3e force field (Genheden & Ryde, 2015). The prime MM-GBSA approach is used to calculate ΔG bind of each ligand which is based on the docking complex interactions. However, the prioritization of lead compound was based on rescoring function of MM-GBSA approach and its binding free energies. These calculations along with docking scores were considered to optimize seven final identified ligand molecules.
2.7. ADMET screening
The seven inhibitors were analyzed for their drug likeliness and pharmacokinetic properties such as ADME/T (Absorption, Distribution, Metabolism, Elimination or Toxicity) (Butina et al., 2002). The QikProp module implemented in the ‘Schrodinger Suite 2019’ was used to estimate the drug likeliness of the inhibitors using pharmacokinetic parameters such as probable H-bonding atoms, human oral absorption (<25%: poor; >above 80%: good) and the IC50 values for blockage of human Ether-a-go-go-Related Gene (hERG) K + channels(< −5: satisfactory) and Lipinski’s rule of five (Lipinski, 2004).
2.8. Molecular dynamics simulation studies
The molecular dynamics (MD) simulation for protein–ligand interaction was done by using ‘Desmond module’ for studying the conformational change of protein in a solvent (https://www.schrodinger.com/desmond). The influence and structural stability of water molecules was investigated by running the protocol for 50 ns after following the MD protocol of minimization, heating, equilibrium and production run. The default protocol with OPLS3e force field of Desmond was used to saturate, minimizing the energy and determination of partial charges (Bowers et al., 2006).
The complexes were immersed in orthorhombic box (10 × 10 × 10 Å) of TIP3P solvent with 15,472 (ID 5817), 15,459 (ID 6799) and 15,467 (72187) water molecules, respectively, buffered at a distance of 10 Å after the determination of topology and atomic coordinates of protein with the edge of the box. A constant temperature of 300 K along with isobaric isothermal ensemble (NPT equilibrium) was used by Nose–Hoover chain thermostat along with Martyn–Tobais–Klein barostat for maintaining pressure at one bar for 50 ns for MD simulations. The resulting trajectories of protein–ligand interactions were recorded at every 50.0 ps interval for analysis. The simulations interaction diagrams, protein structure details, protein–ligand RMSD and individual RMSF figures were generated.
3. Results and discussions
3.1. Protein structure reliability and active site selection
Quality and reliability of three-dimensional structure of target protein is the important parameter for drug designing. The PROCHECK server presents Ramachandran plot displaying allowed and the disallowed regions about backbone dihedrals of protein residues. More than 90% score of most favored regions ensures a model of good quality. The Ramachandran plot (Figure 2(A)) of 6VYO proteins structures reveals that 100% of residues fall within most allowed region, which indicates that the three-dimensional protein structures are reliable (Table 1).
Figure 2.

(A) Ramachandran plots of 6VYO proteins structure describe favored and the disallowed regions of the residues. (B) Various predicted ligand-binding sites of target protein structure (6VYO) with their respective DScore by sitemap module of Schrödinger’s software.
Table 1.
Ramachandran plot statistics showing residues present infavoured and disallowed regions of 6VYO protein structure.
| Properties | Residues | Percentage (%) |
|---|---|---|
| The Most favored regions | 358 | 92.5 |
| Additional allowed regions | 29 | 7.5 |
| Generously allowed regions | 0 | 0.00 |
| Disallowed regions | 0 | 0.00 |
| The end residues (excluding Glycine and Proline) | 8 | |
| Proline | 64 | |
| Glycine | 40 | |
| Total number of residues | 499 | 100 |
This shows minimum steric interactions due to the forbidden psi and the phi angles thereby advocating good worth of stereo chemical quality of the 3D protein structure (Madhavi Sastry et al., 2013). After selection of 3D structure of target protein, active site was predicated for molecular docking. Five active sites were predicted using sitemap modules of Schrodinger’s software (Figure 2(B)).
Table 2 shows the associations of Sites 1, 2, 3, 4 and 5 with their respective amino acid residues grouped into chain A, B, C and D, docking score, size and site score. Out of the five-hotspot sites, the best three sites were selected based on their DScore. Site 3 having 1.045 DScore was selected as main target site for the study. The DScore of Sites 1 and 2 are 1.039 and 0.99, respectively. The site with DScore of more than one was taken as predictive for better study of interaction. The amino acids residues of Sites 1, 2 and 3 are associated with chain A & B, A &D and C & D, respectively. The docking results also showed the better docking scores with Sites 1 and 3 as compared to Site 2 (Figures 3 and 4).
Table 2.
Various active sites of target protein (PDB ID: 6VYO) with their respective DScore, size and site score and associated residues predicated by sitemap module of Schrödinger’s software.
