

Abstract
Lipopolysaccharides (LPS) can lead to a lethal endotoxemia, which is a systemic inflammatory response syndrome (SIRS) characterized by a systemic release of cytokines, such as TNF. Endotoxemia is studied intensely, as a model system of Gram‐negative infections. LPS‐ and TNF‐induced SIRS involve a strong induction of interferon‐stimulated genes (ISGs), some of which cause cell death in the intestinal epithelium cells (IECs). It is well known that glucocorticoids (GCs) protect against endotoxemia. By applying numerous mutant mouse lines, our data support a model whereby GCs, via their glucocorticoid receptor (GR), apply two key mechanisms to control endotoxemia, (i) at the level of suppression of TNF production in a GR monomer‐dependent way in macrophages and (ii) at the level of inhibition of TNFR1‐induced ISG gene expression and necroptotic cell death mediators in IECs in a GR dimer‐dependent way. Our data add new important insights to the understanding of the role of TNF in endotoxemia and the two separate key roles of GCs in suppressing TNF production and activity.
Keywords: glucocorticoids, lipopolysaccharides, systemic inflammatory response syndrome, TNF
Subject Categories: Autophagy & Cell Death, Immunology, Signal Transduction
Glucocorticoids limit the effects of LPS in vivo at two critical control points, namely at the level of macrophages by controlling TNF production, and at the level of intestinal epithelial cells by inhibiting TNFR1‐induced interferon‐stimulated gene expression.
Introduction
Infections of humans by Gram‐negative (GN) bacteria cause huge social and economic health problems. Lipopolysaccharides (LPS) form the major immune‐reactive components of such bacteria. In mammals, LPS is mainly recognized by the pattern recognition receptor (PRR) Toll‐like receptor 4 (TLR4) 1, 2, leading to a signaling cascade. LPS injection in experimental animals can result in a lethal systemic inflammatory response syndrome (SIRS) which is characterized by a fast systemic release of cytokines, such as TNF, interferons (IFNs), IL‐6, and IL‐1β. Mainly, activated macrophages stimulate the production of these cytokines, upon TLR4 activation of transcription factors such as NFκB and AP‐1. LPS‐induced lethal SIRS has been shown to depend on TNF 3.
TNF has two major receptors, known as TNFR1 and TNFR2, encoded by the genes Tnfrsf1a and Tnfrsf1b. Tnfrsf1a −/− mice are resistant against TNF‐induced and LPS‐induced SIRS (the latter also known as endotoxemia) 4, 5, 6. The intestinal epithelial cells (IECs) have been described as the main targets of TNF since IEC‐specific Tnfrsf1a −/− mice (Tnfrsf1a VillinKO) are significantly protected against TNF‐induced lethal SIRS 4, 7. TNF has a broad range of effects on IECs 8, but three mechanisms are thought to be related to TNF‐induced lethal SIRS: (i) ER stress in the intestinal epithelium, (ii) goblet and Paneth cell dysfunction, and (iii) bacterial translocation, of commensal flora from the intestinal lumen to the draining lymph nodes and spleen 9. Different types of TNF‐induced cell death modalities have been observed in the intestine 10, but Paneth cells seem to be quite sensitive for TNF and to necroptosis 11. For example, IEC‐specific deletion of caspase‐8, the key initiator of TNF‐induced apoptosis and suppressor of necroptosis12, leads to inflammatory necroptosis and Paneth cell loss 13. Similarly, deletion of FAS‐associated death domain protein (FADD), which normally recruits caspase 8 and thereby triggers apoptosis 12, in the IECs also leads to spontaneous IEC necroptosis with loss of Paneth cells 14. The loss of Paneth cells is also observed in patients with Crohn's disease, associated with high levels of RIPK3 13.
In mouse models, both endogenous and exogenous glucocorticoids (GCs) have been shown to have strong protective effects against TNF‐induced lethal SIRS and intestinal permeability 9, 15, 16. GCs, such as dexamethasone (DEX), stimulate the glucocorticoid receptor (GR), a transcription factor encoded by the Nr3c1 gene. After ligand binding, GR translocates to the nucleus to interact with DNA and/or with other transcription factors. The best described mechanism of function of GR is the activation of gene transcription (transactivation, TA) in which (mainly) GR homodimers directly bind to DNA (GR responsive elements, GREs), attract nuclear cofactors, and stimulate RNA polymerase 2. GR dimers can also repress gene expression (transrepression, TR) by binding to negative GREs (nGRE), or by binding to alternative genomic elements known as IR‐nGREs 17. Finally, also GR monomers can regulate gene expression in a DNA‐binding or protein–protein dependent way. Recent research indicates that GR homodimers are essential for mediating anti‐inflammatory properties of GCs in acute inflammatory settings, such as in TNF‐ or LPS‐induced SIRS 11, 16, 18. Most of these recent data are based on experiments using GRdim/dim mice, which express a mutant version of GR (A465T) which exhibits reduced GR dimerization and DNA binding, while maintaining an intact monomer profile. GRdim/dim mice are extremely sensitive for TNF‐induced SIRS 16. RNA sequencing of IECs of GRdim/dim mice revealed that these mice fail to control the activity of STAT1, leading to a chronic upregulation of microbiota‐induced interferon‐stimulated genes (ISG) in both unstimulated and TNF‐stimulated conditions 11. Since several ISG genes are involved in induction of necroptotic cell death, the data suggested that the protection by GCs against TNF‐induced SIRS is mainly due to dimer‐dependent repression of the axis STAT1/ISG/cell death in IECs 11.
GCs are well‐known inhibitors of LPS‐induced endotoxemia, by inhibition of TNF production by macrophages. As LPS‐induced SIRS is strongly mediated by TNF, we investigated whether the mechanisms of GC control on TNF‐induced SIRS in IECs are relevant in LPS‐induced SIRS model. Indeed, based on our work, we conclude that GCs limit the in vivo LPS effects at two critical control points, namely at the level of macrophages, i.e., controlling TNF production in a GR‐monomer way, and at the level of inhibition of TNFR1‐induced ISRE gene expression and cell death mediators at the level of IECs in a GR dimer‐dependent way.
Results
Glucocorticoids protect against LPS‐induced lethal SIRS, intestinal permeability, and intestinal expression of ISGs
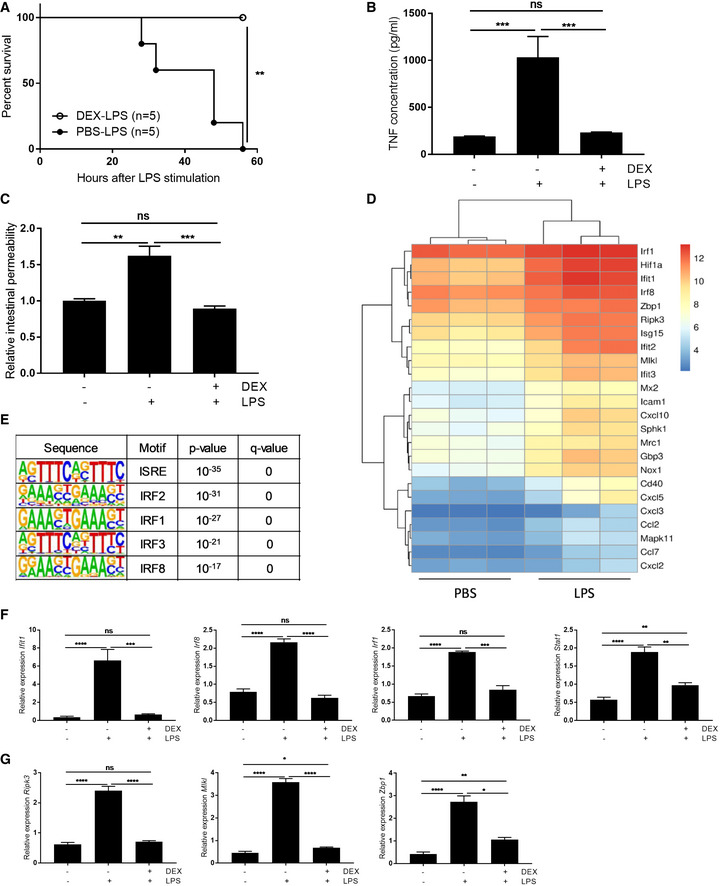
Wild‐type (WT) C57BL/6J mice were treated with 10 mg/kg dexamethasone (DEX) or PBS, followed 30 min later by a single lethal dose of 12.5 mg/kg LPS. Survival was monitored (Fig 1A) and systemic TNF concentration was measured 2 h after LPS injection (Fig 1B). DEX was found to protect mice against LPS‐induced lethality and to suppress the systemic release of TNF. Furthermore, LPS induced intestinal permeability 8 h after LPS challenge, as shown by an increase in systemic FITC‐dextran upon oral gavage. This intestinal leakage was also suppressed by a single injection of DEX 30 min before LPS (Fig 1C). TNF‐induced lethality has been explained by effects on IECs. As TNF is an important mediator in LPS‐induced lethality 3, we studied the effects of LPS on the IECs, by means of an RNA sequencing (RNA‐seq) analysis of IECs of WT mice 6 h after injection with PBS or LPS. In LPS‐challenged mice, 1,419 and 1,257 genes showed significant up‐ and downregulation, respectively, when compared to PBS‐treated mice. Figure 1D shows a heatmap with a selection of LPS‐upregulated genes (complete list of differentially expressed genes in Dataset EV1). This list was chosen because known motif analysis of LPS‐induced genes by HOMER revealed a top‐5 significant enrichment of ISRE, IRF2, IRF1, IRF3, and IRF8 motif‐containing genes, indicating enrichment of interferon‐stimulated genes (Fig 1E). As the motifs are very similar, there is overlap between the sites; each motif is scanned and scored independently. In addition, some genes can have multiple IRF motifs, which are distinct. Of the 1,419 upregulated genes, 711 are recognized as interferon‐induced genes by the interferome database 19, which is a higher frequency than expected by chance (P < 0.001) (list of LPS‐upregulated interferome genes in Dataset EV2). The expression of some of these interferon‐stimulated genes (ISGs) was confirmed via qPCR in which a DEX condition was included. DEX repressed the LPS‐induced expression of ISGs (Fig 1F and G). Thus, intestinal permeability, systemic TNF induction, and IEC‐ISG expression follow similar patterns and are all DEX‐inhibited.
