

Abstract
Inflammasomes are intracellular complexes that form in the cytosol of inflammatory cells. NLRP3 is one of the sensor proteins in the complex that can recognize a wide variety of stimuli ranging from microbial components to environmental particulates. Here, we report that in mouse airway epithelial cells (AECs), inflammasome activation is inhibited by EphA2, a member of the transmembrane tyrosine kinase receptor family, via tyrosine phosphorylation of NLRP3 in a model of reovirus infection. We find that EphA2 depletion markedly enhances interleukin‐1β (IL‐1β) and interleukin‐18 (IL‐18) production in response to the virus. EphA2 −/− mice show stronger inflammatory infiltration and enhanced inflammasome activation upon viral infection, and aggravated asthma symptoms upon ovalbumin (ova) induction. Mechanistically, EphA2 binds to NLRP3 and induces its phosphorylation at Tyr132, thereby interfering with ASC speck formation and blocking the activation of the NLRP3‐inflammasome. These data demonstrate that reovirus employs EphA2 to suppress inflammasome activation in AECs and that EphA2 deficiency causes a pathological exacerbation of asthma in an ova‐induced asthma model.
Keywords: asthma, EphA2, inflammasome, NLRP3, reovirus
Subject Categories: Immunology; Signal Transduction; Post-translational Modifications, Proteolysis & Proteomics
The tyrosine kinase receptor EphA2 functions as a negative regulator of inflammasome activation upon reovirus infection by targeting NLRP3 in airway epithelia. EphA2 also suppresses inflammatory responses in a murine ova‐induced asthma model.

Introduction
Airway epithelial cells (AECs) contain basal, ciliated, secretory (goblet, serous, and Clara cells), and less well‐categorized undifferentiated cells. This cluster of cells is located between the host and the environment and works as a physical barrier against inhaled pathogens, allergens, pollutants, and medications 1, 2. AECs contribute to innate immunity and immune regulation and play a central role in airway inflammatory status. AECs respond to a variety of stimuli by producing numerous mediators ranging from cytokines, to chemokines, to peptides, to liquid and reactive oxygen which attract innate and adaptive immune cells 1. Yet, excessive production of inflammatory cytokines causes unwanted tissue damage and can lead to severe immunological disorders such as asthma 3 and chronic obstructive pulmonary disease (COPD) 4.
Inflammasomes are intracellular complexes consisting of a receptor protein and caspases which mediate the activation of inflammatory responses. Several receptor proteins have been reported in recent years, such as NLRP1 5, NLRP3 6, NLRC4 7, and AIM2 8. Usually, an adaptor protein, ASC, helps inflammasome formation by bridging the upstream inflammasome sensor molecule to caspase‐1, initiating its activation and downstream responses including the cleavage of pro‐inflammatory cytokines IL‐1β and IL‐18. While much effort has been devoted to the role of inflammasomes in immune cells, recent studies have revealed that epithelial cells also express an active inflammasome complex. In lung epithelium, viruses such as influenza A 6, urban particle matter 9, silica particles 10, asbestos 11, and hyperoxia 12 can induce the activation of inflammasomes. Coincidentally, NLRP3 is a common receptor protein of all the above diverse stimuli. Not only have various exogenous activators, ranging from RNA virus 13 to particulates, been found in the environment, many endogenous molecules such as urate crystals 14 and amyloid 15 have also been found to promote the activation of the NLRP3 inflammasome after their accumulation or alteration while the host is undergoing tissue damage or metabolic dysfunction.
Given the essential role of IL‐1β in inflammation, it is likely that dysregulated inflammasome activation is associated with the pathogenesis of inflammatory diseases. Notably, NLRP3 has been linked with a series of diseases including metabolic disorders such as type 2 diabetes, atherosclerosis 16, and inflammatory disorders such as Crohn's disease 17 and Muckle–Wells syndrome 18. Inflammasome activation may also be involved in acute lung inflammation after viral infection and chronic pulmonary diseases 19. Previous studies have indicated that EphA2‐deficient mice treated with Mycoplasma pulmonis infection had increased cytokine production and more leukocyte infiltration 20, while in a bleomycin lung injury model, EphA2‐knockout mice were protected 21. All of these studies suggest EphA2 plays a role in lung inflammation; however, the biological function of EphA2 in AECs against viral infection remains unclear. We assessed its immunological function in AECs upon DNA and RNA virus stimulation.
Results
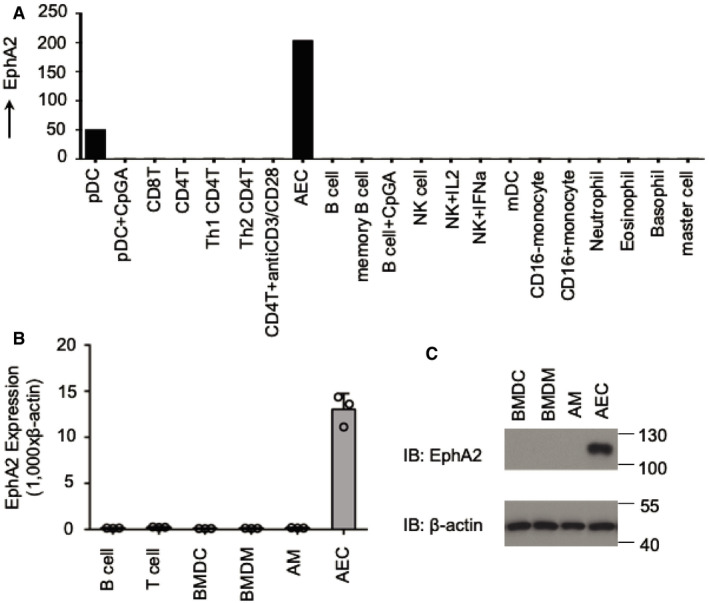
EphA2 is selectively expressed in AECs
Microarray gene expression analysis indicated that EphA2 is selectively expressed in human AECs. Apart from pDCs, there was almost no detectable expression of EphA2 in B cells, T cells, NK cells, mDCs, neutrophils, eosinophils, basophils, master cells, and active cells including pDCs + CpGA, CD4+ T cells + anti‐CD3/CD28, B cells + CpGA, NK cells + IL2, NK cells + IFNa, CD16+ monocytes, and CD16− monocytes (Fig 1A). In the expression profile of human EphA2 in the public database BioGPS (Appendix Fig S1), EphA2 showed specific expression in BDCA4+ dendritic cells, bronchial epithelial cells, and colorectal adenocarcinoma. Immunoblot analysis and real‐time PCR showed that EphA2 had a similar expression pattern in mice. We found that the mRNA levels of EphA2 in AECs were especially high when compared with murine B cells, T cells, bone marrow‐derived dendritic cells (BMDCs), bone marrow‐derived macrophages (BMDMs), and alveolar macrophages (AMs). Immunoblot analysis of EphA2 and β‐actin in BMDCs, BMDMs, AMs, and AECs revealed consistent protein levels with those of mRNA (Fig 1B and C). These data showed that EphA2 was highly expressed in AECs but barely detectable in other immune cells, except for pDCs.
Figure 1. Expression of EphA2 in human and murine cells.
-
AHuman myeloid cells and lymphoid cells were purified from PBMCs using a cell sorter. Total RNA was isolated from these primary cells and primary AECs to perform chip hybridization and microarray analysis.
-
B, CThe profile of EphA2 expression in different cells is indicated. (B) Murine B cells and T cells were purified from spleens using a cell sorter, and total RNA was isolated from murine B cells, T cells, BMDCs, BMDMs, AMs, and AECs. Shown is mean ± SD. Data represent three independent biological replicates. (C) Immunoblot analysis of EphA2 and β‐actin in BMDCs, BMDMs, AMs, and AECs.
Source data are available online for this figure.
