

Abstract
Ezrin, a membrane–cytoskeleton linker protein, plays an essential role in cell polarity establishment, cell migration, and division. Recent studies show that ezrin phosphorylation regulates breast cancer metastasis by promoting cancer cell survivor and promotes intrahepatic metastasis via cell migration. However, it was less characterized whether there are additional post-translational modifications and/or post-translational crosstalks on ezrin underlying context-dependent breast cancer cell migration and invasion. Here we show that ezrin is acetylated by p300/CBP-associated factor (PCAF) in breast cancer cells in response to CCL18 stimulation. Ezrin physically interacts with PCAF and is a cognate substrate of PCAF. The acetylation site of ezrin was mapped by mass spectrometric analyses, and dynamic acetylation of ezrin is essential for CCL18-induced breast cancer cell migration and invasion. Mechanistically, the acetylation reduced the lipid-binding activity of ezrin to ensure a robust and dynamic cycling between the plasma membrane and cytosol in response to CCL18 stimulation. Biochemical analyses show that ezrin acetylation prevents the phosphorylation of Thr567. Using atomic force microscopic measurements, our study revealed that acetylation of ezrin induced its unfolding into a dominant structure, which prevents ezrin phosphorylation at Thr567. Thus, these results present a previously undefined mechanism by which CCL18-elicited crosstalks between the acetylation and phosphorylation on ezrin control breast cancer cell migration and invasion. This suggests that targeting PCAF signaling could be a potential therapeutic strategy for combating hyperactive ezrin-driven cancer progression.
Keywords: ezrin, acetylation, phosphorylation, actin, cell migration
Introduction
Cell migration plays an important role in a wide variety of cellular dynamics, such as embryonic development, wound healing, immune response, and cancer metastasis (Stuelten et al., 2018). During cell migration, the coordination among membrane trafficking, actin skeleton remodeling, and formation of new adhesion complexes is required for protrusive activities at the leading edges of the migrating cells (Inagaki and Katsuno, 2017). The members of the ezrin/radixin/moesin (ERM) family proteins are involved in multiple aspects of cell migration by acting as cross-linkers between dynamic plasma membrane and the actin cytoskeleton in polarized secretory cells (Yao et al., 1993, 1995; Yao and Forte, 2003; Cao et al., 2005; Yao and Smolka, 2019), as well as regulators of signaling molecules that are implicated in cell polarity, adhesion, and migration (Bretscher et al., 2002; Clucas and Valderrama, 2014).
Ezrin is held inactive in the cytoplasm through an intramolecular interaction between its FERM (four-point-one protein/ezrin/ radixin/moesin) domain and C-ERMAD domain, and this interaction blocks its binding sites with plasma membrane and F-actin (Bretscher et al., 2002; Fehon et al., 2010). The conserved threonine residue T567 of ezrin can be phosphorylated in a tissue- and cell-dependent manner by Rho kinase in hepatocellular carcinoma (Chen et al., 2011b), PKCα in breast cancer (Ng et al., 2001), or MST4 in gastrointestinal epithelial cells (ten Klooster et al., 2009; Jiang et al., 2015). Our previous study also showed that ezrin plays an essential role in hepatocellular carcinoma metastasis through Rho kinase-mediated T567 phosphorylation, indicating that ezrin phosphorylation is involved in a wide variety of signaling pathways (Chen et al., 2011b). To date, a dominant view has been proposed that activation of ezrin is in a two-step fashion: (i) the binding with PIP2 induces a favorable conformational change and partial activation of ezrin and (ii) C-terminal T567 phosphorylation follows the conformational change, which made the phosphorylation sites accessible (Liu et al., 2007). However, whether other post-translational modifications (PTMs) are also involved in the regulation of ezrin function, tumor metastasis, or cell migration remains unknown.
In breast cancer cells, acetylation of ezrin is essential for cell migration, and persistent acetylation will perturb its directional cell migration. Here we identified four acetylation sites in the FERM domain of ezrin, which were acetylated by acetyltransferase p300/CBP-associated factor (PCAF). Ezrin acetylation attenuates its binding with plasma membrane PIP2, membrane-associated proteins, and F-actin. Finally, the acetylation of ezrin causes the unfavorable conformational changes and inactivity. Our results elucidate a previously unknown mechanism of ezrin acetylation, regulation, and crosstalk with phosphorylation.
Results
FERM domain of ezrin is acetylated in vivo in response to CCL18 stimulation
Recent study unraveled the importance of ACAP4 protein acetylation in CCL18-elicited breast cancer metastasis (Fang et al., 2006; Chen et al., 2011a; Song et al., 2018). Since our early study revealed the role of ezrin T567 phosphorylation in promoting intra-hepatocellular metastasis in xenografts (Chen et al., 2011b) and ezrin interacts with ACAP4 in context-dependent manner (Ding et al., 2010; Yuan et al., 2017), we sought to examine if there are context-dependent PTMs of ezrin in response to CCL18 stimulation in MDA-MB-231 cells. To this end, endogenous ezrin was isolated from CCL18-stimulated MDA-MB-231 cells and subjected the immunoisolated ezrin for western blotting analysis with a pan anti-acetyl-lysine antibody. As shown in Figure 1A, ezrin isolated from CCL18-stimulated but not control sample exhibited strong acetylation signal (lane 2), indicating that ezrin is acetylated in response to CCL18 stimulation. To further confirm that ezrin is acetylated in breast cancer cells, we carried out acetylome analysis in TSA and NAM treated MDA-MB-231 cells using anti-acetyllysine agarose affinity matrix coupled with mass spectrometric identification of acetylation sites. As a quality control of our experimentation, an acetylation blot indicated that ezrin is acetylated in response to the stimulation of CCL18 in vivo (Figure 1A and B; Supplementary Figure S1B).
Figure 1.