Figure 1. Glucocorticoids protect against LPS‐induced lethality, intestinal permeability, and intestinal expression of ISGs.
-
A–CWT C57BL/6J mice were injected with 12.5 mg/kg LPS, with or without pre‐treatment with 10 mg/kg DEX 30 min before LPS. (A) Lethality was followed up. LPS‐treated mice (n = 5) are depicted as black circles and DEX/LPS‐treated mice (n = 5) are depicted as white circles. P‐values for survival curves were analyzed with Fisher's exact test. (B) Blood samples were taken 2 h after LPS and TNF levels were measured in serum (n = 4–5). (C) Three hours after LPS challenge, dextran labeled with FITC was administered by oral gavage. Five hours after gavaging, blood plasma was collected and fluorescence was measured as a degree of intestinal permeability (λexc/λem = 488/520 nm) (PBS: n = 3, LPS and DEX/LPS: n = 5).
-
D, ERNA sequencing (RNA‐seq) results of IECs of WT mice (n = 3) treated with PBS or 12.5 mg/kg LPS for 6 h. (D) Heatmap with selection of LPS‐induced genes. Color key shows the log2 of normalized counts. (E) HOMER motif analysis of LPS‐induced genes. Motifs with the highest rank and their P‐value and q‐value are displayed.
-
F, GWT mice (n = 5) were treated with PBS or 12.5 mg/kg LPS with or without 10 mg/kg DEX pre‐treatment. Ileum was isolated and gene expression was measured via qPCR.
LPS leads to induction of necroptosis‐stimulating ISG genes in IECs
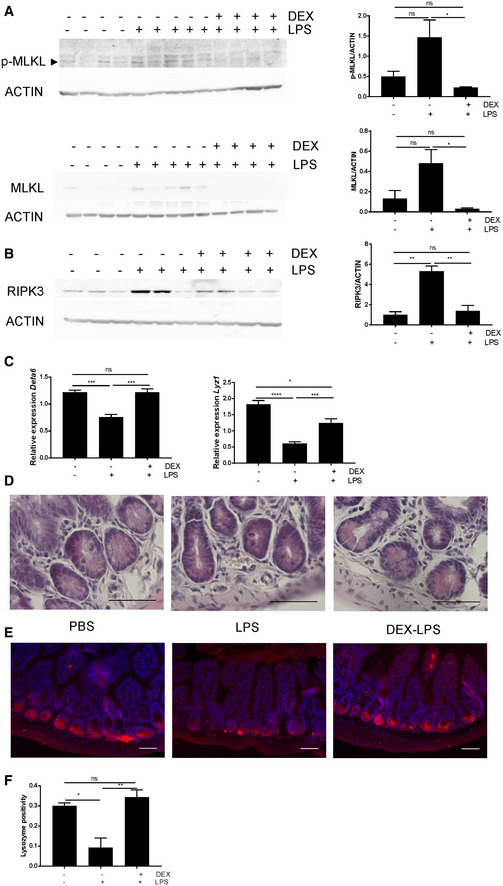
The list of LPS‐induced ISGs contains three genes involved in necroptosis, namely Ripk3, Mlkl, and Zbp1. Increased Ripk3 and Mlkl gene expression may indicate a predisposition to necroptosis 20 (Fig 1G). To study the relevance of the upregulation of these genes, we studied protein expression of RIPK3, MLKL, and phosphorylated MLKL (pMLKL) via Western blot. MLKL is phosphorylated by active RIPK3, leading to conformational changes and activation able to induce necroptosis 21. WT mice were injected with PBS or 12.5 mg/kg LPS with or without DEX pre‐treatment, and ileum samples were collected 6 h later. Protein expression of MLKL and pMLKL and RIPK3 were found to be increased upon LPS injection, and this upregulation was prevented by DEX pre‐treatment (Fig 2A and B). Paneth cells (PC) were shown to be sensitive to necroptosis in a caspase‐8‐deficient background 11, 13, 14. Via qPCR, we first studied the expression of two PC markers: Defa6 and Lyz1 (Fig 2C). We found that LPS caused a reduction in expression of Defa6 and Lyz1 6 h after LPS, which is prevented by DEX treatment 30 min prior to LPS. H&E staining reveals a loss of granules in the crypts 6 h after LPS treatment (Fig 2D). In addition, we observe a reduction in lysozyme expression, confirming the qPCR expression data (Fig 2E and F). Degranulation of PC appeared to go together with luminal extrusion and death of PCs 22.
Figure 2. LPS leads to induction of necroptosis‐stimulating ISG genes in IECs.
-
A–CWT mice (n = 3–5) were treated with PBS or 12.5 mg/kg LPS with or without 10 mg/kg DEX pre‐treatment. Ileum was isolated 6 h after LPS challenge. (A) pMLKL and MLKL protein levels and (B) RIPK3 protein levels were analyzed via Western blot using ACTIN as a loading control. Expression levels were quantified using FIJI and normalized to ACTIN levels. P‐values were calculated using one‐way ANOVA. (C) Gene expression (Defa6, Lyz1) was measured via qPCR. P‐values were calculated using one‐way ANOVA. All bars represent mean ± SEM. ****P < 0.0001; ***P < 0.001; **P ≤ 0.01; *P ≤ 0.05.
-
DH&E staining was performed on ileum samples. Scale bar: 50 μm.
-
ELysozyme expression (in red) was detected via IHC, with Hoechst counterstaining. Scale bars: 50 μm.
-
FQuantification of lysozyme expression. For every picture (n = 3–4 per treatment), a selection of 10 crypts was made. Lysozyme positive signal was quantified in this region of interest (ROI) using an automated script in QuPath. Values are the positive area relative to total area of ROI. P‐values were calculated using one‐way ANOVA. Data are shown as mean ± SEM. ****P < 0.0001; ***P < 0.001; **P ≤ 0.01; *P ≤ 0.05.
TNF and IEC TNFR1 play critical roles in LPS‐induced lethality and ISG induction in IECs
LPS‐induced lethality has been shown to depend on TNF: Tnfrsf1a −/− (hereafter called TNFR1−/−) mice are resistant to TNF‐ and LPS‐induced SIRS compared to control Tnfrsf1a +/+ (TNFR1+/+) littermates 4, 6, the latter confirmed by us here (Fig 3A). To investigate whether the LPS effects on the IECs are indirect effects, mediated by TNF, we applied mice that lack TLR4 or TNFR1 specifically in the IECs: Tlr4 fl/fl Villin‐cre tg/+ (hereafter called TLR4VillinKO) and Tnfrsf1a fl/fl Villin‐cre tg/+ (hereafter called TNFR1VillinKO), respectively. TLR4fl/fl control mice and TLR4VillinKO mice were treated with LPS and lethality was monitored. There was no difference in lethal response between both groups showing no important role for TLR4 expression on IECs during LPS‐induced lethal SIRS (Fig 3B). This is in line with earlier data showing that LPS administration induces high TNF expression in lamina propria cells, but not in IECs, suggesting that TNF produced by non‐epithelial cells is important, rather than direct TLR4 signaling in IECs 23. Next, we treated TNFR1fl/fl and TNFR1VillinKO with LPS and lethality was monitored (Fig 3C). Compared to TNFR1fl/fl mice, TNFR1VillinKO mice were partly protected against LPS, similar to protection of these mice observed in TNF‐induced SIRS 4. Our data suggest that LPS induces TNF production in other cells, which subsequently exerts its detrimental effect on the IECs by binding to TNFR1. Based on our studies, showing that TNF induces an ISG signature in the small intestine 11, we investigated to what extent the intestinal ISG expression observed after LPS challenge is caused by TNF. We made an overlap of LPS‐upregulated and TNF‐upregulated genes, both identified via RNA‐seq of IEC samples taken from WT mice. Of note, RNA‐seq analysis of TNF‐induced SIRS was performed in WT mice with FVB background, while RNA‐seq analysis of LPS‐induced SIRS was performed in WT mice with C57BL/6J mice. Therefore, we only studied overlapping genes and did not focus on TNF‐ or LPS‐specific genes because this could be biased by the mouse background. Strikingly, the overlap was quite big. On the overlap of 692 genes, we performed a HOMER motif analysis and the top 5 motifs were as follows: ISRE, IRF3, IRF1, IRF2, and NFκB (Fig 3D). The overlap of 692 genes contained 351 ISGs, i.e., genes confirmed by the interferon database as interferon‐induced. In this group of 351 genes, we again find Ripk3, Mlkl, and Zbp1. To study whether these and other ISG gene induction by LPS in IECs are mediated by TNF, we treated TNFR1+/+ and TNFR1−/− mice with PBS or an LPS dose which is lethal in the TNFR1+/+ mice and measured the expression of different ISG by qPCR, in the ileum 6 h after injection. In Fig 3E, we display the necroptosis‐relevant ISG genes Ripk3, Mlkl, and Zbp1, and indeed found that the induction levels were less pronounced in TNFR1−/− compared to TNFR1+/+ mice. Similar findings were done in TNFR1VillinKO mice (Fig 3F).