EphA2 inhibits inflammasome‐related cytokine production in AECs
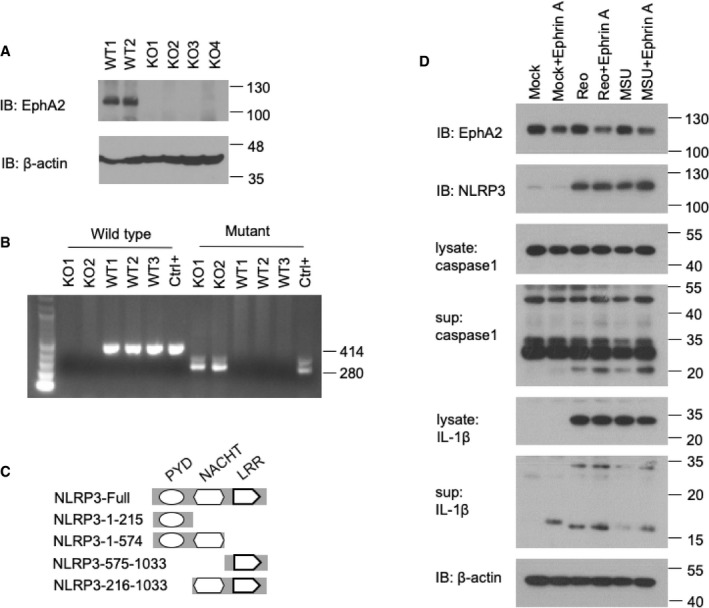
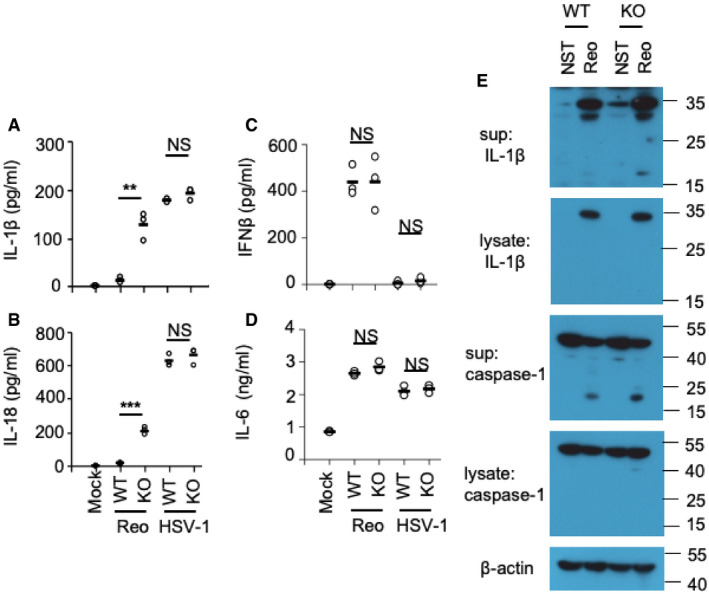
To further determine the role of EphA2 in AECs, we prepared primary lung epithelial cells from wild‐type mice (WT) and EphA2‐knockout (KO) mice after genotyping and immunoblot identification (Fig EV1A and B). We stimulated those epithelial cells for 16 h with the RNA virus reovirus and the DNA virus HSV‐1. Knockout of EphA2 enhanced the production of IL‐1β and IL‐18 significantly in response to the reovirus (Fig 2A and B) but showed a similar amount of interferon‐β (IFN‐β) and pro‐inflammatory cytokine IL‐6 (Fig 2C and D). The deletion of EphA2 had no effect on the production of IL‐1β and IL‐18 in AECs induced by HSV‐1. In regard to the protein level, cleaved active IL‐1β and caspase‐1 were much higher in the supernatant of KO AECs with reovirus infection than WT AECs (Fig 2E). But cleaved active IL‐1β and caspase‐1 were barely detected in the lysate of WT cells and KO AECs. Such data imply that EphA2 has a negative role in IL‐1β and IL‐18 production induced by reovirus in AECs. Since EphA2 was not expressed in BMDMs, we also determined IL‐1β production in BMDMs from both wild‐type and EphA2‐knockout mice. We found knockout of EphA2 had no effect on IL‐1β production in BMDMs in response to the reovirus infection (Fig EV2A). To consolidate these findings, we tested macrophage's response to the reovirus infection upon forced expression of EphA2. We overexpressed EphA2 in the RAW264.7 cell line and stimulated cells with the reovirus. The results showed that overexpression of EphA2, but not in the control vector, dramatically downregulated IL‐1β production in the RAW264.7 cells upon reovirus infection (Fig EV2B and C).
Figure EV1. Identification of EphA2‐knockout mice.
-
A, BGenotyping graph of wild‐type and EphA2‐knockout mice. (A) Immunoblot analysis of EphA2 and β‐actin in lung homogenates from wild‐type (WT) and EphA2‐knockout (KO) mice. (B) PCR analysis of wild‐type (WT) and EphA2‐knockout (KO) mice. 414 bp represents wild‐type, and 280 bp represents EphA2‐knockout.
-
CFull‐length NLRP3 and serial truncations of NLRP3 with deletion of various domains.
-
DImmunoblot analysis of EphA2, NLRP3, IL‐1β, and caspase‐1 in lysates or supernatant of AECs; β‐actin served as a loading control throughout. Mock indicates unstimulated cells. MSU was added for 8 h after 200 ng/ml LPS priming for 12 h. Reovirus was added for 8 h. Ephrin A1–Fc was added 0.4 μg/ml for 4 h before harvest.
Source data are available online for this figure.
Figure 2. EphA2 regulates the production of inflammasome‐related cytokines in AECs.
-
A–DELISA of IL‐1β, IL‐18, IFN‐β, and IL‐6 in the supernatant of AECs, which were stimulated for 16 h with the RNA virus reovirus (MOI = 5) or the DNA virus HSV‐1 (MOI = 5). n = 3 primary samples.
-
EImmunoblot analysis of IL‐1β and caspase‐1 in AEC lysate or supernatant; β‐actin served as a loading control throughout.
Figure EV2. EphA2 has no effect on IL‐1β production in BMDMs in response to reovirus.
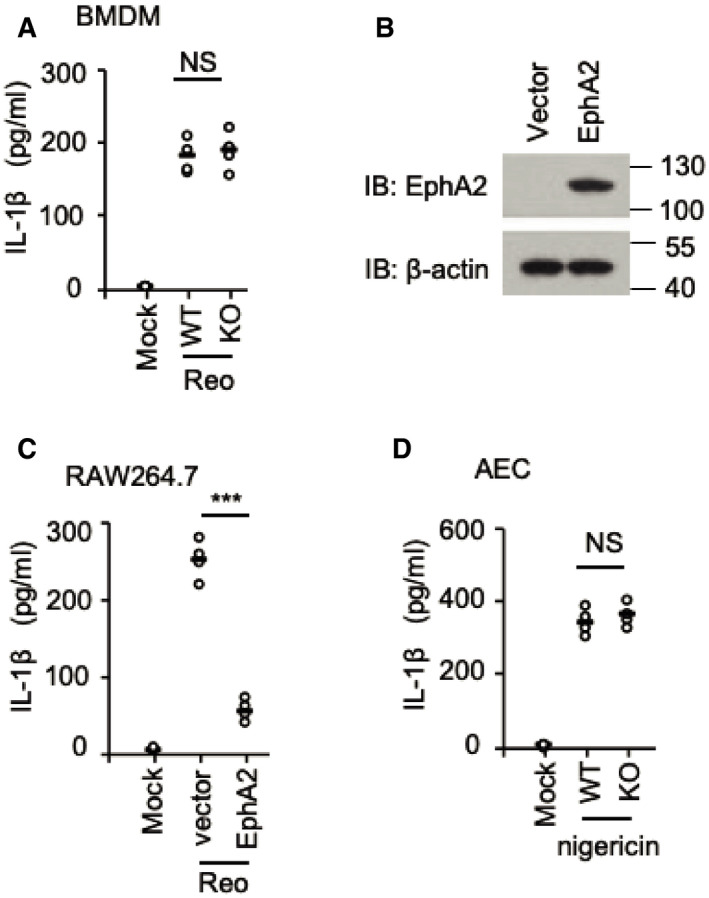
- ELISA of IL‐1β in the supernatant of BMDM cells from wild‐type (WT) and EphA2‐knockout (KO) mice (n = 4 per strain) stimulated for 16 h with the RNA virus reovirus (MOI = 5).
- Immunoblot analysis of EphA2 in lysates of the RAW264.7 cell line; β‐actin served as a loading control throughout. Cells were transfected with HA vector or HA‐EphA2 plasmid.
- ELISA of IL‐1β in the supernatant of the RAW264.7 cells transfected with HA vector or HA‐EphA2 plasmid and stimulated for 16 h with the RNA virus reovirus (MOI = 5).
- ELISA of IL‐1β in the supernatant of AECs from wild‐type (WT) and EphA2‐knockout (KO) mice (n = 4 per strain) treated with LPS (200 ng/ml, 4 h) and nigericin (7.5 mM, 1 h).
Ephrin A1, a ligand of EphA2, was reported to play a role in lung inflammation 22. However, we did not detect endogenous Ephrin A1 in AECs, so we applied additional Ephrin A1 in reovirus‐ or monosodium urate crystal (MSU)‐activated AECs. EphA2 was markedly downregulated by Ephrin A1, and the cleavage of caspase‐1 and IL‐1β was promoted, which is consistent with EphA2‐KO AECs (Fig EV1D).