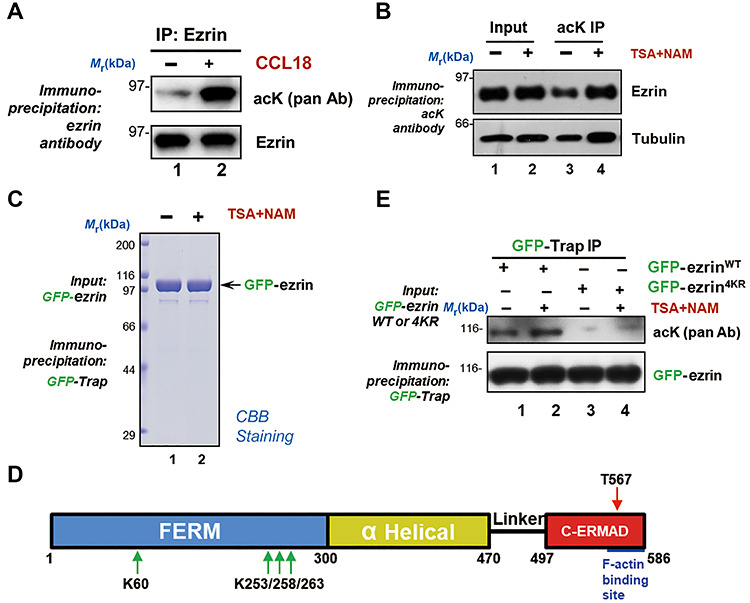
CCL18 stimulation induces ezrin acetylation in breast cancer cells. (A) Ezrin is acetylated in response to CCL18 stimulation. Starved MDA-MB-231 cells were treated with 20 ng/ml CCL18 for 10 min followed by ezrin immunoprecipitation (IP) and subsequent immunoblotting with pan-acK antibody (acK pan Ab). Note that the ezrin band was reacted by pan-acK antibody. (B) MDA-MB-231 cells were treated with DMSO or deacetylase inhibitors 1 μM Trichostatin A (TSA) and 10 mM Nicotinamide (NAM) for 4 h. The whole-cell lysates were immunoprecipitated by anti-acetyllysine agarose. Acetylated ezrin was detected by immunoblotting with ezrin antibody. (C) MDA-MB-231 cells expressing GFP-tagged ezrin were treated with TSA and NAM for 4 h and subjected to immunoprecipitation with GFP-Trap. The bound proteins were lysed with SDS sample buffer and separated by SDS–PAGE. (D) Schematic diagram of ezrin and the position of its acetylation sites. The red arrow indicates the phosphorylation site (T567), which have been reported previously, and the green arrows indicates the acetylation sites, which are located in the ezrin N-terminal FERM domain. (E) MDA-MB-231 cells expressing GFP-tagged ezrin WT or nonacetylatable mutant (4KR) were treated with TSA and NAM for 4 h and subjected to immunoprecipitation with GFP-Trap. Acetylation level of ezrin was detected by western blotting using pan-acK antibody.
To pinpoint the acetylation sites of ezrin in response to CCL18 stimulation, we treated GFP-ezrin transfected MDA-MB-231 cells with CCL18 in addition to TSA and NAM to perform an immunoprecipitation using GFP-Trap (Figure 1C). Mass spectrometric analysis revealed several potential acetylated lysine sites in the FERM domain in MDA-MB-231 cells (Figure 1D). Some of the potential acetylation sites on ezrin have also been reported in previous acetylome databases (Kim et al., 2006; Choudhary et al., 2009; Zhao et al., 2010). Mass spectrometric analysis indicated that four evolutionarily conserved sites (K60, K253, K258, and K263) are reproducibly found in CCL18-stimulated MDA-MB-231 cells (Supplementary Figure S1). To validate the acetylation, we introduced wild type (WT) or nonacetylatable (4KR) ezrin into MDA-MB-231 cells treated with TSA and NAM. Immunoprecipitation with GFP-Trap was conducted to detect the ezrin acetylation level by pan-acetylated lysine (pan-acK) antibody. As show in Figure 1E, WT but not 4KR mutant ezrin was acetylated, suggesting that these four identified sites represent major acetylation sites on ezrin. Therefore, we conclude that ezrin is acetylated in response to CCL18 stimulation in breast cancer cells.
Ezrin is a novel substrate of acetyltransferase PCAF
Lysine acetylation is an important PTM that regulates breast cancer recurrence and metastasis (Rios Garcia et al., 2017; Zhao et al., 2019). Our previous results revealed that Rho kinase-mediated ezrin T567 phosphorylation is essential in hepatocellular carcinoma metastasis (Chen et al., 2011b). However, there is no evidence showing the relationship between acetylation and ezrin in breast cancer cell invasion. The identification of ezrin acetylation prompted us to identify the upstream acetyltransferase. To this end, we performed immunoprecipitation assays, in which HEK293T cells were co-transfected with FLAG-ezrin and GFP-PCAF or GFP-TIP60. The transfected cells were then lysed and incubated with the anti-FLAG beads followed by western blotting analysis. As shown in Figure 2A, the immunoblotting results with anti-GFP antibody confirmed that ezrin interacts with PCAF specifically. To further confirm the above interaction, HEK293T cells were co-transfected with FLAG-PCAF and GFP-ezrin, or GFP followed by immunoprecipitation. Western blotting analysis verified the ezrin–PCAF interaction in vivo (Figure 2B).
Figure 2.