Figure 3. TNF and IEC TNFR1 play critical roles in LPS‐induced lethality and ISG induction in IECs.
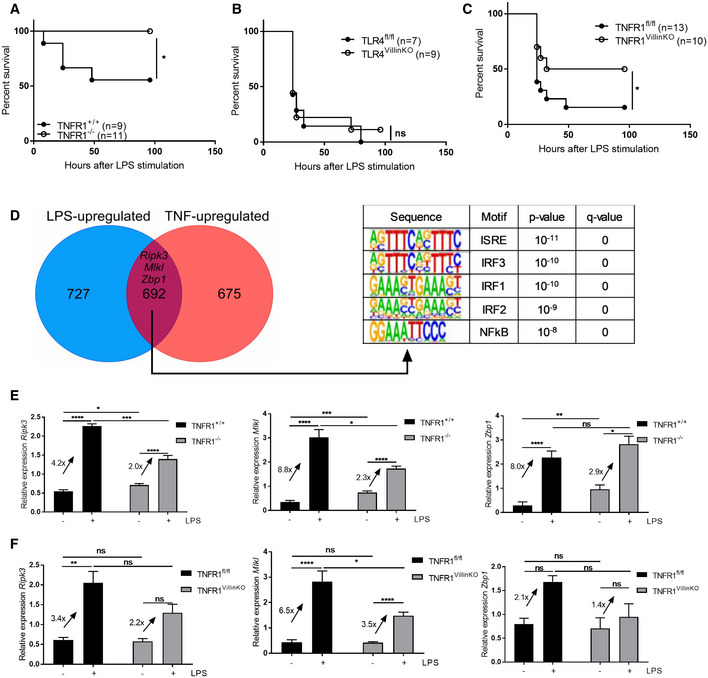
- TNFR1+/+ (n = 9) and TNFR1−/− mice (n = 11) were treated with 12.5 mg/kg LPS and lethality was monitored. P‐values were analyzed with Fisher's exact test.
- TLR4fl/fl mice (n = 7) and TLR4VillinKO (n = 9) mice were treated with 12.5 mg/kg LPS and lethality was monitored. P‐values were analyzed with Fisher's exact test.
- TNFR1fl/fl mice (n = 13) and TNFR1VillinKO (n = 10) mice were treated with 12.5 mg/kg LPS and lethality was monitored. P‐values were analyzed with Fisher's exact test.
- Venn diagram of LPS‐ and TNF‐upregulated genes in IEC, as identified by RNA‐seq (n = 3). HOMER motif analysis was performed on the 692 overlapping genes. Motifs with the highest rank and their P‐value and q‐value are displayed.
- TNFR1+/+ and TNFR1−/− mice (n = 4–6) were treated with 12.5 mg/kg LPS. Ileum was isolated and gene expression was measured via qPCR. Fold changes are shown on the graph.
- TNFR1fl/fl and TNFR1VillinKO mice (n = 4–6) were treated with 12.5 mg/kg LPS. Ileum was isolated and gene expression was measured via qPCR. Fold changes are shown on the graph.
First lethality reducing GR control point: to suppress TNF production in macrophages
As reported earlier, mice lacking the GR‐coding gene (Nr3c1) in macrophages (Nr3c1 fl/fl LysM‐Cre Tg/+ mice, hereafter called GRLysMKO) are highly susceptible to LPS‐induced inflammation and SIRS 24, 25 (Fig 4A). However, these mice could be protected against LPS by a DEX pre‐treatment (Fig 4B); GRLysMKO mice were treated with PBS or 10 mg/kg DEX 30 min before a lethal dose of 2.5 mg/kg LPS, and lethality was monitored. DEX‐treated mice were found to be more resistant to lethal endotoxemia, compared to PBS pre‐treated mice (Fig 4B). As expected, GRLysMKO mice exhibited higher systemic TNF levels 2 h after LPS challenge, compared to Nr3c1 fl/fl (GRfl/fl) mice (Fig 4C). Though DEX is able to protect GRLysMKO mice against LPS‐induced lethality, it is not able to suppress TNF production in these mice (Fig 4D), which suggests that macrophage GR is important in the protection against LPS, but that DEX protection against LPS in GRLysMKO mice involves GR in another tissue and at another level than TNF production control. If we study the expression of Ripk3, Mlkl, and Zbp1 in the ileum by qPCR, induced by an equal dose of 2.5 mg/kg LPS in GRfl/fl and GRLysMKO mice, which is lethal in the latter ones, we observe a higher fold induction of expression of Ripk3, Mlkl, and Zbp1 in the GRLysMKO mice (Fig 4E). In addition, we measured expression of PC marker Defa6 and observed that GRLysMKO, but not GRfl/fl mice show a significant drop in Defa6 gene expression (Fig 4F). Although DEX is unable to repress high systemic TNF concentrations, DEX is able to fully repress the induction of these genes by an adapted dose of LPS (12.5 and 2.5 mg/kg in GRfl/fl and GRLysMKO mice, respectively) in both GRfl/fl and in GRLysmKO mice (Fig 4G). Therefore, we studied the role of IEC GR in the LPS model.
Figure 4. First GR control point: to suppress TNF production in macrophages.
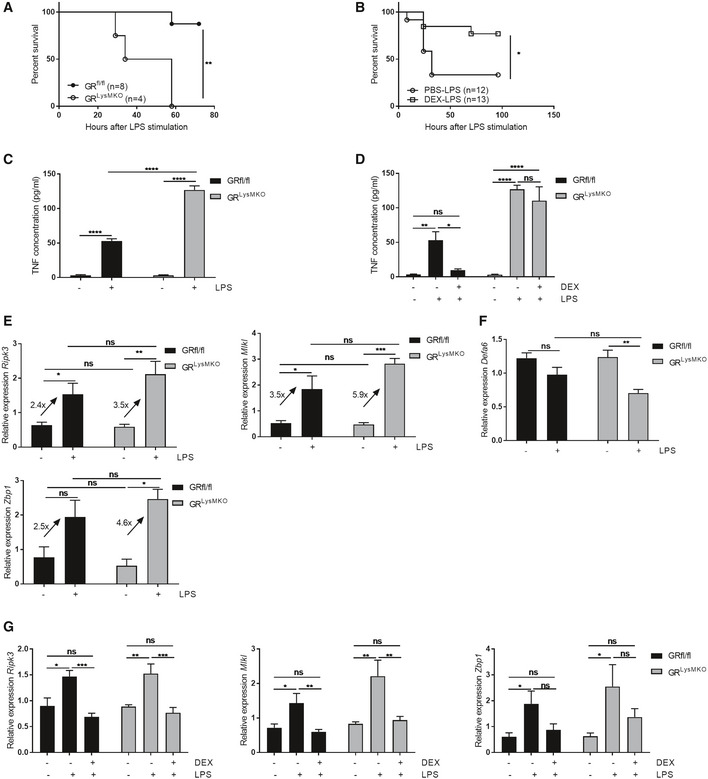
-
AGRfl/fl (n = 8), and GRLysMKO mice (n = 4) were treated with 2.5 mg/kg LPS and lethality was monitored. P‐values were analyzed with Fisher's exact test.
-
BGRLysMKO mice were treated with PBS (n = 12) or 10 mg/kg DEX (n = 13) before a lethal dose of LPS and lethality was monitored. P‐values were analyzed with a Fisher's exact test.
-
CTNF measurement in the serum of GRfl/fl (n = 4) and GRLysMKO mice (n = 3). Mice were treated with 2.5 mg/kg LPS and blood was drawn 2 h later. TNF concentration was measured with ELISA.
-
DTNF measurement in the serum of GRfl/fl and GRLysMKO (n = 4). Mice were treated with PBS or a lethal dose of LPS (12.5 and 2.5 mg/kg for GRfl/fl and GRLysMKO mice, respectively), with or without pre‐treatment with 10 mg/kg DEX and blood was drawn 2 h later. TNF concentration was measured with ELISA.
-
E, FGRfl/fl (n = 4) and GRLysMKO mice (n = 3–4) were treated with 2.5 mg/kg LPS. Ileum was isolated after 6 h and gene expression was measured via qPCR. Fold changes are depicted on the graphs.