EphA2 inhibits inflammasome activation upon reovirus infection in vivo
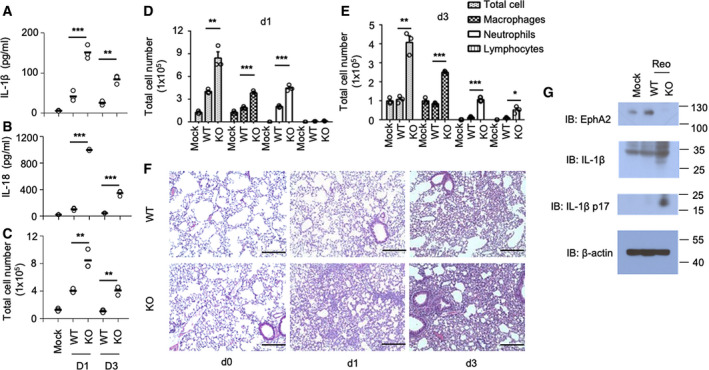
To evaluate the importance of EphA2 in inflammasome activation upon reovirus infection in vivo, we intranasally infected both wild‐type mice and EphA2‐knockout mice with reovirus, which is transmitted nasally in nature. EphA2‐knockout mice produced three‐ to fivefold more IL‐1β and three‐ to sixfold more IL‐18 in the bronchoalveolar lavage fluid (BALF) than the wild‐type mice on day 1 and day 3 post‐infection (Fig 3A and B). There were a total of 2–4 times more infiltration cells in BALF of EphA2‐knockout mice compared with wild‐type mice. The increased cells mainly consisted of macrophages, neutrophils, and lymphocytes (Fig 3C–E).
Figure 3. EphA2 controls airway allergy inflammation in reovirus infection murine model.
-
A, BELISA of IL‐1β and IL‐18 in BALF samples from wild‐type (WT) and EphA2‐knockout (KO) mice (n = 3 per strain) on day 1 and day 3 after intranasal infection with reovirus (4 × 104 PFU per mouse).
-
C–ECell number and category of BALF samples from wild‐type (WT) and EphA2‐knockout (KO) mice (n = 3 per strain) on day 1 and day 3 after intranasal infection with reovirus (4 × 104 PFU per mouse). Shown is mean ± SD.
-
FHematoxylin and eosin (H&E) staining of lung sections from EphA2 +/+ and EphA2 −/− mice intranasally infected with reovirus (4 × 104 PFU per mouse) on day 0, day 1, and day 3. Scale bars represent 200 μm.
-
GImmunoblot analysis of EphA2 and IL‐1β in lung homogenates from wild‐type (WT) and EphA2‐knockout (KO) mice on day 1 and day 3 of intranasal infection with reovirus (4 × 104 PFU per mouse); β‐actin served as a loading control throughout.
Lung histopathology revealed an increase in edema, alveolar wall thickness, alveolar hemorrhage, and cell infiltration in EphA2‐knockout mice compared with the wild‐type mice after viral infection (Fig 3F). Lung tissue for protein detection exhibited a highly induced cleaved IL‐1β in KO lung homogenate compared with wild‐type mice (Fig 3G). These data indicate that EphA2 inhibits inflammasome activation, especially the production of IL‐1β and IL‐18 in the respiratory tract, thus protecting lung tissue from severe inflammatory damage.
EphA2 inhibits ova‐induced airway allergy inflammation
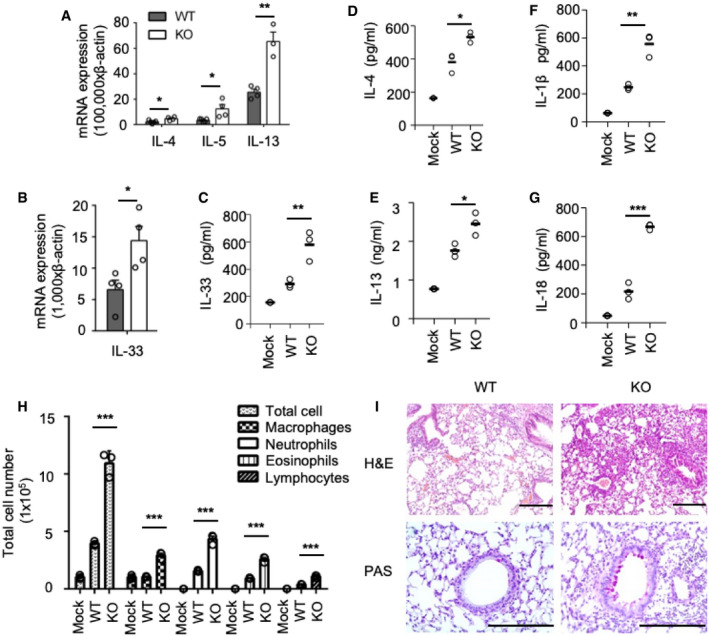
Since EphA2 is highly expressed in airway epithelial cells, we used an ova‐induced asthma model to investigate the role of EphA2 in inflammatory disease. To induce airway chronic inflammation, the mice were intraperitoneally injected with 20 μg ova every 3 days until day 12, followed by intranasal administration of 1% ova in saline once per week, for 6 weeks. Twenty‐four hours after the last treatment, we collected the lung tissue from the wild‐type mice and EphA2‐knockout mice. We found that the mRNA of Th2 cytokines IL‐4, IL‐5, and IL‐13 had increased in the KO lungs (Fig 4A), along with IL‐33, which is a significant product of lung epithelium (Fig 4B). We also detected cytokines from lung homogenates of wild‐type mice and EphA2‐knockout mice and found that KO lungs produced more IL‐33, IL‐4, and IL‐13 (Fig 4C–E). Notably, IL‐1β and IL‐18 in the lung tissue of the KO mice were also increased compared with the WT mice (Fig 4F and G).
Figure 4. EphA2 functions as a negative regulator in ova‐induced airway inflammation.
-
A, BQuantitative RT–PCR analysis of mRNA encoding various cytokines (IL‐4, IL‐5, IL‐13, IL‐33) in lung samples from wild‐type (WT) and EphA2‐knockout (KO) mice (n = 4 per strain) sensitized with ova; results presented relative to β‐actin. Shown is mean ± SD.
-
CELISA of IL‐33 in lung tissue homogenate samples from wild‐type (WT) and EphA2‐knockout (KO) mice (n = 3 per strain) sensitized with ova.
-
D–GELISA of IL‐4, IL‐13, IL‐1β, and IL‐18 in lung tissue homogenate samples from wild‐type (WT) and EphA2‐knockout (KO) mice (n = 3 per strain) sensitized with ova.
-
HCell number and category of BALF samples from wild‐type (WT) and EphA2‐knockout (KO) mice (n = 3 per strain) sensitized with ova. Shown is mean ± SD.
-
IH&E staining and PAS staining of lung sections from wild‐type (WT) and EphA2‐knockout (KO) mice sensitized with ova. Scale bars represent 200 μm.
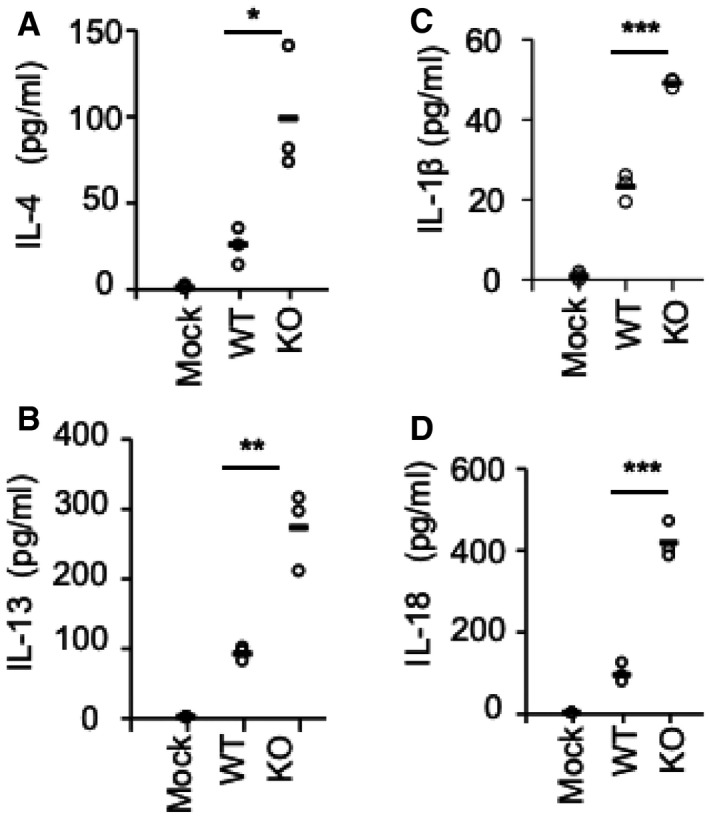
We also collected BALF from those mice and found KO mice produced more IL‐4 and IL‐13 (Fig EV3A and B). IL‐1β and IL‐18 in BALF of EphA2‐knockout mice were also higher than in wild‐type mice, which was similar to the reovirus infection model (Fig EV3C and D). The main components of inflammatory infiltrated cells in BALF were macrophages, neutrophils, eosinophils, and lymphocytes, all of which were clearly increased in KO mice compared with WT mice (Fig 4H).
