

Abstract
Background
Calcific aortic valve disease (CAVD) is a slowly progressive pathologic process associated with significant morbidity and mortality, CAVD is the most common valve heart disease in the elderly and a leading cause of aortic valve stenosis. Multiple steps characterize the process: inflammation, cell apoptosis, lipid deposition, renin-angiotensin system activation, extracellular matrix remodeling, and bone formation. This paper focuses on detecting and analyzing the expression of serum inflammatory factors in CAVD by antibody microarray techniques.
Methods
In this study, a total of 258 patients were included at Tianjin Chest Hospital between January 2017 and December 2018, subjects were divided into three groups: control, coronary artery disease (CAD), and CAVD. Blood samples were collected, and adipokine/cytokine/chemokine serum profiles were measured by antibody arrays.
Results
These data suggest that B-Lymphocyte Chemoattractant (BLC), Interleukin (IL)-12p40, monokine inducible by γ interferon (MIG), and Macrophage inflammatory protein (MIP)-1delta were significantly increased in CAVD compared to control or CAD. Furthermore, Real-time quantified PCR, Western blot assay, and Flow cytometer detection showed that these four cytokines/chemokines were from peripheral blood mononuclear cells.
Conclusions
These findings suggest that BLC, IL-12p40, MIG, and MIP-1delta can be used as a marker to assess CAVD, which could have significant clinical implications.
Keywords: Calcific aortic valve disease (CAVD), coronary artery disease (CAD), antibody microarray
Introduction
Valvular heart disease is a common cause of heart failure and continues to occur in many patients with significant morbidity and mortality. The age-adjusted prevalence of moderate or severe valve diseases was estimated at 2.5% (1,2). Calcific aortic valve disease (CAVD) is the most common valve heart disease in the elderly and a leading cause of aortic valve stenosis (3). Because no effective pharmacological therapy exists (3), CAVD confers a high clinical and economic burden. The disease progression is rapid and, although initially indolent, it results in heart failure and premature death if left untreated (4). Due to rising life expectancy and aging populations in China, the burden of CAVD will significantly increase in the future.
CAVD shares some risk variables with coronary artery disease (CAD), including diabetes, hypertension, metabolic syndrome, dyslipidemia, and smoking (4-6). The pathobiology of CAVD is akin to an athero-inflammatory process (7-9). While CAVD was initially thought to be a degenerative process with passive deposition of calcium phosphate in the valve occurring with age, it now is a complex and actively regulated progress mediated by inflammation, cell apoptosis, lipid deposition, renin-angiotensin system activation, extracellular matrix remodeling, and bone formation (6,7). CAVD and CAD often coexist. CAD is prevalent in >60% of patients undergoing surgical aortic valve replacement (SAVR), and up to 65% of patients undergoing transcatheter aortic valve replacement (TAVR) (8,9). This strong association is believed to be due to the common pathophysiology involving low-density lipoprotein-mediated inflammatory response resulting in an accelerated atherosclerotic procedure as well as shared identical danger elements including age, diabetes, high blood pressure, metabolic disorder, dyslipidemia and cigarette smoking (10,11). However, CAVD mainly destroys endothelial valve cells and stromal cells, aggravating valve calcification, resulting in valve orifice stenosis and obstruction of the left ventricular outflow tract (12). Atherosclerosis destroys vascular endothelial cells and smooth muscle cells, and coronary artery occlusion and severe stenosis are the main causes of harm to patients with atherosclerosis (13,14). Therefore, there should be similarities and differences between them. To better keep an eye on the development of CAVD and show one of the most proper circumstances for surgical intervention, biomarkers can be serially checked. Such biomarkers would undoubtedly stand for objective research laboratory dimensions, as older individuals with CAVD could have irregular signs and symptoms connected with comorbidities, including pulmonary illness or orthopedic impairments.