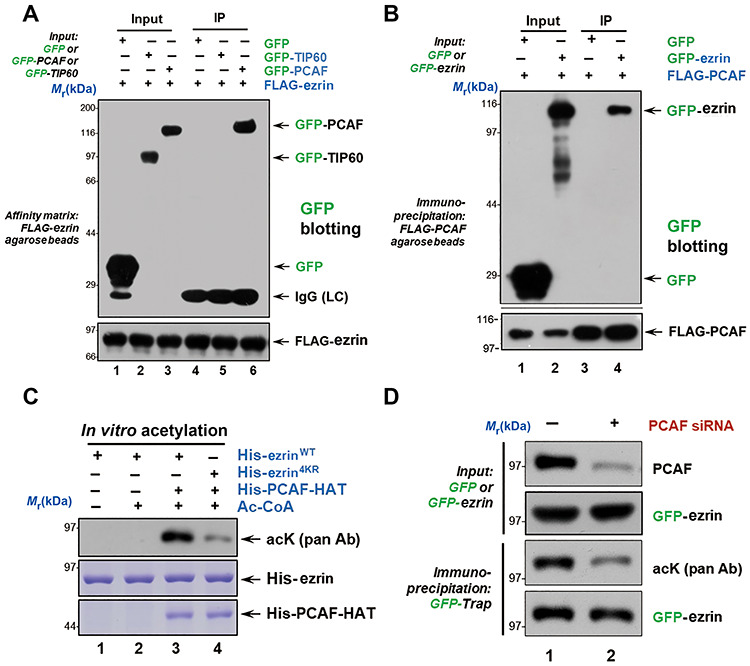
Ezrin is a substrate of acetyltransferase PCAF. (A and B) HEK293T cells were transfected with indicated plasmids and immunoprecipitated with anti-FLAG M2 resin. The immunoprecipitates were detected by GFP blot. (C) His-ezrinWT and His-ezrin4KR proteins were incubated with His-PCAF-HAT and Ac-CoA at 30°C for 1 h. EzrinWT was acetylated in the presence of PCAF and Ac-CoA as seen from anti-acK blot. However, low acetylation level was detected in ezrin4KR. (D) MDA-MB-231 cells depleted of PCAF were transfected with GFP-ezrin and immunoprecipitated with GFP resin. The immunoprecipitates were analyzed by western blotting using PCAF antibody to validate the knockdown efficiency and pan-acK antibody to report the suppression of ezrin acetylation.
To confirm that ezrin–PCAF interaction is not cell line oriented and not due to overexpression of exogenous proteins, we carried out an immunoprecipitation of endogenous ezrin and its associated proteins from MDA-MB-468 cells. As shown in Supplementary Figure 2A, PCAF but not TIP60 was precipitated by an anti-ezrin antibody (lane 2). In addition, ACAP4, a component of ezrin complex, was also recovered in the precipitates (third panel; lane 2). Thus, PCAF is a cognate binding protein of ezrin in breast cancer cells.
To confirm whether the cellular response to CCL18 is not cell line oriented, we carried out similar characterization using another triple negative breast cancer cell line MDA-MB-468. As shown in Supplementary Figure 2B, MDA-MB-468 cells were serum-deprived for 6 h followed by stimulation (20 ng/ml CCL18 for 10 min) or control vehicle. Examination of subcellular localization of endogenous ezrin relative to F-actin revealed a CCL18 stimulation-elicited remodeling of cytoskeleton–membrane, as ruffles were readily apparent on the plasma membrane (bottom panel; arrows).
We next sought to determine whether PCAF directly acetylates ezrin in vitro. The purified His-ezrinWT or His-ezrin4KR proteins were incubated with His-PCAF-HAT in the absence or presence of Ac-CoA as previously described (Xia et al., 2012). Western blotting using pan-acK antibody confirmed that ezrin is a direct substrate of PCAF (Figure 2C, lane 3). To further confirm if ezrin is a substrate of PCAF in cells, MDA-MB-231 cells depleted of PCAF were transfected with GFP-ezrinWT and immunoprecipitated with GFP-Trap. Western blotting against PCAF antibody was used to validate the knockdown efficiency and pan-acK antibody was used to report ezrin acetylation. As shown in Figure 2D, suppression of PCAF dramatically reduced the acetylation level of ezrin (lane 2). Therefore, we conclude that ezrin is a cognate substrate of PCAF in breast cancer cells.
Acetylation induces the translocation of ezrin from plasma membrane to cytoplasm
To determine the spatial-temporal distribution of acetylated ezrin in cells or tumor samples, we generated a site-specific acetylation antibody for K253, K258, and K263. Unfortunately, the modification specific antibodies generated exhibited high cross-reactivity beyond ezrin (data not shown). To solve this problem, we generated ezrin mutants K60Q/R, 4KQ, 4KR, 6KQ and 6KR to examine their subcellular distribution in cells (Supplementary Figure S3). As shown in Figure 3A, GFP-ezrin4KQ was enriched in the cytosol, while GFP-ezrinWT associates with the membrane and F-actin, especially in the leading edges of migrating cells. This is consistent with previous results that ezrin, as a linker protein between F-actin and plasma membrane, involved in the regulation of cell morphology and cytoskeleton–membrane dynamics. The transfected cells were lysed in homogenization buffer (10 mM HEPES pH 7.4, 1 mM EDTA, 250 mM sucrose) to separate cytosol and membrane components by centrifugation at 100000 g for 30 min at 4°C (Figure 3B). As a result, ezrin translocated from membrane to cytosol in cells treated with TSA and NAM (Supplementary Figure S4A and B) and so did ezrin acetylation-mimicking mutant and CCL18-elicition (Figure 3C and D; Supplementary Figure S4C and D). Thus, we conclude that ezrin acetylation induces its translocation from the plasma membrane to cytoplasm.
Figure 3.