Figure EV3. EphA2 inhibits IL‐1β production in BALF .
-
A–DELISA of IL‐4, IL‐13, IL‐1β, and IL‐18 in BALF samples from wild‐type (WT) and EphA2‐knockout (KO) mice (n = 3 per strain) sensitized with ova. Each symbol represents an independent experiment; small horizontal lines indicate the average of triplicates. *P < 0.05, **P < 0.01, and ***P < 0.001 (unpaired t‐test). Data represent three independent biological replicates.
Lung histopathology also revealed increased edema, cell infiltration, and PAS‐positive mucin‐producing cells in the airway epithelium of EphA2‐knockout mice (Fig 4l). Double staining immunohistochemistry assay also revealed that IL‐1β was markedly produced by AECs in our chronic asthma model (Fig EV4). Taken together, these results indicate that EphA2 inhibits inflammatory response and functions as a negative regulator in this ova‐induced chronic asthma model.
Figure EV4. IL‐1β is markedly produced by AEC in a chronic asthma model.
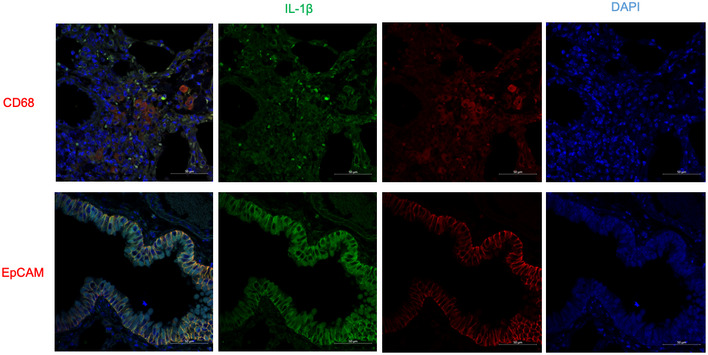
Double staining immunohistochemistry assay of IL‐1β and CD68 or EpCAM in chronic asthma mice lung tissue. IL‐1β was stained with goat anti‐IL‐1β (1:200), followed by Alexa Fluor 488 donkey anti‐goat secondary antibody (green), while CD68 or EpCAM was stained with mouse anti‐CD68 (1:200) or rabbit anti‐EpCAM, followed by Alexa Fluor 594 goat anti‐mouse secondary antibody or Alexa Fluor 594 goat anti‐rabbit secondary antibody (red). DAPI (4′,6‐diamidino‐2‐phenylindole) served as the nuclei marker (blue). Scale bars represent 50 μm.
EphA2 interacts and colocalizes with NLRP3
NLRP3 is the key receptor molecule in RNA virus‐induced inflammasomes. To determine the interaction between EphA2 and NLRP3 in AECs at the endogenous protein level, we used an anti‐EphA2 antibody to pull down NLRP3 in the lysate of reovirus‐activated AECs but failed to in the unstimulated AECs (Fig 5A). This suggests EphA2 interacts with NLRP3 in reovirus‐infected cells due to the higher levels of NLRP3. To further map the binding sites between NLRP3 and EphA2, we analyzed the interactions among Myc‐tagged NLRP3 and hemagglutinin (HA)‐tagged full‐length EphA2 and truncation mutants of EphA2 (Fig 5B). Both full‐length EphA2 and its truncation mutants, except EphA2‐SAM, interacted with NLRP3, suggesting that the tyrosine kinase domain (Pkc) of EphA2 binds to NLRP3 (Fig 5C). Additionally, both full‐length NLRP3 and the LRR domain (aa 575–1,033) of NLRP3 bind to EphA2 (Figs 5D and EV1C). Given NLRP3 is involved in numerous stimulus‐activated inflammasomes, we checked the protein level of EphA2 and NLRP3 in polyinosinic:polycytidylic acid (polyI:C)‐, poly(deoxyadenylic–deoxythymidylic) acid sodium salt (dA:dT)‐, MSU, adenosine triphosphate (ATP)‐, and reovirus‐stimulated AECs. We observed that NLRP3 was induced by all the above stimuli, but that EphA2 was neither induced nor inhibited (Fig EV5A). Additionally, we found wild‐type and EphA2‐knockout AECs produced similar levels of IL‐1β in response to nigericin (Fig EV2D). Besides, we were unable to detect any interaction between EphA2 and NLRP3 under nigericin stimulation, which may explain the stimulus‐specific function of EphA2.
Figure 5. EphA2 binds and colocalizes with NLRP3 in the cytosol.
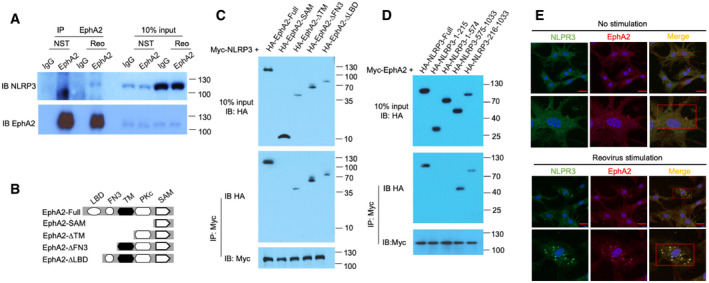
- Immunoblot analysis of proteins precipitated with anti‐EphA2 or IgG (control) from whole‐cell lysates of AECs from wild‐type mice stimulated with reovirus (MOI = 5) for 3 h. Input, 10% of the AEC lysate.
- Full‐length EphA2 and serial truncations of EphA2 with deletion of various domains.
- The indicated plasmids of HA‐tagged full‐length EphA2 and EphA2 truncation mutants were respectively transfected into HEK293T cells along with Myc‐tagged NLRP3, and then, cell lysates were immunoprecipitated with anti‐Myc antibody, followed by Western blot analysis with anti‐HA antibody and anti‐Myc antibody.
- The indicated plasmids of HA‐tagged full‐length NLRP3 and NLRP3 truncation mutants were respectively transfected into HEK293T cells along with Myc‐tagged EphA2, and then, cell lysates were immunoprecipitated with anti‐Myc antibody, followed by Western blot analysis with anti‐HA antibody and anti‐Myc antibody.
- Colocalization of endogenous EphA2 and NLRP3 in AECs. AECs were unstimulated or stimulated with reovirus for 2 h before confocal microscopy analysis. EphA2 was stained with rabbit anti‐EphA2 (1:500), followed by Alexa Fluor 594 goat anti‐rabbit secondary antibody (red), while NLRP3 was stained with mouse anti‐NLRP3 (1:500), followed by Alexa Fluor 488 goat anti‐mouse secondary antibody (green). DAPI (4′,6‐diamidino‐2‐phenylindole) served as the nuclei marker (blue). Scale bars represent 20 μm. The Pearson correlation coefficient and the Mander overlap coefficient in the upper red box are 0.5, and 0.95, respectively, and in the lower red box are 0.78 and 0.94, respectively.
Figure EV5. EphA2 phosphorylates NLRP3 in vitro .
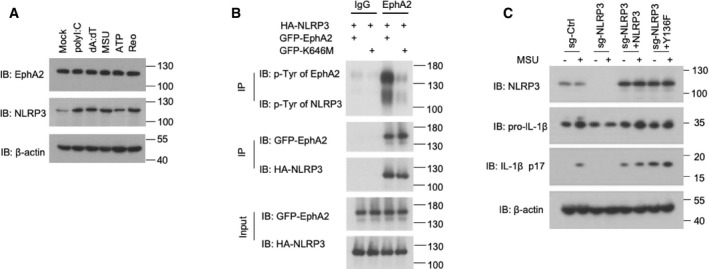
- Immunoblot analysis of EphA2 and NLRP3 in AEC lysate; β‐actin served as a loading control throughout. Mock indicates unstimulated cells. 1 μg/ml polyI:C, 1 μg/ml dA:dT, and 500 ng/ml MSU were added for 10 h after 200 ng/ml LPS priming for 12 h. 5 mM ATP was added for 30 min. Reovirus was added for 10 h.
- Proteins were eluted and analyzed by immunoblot analysis with anti‐p‐Tyr antibody, anti‐HA antibody, and anti‐GFP antibody after in vitro kinase assay.
- Immunoblot analysis of NLRP3, IL‐1β, and cleaved IL‐1β p17 in lysates of the 16HBE cell line; β‐actin served as a loading control throughout. Cells were treated with a CRISPR vector control (sg‐Ctrl) or sgRNA targeting NLRP3 (sg‐NLRP3) with or without overexpression of Myc‐tagged NLRP3 or NLRP3 Y136. 500 ng/ml MSU was added for 3 h after 200 ng/ml LPS priming for 12 h.