| S/no. | Active site | DScore | Size | Site score | Residues |
|---|---|---|---|---|---|
| 1 | Site 1 | 1.039 | 216 | 1.009 | Chain A: 50ALA, 51SER, 53PHE, 54THR, 55ALA, 56LEU, 57THR, 59HIP, 92ARG, 105SER, 107ARG, 109TYR, 149ARG, 156ALA, 157ILE, 158VAL, 160GLN, 161LEU, 162PRO, 163GLN, 164GLY, 165THR, 167LEU, 171PHE, 172TYR, 173ALA Chain B: 52TRP, 53PHE, 73PRO, 74ILE, 75ASN, 76THR, 77ASN, 78SER, 82ASP, 145hie, 146ile, 148thr, 149arg, 150asn, 153ASN, 154ASN, 155ALA, 158VAL, 159LEU, 160GLN, 157ILE |
| 2 | Site 2 | 0.99 | 196 | 0.98 | Chain A: 52TPR, 53PHE, 73PRO, 74ILE, 75ASN 76THR, 77ASN, 112TYR, 114GLY, 115THR, 116GLY, 119ALA, 144ASP, 145HIE, 146ILE, 147GLY, 148THR, 149ARG, 150ASN, 153ASN, 154ASN, 157ILE, 158VAL, 159LEU, 160GLN Chain D: 54THR, 55ALA, 57THR, 59HIE, 90ALA, 91THR, 92ARG, 94ILE, 96GLY, 97GLY, 98ASP, 102LYS, 104LEU, 105SER, 106PRO, 107ARG, 109TYR, 158VAL, 159LEU, 160GLN, 161LEU, 163GLN, 165THR, 166THR, 167LEU, 173ALA |
| 3 | Site 3 | 1.045 | 183 | 1.039 | Chain C: 50ALA, 51SER, 53PHE, 54THR, 55ALA, 56LEU, 57THR, 59HIP, 92ARG,107ARG, 109TYR, 149ARG, 156ALA, 157ILE, 158VAL Chain D: 53TRP, 73PRO, 74ILE, 75ASN, 76THR, 77ASN, 78SER, 82ASP, 145HIE, 146ILE, 147GLY, 148THR, 149ARG, 150ASN, 153ASN, 154ASN, 155ALA, 157ILE, 158VAL, 159LEU, 160GLN |
| 4 | Site 4 | 0.925 | 105 | 0.955 | – |
| 5 | Site 5 | 0.981 | 90 | 0.968 | – |
Figure 3.

Selected four lead molecules from Asinex database and their comparative docking score with Sites 1, 2 and 3 of target protein (6VYO).
Figure 4.

Selected three potential lead molecules from PubChem database and their comparative docking score with Sites 1, 2 and 3 of target protein (6VYO).
3.2. Virtual screening &molecular docking
The selection of most probable drug candidates acquired from virtual screening and molecular docking of Asinex and PubChem datasets was obtained by analyzing scoring functions and the binding free energies-MM/GBSA. The top-scored molecules from both the databases were prioritized based on docking score. Out of 100 XP docking molecules, best top 10 docked ligands were selected from each library at Sites 1, 2 and 3. Tables 3(A-1 to A-10), 4 (C-1 to C-10) and 5 (E-1 to E-10) show the results of binding free energies ΔG bind (kcal/mol), docking scores, number of HBs and HB interactive residues of the ligands from the Lib-A on Site 1, Site 2 and Site 3, respectively.
Table 3.
A-1 to A-10: Top 10 ligands from Lib-A; B-1 to B-10: top 10 ligands from Lib-B and their prime MM-GBSA, docking score, no of H bonds and H bond interactive residues with Site 1 (DScore – 1.39) of target protein (6VYO).
| S/no. | Ligand ID | 2D structures | ΔG bind (kcal/mol) | Docking score | No./H-bond | H-bond interactive residues |
|
|---|---|---|---|---|---|---|---|
| Chain A | Chain B | ||||||
| A-1 | 5817 |  |
−38.79 | −10.61 | 5 | ALA55(2) | ASN77 ASN154 ASN153 |
| A-2 | 6799 |  |
−44.61 | −8.82 | 4 | ALA55 | ASN75 ASN154 ASN153 |
| A-3 | 6797 |  |
−40.21 | −8.61 | 4 | THR57 | ASN75 ASN154 ASN153 |
| A-4 | 8517 |  |
−44.09 | −8.28 | 3 | THR57 ALA55 | ASN75 |
| A-5 | 7059 |  |
−49.82 | −8.25 | 4 | THR57 ALA55 | ASN77 ASN75 |
| A-6 | 8503 |  |
−43.45 | −8.22 | 3 | THR57 ALA55 | ASN75 |
| A-7 | 7025 |  |
−33.96 | −8.00 | 3 | ASN75 THR76 ASN77 | |
| A-8 | 7064 |  |
−37.93 | −7.97 | 5 | THR57 ALA55 | ASN77 THR76 ASN75 |
| A-9 | 5782 |  |
−38.66 | −7.90 | 6 | THR57 ALA55 ARG107 | ASN77 ASN154 ASN153 |
| A-10 | 6801 |  |
−30.21 | −7.77 | 4 | THR57 ALA55 | ASN154 ASN75 |
| B-1 | 72187 |  |
−23.50 | −11.08 | 5 | ARG107(2) | ASN77 ASN150(2) |
| B-2 | 37542 |  |
−40.92 | −8.58 | 5 | ALA55(2) | ASN77 ASN154 ASN155 |
| B-3 | 135413534 |  |
−49.88 | −8.10 | 5 | ARG107 | ASN75(2) ASN153 HIE145 |
| B-4 | 2082 |  |
−43.47 | −7.13 | 2 | ALA55(2) | |
| B-5 | 71616 |  |
−43.00 | −6.76 | 1 | ALA55 | |
| B-6 | 23665637 |  |
−33.66 | −6.75 | 4 | ARG107 | ASN77(2) ASN75 |
| B-7 | 159269 | 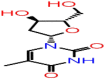 |
−37.04 | −6.73 | 5 | ARG107 THR57(2) | ASN75(2) |
| B-8 | 16124688 |  |
−52.05 | −6.61 | 3 | ASN77 ASN75(2) | |
| B-9 | 131801472 |  |
−14.73 | −6.46 | 1 | ASN75 | |
| B-10 | 60877 | 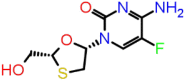 |
−38.34 | −6.38 | 3 | THR57 ALA55 | ASN75 |
While Tables 3(B-1 to B-10), 4(D-1 to D-10) and 5(F-1 to F-10) show the interaction of top 10 ligands from Lib-B on all the three sites (Site 1, Site 2 and Site 3) with their respective binding free energy ΔG bind (kcal/mol), docking scores, number of HBs and HB interactive residues in chain A, B, C and D.