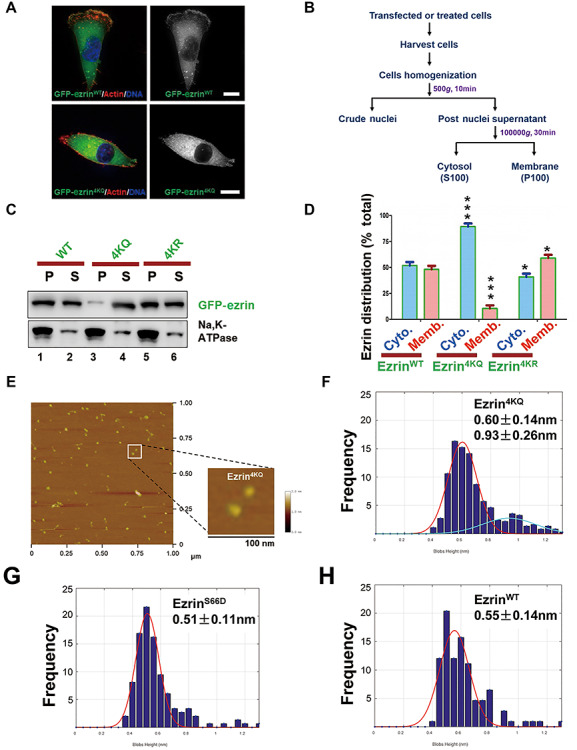
Ezrin acetylation elicits its conformational change and translocation from membrane to cytoplasm. (A) MDA-MB-231 cells were transfected with GFP-ezrin WT or 4KQ (acetylation-mimicking mutant) for 24 h followed by fixation and actin staining. Scale bar, 10 μm. (B) The experimental procedure of cell fractionation assay. (C) HEK293T cells were transfected with GFP-ezrin WT and mutants followed by a cell fractionation assay performed with velocity gradient centrifugation. Cell suspension (S) and pellet (P) were analyzed by western blotting. The plasma membrane fraction was indicated by Na,K-ATPase. (D) Quantitative analysis of cell fractionation assay in C. Data represent mean ± SE from three independent experiments. ***P < 0.001. (E) AFM images of ezrin4KQ molecule structures (low magnification of broader view). Image size, 1000 × 1000 nm. Scale bar, 100 nm in the magnified view of the inset. (F–H) Statistic histogram of single-molecule height of ezrin4KQ, ezrinS66D, and ezrinWT. Analysis was performed for >200 measurements of three independent preparations.
Acetylation of ezrin elicits a conformational change
The open and activated ezrin functions as the membrane–cytoskeleton linker protein to link plasma membrane and F-actin, but the closed and inactive ezrin localizes in the cytoplasm (Bretscher et al., 2002). Therefore, we hypothesize that the dissociation of acetylated ezrin from membrane was elicited by the conformational change. To determine the possible conformational change of ezrin in response to acetylation, we carried out atomic force microscopic (AFM) analyses of recombinant proteins (WT, nonacetylatable ezrin4KR, and acetylation-mimicking ezrin4KQ) as previously reported (Yu et al., 2014). A representative AFM image of ezrin4KQ is shown in Figure 3E.
We obtained images of individual recombinant ezrin protein using scanning AFM as previously described (Liu et al., 2007), which reveals overall elongated structures with different lengths and heights. As expected, the open single globular domain structure of ezrin was enriched in the acetylation-mimicking mutant, ezrin4KQ (Figure 3F), while ezrinS66D exhibits a linear structure with globular domains at both ends (Figure 3G) as reported (Yu et al., 2014), indicating that a conformational change of ezrin is associated with its acetylation. Given the heterogeneity, we measured the height of randomly selected ezrin structures to make unbiased histograms based on the measurement of >200 ezrin molecule structures. As shown in Figure 3F, the majority of ezrin4KQ molecules exhibit a height of 0.60 nm and 0.93 nm (n > 200), whereas ezrin WT molecules exhibit heights of 0.55 nm and ezrinS66D of 0.51 nm (Figure 3G and H, n > 200). It was also in accord with our circular dichroism spectroscopy result (Supplementary Figure S5A and B). Thus, we proposed that ezrin acetylation enhances intramolecular N/C-domain association and the three compact lobes of ezrin and turns the molecular structure of ezrin4KQ to a closed and inactive conformation.
The crosstalk between ezrin acetylation and phosphorylation
A dominant current view is that ezrin activation occurs in two steps: PIP2 binding followed by T567 phosphorylation (Zhou et al., 2005). If the acetylation turns ezrin to be closed and inactive, the ezrin T567 phosphorylation level and PIP2 binding affinity would decrease. To testify this hypothesis, we firstly probed ezrin phosphorylation level in MDA-MB-231 cells expressing GFP-ezrinWT, GFP-ezrin4KR, or GFP-ezrin4KQ with pT567 and pS66 antibodies (Figure 4A). Consistent with our AFM results, ezrin4KQ significantly reduced ezrin T567 phosphorylation which is essential for ezrin activation (Figure 4A and B; Supplementary Figure S6). In contrast, ezrin acetylation preserved S66 phosphorylation, which functions in gastric acid secretion (Zhou et al., 2003). Next, we determined whether ezrin T567 phosphorylation has a crosstalk with acetylation. The immunoprecipitated GFP-ezrin, GFP-ezrinT567A, and GFP-ezrinT567D from the transfected HEK293T cell lysates were subjected to pan-acK immunoblotting analyses. To our surprise, both phosphomimicking and nonphosphorylatable mutants attenuated the acetylation level compared to ezrin WT. However, the phosphomimicking mutant T567D exhibited higher acetylation level than the nonphosphorylatable mutant T567A (Figure 4C and D). The results indicated that in the T567 phosphorylation state, ezrin is prone to be acetylated than in the nonphosphorylation state. Taken together, ezrin acetylation prevented its T567 phosphorylation, showing a crosstalk between them.
Figure 4.
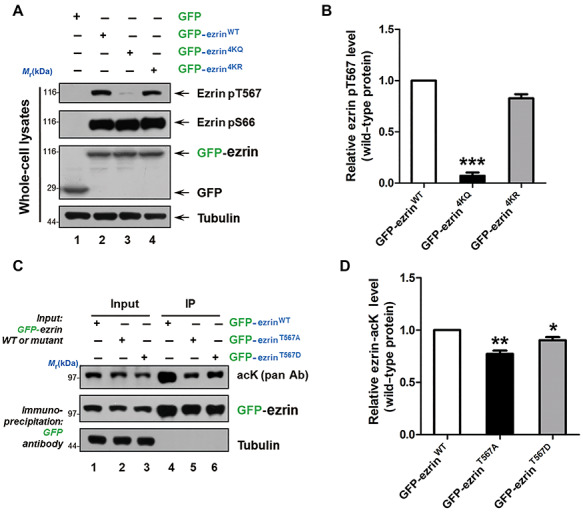
Acetylation of ezrin crosstalks with its phosphorylation. (A) HEK293T cells were transfected with GFP-ezrin WT and mutants for 24 h. Whole-cell lysates were analyzed by western blotting using ezrin pT567 and pS66 antibodies. (B) Quantitative analysis of the relative ezrin pT567 intensity compared to GFP-ezrin in A. Data represent mean ± SE from three independent experiments. ***P < 0.001. (C) HEK293T cells were transfected with GFP-ezrin WT and mutants for 24 h and subjected to immunoprecipitation by GFP-Trap. The acetylation level was detected by anti-acK antibody. (D) Quantitative analysis of the relative ezrin acetylation intensity compared to GFP-ezrin in C. Data represent mean ± SE from three independent experiments. *P < 0.05, **P < 0.01.