Source data are available online for this figure.
Next, we checked the subcellular localization of EphA2 and NLRP3 in AECs. Immunofluorescence imaging revealed endogenous NLRP3 colocalized with EphA2 in the cytosol, and their colocalization was enhanced with reovirus infection (Fig 5E). Intracellular localization of EphA2 is usually found in the plasma membrane and cell junctions, yet in some cell types, EphA2 showed a dispersive distribution, similar to our previous observations 23. Collectively, these data indicate that EphA2 binds to, and colocalizes with, NLRP3 in the cytosol upon reovirus infection.
EphA2 phosphorylates NLRP3 at Tyr132
Since EphA2 has tyrosine kinase activity, we investigated whether NLRP3 is a phosphorylation target of EphA2. We co‐expressed Myc‐NLRP3 with GFP‐EphA2 in HEK293T cells and prepared cell lysates for immunoprecipitation of Myc‐tagged NLRP3. Immunoblot analysis of p‐Tyr demonstrated tyrosine phosphorylation of NLRP3 was strongly induced by overexpression of EphA2, while EphA2 was also strongly tyrosine phosphorylated itself (Fig 6A). To verify whether NLRP3 was directly phosphorylated by EphA2, we purified GFP‐tagged EphA2, kinase‐inactive GFP‐EphA2 mutant (K646M) 24, and HA‐NLRP3 and performed in vitro kinase assay and pull‐down of the lysates with immunoglobulin G (IgG) or EphA2 antibody. Immunoblot analysis of p‐Tyr demonstrated that tyrosine phosphorylation of NLRP3 was induced by the EphA2 protein but not by EphA2‐K646M (Fig EV5B). To determine whether endogenous EphA2 phosphorylated NLRP3 in AECs under reovirus infection conditions, we isolated AECs from wild‐type and EphA2‐knockout mice. After lipopolysaccharide (LPS) priming or reovirus infection, we prepared cell lysates for immunoprecipitation of endogenous NLRP3. Immunoblot analysis of p‐Tyr showed a diminished phosphorylation of NLRP3 in EphA2‐knockout AECs compared with wild‐type AECs (Fig 6B). These data show that EphA2 indeed phosphorylates NLRP3 in an exogenous overexpressed system and in endogenous reovirus‐induced conditions.
Figure 6. EphA2 phosphorylates NLRP3 at Tyr132.
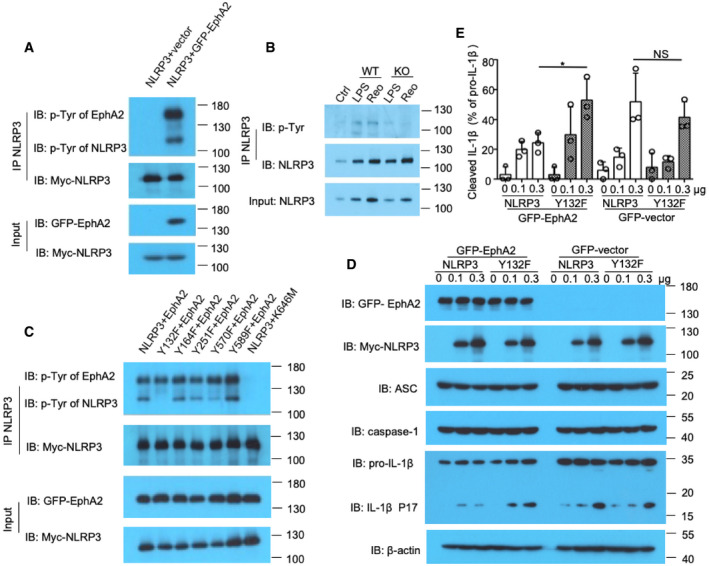
- The indicated plasmids of Myc‐tagged NLRP3 and GFP‐tagged EphA2 were transfected into HEK293T cells, and then, cell lysates were immunoprecipitated with the anti‐Myc antibody, followed by Western blot analysis with anti‐p‐Tyr, anti‐GFP, and anti‐Myc antibodies.
- Immunoblot analysis of p‐Tyr of NLRP3 precipitated with anti‐NLRP3 antibody in AECs without (Ctrl) or with LPS stimulation or reovirus (Reo) infection.
- The indicated plasmids of Myc‐tagged wild‐type NLRP3 or its mutants Y132F, Y164F, Y251F, Y570F, and Y589F were respectively transfected into HEK293T cells along with GFP‐tagged EphA2, and then, cell lysates were immunoprecipitated with the anti‐Myc antibody, followed by Western blot analysis with the anti‐p‐Tyr antibody, anti‐HA antibody, and anti‐Myc antibody.
- Immunoblot analysis of EphA2, ASC, NLRP3, caspase‐1, and IL‐1β in HEK293T cells co‐transfected with 200 ng EphA2, 40 ng ASC, 40 ng caspase‐1, and 200 ng IL‐1β, vector, or wild‐type NLRP3 or Y132F as indicated. The position of protein markers (shown in kDa) is indicated on the right‐hand side.
- Densitometry analysis of (D). *P < 0.05 (unpaired t‐test). NS means no statistical difference. Shown is mean ± SD. Data represent three independent biological replicates.
Source data are available online for this figure.
NLRP3 contains multiple tyrosine residues, five of which (Tyr132, Tyr164, Tyr251, Tyr570, and Tyr589) were predicted as possible phosphorylation sites with high confidence scores by the NetPhos prediction server (Appendix Fig S2). To address the phosphorylation sites of NLRP3, we replaced each of those NLRP3 tyrosine residues noted above individually with phenylalanine residue (Y>F‐NLRP3). We co‐expressed GFP‐tagged EphA2 or GFP‐EphA2‐K646M with Myc‐tagged wild‐type NLRP3 and its mutants (Y132F, Y164F, Y251F, Y570F, Y589F) in HEK293T cells, and then prepared cell lysates. Followed by immunoprecipitation of Myc‐tagged NLRP3, we detected the phosphorylation of NLRP3. The results showed EphA2 phosphorylated wild‐type NLRP3 and its mutants Y164F, Y251F, and Y589F at certain levels, but reduced phosphorylation was detected in the NLRP3 mutant Y570F. No phosphorylation was detected in the NLRP3 mutant Y132F (Fig 6C), thereby demonstrating Tyr132 is the most critical site for EphA2‐mediated phosphorylation of NLRP3.
To verify the function of the NLRP3 mutant Y132F, we transfected HEK293T cells, which do not express NLRP3 inflammasome components, with plasmids of EphA2, ASC, pro‐caspase‐1, pro‐IL‐1β, NLRP3, or Y132F. By doing this, the NLRP3 inflammasome was activated. However, when we replaced NLRP3 with the mutant Y132F, IL‐1β was increasingly cleaved (Fig 6D). We also performed quantitative analysis, and the ratios of cleaved IL‐1β to pro‐IL‐1β confirmed the above results (Fig 6E). However, we could not rule out the possibility of spontaneous activation of inflammasomes in this system. Furthermore, we verified the function of the human NLRP3 mutant Y136F (the ortholog of mouse Y132F) in NLRP3 depleted human airway epithelial cell line 16HBE by reconstitution assay. We found that the reconstitution of Y136F in NLRP3‐KO cells promoted the cleavage of IL‐1β under MSU stimulation (Fig EV5C). However, IL‐1β was also found to be cleaved in NLRP3‐KO cells reconstituted with wild‐type NLRP3 and mutant Y136F even without MSU stimulation. This spontaneous inflammasome activation is possibly due to the high expression of inflammasome components.
NLRP3 Y132 phosphorylation interferes in inflammasome assembly
To investigate how EphA2 regulates inflammasome activation by phosphorylating NLRP3, we examined whether the assembly of inflammasomes was affected in EphA2‐knockout AECs. We prepared AECs from wild‐type and EphA2‐knockout mice, leaving them unstimulated (Mock) or infected them with reovirus for 3 h. We also primed and activated AECs with LPS and ATP for 30 min to serve as a positive control. We found ASC speck formation was markedly increased in EphA2‐knockout AECs than in wild‐type AECs (Fig 7A and B). We then transfected HEK293T cells with plasmids of EphA2, ASC, NLRP3, or Y132F and found the number of ASC speck‐positive cells was higher when transfected with Y132F than with wild‐type NLRP3 among ASC/NLRP3 double‐positive cells (Fig 7C and D). These results show that NLRP3 Y132 phosphorylation interferes with inflammasome assembly, thereby regulating inflammasome activation and IL‐1β cleavage.
Figure 7. NLRP3 Y132 phosphorylation interferes with inflammasome assembly.