It has been observed that many drugs in each category are common with all three sites. Therefore, those drugs, which are common among top 10 with interactions on all three sites, were taken out for the conclusion. Out of top 10 from each target site, four drug-like-moieties having antiviral activities from the Asinex databases and three antiviral drugs from PubChem are found to have great affinity on the three selected hotspot sites based on their docking score. Thus, seven drugs are chosen from the Asinex and PubChem database (Table 6) based on their docking score on all the three sites.
Table 6.
G-1 to G-4: Four selected molecules of Lib-A from top 10 molecules against Sites 1, 2 and 3; H-1 to H-3: three selected molecules of Lib-B from top 10 molecules against Sites 1, 2 and 3.
| Binding Site 1 D score (1.039) |
Binding Site 2 D score (0.99) |
Binding Site 3 D score (1.045) |
|||||
|---|---|---|---|---|---|---|---|
| S/no. | Ligand ID | Docking score | MM-GBSA (kcal/mol) | Docking score | MM-GBSA (kcal/mol) | Docking score | MM-GBSA (kcal/mol) |
| G-1 | 5817 | −10.19 | −38.79 | −6.752 | −47.93 | −10.288 | −45.23 |
| G-2 | 6799 | −8.826 | −44.61 | −6113 | −29.47 | −10.156 | −46.49 |
| G-3 | 6797 | −8.613 | −40.21 | −6.922 | −35.18 | −8.42 | −46.54 |
| G-4 | 7025 | −8.28 | −33.96 | −6.94 | −51.99 | −8.92 | −61.45 |
| H-1 | 72187 | −11.084 | −23.50 | −9.091 | −16.37 | −9.753 | −44.64 |
| H-2 | 135413534 | −8.102 | −49.88 | −5.403 | −39.44 | −8.298 | −48.05 |
| H-3 | 37542 | −8.588 | −40.92 | −6.377 | −47.82 | −7.943 | −44.96 |
Thus, the ligands of Asinex databases with their docking scores on each sites (Site 1, Site 2, Site 3) are 5817 (−10.19, −6.75, −10.29); 6799 (−8.83, −6.11, −10.156); 6797 (−8.61, −6.92, −8.42) and 7025 (−8.28, −6.94, −8.92). While three drugs concluded from PubChem databases and their docking score on each sites are Zidovudine (−11.08, −9.09, −9.75), Valganciclovir (−8.102, −5.40, −8.30) and Ribavirin (−8.59, −6.38, −7.94). Further ranking of these molecules was done by rescoring MM-GBSA functions. It indicates change in free energies of binding of protein − receptor complex and corroborates binding affinities and docking accuracies.
3.3. Lead optimization through binding free energy, HB interaction and ADMET properties
The binding free energies (ΔG bind) are also in the acceptable range between −33.96 and −61.45 kcal/mol of all four ligands from Lib A. The range of selected three ligands from Lib B is between −16.37 and −49.88 and each ligand molecule interacted with all three sites through different amino acid residues by H-bond interactions as well as other chemical interactions.
Thus, the four ligands are from Asinex databases and their binding free energies (ΔG bind) on each sites (Site 1, Site 2, Site 3) are 5817 (−38.79, −47.93, −45.23); 6799 (−44.61, −29.47, −46.49); 6797(−40.21, −35.18, −46.54) and 7025 (−33.96, −51.99, −61.45). However, three drugs accomplished from PubChem databases and their binding free energies (ΔG bind) on each sites (Site 1, Site 2, Site 3) are Zidovudine (−23.50, −16.37, −44.64), Valganciclovir (−49.88, −39.44, −48.05) and Ribavirin (−40.92, −47.82, −44.96) (Table 6). Further ranking of these molecules is done by rescoring MM-GBSA functions. It indicates change in free energies of binding of protein–receptor complex and corroborates binding affinities and docking accuracies. The number of HBs interaction, docking score and binding free energies with that site (Tables 3–5) depict the binding capacity of a ligand to the target.
Table 4.
C-1 to C-10: Top 10 ligands from Lib-A; D-1 to D-10: top 10 ligands from Lib-B and their Prime MM-GBSA, docking score, no. of H bonds and H bond interactive residues with Site 2 (DScore – 0.99) of target protein (6VYO).