Methods
Serum sample collection
In the current study, a total of 258 patients were included at Tianjin Chest Hospital between January 2017 and December 2018. Informed consent was obtained from all individual participants included in the study. Table 1 shows the detailed characteristics of the patients. CAVD patient group (n=86): criteria for transthoracic, echocardiography diagnosis of aortic stenosis: adopt the guidelines for diagnosis and treatment of valvular heart disease issued by the American heart association/American College of Cardiology (AHA/ACC) in 2014. Severe stenosis: aortic valve orifice area <1.0 cm2, maximum jet velocity >4.0 m/s, Average cross-valve pressure difference >40 mmHg (congenital, aortic valve defoliation deformity, rheumatic valve disease; severe infection; clear history of myocardial infarction; severe mitral or aortic regurgitation; moderate to severe renal insufficiency; current or chronic liver disease; connective tissue disease is active; recent history of bleeding; long-term use of cortisol hormones or non-steroidal anti-inflammatory drugs; incomplete data or poor compliance were excluded). CAD patients group (n=86); the diagnostic criteria of CAD proposed by the Coronary Artery Surgery Study (CASS) were as follows: coronary angiography showed positive coronary artery stenosis ≥50% of the left main coronary artery or stenosis ≥70% of the left anterior descending branch, left spiral branch and right coronary artery (combined with various heart valve diseases; severe infection; severe renal insufficiency; severe hepatic insufficiency; connective tissue disease is active; recent history of bleeding; malignant tumor; incomplete data or poor compliance were excluded). The control group (n=86): healthy people (valvular heart disease; coronary atherosclerotic heart disease; severe infection; neoplastic disease; other organs combined with organic functional insufficiency; incomplete data or poor compliance were excluded). The study was conducted in accordance with the Declaration of Helsinki and was approved by the ethics committee of Tianjin Chest Hospital.
Table 1. Demographic comparison between different groups.
| Variable | Control | CAVD | CAD | χ2 | P |
|---|---|---|---|---|---|
| Age (years) | 76.00 (7.00) | 78.00 (10.75) | 75.00 (7.00) | 23.769 | 0.010 |
| Hypertension | 67.90% | 88.90%* | 66.30% | 9.429 | 0.024 |
| Diabetes mellitus | 19.20% | 50.00%* | 42.30%* | 18.542 | 0.000 |
| Smoke | 26.90% | 63.90%* | 48.10%* | 19.412 | 0.000 |
| Man | 60.26% | 55.60% | 72.10% | 4.610 | 0.203 |
Data are shown as n (in %) or mean ± SD. *, statistically significant (P<0.05, vs. control). CAVD, calcific aortic valve disease; CAD, coronary artery disease.
Human inflammation array
Serum was diluted 1:2 and probed for cytokine profile using the RayBio Human Inflammation Array kit according to the standard hybridization process and kit provided by Raybiotech. Briefly, after drying the chip for 2 hours, we added 100 L of blocking buffer to each hole and closed it at room temperature for 30 minutes. We completely removed the blocking buffer from each hole. Then, we add the corresponding standard diluted sample diluent 100 L, 4 °C, for each hole overnight. Then add 150 L 1× Wash Buffer I and gently shake at room temperature for 5 times, 5 minutes each time. Put the chip into the washing box and add enough 1× Wash Buffer I for washing twice, each time for 10 minutes. Then add 150 L 1× Wash Buffer II and wash twice, 5 minutes each time. The biotin-antibody cocktail was removed, centrifuged, and diluted with 1,400 g/L Sample Diluent. The biotin-antibody cocktail was diluted with 80 L per well at room temperature and incubated for 2 hours. The biotin-antibody cocktail is then completely removed from each hole and washed. Remove cy3-streptavidin, centrifuge, and add 1,400 mL 1× blocking buffer, gently shaken, and mix.80 L diluted cy3-streptavidin was added to each well, and the aluminum foil was shielded from light, at room temperature, and incubated for 1 hour. Then remove cy3-streptavidin. Finally, the chip was scanned at 532 nm by the Agilent SureScan Dx Microarray Scanner.
Cell preparation
Human peripheral blood mononuclear cells are isolated by Lymphocyte Separation Medium (17-829F, Lonza) density gradient centrifugation. Monocytes were isolated using the Human Peripheral Blood Monocyte Isolation kit (Tianjin Haoyang Biological Products Technology Co., Ltd.) according to the manufacturer’s protocol and cultured in RPMI-1640 medium supplemented with 10% FBS and 1% P/S (penicillin and streptomycin). All cell lines are grown at 37 °C in a 5% CO2 atmosphere. A comparison of hs-CRP and NT-pro BNP between the distinct groups in peripheral blood mononuclear cells are shown in Table 2.