Ezrin acetylation attenuates its binding affinity with PIP2
According to the fact that the acetylation of ezrin could induce its translocation from plasma membrane to cytoplasm, we next examined whether the acetylation would regulate the interaction between ezrin and PIP2, a component of plasma membrane. Based on the structures of Spodoptera frugiperda moesin (sfMoesin, PDB ID: 2I1K) (Li et al., 2007) and radixin–IP3 (PDB ID: 1GC6) (Hamada et al., 2000), we drew a structural model of ezrin–PIP2 interaction. We found that K253, K258, and K263 sites locate in the interface of ezrin FERM and C-ERMAD domain, which are essential for T567 phosphorylation and ezrin unfolding (Figure 5A). In addition, K60, which locates at the interface of ezrin and the plasma membrane lipid, is essential for the first step of ezrin activation (Figure 5B). Therefore, we speculated that ezrin acetylation perturbed the association between ezrin and plasma membrane. To test the direct association of ezrin and PIP2, we conducted PIP strip assay (Figure 5C) using purified His-ezrin WT and mutants, which were verified by Coomassie Brilliant Blue (CBB) staining (Figure 5D). As shown in Figure 5E, ezrinWT interacts with most of the membrane lipids, whereas the lipid binding affinity of ezrin4KQ dramatically decreased. In accordance with our hypothesis, the acetylation and phosphorylation-mimicking double mutant (ezrin4KQ + T567D) partially rescued the binding affinity. In particular, the binding activity with PIP2, which is critical in ezrin activation, was prevented by acetylation (Figure 5E and F). Therefore, we conclude that ezrin acetylation attenuates the direct association of ezrin and the plasma membrane lipid PIP2.
Figure 5.
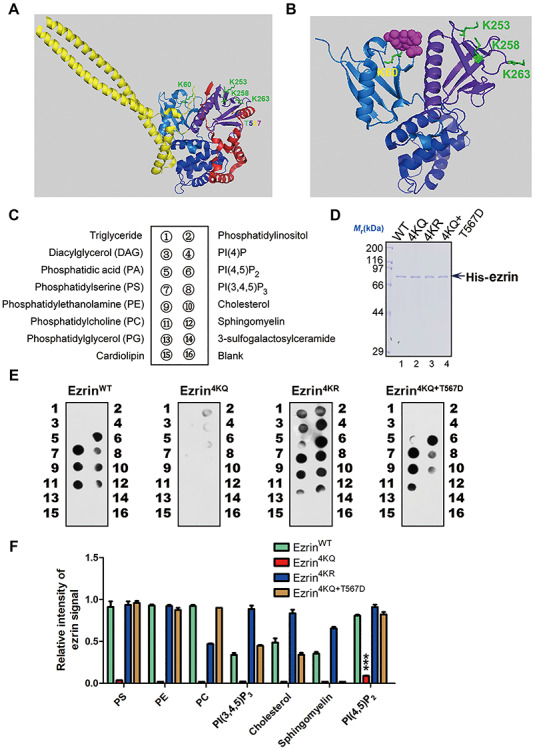
Acetylation at FERM domain attenuates ezrin–membrane association. (A) Structural analysis of ezrin acetylation sites. The corresponding acetylation sites in full-length sfMoesin (PDB ID: 2l1K) and the four lysine residues were labelled and highlighted in green to mimic the ezrin acetylation. The phosphorylation site T567 was labelled in rainbow. For the domain structure, the three-leaf clover domain structure of the FERM domain (F1 for marine, F2 for blue, and F3 for purple-blue), α helical region (yellow), and C-ERMAD domain (red) was applied. (B) Schematic representation of the radixin FERM domain bound to IP3 reveals most of the acetylation sites (K60, K253, K258, and K263) located on the interaction interface of FERM and IP3 (PDB ID: 1GC6). (C) The diagram of Echelon PIP strip and the membrane was spotted with 15 different biologically active lipids as indicated. (D) His-ezrinWT, His-ezrin4KR, His-ezrin4KQ, and His-ezrin4KQ + T567D proteins were purified from bacteria. (E) Purified His-ezrin WT and mutants were subjected to PIP strip assay to detect its binding affinity of PIP2. (F) Quantitative analysis of the binding affinity of ezrin WT and mutants as in E. Data represent mean ± SE from three independent experiments. ***P < 0.001.
Ezrin acetylation reduces its binding with membrane-associated proteins and F-actin
The membrane association property of ezrin is not only dependent on its direct binding to plasma membrane via PIP2, but also mediated by other membrane-associated proteins indirectly. Ezrin binds to PDZ domain-containing proteins, such as Na+/H+ exchange regulatory factor 2 and ERM binding protein 50 kDa (EBP50) (Reczek et al., 1997). It also binds to transmembrane receptors such as EGFR and PDGFR, and some co-factors such as CD44, ICAM1, and ICAM2 (Reczek et al., 1997; Heiska et al., 1998). To evaluate the binding ability of acetylated ezrin with membrane-associated proteins, we purified GST-tagged EBP50 C-terminal (GST-EBP50-CT) and ICAM2 C-terminal (GST-ICAM2-CT), which are responsible for ezrin binding in vitro. The truncations were used as baits to pull down His-ezrin WT and mutants. The results showed that ezrin mutants significantly reduced the binding affinity of ezrin–ICAM2 (Figure 6A and B) and ezrin–EBP50 (Figure 6C and D). Ezrin is a membrane–cytoskeleton linker protein, which interacts with cytoplasm β-actin rather than skeletal α-actin (Yao et al., 1996). Thus, we also detected the ezrin and F-actin binding by an actin co-sedimentation assay. The results showed less F-actin co-sedimentated with ezrin4KQ (Figure 6E and F) compared with WT and other mutants. Thus, we conclude that acetylation of ezrin also attenuates its direct binding with F-actin.