| S/no. | Asinex ID | 2D structures | ΔG bind (kcal/mol) | Docking score | No./H-bond | H-bond interactive residues |
|
|---|---|---|---|---|---|---|---|
| Chain A | Chain D | ||||||
| C-1 | 6065 |  |
−27.76 | −8.41 | 6 | ASN77 ASN75 ASN154 ALA155 | ARG92 ALA173 |
| C-2 | 3686 |  |
−26.39 | −7.96 | 2 | ASN154 | ARG92 |
| C-3 | 6797 | Mentioned in Table 3A-3 | −35.18 | −6.92 | 3 | ASN75(2) ASN154 | |
| C-4 | 5817 | Mentioned in Table 3A-1 | −47.93 | −6.75 | 5 | ASN77 ASN75(2) | ALA55, ARG149 |
| C-5 | 7093 | −49.36 | −6.63 | 4 | ASN77(2) ASN154 | ALA55 | |
| C-6 | 7025 | Mentioned in Table 3A-7 | −51.99 | −6.39 | 3 | ASN77 ASN75 THR76 | |
| C-7 | 5584 | −33.13 | −6.37 | 1 | ARG92 | ||
| C-8 | 6806 | −33.00 | −6.21 | 5 | ASN77 ASN154 | ALA173 ALA55(2) | |
| C-9 | 6799 | Mentioned in Table 3A-2 | −29.47 | −6.11 | 4 | ALA155(2) ASN77 | ARG107 |
| C-10 | 7064 | Mentioned in Table 3A-8 | −53.01 | −6.09 | 4 | ASN77(2) ALA155 | ARG92 |
| D-1 | 72187 | Mentioned in Table 3B-1 | −16.37 | −9.09 | 7 | ASN154 ASN150 ARG107(2) | ALA55 ARG92(2) |
| D-2 | 6437075 | 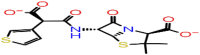 |
−23.45 | −6.55 | 4 | ASN154(2) | ARG107 ARG92 |
| D-3 | 37542 | Mentioned in Table 3B-2 | −47.82 | −6.37 | 4 | ASN77(2) | ARG92, ARG107 |
| D-4 | 23674512 | 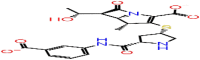 |
−60.99 | −5.86 | 4 | ASN154 ILE146 | ARG107 ARG92 |
| D-5 | 159269 | Mentioned in Table 3B-7 | −36.31 | −5.56 | 4 | ASN75(2) HIE145 | ARG92 |
| D-6 | 18283 |  |
−43.15 | −5.49 | 3 | ASN75 | ARG92 SER105 |
| D-7 | 135413534 | Mentioned in Table 3B-3 | −39.44 | −5.40 | 4 | ASN75 ASN77 | ALA173 ARG92 |
| D-8 | 6256 | 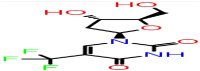 |
−30.87 | −5.37 | 3 | ASN75 HIE145 | ARG92 |
| D-9 | 6321416 |  |
−57.63 | −5.37 | 4 | ASN77 ALA155 | ARG107 ALA55 |
| D-10 | 3672 | 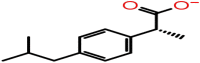 |
−28.95 | −5.17 | 1 | ARG92 | |
Table 5.
E-1 to E-10: Top 10 ligands from Lib-A; F-1 to F-10: top 10 ligands from Lib-B and their prime MM-GBSA, docking score, no. of H bonds and H bond interactive residues with Site 3 (DScore – 1.045) of target protein (6VYO).
| S/no. | Asinex ID | 2D structures | ΔG bind (kcal/mol) | Docking bnjkop[score | No./H-bond | H-bond interactive residues |
|
|---|---|---|---|---|---|---|---|
| Chain C | Chain D | ||||||
| E-1 | 5817 | Mentioned in Table 3A-1 | −45.23 | −10.28 | 4 | ALA55(2) | ASN154 ASN154 |
| E-2 | 6799 | Mentioned in Table 3A-2 | −46.49 | −10.15 | 5 | ALA55 | ASN75(2) ASN153 ASN154 |
| E-3 | 7066 |  |
−42.79 | −9.96 | 4 | ALA55 THR57 ALA156 | ASN75 |
| E-4 | 6993 |  |
−32.06 | −9.29 | 3 | ARG149 SER51 | ASN154 |
| E-5 | 6061 |  |
−43.53 | −9.17 | 6 | ALA55(2) ARG107 THR57 | ASN15 ASN154 |
| E-6 | 7025 | Mentioned in Table 3A-7 | −61.45 | −8.92 | 5 | ALA55 ALA156 ARG107 | ASN154 ASN75 |
| E-7 | 6797 | Mentioned in Table 3A-3 | −46.54 | −8.24 | 5 | ALA55, THR57 | ASN153 ASN154 ASN75 |
| E-8 | 5802 |  |
−50.28 | −8.03 | 4 | ALA55(2), THR57 | ASN75 |
| E-9 | 8503 |  |
−35.38 | −7.70 | 3 | ALA55, THR57 | ASN75 |
| E-10 | 5584 | Mentioned in Table 4C-7 | −63.61 | −7.66 | 1 | ASN153 | |
| F-1 | 72187 | Mentioned in Table 3B-1 | −44.64 | −9.75 | 6 | ALA55, ARG107 | ASN75, ASN153, ASN150(2) |
| F-2 | 135413534 | Mentioned in Table 3B-3 | −48.05 | −8.29 | 6 | ALA55 | ASN75(2), ASN77, ASN154 |
| F-3 | 37542 | Mentioned in Table 3B-2 | −44.96 | −7.94 | 7 | ARG107, THR57, TYR109 | ASN77, HIS145, ASN154(2) |
| F-4 | 60871 | 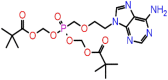 |
−47.81 | −7.60 | 3 | ARG107, TYR111 | ASN154 |
| F-5 | 464205 | 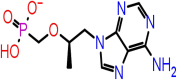 |
−38.84 | −6.96 | 6 | ARG107, ALA55, TYR109 | ASN75(2), ASN154 |
| F-6 | 71616 |  |
−45.34 | −6.86 | 1 | ASN154 | |
| F-7 | 135526609 |  |
−44.02 | −6.86 | 3 | THR57, ALA55, ARG107 | |
| F-8 | 6256 | Mentioned in Table 4D-8 | −42.51 | −6.83 | 3 | ARG107 | HIS145, ASN75 |
| F-9 | 90643431 | 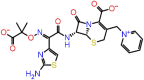 |
−46.43 | −6.72 | 4 | SER51, ARG107(2) | ASN153 |
| F-10 | 16124688 |  |
−52.76 | −6.46 | 3 | ALA55, TYR109 | ASN154 |
The chemical interaction of the top four ligands (Asinex ID: 5817; 6799, 6797 and 7025) from Lib A is shown in Supplementary Figures S1–S3(A–D) with Sites 1, 2 and 3. Asinex Id: 5817 interacts with all the three sites with variable number of HB interactions like 5, 4 and 4, respectively. Their interactive residues are five amino acids (Chain A: ALA55 (2); Chain B: ASN77, ASN154, ASN153) with Site 1; 4AA (Chain A: ASN77, ASN75 (2); Chain D: ALA55, ARG149) with Site 2 and four amino acids (Chain C: ALA55 (2); Chain D: ASN153, ASN154) with Site 3.