-
GGRfl/fl and GRLysMKO mice (n = 4) were treated with a PBS or LPS (12.5 and 2.5 mg/kg for GRfl/fl and GRLysMKO mice, respectively) with or without 10 mg/kg DEX pre‐treatment of 30 min. Ileum was isolated after 6 h and gene expression was measured via qPCR. Fold changes are depicted on the graphs.
Second essential GR control point: to suppress TNF‐induced ISG expression in intestinal epithelium
We recently provided evidence for an important protective role of GR in the protection against TNF‐induced intestinal permeability, cell death, and eventually lethality, based on studies with Nr3c1 fl/fl VillinCre Tg/+ (GRVillinKO) mice and GRdim/dim mice, which show a higher sensitivity for TNF and cannot be protected by exogenous GCs 11. In analogy, we studied the role of GR in the IECs during LPS‐induced endotoxemia. GRVillinKO and control GRfl/fl mice were injected with 5 mg/kg LPS, and lethality was monitored. GRVillinKO mice were significantly more sensitive to LPS than the control mice (Fig 5A). In contrast to TNF‐induced SIRS, a pre‐treatment with 10 mg/kg DEX can protect GRVillinKO mice against a lethal dose of LPS (Fig 5B). Two hours after LPS injection, we measured systemic TNF concentrations and found no significant difference in LPS‐induced TNF concentration, which is in line with the idea that TNF is mainly produced by macrophages (Fig 5C). When TNF concentration was measured in mice that had been pre‐treated with 10 mg/kg DEX 30 min before a lethal dose of LPS, we found that GRVillinKO mice exhibited an equal DEX‐induced repression of systemic TNF concentration compared to WT littermates (Fig 5D). These data confer an explanation for the lack of protection by DEX against TNF‐induced rather than TNF‐mediated effects in GRVillinKO mice. Our data also confirm the role of TNF in the LPS‐induced lethality. In addition, the high sensitivity of GRVillinKO mice for LPS was also partly reverted by a neutralizing monoclonal antibody against TNFR1 (Fig 5E). We recently found that the IECs of unstimulated GRdim/dim mice 11 over‐express hundreds of ISGs. Unstimulated IECs of GRVillinKO mice (i.e., without LPS treatment) exhibit a similar phenotype, as is displayed in Fig 5F, which shows three genes of immediate interest to this study. Challenging GRfl/fl and GRVillinKO mice with an equal dose of 5 mg/kg LPS leads to higher fold induction in expression of Ripk3 in the mutant mice (Fig 5G). We hypothesized that a minimal higher expression of Ripk3 is enough to induce necroptosis, because of the higher expression of Mlkl and Zbp1 under basal condition. Our data suggest that IEC GR controls LPS‐induced lethal SIRS by dampening the use of the expression of ISG genes by TNF to induce necroptosis in these cells. Figure 5H displays Defa6 gene expression, which is significantly reduced after LPS treatment in GRVillinKO mice, but not GRfl/fl mice.
Figure 5. Second GR control point: to suppress TNF‐induced ISG expression in intestinal epithelium.
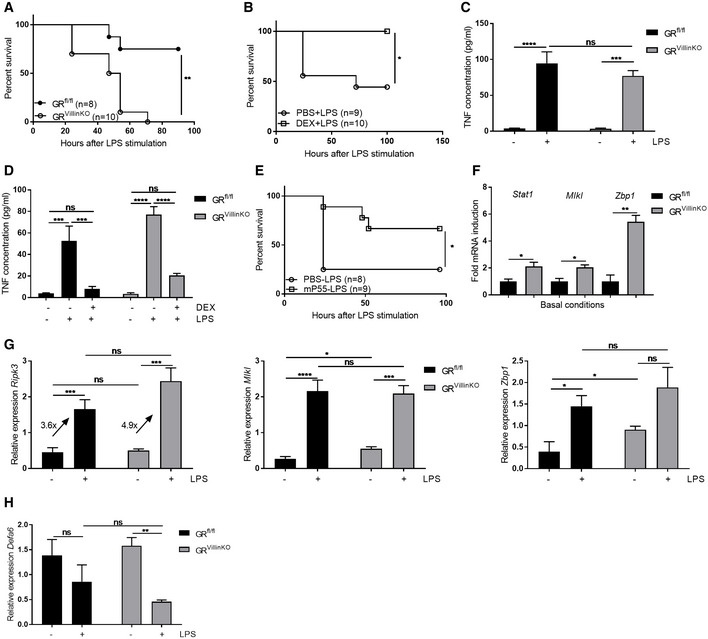
-
AGRfl/fl (n = 8) and GRVillinKO mice (n = 10) were treated with 5 mg/kg LPS and lethality was monitored. P‐values were analyzed with Fisher's exact test.
-
BGRVillinKO mice were treated with PBS (n = 9) or 10 mg/kg DEX (n = 10) before 5 mg/kg LPS and lethality was monitored. P‐values were analyzed with Fisher's exact test.
-
CTNF measurement in the serum of GRfl/fl (n = 4) and GRVillinKO mice (n = 4). Mice were treated with 5 mg/kg LPS and blood was drawn 2 h later.
-
DTNF measurement in the serum of GRfl/fl (n = 4) and GRVillinKO mice (n = 4). Mice were treated with PBS or LPS (12.5 and 5 mg/kg for GRfl/fl and GRVillinKO mice, respectively) with or without pre‐treatment with 10 mg/kg DEX for 30 min. Blood was taken 2 h after LPS challenge, and TNF concentrations were measured with ELISA.
-
EGRVillinKO mice were treated with PBS (n = 8) or a neutralizing P55 (TNFR1) antibody (n = 9) 2 h before and 8 h after 5 mg/kg LPS and lethality was monitored. P‐values were analyzed with Fisher's exact test.
-
FIleum was isolated from non‐stimulated GRfl/fl (n = 4) and GRVillinKO mice (n = 4), and gene expression was measured via qPCR. Expression levels are shown as fold induction with GRfl/fl expression values set at 1. P‐values were calculated using Student's t‐test.
-
G, HGRfl/fl (n = 4) and GRVillinKO mice (n = 4) were treated with 5 mg/kg LPS. Ileum was isolated and gene expression was measured via qPCR.
GR dimers play an essential role in protection against LPS‐induced lethality and repression of ISGs in IECs
GRdim/dim mice display a significant increase in sensitivity toward TNF‐induced lethality, and DEX is unable to protect these mice against TNF‐induced lethal SIRS 11. Furthermore, it has been described that these mice display a higher sensitivity for LPS‐induced SIRS 25, 26. We studied whether these mice could be of further support for our double control mechanism of GR in endotoxemia. We treated GRwt/wt and GRdim/dim mice with increasing doses of LPS, survival was monitored, and an LD50 was determined (Fig 6A). GRdim/dim mice displayed a higher sensitivity to LPS compared to GRwt/wt mice with LD50 values of, respectively, 325 and 75 μg per mouse. Although DEX was unable to protect GRdim/dim mice against lethal doses of TNF, we observed that DEX conferred protection of GRdim/dim mice against LPS‐induced lethality (Fig 6B). As was already shown in the past, TNF was induced to the same extent in both genotypes 25 when challenged with LPS (Fig 6C). Here, we found that DEX remained able to repress LPS‐induced systemic TNF concentration (Fig 6D), suggesting that GR‐induced repression of TNF is a GR monomer‐specific effects which explains why we could protect the GRdim/dim mice against LPS with DEX. The critical role of TNF in endotoxemia in GRdim/dim mice was proven with a neutralizing antibody against TNFR1, since we could significantly prolong survival (Fig 6E).
Figure 6. GR dimers play an essential role in protection against LPS‐induced lethality and suppression of ISG expression in IECs.
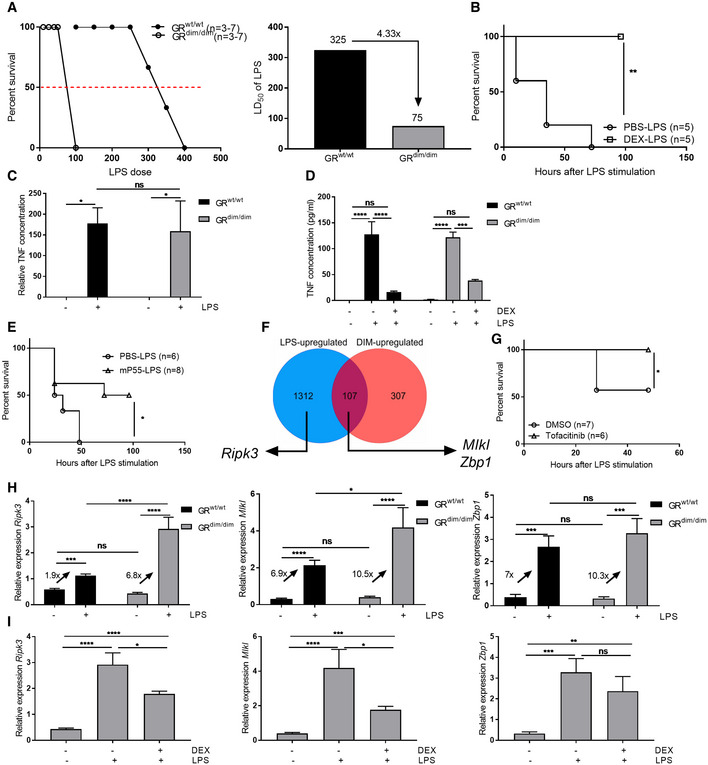
- Left: LPS dose–response curves of GRwt/wt (n = 3–7 per LPS dose) and GRdim/dim mice (n = 3–7 per LPS dose) treated with 1.5 mg/kg LPS. Right: LD50 values of LPS are depicted on top of each bar for each group.