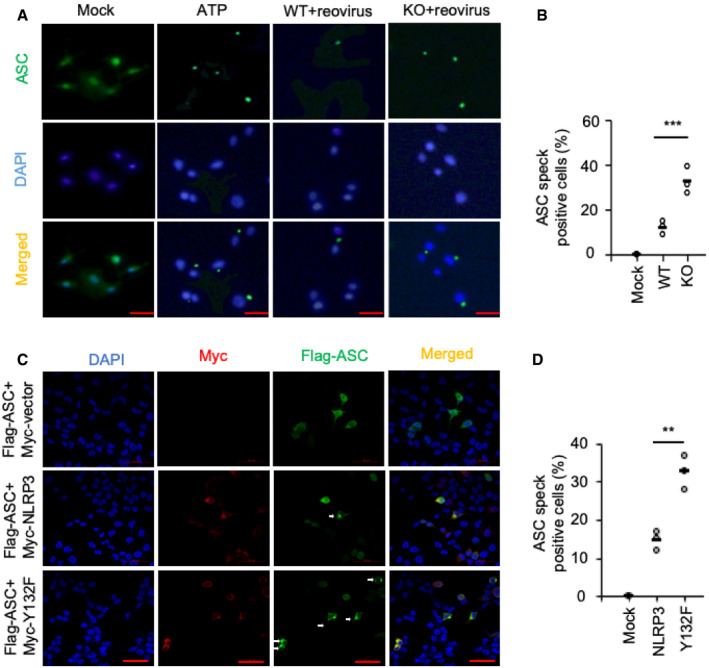
-
A, BConfocal microscopy analysis of AECs uninfected or infected with reovirus (MOI = 20) for 3 h. AECs primed with LPS and stimulated with ATP for 30 min severed as a positive control. ASC was stained with mouse anti‐ASC (1:200), followed by Alexa Fluor 488 goat anti‐mouse secondary antibody (green). DAPI served as the nuclei marker (blue).
-
C, DConfocal microscopy analysis of HEK293T cells transfected with Flag‐ASC and Myc‐tagged vector or Myc‐tagged NLRP3 or Myc‐tagged NLRP3 Y132F. Cells were stained with anti‐FLAG (1:200) and anti‐Myc (1:200), followed by Alexa Fluor 488 goat anti‐rabbit secondary antibody (green) or Alexa Fluor 594 goat anti‐mouse secondary antibody (red). DAPI served as the nuclei marker (blue).
Discussion
In this study, we found that EphA2 functions as a negative regulator of inflammasome activation upon reovirus infection through targeting NLRP3, thus guarding against an overzealous self‐destructive immune response. EphA2 also suppresses inflammatory response in a murine ova‐induced asthma model.
We found that human and mouse AECs selectively expressed EphA2, which phosphorylated NLRP3 upon reovirus activation and then inhibited the production of IL‐1β and IL‐18 and suppressed local inflammatory responses. We provide evidence that EphA2 interacts with NLRP3 and induces its phosphorylation at the site Tyr132.
IL‐1β is an emerging therapeutic target for autoimmune diseases, and there are FDA‐approved drugs for IL‐1 blockade against autoimmune diseases such as rheumatoid arthritis, and extensive clinical research is ongoing 25. Previously, it was speculated that EphA2 played a role in an LPS‐induced inflammatory lung injury model 26, here, we elucidated the exact function of EphA2 in a reovirus‐induced NLRP3 inflammasome and confirmed that EphA2 negatively regulates the inflammatory response in an ova‐induced chronic asthma disease model.
NLRP3 plays a pivotal role in RNA virus‐induced inflammasome activation, in regard to not only influenza A, but also RSV 27, VSV, HCV, and the Sendai virus, which activate NLRP3 inflammasome signaling 28. Here, we confirmed reovirus, a double‐stranded RNA (dsRNA) virus, that infects the respiratory tract in nature, strongly induces NLRP3 activation and accumulation thereby triggering the formation of inflammasomes.
Until now, multiple mechanisms have been explored to modulate the function of NLRP3 for generating an appropriate inflammatory response. Evidence was presented that NLRP3 is subject to regulation at the mRNA level and that miR‐233 decreases NLRP3 mRNA abundance and inhibits NLRP3‐inflammasome activation 29. Another mechanism is interferon‐γ (IFN‐γ), which specifically inhibits assembly of the NLRP3 inflammasome via thiol nitrosylation 30. It was reported that BRCC3 promotes inflammasome activation by deubiquitinating NLRP3 31. In contrast, MARCH7, an E3 ubiquitin ligase, promotes the degradation of NLRP3, followed by a second messenger cyclic adenosine monophosphate (cAMP) binding to NLRP3 32.
Over the last 2 years, a series of articles have shed light on the phosphorylation of NLRP3 as a crucial mechanism for modulating the function of NLRP3. It has been reported that PKA directly phosphorylated the inflammasome receptor NLRP3 at S295 and attenuated its ATPase function 33. Further studies clarified that JNK1 mediates NLRP3 phosphorylation at S194, which is a critical priming event and is essential for NLRP3 inflammasome activation 34. While the mutations in NLRP3‐encoding residues adjacent to S295 have been speculatively linked to the inflammatory disease cryopyrin‐associated periodic syndromes (CAPS), it has also been reported that blocking S194 phosphorylation prevented NLRP3 inflammasome activation in CAPS. Evidence shows that inflammatory bowel disease (IBD) is another human pathological condition that is possibly related to NLRP3 phosphorylation. It has also been shown that PTPN22 deficiency may lead to increased NLRP3 phosphorylation at Tyr861, reduced levels of mature IL‐1β in a murine model and finally, pronounced colitis 35.
EphA2 represents the first tyrosine kinase to induce NLRP3 tyrosine phosphorylation at Tyr132, which is identified as a novel phosphorylation site of NLRP3. The activation of inflammasomes is critical for protecting the host from invading microorganisms such as influenza A and RSV. However, uncontrolled production of IL‐1β and cytokines induced by viruses may lead to severe inflammatory disorders, such as IBD and asthma. The identification of EphA2 as a negative regulator of NLRP3 will have important implications for understanding excessive inflammasome activation control and the pathogenesis of human autoimmune disease prevention.
Materials and Methods
Mice
EphA2‐knockout (EphA2 −/−) mice were purchased from Jackson Lab. Bone marrows were taken from wild‐type and EphA2 −/− mice to prepare BMDCs and BMDMs. All animals were maintained in a specific pathogen‐free facility at Houston Methodist Research Institute in Houston, Texas. Animal use and care were approved by the Houston Methodist Animal Care Committee in accordance with the institutional animal care and use committee guidelines.
AEC isolation
Age‐ and sex‐matched wild‐type and EphA2‐knockout mice were euthanized by CO2 asphyxiation. Heart and lungs were exposed by cutting away the ribs, followed by puncturing the right ventricle of the heart and perfusion of the lung with phosphate‐buffered saline (PBS) until free of blood. Lung tissues were cut into small pieces and digested with Dispase II (2 U/ml), collagenase IV (25 U/ml), 2.5 mM Ca2+, and DNase in a basic non‐serum medium for 1 h at 37°C. Cell suspensions were filtered through an EASY strainer (Greiner Bio‐One) with 70‐μm pores. AECs were then seeded and cultured. The proteins in the supernatants of AECs were precipitated by 20% trichloroacetic acid (TCA).
Chronic allergy lung inflammation model
Groups of 6‐week‐old sex‐matched wild‐type mice and EphA2‐deficient mice were intraperitoneally injected with 20 μg ova (Sigma) dissolved in PBS on day 0, day 3, day 6, day 9 and day 12. Mice were challenged intranasally for 6 weeks with 1% ova from day 19. All mice were euthanized for analysis 1 day after the final challenge. BALF was prepared and the total number of BAL cells was determined by trypan blue exclusion. Differential cell counts were assessed by cytospins and stained with a Hema 3 staining kit (Fisher Scientific) and a modified Wright–Giemsa stain. Tissue samples were stained with hematoxylin and eosin (H&E) or with periodic acid–Schiff (PAS) for analysis 36.
Reagents
Lipofectamine 3000 was purchased from Invitrogen, Dispase II from Sigma, collagenase type IV from Gibco, and DNase from Roche.