The HB interaction of Asinex ID: 6799 and their interactive residues are four HB and 4AA (Chain A: ALA55; Chain B: ASN75, ASN154, ASN153) with Site 1; 4HB and 4AA (Chain C: ALA155 (2), ASN (77); Chain D: ARG107) with Site 2 and 5HB and 5AA (Chain C: ALA55 and Chain D: ASN75 (2), ASN153, ASN154) with Site 3.
Ligands 6797 and their HB interaction with Sites 1, 2 and 3 are four HB, 4AA (Chain A: ASN57 and Chain B: ASN75, ASN153, ASN154); 3HB, 3AA (Chain A: ASN75 (2), ASN154) and 5HB and 5AA (Chain C: ALA55, THR57 and Chain D: ASN153, ASN154, ASN75), respectively.
Ligands 7025 and their HB interaction with Sites 1, 2 and 3 are three HB, 3AA (Chain B: ASN75, ASN76, ASN77); 3HB, 3AA (Chain A: ASN75, ASN76, ASN77) and 5HB and 5AA (Chain C: ALA55, ALA156, ARG107 and Chain D: ASN154, ASN75), respectively.
Supplementary Figures S4–S6(A–C) depict the chemical interaction of top three ligands (PubChem IDs: 72187, 135413534, 37542) from the PubChem database with Sites 1, 2 and 3 of N proteins. However PubChem ID: 72187 interacts with all the three sites with variable number of HB interactions numbering 5HBs with Site 1, 7HBs with Site 2 and 6HBs with site 3. The interaction of this ligand with H bond interactive amino acids are ARG107(2) in Chain A and ASN77, ASN150(2) in Chain B at Site 1, ASN154, ASN150 in Chain A and ALA55, ARG92(2), ARG(2) in Chain D with Site 2 and ALA55, ARG107 in Chain C & ASN75, ASN153, ASN150(2) in Chain D with Site 3.
In addition, PubChem ID: 135413534 also has activity on all the three sites. The number of HB interactions on Site 1, Site 2 and Site 3 are 5, 4 and 6, respectively. The HB interactive amino acid with Site 1, Site 2 and Site 3 corresponds to five amino acid (ARG107 in Chain A and ASN75 (2), ASN153, HIE145 in Chain B), four AA (ASN75, ASN77 in Chain A and ALA173, ARG92 in Chain B) and six AA (ALA55 in Chain C and ASN75 (2), ASN154(2), ASN77), respectively.
The ADME/T property values of all the four molecules from Asinex database molecules satisfied the acceptable ranges in term of Lipinski’s rule of five 5 (Lipinski, 2004) and other pharmacokinetic parameters such as probable H-bonding atoms, human oral absorption (<25%: poor; >above 80%: good) and the IC50 values for blockage of human Ether-a-go-go-Related Gene (hERG) K + channels (< −5 satisfactory) (Sato et al., 2018). Finally, four lead optimized molecules are predicted to have degree of drug likeliness. Table 7A shows that molecular weights of the selected compounds are in the range from 325 to 420 D, hydrogen bond acceptance from 9 to 10 and hydrogen bond donor between 2 and 4. It also shows that the compounds have rotatable bonds in the range of 4–8 while their polar surface areas ranges from 144.664 to 209.137. Furthermore, Table 7A mentions the detailed results including Lipinski rule of 5, Veber’s rule and other pharmacokinetics parameters for these four compounds. Whereas, there is no need of ADMET prediction for three FDA approved drug molecules as they are already approved as anti-HIV as well as anti-Herpes drugs (Table 7B).
Table 7A.
The drug likeliness properties of the antiviral small molecules predicted by QikProp module.
| S/no. | Ligand ID | Mol./weight | H-bond acceptor | H-bond donor | Oral absorption | % Human oral absorption | QP Log HERG | Lipinski rule of 5 | Veber's rule |
|
|---|---|---|---|---|---|---|---|---|---|---|
| Rotatable bond count | Polar surface area (PSA) | |||||||||
| 1 | 5817 | 383.4 | 10.95 | 4 | 2 | 34.355 | −0.458 | 0 | 8 | 184.047 |
| 2 | 6799 | 383.4 | 9.25 | 3 | 2 | 34.546 | −0.391 | 0 | 4 | 166.824 |
| 3 | 6797 | 325.321 | 9.25 | 3 | 2 | 36.852 | −0.27 | 0 | 4 | 209.137 |
| 4 | 7025 | 419.436 | 10.45 | 2.25 | 1 | 17.254 | −3.868 | 1 | 7 | 144.664 |
Table 7B.