- GRdim/dim mice were treated with PBS (n = 5) or 10 mg/kg DEX (n = 5) before 1.5 mg/kg LPS and survival was monitored. P‐values were analyzed with Fisher's exact test.
- TNF measurement in the serum of GRwt/wt (n = 5) and GRdim/dim mice (n = 5). Mice were treated with 1.5 mg/kg LPS, and blood was drawn 2 h later. TNF concentration was measured with ELISA.
- TNF measurement in the serum of GRwt/wt and GRdim/dim mice (n = 4) treated with PBS or LPS (12.5 and 1.5 mg/kg in GRwt/wt and GRdim/dim mice, respectively) with or without pre‐treatment with 10 mg/kg DEX. Blood was taken 2 h after LPS challenge, and TNF concentrations were measured with ELISA.
- GRdim/dim mice were treated with PBS (n = 6) or a neutralizing P55 (TNFR1) antibody (n = 8) 2 h before and 8 h after 1.5 mg/kg LPS and lethality was monitored. P‐values were analyzed with Fisher's exact test.
- Venn diagram of LPS‐ and DIM‐upregulated genes (in basal condition) in ileum, as identified by RNA‐seq (n = 3).
- Tofacitinib (n = 6) or vehicle (n = 7) was given orally to GRdim/dim mice. After 1.5 mg/kg LPS injection survival was monitored and P‐values were analyzed with Fisher's exact test.
- GRwt/wt (n = 4) and GRdim/dim (n = 4) mice were treated with 1.5 mg/kg LPS. Ileum was isolated and gene expression was measured via qPCR. Fold changes are depicted on the graphs.
- GRdim/dim mice (n = 4) were treated with PBS or 1.5 mg/kg LPS with or without 10 mg/kg DEX pre‐treatment. Ileum was isolated and gene expression was measured via qPCR. Fold changes are depicted on the graphs.
Our data obtained with TNF‐challenged GRdim/dim mice strongly support a detrimental role for ISG‐mediated necroptosis in IECs as a driving force of lethality 11. To study whether this TNF‐mediated pathway is relevant in LPS‐induced lethal SIRS, we made an overlap of LPS‐upregulated genes in IECs (6 h after LPS) and genes that are upregulated in the IECs of GRdim/dim mice in basal conditions to identify genes that are potentially involved in hypersensitivity of GRdim/dim mice (hence genes controlled by GR dimers) and genes that are induced by LPS, and we focused on the necroptosis‐inducing genes mentioned before. Genes Mlkl and Zbp1 belong to the overlapping genes, while Ripk3 is upregulated after LPS, but is not upregulated under basal conditions in GRdim/dim mice (Fig 6F). This suggests that Mlkl and Zbp1 are likely more responsible for sensitizing IECs for necroptosis, but that a second trigger, viz. Ripk3 upregulation, is provided by LPS (mediated by TNF) for actual induction of necroptosis. To test the role of the intestinal ISG‐response, we administered the JAK/STAT inhibitor tofacitinib to GRdim/dim mice. Mice received tofacitinib (100 mg/kg) or vehicle, orally twice a day for 2 days before LPS challenge, and twice (1 h before and 8 h after) on the day of LPS injection. Tofacitinib significantly protected GRdim/dim mice against a lethal dose of LPS (Fig 6G). We measured LPS‐induced expression of ISGs Ripk3, Mlkl, and Zbp1 after LPS stimulation and measured higher induction in GRdim/dim as compared to GRwt/wt mice (Fig 6H). As mentioned, GRdim/dim mice could be protected against lethal doses of LPS by a pre‐treatment with DEX, since DEX is able to suppress TNF levels in the macrophages via GR monomers. This effect is reflected on the level of IEC gene expression of Ripk3, Mlkl, and Zbp1, which is indeed lower when GRdim/dim mice are pre‐treated with DEX (Fig 6I).
Discussion
LPS strongly induces inflammation in Gram‐negative infections in human patients and is considered as an essential mediator in numerous health challenges. To understand the mechanism of LPS‐induced pathologies, and the inborn feedback mechanisms, LPS injection in experimental animals is often applied, as it leads to a potentially lethal SIRS, mainly TLR4‐mediated 27. Despite the decades of studies on the mechanism of LPS‐induced lethal systemic inflammatory response syndrome (SIRS), many aspects are still poorly known, for example how the major feedback system involving glucocorticoids (GCs) control the endotoxemia pathway. In this study, we demonstrate that GCs limit the in vivo LPS effects by stimulating GR at two critical control points, namely at the level of macrophages, i.e., controlling TNF production in a GR‐monomer way, and at the level of inhibition of TNFR1‐induced ISG gene expression and cell death mediators at the level of intestinal epithelial cells (IECs) in a GR dimer‐dependent way.
RNA sequencing of IECs of mice injected with LPS revealed an almost exclusive increase of expression of interferon‐stimulated genes (ISGs) in the IECs, accompanied by the induction of necroptosis‐related proteins: RIPK3 and (phopho‐)MLKL. It is classically accepted that LPS administration induces production of TNF in (intestinal) immune cells, which in turn acts on the IECs, rather than working directly via TLR4 signaling in the epithelium 6, 23, 28. Using TLR4VillinKO mice, we confirm this idea in our model system, and using TNFR1VillinKO mice, we suggest that the induction of ISG genes by LPS critically depends on TNF signaling via TNFR1 expression on IECs.
There are different explanations on how TNF can induce the expression of these ISGs. One possible explanation is that LPS and/or TNF induce IFN signaling, which in turn induce ISGs and directly induce necroptosis. It has also been shown that TNF can directly activate JAK‐STAT signaling and STAT target genes in primary macrophages 29. TNF induces the expression of interferon‐response genes typically activated by interferon‐stimulated gene factor 3 (ISGF3, consisting of STAT1, STAT2, and IRF9), or by STAT1 alone. These genes include genes coding for proteins important for antiviral response, but also proteins that prime cells for augmenting subsequent response to microbial products or inflammatory cytokines. In addition, it has been demonstrated that type I interferons can induce necroptosis in macrophages during infections with Salmonella typhimurium 30. Other in vitro studies found that both type I and type III IFN transcriptionally activate the RNA‐responsive protein kinase PKR, which is able to interact with RIPK1 and RIPK3, resulting in formation of the “PKR necrosome”. This induces the phosphorylation of RIPK3 to finally execute necroptosis 31.
IFN signaling may also prime the cells to respond to TNF, i.e., it licenses TNF‐mediated necroptosis by providing increased expression of important mediators, such as MLKL and ZBP1. In the small intestine, a low constitutive expression of ISGs is observed. Sarhan et al 32 showed that during homeostasis, DNA and mitochondrial stress activate cDAS/STING signaling leading to this constitutive IFN signaling. This sustains the expression of many ISGs, such as Mlkl which must be expressed sufficiently to facilitate oligomerization and cell death. Necroptotic cell death is normally held in check by caspase‐8. However, caspase‐8 and FLIP do not suppress necroptosis under certain pathophysiological conditions 12. The reason for this is unclear, but a possible explanation is that under specific conditions, the expression levels of RIPK3 and MLKL are modulated, altering the tissue sensitivity to necroptosis. The abundance of these constitutive IFN‐regulated effectors of necroptosis (CIRENs) determines the rapidity of necroptosis 33. MLKL was identified to be a CIREN 32, 34, 35, but further studies are needed to identify more proteins. Our data suggest that ZBP1 can also be characterized as a CIREN. ZBP1 (also known as DAI of DLM‐1) is a Z‐DNA‐binding protein that can be induced by IFNs 36 and (like RIPK1 and RIPK3) contains a RIP homotypic interaction motif (RHIM) domain, by which it can associate with RIPK1 and RIPK3 to participate in necroptosis. ZBP1 has indeed been shown to play a role in RIPK1 RHIM mutation‐induced necroptosis by interacting with RIPK3 37, 38. In addition, ZBP1−/− mice are protected against SIRS induced by TNF and IFN‐γ 39.
Paneth cells have been shown to be very sensitive to necroptosis in a caspase‐8‐deficient background. IEC‐specific deletion of caspase‐8 leads to inflammatory necroptosis and Paneth cell loss 13. Similarly, deletion of FADD in the IECs also leads to spontaneous IEC necroptosis with loss of Paneth cells 14. The loss of Paneth cells is also observed in patients with Crohn disease, associated with high levels of RIPK3 13. Injection of TNF in mice also leads to higher expression of RIPK3, specifically in the crypts 11. In the current manuscript, we also show that LPS induces a marked reduction of cells with typical PC secretory granules and lysozyme expression at the crypt base of the ileum and qPCR showed diminished expression of PC‐specific genes Defa6 and Lyz1. Our data thus suggest that LPS leads to PC necroptosis; however, further investigation is needed to test if upregulation of cell death mediators actually leads to the occurrence of necroptosis, specifically in the PCs.