The following antibodies were used for immunoprecipitation: anti‐NLRP3 (1:100) (AG‐20B‐0014‐C100; AdipoGen) and anti‐EphA2 (1:100) (sc‐398832; Santa Cruz). The following antibodies were used for immunoblot analysis: anti‐NLRP3 (1:1,000) (AG‐20B‐0014‐C100; AdipoGen), anti‐EphA2 (1:1,000) (sc‐398832; Santa Cruz), anti‐EphA2 (1:1,000) (clone D4A2; 6997; Cell Signaling), anti‐p‐Tyr (1:1,000) (sc‐7020; Santa Cruz), anti‐p‐Tyr (CST; P‐Tyr‐100, 9411), anti‐caspase‐1 (1:1,000) (AG‐20B‐0042‐C100; AdipoGen), anti‐caspase‐1 (1:1,000) (AG‐20B‐0048‐C100; AdipoGen), anti‐IL‐1β (1:1,000) (AF‐401‐NA; R&D Systems), anti‐IL‐1β (1:1,000) (AF‐201‐NA; R&D Systems), anti‐cleaved‐IL‐1β (Asp116) (1:1,000) (CST; D3A3Z, 83186), anti‐ASC (1:1,000) (CST; D2W8U, 67824), anti‐HA (1:5,000) (clone HA‐7, H6533; Sigma), anti‐β‐actin (1:20,000) (clone AC‐15, A3854; Sigma), anti‐Myc (1:5,000) (ab1326; Abcam), anti‐GFP (1:1,000) (66002‐1; Proteintech), anti‐mouse (1:3,000) (115‐035‐174; Jackson), and anti‐rabbit (1:3,000) (211‐032‐171; Jackson).
The following antibodies were used for confocal microscopy: anti‐EphA2 (1:500) (clone D4A2; 6997; Cell Signaling), anti‐NLRP3 (1:500) (AG‐20B‐0014‐C100; AdipoGen), anti‐IL‐1β (1:200) (AF‐401‐NA; R&D Systems), anti‐CD68 (1:200) (ab31630; Abcam), anti‐EpCAM (1:200) (ab221552; Abcam), anti‐FLAG (1:200) (20543‐1‐AP; Proteintech), anti‐Myc (1:200) (sc‐40; Santa Cruz), anti‐ASC (1:200) (sc‐514414; Santa Cruz), Alexa Fluor 488 goat anti‐rabbit secondary antibody (A‐11034; Thermo Fisher Scientific), Alexa Fluor 594 goat anti‐rabbit secondary antibody (A‐150088; Thermo Fisher Scientific), Alexa Fluor 488 donkey anti‐goat secondary antibody (A‐150129; Thermo Fisher Scientific), and Alexa Fluor 594 goat anti‐mouse secondary antibody (A‐11005; Thermo Fisher Scientific). Anti‐HA and anti‐Myc beads were from Sigma.
Protein A/G agarose beads were purchased from Thermo Scientific; the IFN‐β ELISA kit from PBL InterferonSource; IL‐6, IL‐1β, IL‐18, IL‐4, IL‐13, and IL‐33 ELISA kits from eBioscience; recombinant mouse Ephrin A1–Fc Chimera Protein from R&D; and the HA peptide from APExBIO.
Cell cultures
Murine BMDCs, BMDMs, and AMs were isolated and maintained in RPMI‐1640 medium supplemented with 10% heat‐inactivated FCS and 1% penicillin–streptomycin (Invitrogen‐Gibco) as previously described 37. AECs were grown in BEBM (Lonza, Cat. No. CC3171), with an enhanced bullet kit (Lonza, Cat. No. CC3170) and 1% penicillin–streptomycin (Invitrogen‐Gibco) to create the basal epithelial growth media (BEGM). HEK293T cells and RAW264.7 cells were grown in DMEM supplemented with 10% heat‐inactivated FCS and 1% penicillin–streptomycin. The 16HBE cells were grown in RPMI‐1640 medium supplemented with 10% heat‐inactivated FCS and 1% penicillin–streptomycin.
RNA extraction and real‐time PCR
Total cellular and tissue RNAs were extracted from cultured cells or lung tissue using the RNeasy Mini Kit (Sigma) according to the manufacturer's instructions. cDNA was synthesized from 1 μg of the total RNA using a reverse transcriptase kit (Applied Biosystems). The mRNA level was evaluated by qRT–PCR using the SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad) and was analyzed on Bio‐Rad CFX96. All gene expressions were normalized to the housekeeping gene β‐actin and used as an internal standard. The following primers were used:
IL‐4 forward, 5′‐AGATGGATGTGCCAAACGTCCCAt‐3′, and reverse, 5′‐AATATGCGAAGCACCTTGGAAGCC‐3′; IL‐5 forward, 5′‐TCACCGAGCTCTGTTGACAA‐3′, and reverse, 5′‐CCACACTTCTCTTTTTGGCG‐3′; IL‐13 forward, 5′‐TCACCGAGCTCTGTTGACAA‐3′, and reverse, 5′‐CCACACTTCTCTTTTTGGCG‐3′; EphA2 forward, 5′‐GGACTGCCAGCGTCAGTATT‐3′, and reverse, 5′‐CTTGCGGTAGGTGACTTCGT‐3′; and β‐actin forward, 5′‐TGGTTACAGGAAGTCCCTCAC‐3′, and reverse, 5′‐ACAGAAGCAATGCTGTCACCTT‐3′.
Virus infection
Reovirus (Reovirus type 3 strain Dearing, T3D) was purchased from American Type Culture Collection (VR‐824). For the reovirus infection in mice, 6‐week‐old sex‐matched mice were infected intranasally with 4 × 104 PFU of reovirus in PBS. Mice were euthanized at various time points following infection and tissues were collected for analysis as previously described 38.
Histology
Lungs were removed from naive or infected wild‐type and EphA2 −/− mice and washed using PBS before being fixed with 4% formaldehyde for 48 h. Tissues were embedded in paraffin and processed using standard techniques. Longitudinal 5‐μm sections were stained with H&E or PAS.
In vitro pull‐down and immunoblot analysis
For endogenous immunoprecipitation assay, AECs were lysed in IP lysis buffer (87788; Fisher Scientific) with a protease and phosphatase inhibitor cocktail for 30 min. Primary antibodies were added into precleaned protein A/G agarose beads and rotated for 30 min at 4°C and washed with PBS three times. After collecting the supernatants of lysates by centrifugation (15,000 g for 10 min at 4°C), supernatants were added to protein A/G agarose beads, and suspended and incubated with constant rotation for 2 h at 4°C. The precipitates were washed three times using cold IP lysis buffer. After the final washing, the beads were resolved on SDS–PAGE. For the preparation of purified NLRP3 and EphA2, HEK293T cells were transfected with expression plasmids encoding full‐length or truncated versions of HA‐(N‐terminal), GFP‐(C‐terminal) or Myc‐tagged (N‐terminal) mice NLRP3 or mice EphA2. Lysates were prepared from the transfected cells, followed by incubation with anti‐HA or anti‐Myc beads for 1 h at 4°C. Proteins were eluted from the beads after they were washed six times with IP lysis buffer. Proteins and beads were analyzed by immunoblot analysis with anti‐HA or anti‐Myc antibody.
In vitro kinase assay
GFP‐EphA2, GFP‐EphA2‐K646M, and HA‐NLRP3 were expressed in HEK293T cells and lysed in kinase buffer with a protease and phosphatase inhibitor cocktail. An IgG or EphA2 antibody was added into precleaned protein A/G agarose beads and rotated for 30 min at 4°C and washed with PBS five times. After collecting the GFP‐EphA2 supernatants by centrifugation (15,000 g for 10 min at 4°C), GFP‐EphA2 was added to protein A/G agarose beads, suspended and incubated with constant rotation for 2 h at 4°C. The precipitates were washed five times using cold IP lysis buffer, discarding all the supernatants and leaving the anti‐EphA2‐conjugated protein A/G as kinase. HA‐NLRP3 was purified by HA beads and eluted by HA peptides, and the eluted protein was kept as substrate. 200 μM ATP was added into the substrate and kinase and shaken for 2 h at 30°C. Proteins were eluted from the beads after they were washed six times with IP lysis buffer and were analyzed by immunoblot analysis with anti‐p‐Tyr, anti‐HA, or anti‐EphA2.
Confocal microscopy
AECs were stimulated with reovirus for 3 h. For ASC speck immunofluorescence, both wild‐type (WT) and EphA2‐KO AECs were left uninfected (Mock) or infected with reovirus at a multiplicity of infection (MOI) of 20 for 3 h, or primed with LPS (1 mg/ml) for 2 h and stimulated with ATP (3 mM) for 30 min. After stimulation or infection, AECs were then fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X‐100 and then blocked for 30 min with 5% BSA, incubated with rabbit anti‐EphA2 (1:500) (clone D4A2; 6997; Cell Signaling), mouse anti‐NLRP3 (1:500) (AG‐20B‐0014‐C100; AdipoGen), or rabbit anti‐ASC (1:200) (CST; D2W8U, 67824) for 2 h, followed by Alexa Fluor 488 goat anti‐rabbit secondary antibody and Alexa Fluor 594 goat anti‐mouse secondary antibody for 1 h, and then examined with confocal microscopy. Images of zoomed single cells were quantified using ZEISS 880.
Statistical analysis
A two‐tailed unpaired Student's t‐test was used for statistical analysis with Microsoft Excel and GraphPad Prism software. Differences in P‐values < 0.05 were considered statistically significant.