Potential FDA approved drug molecules, their indication and mechanism of action.
| S/no. | Name of drug molecules | PubChem Id | Indication | Mechanism of action |
|---|---|---|---|---|
| 1 | Zidovudine | 72187 | Antiviral drug used for the treatment of human immunodeficiency virus (HIV) infections | Nucleoside reverse transcriptase inhibitor (NRTI) with activity against Human Immunodeficiency Virus Type 1 (HIV-1) (https://www.drugbank.ca/drugs/DB00495) |
| 2 | Valganciclovir | 135413534 | Antiviral drug used for the treatment of cytomegalovirus infections | Inhibition of viral DNA synthesis by incorporation with viral DNA& inhibits viral DNA polymerases (https://www.drugbank.ca/drugs/DB01610) |
| 3 | Ribavirin | 37542 | Used for the treatment of chronic Hepatitis C virus (HCV) infection | Inhibits of viral RNA and protein synthesis by inhibition of the enzyme RNA dependent RNA polymerase (https://www.drugbank.ca/drugs/DB00811) |
3.4. MD simulation
Three best docked compounds Asinex 5817, 6799 and PubChem 72187 (Zidovudine) were subjected to MD simulations for 50 ns. The RMSD and RMSF values were used to observe the stability of protein–ligand complexes as compared to unbound protein structure. The RMSD plot of 5817, 6799 and 72187 are depicted in Figure 5(A, B & C), respectively. Figure 5(A) elaborates the interaction of protein with the ligand Asinex 5817. It shows main bimodal fluctuations at 3.4 Å during 12–18 ns interval and another at 3.4 Å for 30–36 ns. However, Figure 6A, B and C show the favorable residual interactions which remained for more than 30% of the time. The hydrogen bonds with more than 70% duration of interactions were considered as strong bonds. The interaction with 40–70% was considered as medium while less than 40% was clubbed into weak hydrogen interactions. The medium strength of interaction of ligands were seen with D: ASN75 (66%) and C: TYR109 (45%). It also has D: TRP 52 linked to the center of the benzene ring with Pi–Pi interaction stocking at 72%. Other residues such as C: ARG107, C: THR57, D: ASP82 and D: ALA155 showed water bridges links.
Figure 5.

RMSD trajectories of Site-3 of protein-ligand complexes during 50 ns. (A) 5817, (B) 6799 and (C) 72187 (Zidovudine).
Figure 6.
(A) (A-1) The bar charts of protein–ligand interaction fraction over the course of trajectory. (A-2) Protein residue interactions with ligand 5817. (B) (B-1) The bar charts of protein–ligand interaction fraction over the course of trajectory. (B-2) Protein residue interactions with ligand 6799. (C) (C-1) The bar charts of protein–ligand interaction fraction over the course of trajectory. (C-2) Protein residue interactions with ligand 72187 (Zidovudine).



Figure 7.

Binding free energy between protein–ligand (5817, 6799, 72187) complex was predicted by using MM-GBSA method. The values of binding free energy was calculated per 5 ns.
While Figure 5(B) demonstrates the fluctuations in the region 3.5 Å from 6 to 10 ns and 4.8 Å from 28 to 50 ns. This interaction remained stable and sustained for a longer period. The ligand atom interaction with strong hydrogen bonds were seen in D: ASN75 (86% on NH and 99% on O), C: ALA55 (95%), medium interaction in C: TYR109 (44%) and weak interactions in C: ARG107 (36%) and C: ARG92 (38%). While residues D: ASN77 (38%) and C: ARG 149 (32%) showed water bridges (Figure 6(B)). Figure 5(C) shows the interaction of Zidovudine with the target protein. It shows a very stable interactions and minimal fluctuation. Its increase started to 2.4 Å on 2 ns and remained stable up to 3 Å from 13 to 50 ns. Its interactions are also complex and multiple bond formations. Majority of the residues form the hydrogen bonds and maximum interactions were seen with the residue ASN. Only three residues (D: THR148, D: ASN153, D: ASN77) exclusively forms the water bridges and one residue form the Van der Waals interaction (C: ARG107). All other residues show hydrogen bond interactions with long period of time including three strong, two medium and two weak hydrogen interactions such as C: ARG107 having variable percentage with three oxygen atom and one water linkage, C: ALA55 (71% with N), C: TYR109 (33%), D: ASN75 (95% O and 45% NH), D: ASN 154 (83%, 33% for two different O atom and one water linkage) and D: ASN150 (more than 50% with two O and two water molecules) (Figure 6(C)).
The RMSF fluctuation is also very low in case of Zidovudine (0.8 minimum to 4.3) as compared to two other ligands [5817 (minimum 0.8–5.8) and 6799 (minimum 1.2–6.2). Figure 7 shows comparative binding free energy (ΔG bind) which were calculated at every 5 ns of all three ligands. The ΔG bind value for the three ligands 5817, 6799 and 72187 calculated over 50 ns are −51.96, −64.36 and −59.43, respectively. The binding free energy (ΔG bind) are very significant in all the three ligands. It can be seen (Figure 6(A, B and C)) that compound 5817, 6799 and 72187 formed more stable hydrogen bonds with the residues of binding Site 3 of target protein.
4. Conclusion
We identified a total of seven hits in the first step after molecular docking, four from Asinex and three from PubChem database, which can inhibit the structural N proteins of SARS-CoV-2. Many studies are targeting different proteins including main protease (Mahanta et al., 2020; Mittal et al., 2020) and spike proteins (Basit et al., 2020; de Oliveira et al., 2020). However, the homologous proteins of NTD has been used in one earlier study that has shown theophylline and pyrimidine groups of drugs as promisable interactions (Sarma et al., 2020). However, our study is the first study that targeted binding potential of known drugs and drug like moieties on the crystallized novel proteins structure 6VYO. There are 4–5 significant hydrogen bond interactions, on an average, associated with each drug molecule. It has interestingly been found that most of the hydrogen bonds interactions of all the seven drugs mainly are associated with ASN residues.