Table 2. Comparison of peripheral blood inflammatory cells, hs-CRP and NT-pro BNP between different group.
| Variable | Control | CAVD | CAD | χ2 | P |
|---|---|---|---|---|---|
| WBC (×109/L) | 5.83 (1.44) | 6.39 (2.38) | 6.06 (2.23) | 4.633 | 0.201 |
| N% | 61.35 (13.10) | 68.80 (11.70)* | 62.90 (12.43) | 16.821 | 0.001 |
| N (×109/L) | 3.65 (1.33) | 4.65 (2.62)* | 3.83 (1.89) | 11.364 | 0.010 |
| L (×109/L) | 1.77 (0.77) | 1.41 (0.89)* | 1.65 (0.93) | 10.427 | 0.015 |
| NLR | 2.07 (1.20) | 3.18 (2.45)* | 2.29 (1.61) | 24.809 | 0.000 |
| Hs-CRP (mg/L) | 1.00 (1.77) | 3.15 (13.41)* | 2.27 (3.92)* | 21.207 | 0.000 |
| NT-pro BNP (pg/mL) | 105.00 (58.18) | 1,849.50 (5,015.83)* | 302.50 (387.18)* | 115.916 | 0.000 |
Data are shown as n (in %) or mean ± SD. *, statistically significant (P<0.05, vs. control). CAVD, calcific aortic valve disease; CAD, coronary artery disease; WBC, white blood cell count; N, neutrophil count; L, lymphocyte count; NLR, neutrophil-to-lymphocyte ratio; Hs-CRP, hypersensitive c-reactive protein; NT-proBNP, amino-terminal B-type natriuretic peptide.
ELISA assays
The CXCL13/B-Lymphocyte Chemoattractant (BLC)-1, interleukin (IL)-12p40, CXCL9/monokine inducible by γ interferon (MIG) and CCL15/Macrophage inflammatory protein (MIP)-1delta serum levels were measured using ELISA kit in accordance with the manufacturer’s instructions. according to the manufacturer’s instructions (R&D Systems, USA).
Real-time quantified PCR (qRT-PCR)
Complete RNA was isolated with Trizol reagent (Takara Bio Inc., Japan) and was reverse recorded. The GAPDH was made use of as an internal control for determining the mRNA material. After drawing the standard curve of the PCR reaction, the expression level of mRNA is computed. The outcomes were analyzed by using the organic system 7300 rapid real-time PCR system. The primer pairs used in this study are listed in Table 3.
Table 3. Primers sequences for RT-PCR.
| Gene | 5'-3' primer sequence |
|---|---|
| CXCL13/BLC | F-5'-TCTTTATCCCTAGACGCT-3' |
| R-5'-TTTTCTCAATACTTCCATCA-3' | |
| IL-12p40/IL12B | F-5'-AGTGGAGTGCCAGGAGGACA-3' |
| R-5' TTCTTGGGTGGGTCAGGTTT-3' | |
| CXCL9/MIG | F-5'-TTGCTGGTTCTGATTGGAGTG-3' |
| R-5'-TCTTTCAAGGATTGTAGGTGGAT-3' | |
| GAPDH | F-5'-ATGACATCAAGAAGGTGGTGAAGCAGG-3' R-5'GCGTCAAAGGTGGAGGAGTGGGT-3' |
RT-PCR, real-time quantified PCR.
Western blot assay
Total protein was separated from human peripheral blood mononuclear using a protein extraction kit (Bio-Rad, USA). Then protein samples (20–100 µg) were resolved using SDS-PAGE and then moved to polyvinylidene fluoride (PVDF) membrane (Millipore, USA) with being probed with GAPDH (Proteintech), CXCL13/BLC (RRIDs: AB_1191734), IL-12p40/IL12B (RRIDs: AB_1156284), CXCL9/MIG (RRIDs: AB_2086730) followed by appropriate secondary antibody. All Western blots were repeated at least three times.