Figure 6.
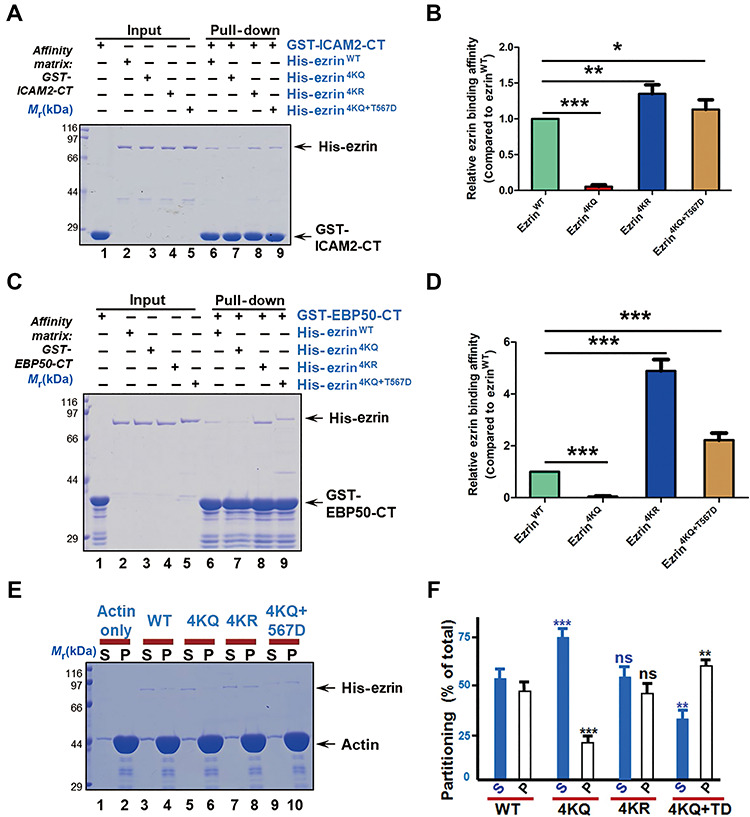
Acetylation of ezrin attenuates its association with plasma membrane and actin. (A) GST-ICAM2-CT was used as affinity matrix to absorb purified His-tagged ezrin WT or mutants. Binding activity was analyzed by CBB staining. (B) Quantitative analysis of the binding affinity to ICAM-CT of ezrin WT and mutants as in A. Data represent mean ± SE from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. (C) GST-EBP50-CT was used as affinity matrix to absorb purified His-tagged ezrin WT or mutants. Binding activity was analyzed by CBB staining. (D) Quantitative analysis of the binding affinity to EBP50-CT of ezrin WT and mutants as in C. Data represent mean ± SE from three independent experiments. ***P < 0.001. (E) Actin co-sedimentation assay of ezrin WT and mutants. Equal volumes of supernatant (S) and pellet (P) fractions were resolved by SDS–PAGE and visualized by CBB staining. (F) Quantitative analysis of the binding affinity to actin of ezrin WT and mutants as in E. Data represent mean ± SE from three independent experiments. *P < 0.05, ***P < 0.001, ns indicates not significant.
The dynamics of ezrin acetylation is essential for directional cell migration
ERMs are involved in many tumor metastasis processes, such as cell polarization, cell–cell communication and cell–extracellular matrix (ECM) communication (Clucas and Valderrama, 2014). Loss of ERMs apical structures contributes to tumor development and metastasis by causing cell depolarization, loss of contact-dependent inhibition of proliferation, and increased motility and invasiveness (McClatchey, 2003). In addition, ezrin is a key component in tumor metastasis as a conduit for signals between metastasis-associated cell-surface molecules and signal transduction components (Hunter, 2004). PKCα in breast cancer (Ng et al., 2001) and Rho kinase in hepatocellular carcinoma (Chen et al., 2011b) mediate ezrin T567 phosphorylation, which promotes tumor metastasis. The crosstalk between ezrin acetylation and T567 phosphorylation suggests a role of dynamic ezrin acetylation in cell migration. To validate this hypothesis, we transfected GFP-ezrinWT, GFP-ezrin4KR, or GFP-ezrin4KQ into MDA-MB-231 cells depleted of endogenous ezrin and examined the migration of single cells. MDA-MB-231 cells were starved and then stimulated with serum. Acetylation-mimicking mutant ezrin4KQ resulted in defects of directionally persistent cell migration (Figure 7A, middle panel).
Figure 7.