Synthetic GCs are very widely applied in medical practice, especially to combat inflammation. It is surprising though how little is known about GCs mechanisms and relevant cell types. Protective effects of GCs against LPS is traditionally associated with a block of TNF production in macrophages 24. Our work suggests that there is a second, equally important check‐point of GCs, clearly visible in the LPS lethal SIRS model. When mice were pre‐treated with DEX, the LPS‐induced expression of ISGs, upregulation of RIPK3 and (phopho‐)MLKL protein expression, and reduction in Paneth cells is prevented. Recently, we found that DEX, via GR dimers, limits the expression of ISGs, possibly through inhibition of Stat1 gene expression, in IECs 11. Hence, during LPS‐induced SIRS, at least two tissue compartments seem to be important, namely the macrophages, which produce cytokines such as TNF, and the IECs of the small intestine, where TNF induces tissue damage. GCs (both endogenous and exogenous) can confer protective inhibitions at both sites. In macrophages, GCs will suppress the TNF production in a GR monomer‐dependent way. In the IECs, GCs suppress the TNF‐induced expression of ISGs and necroptosis mediators in a GR dimer‐dependent way. A neutralizing antibody against mouse TNFR1 could only partially protect GRVillinKO and GRdim/dim mice against lethal doses of GR, suggesting that also other cytokines may be important. For example, also suppression if IL‐1β by GR dimers in macrophages was shown to be important for survival in LPS‐induced shock. GRdim/dim mice could be partially protected against LPS with a recombinant IL‐1R antagonist 25. In addition, there is still higher LPS‐induced expression of ISGs in TNFR1−/− and TNFR1VillinKO mice, compared to basal conditions, possibly because upon LPS stimulation, macrophages will also produce interferons which induce ISG expression in the IECs.
DEX suppresses the expression of ISGs and subsequently protects against LPS‐induced cell death. In addition, also the JAK/STAT inhibitor tofacitinib can protect GRdim/dim mice against LPS‐induced shock. Recently, it was shown that tofacitinib can also attenuate expression of cell death mediators and the accompanying loss of Paneth cells after injection of IFN‐λ 40. These data illustrate the potential of inhibiting the IFN‐induced signaling to protect against LPS‐induced intestinal cell death.
In conclusion, we provide evidence that LPS‐induced lethal SIRS is controlled by GCs at two critical check‐points. LPS will activate macrophages that subsequently produce inflammatory cytokines such as TNF, IL‐1β, and interferons. TNF, working via TNFR1, will profit from the constitutive interferon response in cells of the small intestine, including Paneth cells, to induce these ISGs further and via the genes coding for RIPK3, MLKL, and ZBP1 potentially cause necroptotic cell death (Fig 7). The actual occurrence of necroptosis in the IECs still needs further investigation.
Figure 7. Graphical abstract.
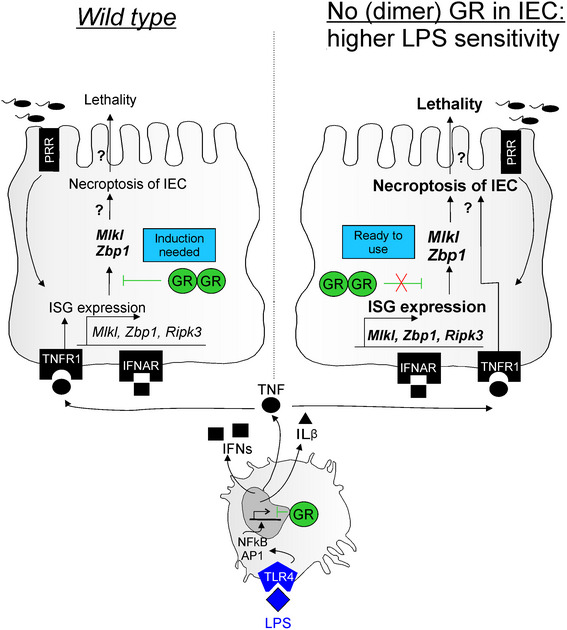
In the intestinal epithelial cells (IECs) of the small intestine, a constitutive expression of interferon‐stimulated genes (ISGs) is suppressed by the presence of dimer GR. During LPS‐induced endotoxemia, LPS has indirect effects on the IECs: TLR4 binding on macrophages results in production of diverse cytokines, such as IFNs, IL‐1β, and TNF. TNF performs its effects on IECs via binding to TNFR1. TNF induces the expression of ISGs, such as necroptosis mediators: Mlkl, Zbp1 and Ripk3. High expression of these mediators can potentially lead to cell death of the IEC and eventually lethality of the animal. When no (dimer) GR is present in the IECs, there is no dampening of the constitutive ISGs signature. We hypothesize that necroptosis proteins such as MLKL and ZBP1 are in this case “ready to use” and that TNFR1 signaling by TNF can directly lead to necroptosis, death of the cell and eventually lethality. This hypothesis still needs further investigation.
We suggest different therapeutic possibilities to influence these LPS‐induced effects. First, GCs protect against LPS via two important control points, i.e., suppression of TNF in the macrophages and suppression of TNF‐induced effects in the IECs of the ileum. We show that the first control point depends on GR monomers and the second control point depends critically on GR dimers. As a result, classical glucocorticoids, such as dexamethasone, with GR monomer and GR dimer effects would be preferred as a therapeutic tool above GR monomer‐ or dimer‐specific GCs. However, with regard to side‐effects, it would also be of therapeutic benefit to target selective dimerizing GR agonists and modulators (SEDIGRAMs) 41, 42 to the small intestine, since even in the absence of the first control point, the second control point can cause significant protection. In addition, neutralization of TNFR1 and suppression of the IFN signaling by using JAK/STAT inhibitor, at the level of IECs can be considered to yield therapeutic benefit.
Materials and Methods
Mice
C57BL/6J WT mice were purchased from Janvier. GRdim/dim mice were generated by Reichardt et al 43 and kept on an FVB/N background. Heterozygous GRdim/wt mice were intercrossed to generate GRwt/wt and GRdim/dim homozygous mutant mice. Nr3c1 fl/fl mice 44 were crossed with Villin‐Cre and LysM‐Cre transgenic mice, and the offspring intercrossed to generate Nr3c1 fl/fl VillinCre Tg/+ (GRVillinKO), Nr3c1 fl/fl LysMCre Tg/+ (GRLysMKO), and Nr3c1 fl/fl (GRfl/fl) mice, all in a C57BL6/J background. Tnfrsf1a fl/fl mice and Tlr4 fl/fl mice were crossed with Villin‐Cre transgenic mice, and offspring intercrossed to generate Tnfrsf1a fl/fl VillinCre Tg/+ (TNFR1VillinKO) 4, Tnfrsf1a fl/fl (TNFR1fl/fl), Tlr4 fl/fl VillinCre Tg/+ (TLR4VillinKO), and Tlr4 fl/fl mice (TLR4fl/fl), all in a C57Bl6/J background. All offspring were genotyped by PCR on genomic DNA isolated from toe biopsies. Mice were kept in individually ventilated cages under a 12‐h dark/light cycle in a conventional and specific pathogen‐free (SPF) animal facility and received food and water ad libitum. All mice were used at 8–12 weeks of age and gender‐matched in each experiment. Group sizes were determined based on previous experiments. All animal experiments were approved by the ethical committee for animal welfare of the Faculty of Sciences, Ghent University.
Reagents
LPS from Salmonella abortus equi was purchased from Sigma (L‐5886). Recombinant mouse TNF was produced in Escherichia coli and purified to homogeneity in our laboratories. TNF had a specific activity of 2.15 × 108 IU/mg and no detectable endotoxin contamination. The neutralizing hamster anti‐mp55TNFR IgG1 antibody (55R170) was bought from R&D Systems. It was pure and contained no detectable LPS contamination. FD4 fluorescein isothiocyanate (FITC)–dextran (FD4‐1G, Mw 3,000–5,000 Da) was purchased from Sigma‐Aldrich NV. For in vivo DEX injection, we used rapidexon (Medini N.V.). Recombinant TNF, DEX, and FITC‐dextran were diluted in pyrogen‐free PBS. Tofacitinib (CP‐690550) Citrate (S5001; Shelleckchem Inc.) was diluted in 2% Tween 80 with 0.5% methylcellulose diluted in PBS.
Injections and sampling
All injections were given intraperitoneally. Injection volumes were always adapted to the bodyweight of the mice. In lethality experiments, mice were monitored by measuring rectal body temperature. Mice with a body temperature below 28°C were euthanized using cervical dislocation. FITC‐dextran (25 mg/ml) and tofacitinib were administered via oral gavage. Blood was taken via cardiac puncture after sedation of the mice with a ketamine/xylazine solution (Sigma‐Aldrich NV) or via retro‐orbital bleeding after sedation with isoflurane. To obtain mouse serum, samples were allowed to clot overnight at 4°C. The next day the clot was removed, and samples were centrifuged at 22,172 g for 3 min. Serum samples were stored at −20°C. For sampling of ileum and IECs, the mice were killed by cervical dislocation at indicated time points. IEC samples for RNA isolation were prepared with following protocol: ileum was extensively flushed with PBS and incubated for 5 min on ice in lysis buffer (732 6802; Bio‐Rad) supplemented with 2‐mercaptoethanol. Next, samples were snap‐frozen in liquid nitrogen.