Statistical analysis
Analytical analyses were executed with GraphPad Prism 5. The data in this research exist as the mean ± standard error shows (SEM) from at least three independent experiments. The differences among the group’s one-way variation analysis and Duncan multiple range examinations were used to determine considerably distinct groups. P values less than 0.05 were taken into consideration as considerable distinctions for all statistical estimations.
Results
Cytokine and chemokine serum changes
Serum from patients in each group was revealed to Raybiotech inflammation antibody varieties. Forty proteins measured by the range were clear—the specified modifications in these proteins in CAVD and CAD as control group. In the CAVD group, 4 proteins were dramatically different from the control as well as the CAD group (Figure 1). Chemokines are classified into one of four families according to the arrangement of the N-terminal cysteine residues, that is, CXC, CC, CX3C, and C chemokines. The CXC chemokine family has the first two cysteine residues separated by one non-conserved amino acid residue (X), while the CC family has two cysteines in juxtaposition (15).
Figure 1.

RayBio human inflammatory cytokine antibody array was used to detect a panel of inflammatory cytokines among the three groups.
Validation of array data by ELISA
We confirmed the pattern of serum changes of four cytokines discovered on the Raybiotech antibody choice, making use of the ELISA. Consistent with data generated using the antibody arrays, serum BLC, IL-12p40, MIG, and MIP-1delta were not different comparing control to CAD. However, these cytokines were significantly increased in the CAVD compared to control or CAD, consistent with the antibody array data (Figure 2).
Figure 2.

Serum markers of cytokines associated with CAVD as measured by enzyme-linked immunosorbent assay. *, P<0.05 (CAVD compared to control and CAD). CAVD, calcific aortic valve disease; CAD, coronary artery disease.
qPCR detection of peripheral blood monocytes
We used qPCR to detect the BLC, IL-12p40, and MIG in monocytes to verify whether the above abnormal cytokines originated from peripheral blood monocytes. As Figure 3 showed, the mRNA levels of CXCL13/BLC, IL-12p40/IL-12B, and CXCL9/MIG in the CAD and CAVD groups were significantly increased compared with the control group. Their mRNA levels were much higher than those in the CAD group.
Figure 3.
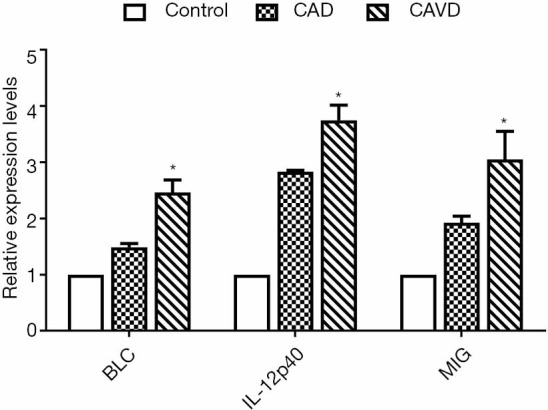
The relative BLC, IL-12p40, and MIG expression in monocytes. *, P<0.05 (CAVD compared to control and CAD). CAVD, calcific aortic valve disease; CAD, coronary artery disease.
WB and FCM detection of peripheral blood monocytes
As shown in Figure 4A, the relative protein expression levels of T BLC, IL-12p40, and MIG in the CAVD group are significantly higher than those in the control or CAD group. The flow cytometry assay also confirmed comparable results (Figure 4B).
Figure 4.

The relative protein level of BLC, IL-12p40, and MIG in monocytes detected by WB (A) and FCM (B). *, P<0.05 (CAVD compared to control and CAD). CAVD, calcific aortic valve disease; CAD, coronary artery disease.
Discussion
Until now, the pathogenesis of CAVD has not yet been fully understood. From comparable risk factors and functional processes of aortic valve sclerosis and atherosclerosis, the related pathogenesis of both entities is probable (16). While the pathogenesis of CAVD is still incompletely defined, there is evidence it is a multifaceted process, including a combination of hereditary as well as systemic elements that incorporate to initiate condition (17). An earlier analysis of human diseased valve explants compared to healthy controls revealed many upregulated pathological pathways that separate CAVD from a non-diseased state on the cellular range (18). Because elderly calcified aortic valve disease, as well as coronary artery atherosclerosis, have similar pathological changes, this study checked out product inflammatory cytokine differences that may contribute to the discovery of CAVD.