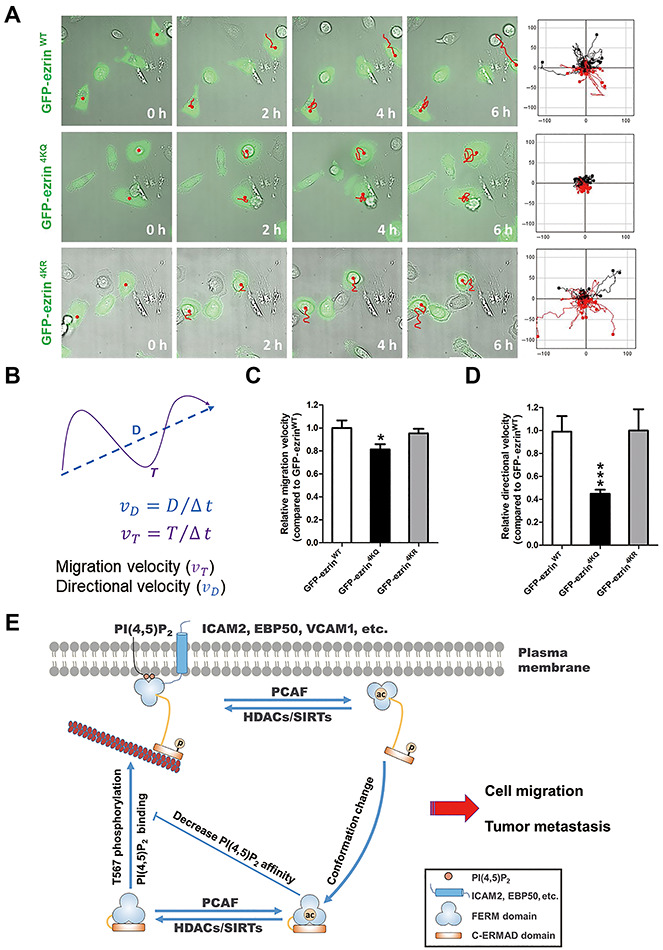
Ezrin acetylation is essential for directional cell migration. (A) MDA-MB-231 cells transfected with GFP-ezrin WT, 4KR (nonacetylatable mutant), or 4KQ (acetylation-mimicking mutant) were treated as described in ‘Materials and methods’ and then imaged at 10 min intervals. Migration tracks of cells are shown as red lines. Scale bar, 20 μm. (B) Diagram of migration path length. The total distance between starting and ending points (T) and the actual trajectory (D) are indicated. Migration velocity (υT) and directional migration velocity (υD) were calculated as illustrated. υT is the ratio of total track distance to total time, and υD is the ratio of direct distance between starting and ending points to total time. (C and D) Quantitative analysis of the migration velocity (C) and directional migration velocity (D) of cells expressing GFP-ezrin WT, 4KR, or 4KQ as in A. Data represent mean ± SE from three independent experiments. *P < 0.05, ***P < 0.001. (E) Working model accounting for ezrin acetylation in cell migration. Ezrin is activated through PIP2 binding and phosphorylation of Thr567, which reduces the binding affinity of the N-terminal FERM domain with the C-ERMAD. This open and activated state either anchors to the membrane lipid PIP2 or binds to membrane-associated proteins, such as ICAM2 and EBP50. Ezrin functions as membrane–cytoskeleton linker protein; therefore, the open and activated ezrin also binds to F-actin directly. In addition, ezrin is acetylated by the lysine acetyltransferase PCAF, which elicits a conformational change and a return to the inactivated molecular state. Ezrin acetylation reduces its binding affinity with PIP2 and other membrane-associated proteins; hence, F-actin finally disassociates from the plasma membrane to cytoplasm. Unlike phosphorylation, acetylation negatively regulates ezrin function and crosstalks with phosphorylation in a complicated pattern.
To quantitatively evaluate ezrin acetylation effects on cell migration, the migration velocity (υT) and directional migration velocity (υD) of MDA-MB-231 cells were calculated as described previously (Zhang et al., 2013). υT is the ratio of the total track distance to the total time, representing the cell migration velocity. υD is the ratio of the direct distance (from starting point to ending point) to the total time, representing the directional migration velocity (Figure 7B). In accordance to the cell migration tracks, the υT of GFP-ezrin4KR-expressing cells was similar to that of the GFP-ezrinWT-expressing cells, but it decreased in GFP-ezrin4KQ-expressing cells (Figure 7C). To our surprise, υD was dramatically reduced in GFP-ezrin4KQ-expressing cells (Figure 7D), indicating that dynamic acetylation of ezrin functions in directional migration of MDA-MB-231 cells. Thus, we conclude that the dynamics of ezrin acetylation is required for directionally persistent migration of MDA-MB-231 cells.
Discussion
Ezrin is an important membrane–cytoskeleton linker essential for cell fate decision. This study reveals that ezrin, which is a cognate substrate of PCAF, is acetylated in response to CCL18 stimulation. This acetylation resides in the FERM domain, which is critical to ezrin by virtue of its membrane targeting function regulated by the PI(4,5)P2. The acetylation of ezrin reduces its binding affinity to PIP2, membrane-associated proteins, and F-actin, which leads to a dynamic translocation of ezrin from plasma membrane to the cytosol. Importantly, this acetylation and translation of ezrin suppressed ezrin phosphorylation at Thr567 via an intramolecular interaction (Figure 7E). Thus, dynamic acetylation of ezrin provides a novel link between CCL18 stimulation and membrane dynamics in directional cell migration and invasion. PCAF signaling could be a potential therapeutic strategy for combating hyperactive ezrin-driven cancer metastasis.
Lysine acetylation is a conserved protein PTM linking cell metabolism and cellular signaling to epigenetic modification (Choudhary et al., 2014). A large number of lysine acetylations have been identified using large-scale mass spectrometry-based proteomics. Crosstalks among different PTMs can add another level of regulation to various cellular processes, in addition to signaling transduction and proteins’ conformational change. As acetylation changes the protein charge in some restricted regions, it can influence protein modification at nearby sites, recruit other PTM regulating enzymes, or compete with other lysine PTMs, such as methylation and ubiquitylation (Lee et al., 2010). The acetyltransferase, p300, acetylates SMAD7 N-terminus, preventing its ubiquitylation by the ubiquitin ligase SMURF1 (Gronroos et al., 2002).
Histone lysine acetylation is essential for regulation of gene transcription. Nonhistone protein acetylation in cancer cells involves in cell cycle regulation (Xia et al., 2012), cellular metabolism (Rios Garcia et al., 2017), and cell migration (Song et al., 2018). Now we identified that acetylation of ezrin FERM domain causes its dissociation from plasma membrane and F-actin cytoskeleton. Our single cell migration tracking assay revealed its essential role in directional persistent cell migration. Many regulators involve in directional cell migration, such as microtubule plus-end tracking protein-mediated microtubule dynamics, PKCα regulation, Par complex-mediated material polar transportation, and actin cytoskeleton dynamics-mediated pseudopodia formation (Petrie et al., 2009). Previous studies have demonstrated the involvement of microtubule plus-end tracking protein DDA3 and SKAP in directional cell migration (Zhang et al., 2013; Cao et al., 2015), and the interaction of ezrin, Cdc42, and PKCα (Legg et al., 2002; Zhou et al., 2003). Recent study showed that ezrin stabilizes a multiprotein complex that maintains active HER2 at the cell surface of breast cancer cells (Jeong et al., 2019). It would be of great interest to examine whether ezrin acetylation alters its association with the aforementioned actin cytoskeleton regulators and destabilizes HER2 at the plasma membrane.