Immunoblot and antibodies
Ileum was excised from euthanized mice, flushed with PBS and snap‐frozen in liquid nitrogen. Samples were lysed using RIPA buffer (10% SDS, 5 M NaCl, 1.5 M Tris pH 8, 10% NP40, 0.5% NaDOC, 0.5 M EDTA), supplemented with protease and phosphatase inhibitor cocktails from Roche). Protein samples containing 50 μg protein were separated by electrophoresis in a 10% gradient SDS polyacrylamide gel and transferred to nitrocellulose membranes (pore size 0.45 μm). After blocking the membranes with 1:2 dilution of Starting Block/PBST 0.1% (Thermo Fisher Scientific), membranes were incubated overnight at 4°C with primary antibody. Blots were washed with PBST 0.1% and then incubated for 1 h at room temperature with secondary antibody. Immunoreactive bands were detected using ECL and quantified using ImageJ (FIJI). Antibodies for immunoblot analysis: anti‐MLKL phospho S345 (ab196436; abcam), anti‐MLKL (MABC604; Sigma), anti‐RIP3 (ADI‐905‐242‐100, ENZO life sciences), anti‐β‐actin (MA5‐15739, Pierce), anti‐rabbit antibody (926‐32211; Li‐cor), and anti‐mouse antibody (926‐32220; Li‐cor).
Histology and immunohistochemistry
Excised ileum was fixed in 4% paraformaldehyde overnight at 4°C, dehydrated and embedded in paraffin. Tissue sections of 5 μm were cut and stained with H&E using standard protocols. Pictures were taken with an Olympus Bx51 light microscope. For lysozyme staining, tissue sections were dewaxed, incubated in antigen retrieval solution (VEC.H‐3300, Vector) at boiling temperature for 20 min in a Pick cell cooking unit, and cooled down for 2.5 h. Blocking buffer (5% donkey serum in PBT, i.e., PBS, 0.1% bovine serum albumin (BSA) and 1% Triton X‐100) was added to the slides for 1 h at room temperature. Primary antibody against lysozyme (sc‐27958, Santa Cruz) was diluted 1:50 in 5% donkey serum in PBT and incubated overnight at 4°C. Slides were then incubated with secondary antibody (donkey anti‐goat Dyl594, abcam, 1:1,000 in PBS). Counterstaining was done with Hoechst reagent (Sigma‐Aldrich NV, 1:1,000 in PBS). Fluorescence microscopy was performed using a Olympus CellM. For quantification, fluorescence microscopy was performed using Zeiss slide scanner. For every picture, a selection was made of 10 crypts and positive signal was quantified using an automated script in QuPath.
RNA sequencing
Total RNA was isolated with Aurum total RNA mini kit (732‐6820; Bio‐Rad) according to the manufacturer's instructions. RNA concentration was measured and RNA quality was checked with the Agilent RNA 6000 Pico Kit (Agilent Technologies) and sequenced on a Illumina Genome Analyzer IIx. Data were mapped to the mouse (mm10) reference genome transcriptome with hisat2 45, 46. Only uniquely mapped reads were retained. Gene level read counts were obtained with featureCounts (subread package) 47. Differential gene expression was assessed with the DESeq2 package, and the false discovery rate (FDR) was set at the 1% level. Motif finding for multiple motifs or de novo motif finding was performed using the HOMER software. We used the promoter region (start offset: −500, end offset: default) to search for known motifs and de novo motifs of length 8, 10, 12, 14, and 16. Searching for specific motifs in a set of sequences was done with the meme suite. Visualizations were made using the R software. The original RNA‐seq data were deposited in the NCBI's Gene Expression Omnibus database (TNF: GEO GSE113691, LPS: GEO GSE139903).
qPCR
RNA was isolated with Aurum total RNA mini kit (732‐6820; Bio‐Rad) according to the manufacturer's instructions. RNA concentration was measured with the Nanodrop 1000 (Thermo Fisher Scientific), and 1 μg RNA was used to prepare cDNA with Sensifast cDNA Synthesis Kit (BIO‐650504, Bioline). qPCR was performed using the Roche LightCycler480 system (Applied Biosystems). The best‐performing housekeeping genes were determined by Genorm 48. Results are given as relative expression (scaled to average) values normalized to the geometric mean of the housekeeping genes. Primers used for qPCR are depicted in Table 1.
Table 1.
Primer sequences used for qPCR
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| Actin | GCTTCTAGGCGGACTGTTACTGA | GCCATGCCAATGTTGTCTCTTAT |
| Rpl | CCTGCTGCTCTCAAGGTT | TGGTTGTCACTGCCTGGTACTT |
| Ubc | AGGTCAAACAGGAAGACAGACGTA | TCACACCCAAGAACAAGCACA |
| Gapdh | TGAAGCAGGCATCTGAGGG | CGAAGGTGGAAGAGTGGGAG |
| Ifit1 | CTGAGATGTCACTTCACATGGAA | GTGCATCCCCAATGGGTTCT |
| Irf8 | AGACCATGTTCCGTATCCCCT | CACAGCGTAACCTCGTCTTCC |
| Irf1 | ATGGAAACCCCGAAACCGC | GATATTTCCAGTGGCCTGGA |
| Stat1 | TCACAGTGGTTCGAGCTTCAG | GCAAACGAGACATCATAGGCA |
| Ripk3 | GTGCTACCTACACAGCTTGGA | CCCTCCCTGAAACGTGGAC |
| Mlkl | AATTGTACTCTGGGAAATTGCCA | TCTCCAAGATTCCGTCCACAG |
| Zbp1 | AAGAGTCCCCTGCGATTATTTG | TCTGGATGGCGTTTGAATTGG |
| Defa6 | CCAGGCTGATCCTATCCAAA | GTCCCATTCATGCGTTCTCT |
| Lyz1 | ATGGAATGGCTGGCTACTATGG | ACCAGTATCGGCTATTGATCTGA |
TNF measurement
TNF was quantified using an ELISA kit according to the manufacturer's protocol (88‐7324‐88, e‐bioscience).
Intestinal permeability
An in vivo permeability assay was performed using FITC‐dextran as described previously 4. Three hours after LPS challenge, 200 μl FITC‐dextran (25 mg/ml in PBS) was administered by oral gavage. Five hours later, blood was collected in an EDTA‐coated tube and centrifuged at 1,018 g for 20 min at 4°C. Plasma was collected and fluorescence was measured (λexc/λem = 488/520 nm).
Statistics
Statistics on survival curves was performed using Fisher's exact test in order to determine differences between final outcomes. Statistical significance between groups was calculated using Student's t‐test, one‐way ANOVA or two‐way ANOVA with 95% confidence intervals. All data are represented as mean ± SEM and analyzed using GraphPad Prism software. P < 0.05 was considered as significant.
Study approval
All animal experiments were approved by the ethical committee of the Faculty of sciences, Ghent University.
Data availability
The original RNA‐seq data were deposited in the NCBI's Gene Expression Omnibus database (TNF: GEO GSE113691; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113691, LPS: GEO GSE139903; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139903).
Author contributions
KVL, ST, MB, JS, ME, JV, TV, CW, KDB, and LVW performed experiments and data analysis. KVL assembled figure panels and wrote the manuscript. CL managed the research, received funding, and edited the text and figures during assembly of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Dataset EV1
Dataset EV2
Review Process File
Acknowledgements
The authors wish to thank Prof. Jan Tuckermann for providing GRdim/dim mice and GRfl/fl mice, Prof. George Kollias for providing TNFR1fl/fl mice and Prof. Charlotte Scott for the TLR4fl/fl mice. We thank Joke Vanden Berghe and animal house caretakers for animal care. We acknowledge the VIB Nucleomics Core for RNA sequencing analysis. We are grateful to Kelly Lemeire for performing IHC on tissue sections. We would like to thank the VIB BioImaging Core for training, support and access to the instrument park. Research in these laboratories was funded by the Research Foundation Flanders (FWO Vlaanderen), University Ghent and VIB.