A certain obstacle in determining hereditary determinants of CAVD is that half of the patients with CAVD likewise were diagnosed with CAD (19-21). Therefore, treatment is called for in genetic fits of aortic valve stenosis to decrease the possibility that any organization observed is not merely a proxy for an association with CAD. After the usual co-occurrence and shared key danger elements of the two illnesses, Helgadottir and coworkers recently examined typical CAD linked variations in CAVD instances (21,22). A combined genetic risk score of 71 CAD variants was associated with the risk of CAVD (22). However, the significant result completely disappeared after adjusting for CAVD (22). In this study, we found the systemic production of adipokines, cytokines, and chemokines in response to alterations in CAVD using antibody array technology. Antibody ranges stand for among the high-throughput strategies that can spot several proteins simultaneously (23). Antibody arrays are high-throughput tools that improve the functional characterization of molecular bases for disease (24).
Our study shows significant changes in cytokine/chemokine profiles in CAD and CAVD patients compared to healthy individuals. It is assumed that the imbalance between pro-inflammatory and anti-inflammatory factors leads to a state of chronic, low-grade inflammation, which in turn can lead to type 2 diabetes and cardiovascular disease. In the CAVD group BLC, IL-12p40, MIG, and MIP-1delta were significantly different from the control and CAD group (14). Chemokines, or chemoattractant cytokines, are produced by many cell types in reaction to stimulus as well as also play a significant role in persistent and acute inflammation (14,25). Previous studies have confirmed that inflammatory cells and their secreted cytokines, including TNF-α, TGF-β, and VEGF, are related to the occurrence and development of CAVD (25). The CXC chemokines BLC, MIG, and MIP-1delta all increased in CAVD. This pattern would suggest a change in the chemokine gradient to greater circulating, chemokine degrees as well as may suggest boosted chemokines in serum from clients. Clinical applications consist of disease marker discovery for possible medical diagnosis, diagnosis, and adjustments related to disease development as well as development.
Conclusions
These findings suggest that BLC, IL-12p40, MIG, and MIP-1delta can be used as a marker to assess CAVD, which could have significant clinical implications.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: The Tianjin Science and Technology Commission funded this study (grant number 16ZXMJSY00160, 18ZXDBSY00160).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsink (as revised in 2013). The Tianjin Chest Hospital Ethics committee approved the study [No. IRB-SOP-016(F)-001-02]. Informed consent was obtained from all individual participants included in the study.
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-20-4463
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-4463). The authors have no conflicts of interest to declare.
(English Language Editor: J. Chapnick)
References
- 1.Nkomo VT, Gardin JM, Skelton TN, et al. Burden of valvular heart diseases: a population-based study. Lancet 2006;368:1005-11. 10.1016/S0140-6736(06)69208-8 [DOI] [PubMed] [Google Scholar]
- 2.Wang K, Liu Y, Huang S, et al. Does an imbalance in circulating vascular endothelial growth factors (VEGFs) cause atrial fibrillation in patients with valvular heart disease? J Thorac Dis 2019;11:5509-16. 10.21037/jtd.2019.11.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iung B, Vahanian A. Epidemiology of acquired valvular heart disease. Can J Cardiol 2014;30:962-70. 10.1016/j.cjca.2014.03.022 [DOI] [PubMed] [Google Scholar]
- 4.Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC Guideline for the Management of Patients With Valvular Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129:e521-643. [DOI] [PubMed] [Google Scholar]