Author contributions
AZ, JX, and TX designed and did most of the experiments; HZ and MF helped with the experiments; SL and YD contributed reagents and materials; AZ wrote the manuscript; XCL revised the manuscript; and ZZ and M‐SZ supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Laurie Minze at Houston Methodist Research Institute for her excellent operational support. We thank our language editor Mr. Christopher Lavender of Sun Yat‐sen University Cancer Center for providing assistance in editing this manuscript. M.Z. is supported by the National Key Research and Development Program of China (2017YFA0505600, 2017YFC0908503, and 2016YFA0502100), the National Natural Science Foundation of China (81520108022, 81621004, and 81830090), and the Guangdong Province Key Research and Development Program (2019B020226002). XCL is supported by the National Institutes of Health grant R01AI080779. Z.Z. is supported by the Lupus Research Alliance Grant 519418.
EMBO Reports (2020) 21: e49666
See also: Z Zhang & R Ricci (July 2020)
Contributor Information
Zhiqiang Zhang, Email: zzhang@houstonmethodist.org.
Mu‐Sheng Zeng, Email: zengmsh@sysucc.org.cn.
Data availability
Primary datasets have been generated and deposited in the Research Data Deposit (RDD) bank (http://www.researchdata.org.cn) including the data of GSE1133 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE1133), microarray analysis, ELISAs, and confocal microscopy statistical analysis, with the approval RDD number RDDB2020000841.
References
- 1. Proud D, Leigh R (2011) Epithelial cells and airway diseases. Immunol Rev 242: 186–204 [DOI] [PubMed] [Google Scholar]
- 2. Rock JR, Randell SH, Hogan BLM (2010) Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech 3: 545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holgate ST (2011) The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev 242: 205–219 [DOI] [PubMed] [Google Scholar]
- 4. Chung KF, Adcock IM (2008) Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J 31: 1334–1356 [DOI] [PubMed] [Google Scholar]
- 5. Guarda G, Dostert C, Staehli F, Cabalzar K, Castillo R, Tardivel A, Schneider P, Tschopp J (2009) T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature 460: 269–273 [DOI] [PubMed] [Google Scholar]
- 6. Allen Ic SMAMCB (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30: 556–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA (2007) Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med 204: 3235–3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 458: 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirota JA, Hirota SA, Warner SM, Stefanowicz D, Shaheen F, Beck PL, MacDonald JA, Hackett T‐L, Sin DD, Van Eeden S et al (2012) The airway epithelium nucleotide‐binding domain and leucine‐rich repeat protein 3 inflammasome is activated by urban particulate matter. J Allergy Clin Immunol 129: 1116–1125.e6 [DOI] [PubMed] [Google Scholar]
- 10. Peeters PM, Perkins TN, Wouters EF, Mossman BT, Reynaert NL (2013) Silica induces NLRP3 inflammasome activation in human lung epithelial cells. Part Fibre Toxicol 10: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hillegass JM, Miller JM, MacPherson MB, Westbom CM, Sayan M, Thompson JK, Macura SL, Perkins TN, Beuschel SL, Alexeeva V et al (2013) Asbestos and erionite prime and activate the NLRP3 inflammasome that stimulates autocrine cytokine release in human mesothelial cells. Part Fibre Toxicol 10: 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fukumoto J, Fukumoto I, Parthasarathy PT, Cox R, Huynh B, Ramanathan GK, Venugopal RB, Allen‐Gipson DS, Lockey RF, Kolliputi N (2013) NLRP3 deletion protects from hyperoxia‐induced acute lung injury. Am J Physiol Cell Physiol 305: C182–C189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang X, Jiang W, Yan Y, Gong T, Han J, Tian Z, Zhou R (2014) RNA viruses promote activation of the NLRP3 inflammasome through a RIP1‐RIP3‐DRP1 signaling pathway. Nat Immunol 15: 1126–1133 [DOI] [PubMed] [Google Scholar]
- 14. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241 [DOI] [PubMed] [Google Scholar]
- 15. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid. Nat Immunol 9: 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haneklaus M, O'Neill LA (2015) NLRP3 at the interface of metabolism and inflammation. Immunol Rev 265: 53–62 [DOI] [PubMed] [Google Scholar]
- 17. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, Baba N, Libioulle C, Belaiche J, Bitton A et al (2009) Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat Genet 41: 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz‐Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A et al (2015) A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Nardo D, De Nardo CM, Latz E (2014) New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am J Pathol 184: 42–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Okazaki T, Ni A, Baluk P, Ayeni OA, Kearley J, Coyle AJ, Humbles A, McDonald DM (2009) Capillary defects and exaggerated inflammatory response in the airways of EphA2‐deficient mice. Am J Pathol 174: 2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carpenter TC, Schroeder W, Stenmark KR, Schmidt EP (2012) Eph‐A2 promotes permeability and inflammatory responses to bleomycin‐induced lung injury. Am J Respir Cell Mol Biol 46: 40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hong JY, Shin MH, Douglas IS, Chung KS, Kim EY, Jung JY, Kang YA, Kim SK, Chang J, Kim YS et al (2016) Inhibition of EphA2/EphrinA1 signal attenuates lipopolysaccharide‐induced lung injury. Clin Sci 130: 1993–2003 [DOI] [PubMed] [Google Scholar]
- 23. Zhang H, Li Y, Wang H‐B, Zhang A, Chen M‐L, Fang Z‐X, Dong X‐D, Li S‐B, Du Y, Xiong D et al (2018) Ephrin receptor A2 is an epithelial cell receptor for Epstein Barr virus entry. Nat Microbiol 3: 164–171 [DOI] [PubMed] [Google Scholar]
- 24. Tanaka M, Kamata R, Sakai R (2005) EphA2 phosphorylates the cytoplasmic tail of Claudin‐4 and mediates paracellular permeability. J Biol Chem 280: 42375–42382 [DOI] [PubMed] [Google Scholar]
- 25. Arranz L, Arriero MDM, Villatoro A (2017) Interleukin‐1 as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev 31: 306–317 [DOI] [PubMed] [Google Scholar]
- 26. Hong JY, Shin MH, Chung KS, Kim EY, Jung JY, Kang YA, Kim YS, Kim SK, Chang J, Park MS (2015) EphA2 receptor signaling mediates inflammatory responses in lipopolysaccharide‐induced lung injury. Tuberc Respir Dis 78: 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han M, Bentley JK, Rajput C, Lei J, Ishikawa T, Jarman CR, Lee J, Goldsmith AM, Jackson WT, Hoenerhoff MJ et al (2019) Inflammasome activation is required for human rhinovirus‐induced airway inflammation in naive and allergen‐sensitized mice. Mucosal Immunol 12: 958–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu J, Wu Y, Wang J (2017) Activation and role of NACHT, LRR, and PYD domains‐containing protein 3 inflammasome in RNA viral infection. Front Immunol 8: 1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid‐Burgk JL, Hornung V (2012) NLRP3 inflammasome activity is negatively controlled by miR‐223. J Immunol 189: 4175–4181 [DOI] [PubMed] [Google Scholar]
- 30. Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, Sassetti CM (2013) Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome dependent processing of IL‐1. Nat Immunol 14: 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Py BF, Kim MS, Vakifahmetoglu‐Norberg H, Yuan J (2013) Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49: 331–338 [DOI] [PubMed] [Google Scholar]
- 32. Yan YJWLL (2015) Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160: 62–73 [DOI] [PubMed] [Google Scholar]
- 33. Mortimer L, Moreau F, MacDonald JA, Chadee K (2016) NLRP3 inflammasome inhibition is disrupted in a group of auto‐inflammatory disease CAPS mutations. Nat Immunol 17: 1176–1186 [DOI] [PubMed] [Google Scholar]
- 34. Song N, Liu Z‐S, Xue W, Bai Z‐F, Wang Q‐Y, Dai J, Liu X, Huang Y‐J, Cai H, Zhan X‐Y et al (2017) NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell 68: 185–197.e6 [DOI] [PubMed] [Google Scholar]
- 35. Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, Scharl S, Gutte PM, Grütter MG, Beer H‐D et al (2016) NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest 126: 1783–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao X, Balasubramanian S, Liu W, Chu X, Wang H, Taparowsky EJ, Fu Y‐X, Choi Y, Walsh MC, Li XC (2012) OX40 signaling favors the induction of TH9 cells and airway inflammation. Nat Immunol 13: 981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng M‐S, Zhang Z (2017) TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun 8: 945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu Y‐J (2013) The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double‐stranded DNA. Nat Immunol 14: 172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
Primary datasets have been generated and deposited in the Research Data Deposit (RDD) bank (http://www.researchdata.org.cn) including the data of GSE1133 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE1133), microarray analysis, ELISAs, and confocal microscopy statistical analysis, with the approval RDD number RDDB2020000841.