EMBO Reports (2020) 21: e49762
References
- 1. Beutler B, Du X, Poltorak A (2001) Identification of Toll‐like receptor 4 (Tlr4) as the sole conduit for LPS signal transduction: genetic and evolutionary studies. J Endotoxin Res 7: 277–280 [PubMed] [Google Scholar]
- 2. Lewis AJ, Seymour CW, Rosengart MR (2016) Current murine models of sepsis. Surg Infect 17: 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beutler B, Rietschel ET (2003) Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol 3: 169–176 [DOI] [PubMed] [Google Scholar]
- 4. Van Hauwermeiren F, Armaka M, Karagianni N, Kranidioti K, Vandenbroucke RE, Loges S, Van Roy M, Staelens J, Puimege L, Palagani A et al (2013) Safe TNF‐based antitumor therapy following p55TNFR reduction in intestinal epithelium. J Clin Invest 123: 2590–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM (1998) TNF receptor‐deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol 160: 943–952 [PubMed] [Google Scholar]
- 6. Williams JM, Duckworth CA, Watson AJM, Frey MR, Miguel JC, Burkitt MD, Sutton R, Hughes KR, Hall LJ, Caamaño JH et al (2013) A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Dis Model Mech 6: 1388–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puimège L, Hauwermeiren FV, Steeland S, Ryckeghem SV, Vandewalle J, Lodens S, Dejager L, Vandevyver S, Staelens J, Timmermans S et al (2015) Glucocorticoid‐induced microRNA‐511 protects against TNF by down‐regulating TNFR1. EMBO Mol Med 7: 1004–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Looveren K, Libert C (2018) Should we target TNF receptors in the intestinal epithelium with glucocorticoids during systemic inflammation? Expert Opin Ther Targets 22: 1029–1037 [DOI] [PubMed] [Google Scholar]
- 9. Van Hauwermeiren F, Vandenbroucke RE, Grine L, Lodens S, Van Wonterghem E, De Rycke R, De Geest N, Hassan B, Libert C (2015) TNFR1‐induced lethal inflammation is mediated by goblet and Paneth cell dysfunction. Mucosal Immunol 8: 828–840 [DOI] [PubMed] [Google Scholar]
- 10. Negroni A, Cucchiara S, Stronati L (2015) Apoptosis, necrosis, and necroptosis in the gut and intestinal homeostasis. Mediators Inflamm 2015: 250762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ballegeer M, Looveren KV, Timmermans S, Eggermont M, Vandevyver S, Thery F, Dendoncker K, Souffriau J, Vandewalle J, Wyngene LV et al (2018) Glucocorticoid receptor dimers control intestinal STAT1 and TNF‐induced inflammation in mice. J Clin Invest 128: 3265–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weinlich R, Oberst A, Beere HM, Green DR (2017) Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18: 127–136 [DOI] [PubMed] [Google Scholar]
- 13. Günther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF et al (2011) Caspase‐8 regulates TNF‐α‐induced epithelial necroptosis and terminal ileitis. Nature 477: 335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Welz P‐S, Wullaert A, Vlantis K, Kondylis V, Fernández‐Majada V, Ermolaeva M, Kirsch P, Sterner‐Kock A, Gv Loo, Pasparakis M (2011) FADD prevents RIP3‐mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477: 330 [DOI] [PubMed] [Google Scholar]
- 15. Bogaert T, Vandevyver S, Dejager L, Hauwermeiren F, Pinheiro I, Petta I, Engblom D, Kleyman A, Schütz G, Tuckermann J et al (2011) Tumor necrosis factor inhibits glucocorticoid receptor function in mice: a strong signal toward lethal shock. J Biol Chem 286: 26555–26567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vandevyver S, Dejager L, Bogaert TV, Kleyman A, Liu Y, Tuckermann J, Libert C (2012) Glucocorticoid receptor dimerization induces MKP1 to protect against TNF‐induced inflammation. J Clin Invest 122: 2130–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P (2011) Widespread negative response elements mediate direct repression by agonist‐liganded glucocorticoid receptor. Cell 145: 224–241 [DOI] [PubMed] [Google Scholar]
- 18. Kleiman A, Tuckermann JP (2007) Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Mol Cell Endocrinol 275: 98–108 [DOI] [PubMed] [Google Scholar]
- 19. Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ (2013) Interferome v2.0: an updated database of annotated interferon‐regulated genes. Nucleic Acids Res 41: D1040–D1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jouan‐Lanhouet S, Riquet F, Duprez L, Berghe TV, Takahashi N, Vandenabeele P (2014) Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol 35: 2–13 [DOI] [PubMed] [Google Scholar]
- 21. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X et al (2012) Mixed lineage kinase domain‐like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227 [DOI] [PubMed] [Google Scholar]
- 22. Farin HF, Karthaus WR, Kujala P, Rakhshandehroo M, Schwank G, Vries RG, Kalkhoven E, Nieuwenhuis EE, Clevers H (2014) Paneth cell extrusion and release of antimicrobial products is directly controlled by immune cell‐derived IFN‐gamma. J Exp Med 211: 1393–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Günther C, Buchen B, He G‐W, Hornef M, Torow N, Neumann H, Wittkopf N, Martini E, Basic M, Bleich A et al (2014) Caspase‐8 controls the gut response to microbial challenges by Tnf‐α‐dependent and independent pathways. Gut 64: 601–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ (2007) Macrophage glucocorticoid receptors regulate Toll‐like receptor 4‐mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 109: 4313–4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kleiman A, Hübner S, Parkitna JM, Neumann A, Hofer S, Weigand MA, Bauer M, Schmid W, Schütz G, Libert C et al (2011) Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin‐1 in macrophages. FASEB J 26: 722–729 [DOI] [PubMed] [Google Scholar]
- 26. Silverman MN, Mukhopadhyay P, Belyavskaya E, Tonelli LH, Revenis BD, Doran JH, Ballard BE, Tam J, Pacher P, Sternberg EM (2013) Glucocorticoid receptor dimerization is required for proper recovery of LPS‐induced inflammation, sickness behavior and metabolism in mice. Mol Psychiatry 18: 1006–1017 [DOI] [PubMed] [Google Scholar]
- 27. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C et al (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282: 2085–2088 [DOI] [PubMed] [Google Scholar]
- 28. Ruder B, Atreya R, Becker C (2019) Tumour necrosis factor alpha in intestinal homeostasis and gut related diseases. Int J Mol Sci 20: E1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yarilina A, Park‐Min KH, Antoniv T, Hu X, Ivashkiv LB (2008) TNF activates an IRF1‐dependent autocrine loop leading to sustained expression of chemokines and STAT1‐dependent type I interferon‐response genes. Nat Immunol 9: 378–387 [DOI] [PubMed] [Google Scholar]
- 30. Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S (2012) Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol 13: 954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, Rall GF, Degterev A, Balachandran S (2013) Interferon‐induced RIP1/RIP3‐mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci USA 110: E3109–E3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, Ilyukha V, Sorokin M, Buzdin A, Fitzgerald KA et al (2019) Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ 26: 332–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sarhan M, Land WG, Tonnus W, Hugo CP, Linkermann A (2018) Origin and consequences of necroinflammation. Physiol Rev 98: 727–780 [DOI] [PubMed] [Google Scholar]
- 34. Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG et al (2015) Characterization of RIPK3‐mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ 23: 76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN et al (2014) RIPK1 blocks early postnatal lethality mediated by caspase‐8 and RIPK3. Cell 157: 1189–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fu Y, Comella N, Tognazzi K, Brown LF, Dvorak HF, Kocher O (1999) Cloning of DLM‐1, a novel gene that is up‐regulated in activated macrophages, using RNA differential display. Gene 240: 157–163 [DOI] [PubMed] [Google Scholar]
- 37. Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin‐McNulty B, Carano RAD, Cao TC, Bruggen NV, Bernstein L et al (2016) RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ 23: 1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin J, Kumari S, Kim C, Van T‐MM, Wachsmuth L, Polykratis A, Pasparakis M (2016) RIPK1 counteracts ZBP1‐mediated necroptosis to inhibit inflammation. Nature 540: 124–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang D, Liang Y, Zhao S, Ding Y, Zhuang Q, Shi Q, Ai T, Wu S‐Q, Han J (2019) ZBP1 mediates interferon‐induced necroptosis. Cell Mol Immunol 17: 356–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gunther C, Ruder B, Stolzer I, Dorner H, He GW, Chiriac MT, Aden K, Strigli A, Bittel M, Zeissig S et al (2019) Interferon lambda promotes paneth cell death via STAT1 signaling in mice and is increased in inflamed ileal tissues of patients with Crohn's disease. Gastroenterology 157: 1310–1322.e13 [DOI] [PubMed] [Google Scholar]
- 41. De Bosscher K, Beck IM, Ratman D, Berghe WV, Libert C (2016) Activation of the glucocorticoid receptor in acute inflammation: the SEDIGRAM concept. Trends Pharmacol Sci 37: 4–16 [DOI] [PubMed] [Google Scholar]
- 42. Vandewalle J, Luypaert A, Bosscher K, Libert C (2018) Therapeutic mechanisms of glucocorticoids. Trends Endocrinol Metab 29: 42–54 [DOI] [PubMed] [Google Scholar]
- 43. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P et al (1998) DNA binding of the glucocorticoid receptor is not essential for survival. Cell 93: 531–541 [DOI] [PubMed] [Google Scholar]
- 44. Tronche F, Kellendonk C, Reichardt HM, Schutz G (1998) Genetic dissection of glucocorticoid receptor function in mice. Curr Opin Genet Dev 8: 532–538 [DOI] [PubMed] [Google Scholar]
- 45. Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL (2016) Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11: 1650–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- 48. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: RESEARCH0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset EV1
Dataset EV2
Review Process File
Data Availability Statement
The original RNA‐seq data were deposited in the NCBI's Gene Expression Omnibus database (TNF: GEO GSE113691; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113691, LPS: GEO GSE139903; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139903).