Thus, four Asinex ligands (5817, 6799, 6797, 7025) and three FDA approved drug molecules (PubChem ID: 72187, 135413534, 37542) were found to have good docking score on each site (Site 1, Site 2 and Site 3). The drug like moieties from Asinex have antiviral activities and drugs from PubChem databases are Zidovudine, Valganciclovir and Ribavirin. Thus, anti-HIV drugs as well as anti-Herpes drugs are active on this protein. Furthermore, second-step analysis was done by MD simulation to find final three drugs. Out of those, Zidovudine has stronger and more stable interaction with this protein structure. Therefore, the drugs especially Zidovudine can be further explored in the clinical trial as the potential treatment against the SARS CoV-2.
Supplementary Material
Glossary
Abbreviations
- CoV
coronavirus
- COVID-19
coronavirus disease 2019
- FDA
food and drug administration
- HB
hydrogen bond
- HTVS
high throughput virtual screening
- MD
molecular dynamics
- MERS
Middle East Respiratory Syndrome
- PDB
Protein Data Bank
- RNA
ribonucleic acid
- SARS
Sudden Acute Respiratory Syndrome
- SP
standard precision
- WHO
World Health Organization
- XP
extra precision
Acknowledgements
Special thanks to the Schrodinger corporations for their help.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Aanouz I., Belhassan A., El-Khatabi K., Lakhlifi T., El-Ldrissi M., & Bouachrine M. (2020). Moroccan medicinal plants as inhibitors against SARS-CoV-2 main protease: Computational investigations. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1758790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basit A., Ali T., & Rehman S. U. (2020). Truncated human Angiotensin Converting Enzyme 2; A potential inhibitor of SARS-CoV-2 spike glycoprotein and potent COVID-19 therapeutic agent. Journal of Biomolecular Structure and Dynamics, 1–17. 10.1080/07391102.2020.1768150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi S., Poma A. B., & Kolandaivel P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K. J., Chow E., Xu H., Dror R. O., Eastwood M. P., Gregersen B. A., Klepeis J. L., Kolossváry I., Moraes M. A., Sacerdoti F. D., Salmon J. K., Shan Y., &Shaw D. E. (2006). Scalable algorithms for molecular dynamics simulations on commodity clusters [Paper presentation]. Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, SC’06, New York, New York, USA (p. 84). ACM Press. 10.1145/1188455.1188544 [DOI]
- Brian D. A., & Baric R. S. (2005). Coronavirus genome structure and replication In Enjuanes L. (Ed.), Coronavirus replication and reverse genetics. Current topics in microbiology and immunology. Springer; 10.1007/3-540-26765-4_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butina D., Segall M. D., & Frankcombe K. (2002). Predicting ADME properties in silico: Methods and models. Drug Discovery Today, 7(11), S83–S88. 10.1016/S1359-6446(02)02288-2 [DOI] [PubMed] [Google Scholar]
- Chang C.-K., Michael Chen C.-M., Chiang M.-H., Hsu Y.-I., & Huang T.-H. (2013). Transient oligomerization of the SARS-CoV N protein – Implication for virus ribonucleoprotein packaging. PLoS One, 8(5), e65045 10.1371/journal.pone.0065045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T., Wu D., Chen H., Yan W., Yang D., Chen G., Ma K., Xu D., Yu H., Wang H., Wang T., Guo W., Chen J., Ding C., Zhang X., Huang J., Han M., Li S., Luo X., Zhao J., & Ning Q. (2020). Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ (Clinical Research ed.), 368, m1091 10.1136/bmj.m1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y., Ulasli M., Schepers H., Mauthe M., V’kovski P., Kriegenburg F., Thiel V., de Haan C. A. M., & Reggiori F. (2019). Nucleocapsid protein recruitment to replication–transcription complexes plays a crucial role in coronaviral life cycle. Journal of Virology, 94(4), e01925 10.1128/JVI.01925-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira O. V., Rocha G. B., Paluch A. S., & Costa L. T. (2020). Repurposing approved drugs as inhibitors of SARS-CoV-2S-protein from molecular modeling and virtual screening. Journal of Biomolecular Structure & Dynamics, 1–14. 10.1080/07391102.2020.1772885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinesh D. C., Chalupska D., Silhan J., Veverka V., & Boura E. (2020). Structural basis of RNA recognition by the SARS-CoV-2 nucleocapsid phosphoprotein. BioRxiv. 10.1101/2020.04.02.022194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L., Zhao G., Lin Y., Chan C., He Y., Jiang S., Wu C., Jin D.-Y., Yuen K.-Y., Zhou Y., & Zheng B.-J. (2008). Priming with rAAV encoding RBD of SARS-CoV S protein and boosting with RBD-specific peptides for T cell epitopes elevated humoral and cellular immune responses against SARS-CoV infection. Vaccine, 26(13), 1644–1651. 10.1016/j.vaccine.2008.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. (2020. a). Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1761881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. (2020. b). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure & Dynamics, 1–15. 10.1080/07391102.2020.1761882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen A. D., Al-Obaidi A., Şahin A. T., & Yelekçi K. (2020). Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1758791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi S. K., Raja K., Sebastine I., & Joseph J. (2020). Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1760136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr A. R., & Perlman S. (2015). Coronaviruses: An overview of their replication and pathogenesis In Coronaviruses: Methods and Protocols. (Vol. 1282, pp. 1–23). Springer; 10.1007/978-1-4939-2438-7_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eurosurveillance Editorial Team. (2020). Note from the editor: World Health Organization declares novel coronavirus (2019-nCoV) sixth public health emergency of international concern. Euro Surveill, 25(5), 200131e. doi: 10.2807/1560-7917.ES.2020.25.5.200131e. [DOI] [PMC free article] [PubMed]
- Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., & Mainz D. T. (2006). Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes . Journal of Medicinal Chemistry, 49(21), 6177–6196. 10.1021/jm051256o [DOI] [PubMed] [Google Scholar]
- Genheden S., & Ryde U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opinion on Drug Discovery, 10(5), 449–461. 10.1517/17460441.2015.1032936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E., Jang G. M., Bouhaddou M., Xu J., Obernier K., O’Meara M. J., Guo J. Z., Swaney D. L., Tummino T. A., Huettenhain R., Kaake R. M., Richards A. L., Tutuncuoglu B., Foussard H., Batra J., Haas K., Modak M., Kim M., Haas P., & Krogan N. J. (2020). A SARS-CoV-2-human protein–protein interaction map reveals drug targets and potential drug-repurposing. BioRxiv. 10.1101/2020.03.22.002386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M. K., Vemula S., Donde R., Gouda G., Behera L., & Vadde R. (2020). In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1751300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. A. (2009). Identifying and characterizing binding sites and assessing druggability. Journal of Chemical Information and Modeling, 49(2), 377–389. 10.1021/ci800324m [DOI] [PubMed] [Google Scholar]
- Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., & Banks J. L. (2004). Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. Journal of Medicinal Chemistry, 47(7), 1750–1759. 10.1021/jm030644s [DOI] [PubMed] [Google Scholar]
- Hasan A., Paray B. A., Hussain A., Qadir F. A., Attar F., Aziz F. M., Sharifi M., Derakhshankhah H., Rasti B., Mehrabi M., Shahpasand K., Saboury A. A., & Falahati M. (2020). A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1754293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., … Cao B. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet, 395(10223), 497–506. 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam R., Parves R., Paul A. S., Uddin N., Rahman M. S., Mamun A. A., Hossain M. N., Ali M. A., & Halim M. A. (2020). A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. Journal of Biomolecular Structure & Dynamics, 1–20. 10.1080/07391102.2020.1761883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S., Yang M., Hong Z., Zhang L., Huang Z., Chen X., He S., Zhou Z., Zhou Z., Chen Q., Yan Y., Zhang C., Shan H., & Chen S. (2020). Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. BioRxiv. 10.1101/2020.03.06.977876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. Y., Liu C. L., Chang Y. M., Zhao J., Perlman S., & Hou M. H. (2014). Structural basis for the identification of the N-terminal domain of coronavirus nucleocapsid protein as an antiviral target. Journal of Medicinal Chemistry, 57(6), 2247–2257. 10.1021/jm500089r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A. (2004). Lead- and drug-like compounds: The rule-of-five revolution. Drug Discovery Today: Technologies, 1(4), 337–341. 10.1016/j.ddtec.2004.11.007 [DOI] [PubMed] [Google Scholar]
- Lo Y. S., Lin S. Y., Wang S. M., Wang C. T., Chiu Y. L., Huang T. H., & Hou M. H. (2013). Oligomerization of the carboxyl terminal domain of the human coronavirus 229E nucleocapsid protein. FEBS Letters, 587(2), 120–127. 10.1016/j.febslet.2012.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., & Sherman W. (2013). Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-Aided Molecular Design, 27(3), 221–234. 10.1007/s10822-013-9644-8 [DOI] [PubMed] [Google Scholar]
- Mahanta S., Chowdhury P., Gogoi N., Goswami N., Borah D., Kumar R., Chetia D., Borah P., Buragohain A. K., & Gogoi B. (2020). Potential anti-viral activity of approved repurposed drug against main protease of SARS-CoV-2: An in silico based approach. Journal of Biomolecular Structure and Dynamics, 1–15. 10.1080/07391102.2020.1768902 [DOI] [PubMed] [Google Scholar]
- McBride R., van Zyl M., & Fielding B. C. (2014). The coronavirus nucleocapsid is a multifunctional protein. Viruses, 6(8), 2991–3018. 10.3390/v6082991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal L., Kumari A., Srivastava M., Singh M., & Asthana S. (2020). Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. Journal of Biomolecular Structure and Dynamics, 1–26. 10.1080/07391102.2020.1768151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant S., Singh M., Ravichandiran V., Murty U. S. N., & Srivastava H. K. (2020). Peptide-like and small-molecule inhibitors against Covid-19. Journal of Biomolecular Structure and Dynamics, 1–15. 10.1080/07391102.2020.1757510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules C. I., Marston H. D., & Fauci A. S. (2020). Coronavirus infections-more than just the common cold. JAMA – Journal of the American Medical Association, 323(8), 707 10.1001/jama.2020.0757 [DOI] [PubMed] [Google Scholar]
- Sarma P., Sekhar N., Prajapat M., Avti P., Kaur H., Kumar S., Singh S., Kumar H., Prakash A., Dhibar D. P., & Medhi B. (2020). In silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure & Dynamics, 1–11. 10.1080/07391102.2020.1753580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Yuki H., Ogura K., & Honma T. (2018). Construction of an integrated database for hERG blocking small molecules. PLoS One, 13(7), e0199348 10.1371/journal.pone.0199348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon A., Le N. T.-T., Selisko B., Eydoux C., Alvarez K., Guillemot J.-C., Decroly E., Peersen O., Ferron F., & Canard B. (2020). Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 exonuclease active-sites. Antiviral Research, 178, 104793 10.1016/j.antiviral.2020.104793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surjit M., Liu B., Chow V. T. K., & Lal S. K. (2006). The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin–cyclin-dependent kinase complex and blocks S phase progression in mammalian cells . The Journal of Biological Chemistry, 281(16), 10669–10681. 10.1074/jbc.M509233200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., & Tan W. (2020). A novel coronavirus from patients with pneumonia in China, 2019. The New England Journal of Medicine, 382(8), 727–733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.