- 5.Ross J, Jr, Braunwald E. Aortic stenosis. Circulation 1968;38:61-7. 10.1161/01.CIR.38.1S5.V-61 [DOI] [PubMed] [Google Scholar]
- 6.Mathieu P, Boulanger MC. Basic mechanisms of calcific aortic valve disease. Can J Cardiol 2014;30:982-93. 10.1016/j.cjca.2014.03.029 [DOI] [PubMed] [Google Scholar]
- 7.Kostyunin AE, Yuzhalin AE, Ovcharenko EA, et al. Development of calcific aortic valve disease: Do we know enough for new clinical trials? J Mol Cell Cardiol 2019;132:189-209. 10.1016/j.yjmcc.2019.05.016 [DOI] [PubMed] [Google Scholar]
- 8.Kvidal P, Bergstrom R, Horte LG, et al. Observed and relative survival after aortic valve replacement. J Am Coll Cardiol 2000;35:747-56. 10.1016/S0735-1097(99)00584-7 [DOI] [PubMed] [Google Scholar]
- 9.El Sabbagh A, Nishimura RA. Clinical Conundrum of Coronary Artery Disease and Aortic Valve Stenosis. J Am Heart Assoc 2017;6:e005593. 10.1161/JAHA.117.005593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stewart BF, Siscovick D, Lind BK, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol 1997;29:630-4. 10.1016/S0735-1097(96)00563-3 [DOI] [PubMed] [Google Scholar]
- 11.Freeman RV, Otto CM. Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies. Circulation 2005;111:3316-26. 10.1161/CIRCULATIONAHA.104.486738 [DOI] [PubMed] [Google Scholar]
- 12.Dutta P, Lincoln J. Calcific Aortic Valve Disease: a Developmental Biology Perspective. Curr Cardiol Rep 2018;20:21. 10.1007/s11886-018-0968-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci (Lond) 2018;132:1243-52. 10.1042/CS20180306 [DOI] [PubMed] [Google Scholar]
- 14.Fenton JI, Nunez NP, Yakar S, et al. Diet-induced adiposity alters the serum profile of inflammation in C57BL/6N mice as measured by antibody array. Diabetes Obes Metab 2009;11:343-54. 10.1111/j.1463-1326.2008.00974.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez EJ, Lolis E. Structure, function, and inhibition of chemokines. Annu Rev Pharmacol Toxicol 2002;42:469-99. 10.1146/annurev.pharmtox.42.091901.115838 [DOI] [PubMed] [Google Scholar]
- 16.Weiss RM, Miller JD, Heistad DD. Fibrocalcific aortic valve disease: opportunity to understand disease mechanisms using mouse models. Circ Res 2013;113:209-22. 10.1161/CIRCRESAHA.113.300153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Brien KD. Pathogenesis of calcific aortic valve disease: a disease process comes of age (and a good deal more). Arterioscler Thromb Vasc Biol 2006;26:1721-8. 10.1161/01.ATV.0000227513.13697.ac [DOI] [PubMed] [Google Scholar]
- 18.Bossé Y, Miqdad A, Fournier D, et al. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet 2009;2:489-98. 10.1161/CIRCGENETICS.108.820795 [DOI] [PubMed] [Google Scholar]
- 19.Iung B, Vahanian A. Epidemiology of valvular heart disease in the adult. Nat Rev Cardiol 2011;8:162-72. 10.1038/nrcardio.2010.202 [DOI] [PubMed] [Google Scholar]
- 20.Rajamannan NM, Evans FJ, Aikawa E, et al. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 2011;124:1783-91. 10.1161/CIRCULATIONAHA.110.006767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trenkwalder T, Nelson CP, Musameh MD, et al. Effects of the coronary artery disease associated LPA and 9p21 loci on risk of aortic valve stenosis. Int J Cardiol 2019;276:212-7. 10.1016/j.ijcard.2018.11.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helgadottir A, Thorleifsson G, Gretarsdottir S, et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat Commun 2018;9:987. 10.1038/s41467-018-03252-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez-Carbayo M. Antibody arrays: technical considerations and clinical applications in cancer. Clin Chem 2006;52:1651-9. 10.1373/clinchem.2005.059592 [DOI] [PubMed] [Google Scholar]
- 24.Barry R, Diggle T, Terrett J, et al. Competitive assay formats for high-throughput affinity arrays. J Biomol Screen 2003;8:257-63. 10.1177/1087057103008003003 [DOI] [PubMed] [Google Scholar]
- 25.Turner MD, Nedjai B, Hurst T, et al. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta 2014;1843:2563-82. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as