There is a strong correlation between increased cytoplasmic ezrin and poor cancer survival in many kinds of primary cancer cells, such as head and neck cancer, gastric cancer, and breast carcinomas (Sarrio et al., 2006). Global RNA analysis of head and neck cancer reveals a positive correlation between cytoplasmic ezrin and RNA expression, including cell death, proliferation, invasion, and differentiation (Schlecht et al., 2012). It would be of great interest, in follow-up studies, to uncover the link between ezrin acetylation and cancer prognosis.
The newly established organoid system is a cell-derived in vitro 3D organ model and allows the study of biological processes, such as cell behavior, tissue repair, and organ structures (Sato et al., 2009; Liu et al., 2019). To date, a variety of organoids derived from epithelial tissue such as breast, have been established. Such a preclinical model complies with the urgent need to efficiently translate basic cancer research into novel treatments for cancer patients. In combination with breast cancer patient-derived organoids technology (Sachs et al., 2018), the newly developed adaptive optics lattice light sheet microscopy technology (AO-LLSM), by which high-resolution live cell images of subcellular processes and physiologically relevant 3D environment within whole transparent organisms were taken (Liu et al., 2018), promises a new insight into studying the function of protein PTMs during tumor metastasis in organoids.
The present results demonstrate that acetylation at the FERM of ezrin regulates the dynamic interaction of ezrin with the plasma membrane in response to chemokine CCL18 stimulation. During breast cancer cell migration and invasion, this dynamic membrane association/dissociation by the ezrin FERM domain regulates HER2 activity at the plasma membrane and promotes the directional motility of MDA-MB-231 cells in cell migration and invasion. Thus, the results provide insights on the regulation of ezrin signaling in the CCL18 signaling pathway during breast cancer cell migration. Given the fact that hyperactive ezrin-driven cancer metastasis is seen in many solid tumors, strategies targeting PCAF signaling could be potential therapeutic strategies interrogating tumor progression and metastasis.
Materials and methods
Plasmids
EGFP- and FLAG-tagged ezrin plasmids were produced as described before (Yu et al., 2014). Ezrin was subcloned into pET-22b (+) (Novagen) to generate His-ezrin. Site-specific mutants of His- or EGFP-tagged ezrin were generated by PCR-based, site-directed mutagenesis kit from Vazyme (C212) according to the manufacturer’s instructions. EBP50 EB domain (amino acids 266–358) and ICAM2 (last 28 amino acids) were subcloned and inserted into pGEX-6p-1 (GE Healthcare) to generate GST-EBP50-CT and GST-ICAM2-CT plasmids. PCAF and TIP60 plasmids were produced as described (Mo et al., 2016). BGI, Sangon Biotech, General Biosystems, or Tsingke Biological Tech sequenced all of the plasmids.
Cell culture and transfection
HEK293T and LLC-PK1 cells (American Type Culture Collection) were maintained as sub-confluent monolayers in advanced DMEM (Invitrogen) with 10% fetal bovine serum (HyClone), 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen) at 37°C with 8% CO2. MDA-MB-231 and MDA-MB-468 cells (American Type Culture Collection) were maintained as sub-confluent monolayers in L15 medium (Invitrogen) with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C without CO2 (Adams Jr et al., 2016). Cells were transfected with plasmids or siRNA duplex using Lipofectamine 3000 according to the manufacturer’s protocol (Invitrogen).
Antibodies and siRNAs
Antibodies against ezrin, ezrin pT567, acetylated-Lysine, and α-tubulin (DM1A) were purchased from Cell Signaling Technology. Anti-Na,K-ATPase and anti-GFP antibodies were purchased from Santa Cruz Biotechnology. Anti-FLAG-tag (M2) antibody was from Sigma. Phalloidin was purchased from Invitrogen, and ezrin pS66 antibody was described before (Yu et al., 2014; Fang et al., 2017). The PCAF siRNA and ezrin siRNA sequence were described before (Ding et al., 2010; Xia et al., 2012).
Recombinant protein purification
GST-tagged proteins were produced in Escherichia coli strain BL21 or Rosetta (DE3) as previously described (Xia et al., 2012). Briefly, the plasmids were transformed into E. coli strain, and protein expression was induced with 0.2 mM IPTG at 16°C. Bacterially expressed GST-tagged proteins were suspended and lysed by sonication in PBS containing 1 μg/ml phenylmethylsulfonylfluoride (PMSF), followed by incubation with glutathione-Sepharose 4B (GE Healthcare Life Science) for 1.5 h at 4°C. Bacteria expressing His-tagged recombinant proteins were resuspended and lysed by sonication in lysis buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole) supplemented with 1 μg/ml PMSF. The preparation was incubated with Ni-NTA resin (Qiagen) for 1.5 h at 4°C. The agarose was washed three times in wash buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 40 mM imidazole) and eluted with elution buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 200 mM imidazole). All purification procedures were performed at 4°C, and protease inhibitor cocktail (Sigma) was added to prevent protein degradation. All purified proteins were analyzed and confirmed with SDS–PAGE.
Immunoprecipitation
HEK293T cells or MDA-MB-231 cells were collected and lysed in IP buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM MgCl2, 1 mM EGTA, 0.2% Triton X-100, 1 mM DTT, 10% glycerol) supplemented with Protease Inhibitor Cocktail (Sigma). Cell lysates were clarified by centrifugation at 12000 rpm for 20 min at 4°C. For different purposes, clarified cell lysates were incubated with acetyl lysine antibody coupled agarose (Immunechem), GFP-Trap A (Chromotek), and FLAG-M2 resin (Sigma) for 4 h before washing, respectively. The binding fraction was washed with IP buffer five times before being resolved by SDS–PAGE and immunoblotted with indicated antibodies.
Pull-down assay
The pull-down assay was performed as previously described (Akram et al., 2018). Briefly, purified GST-tagged protein-bound Sepharose beads were used as affinity matrix to incubate with purified His-tagged proteins. After incubation for 3 h, the beads were washed three times with PBS containing 0.2% Triton X-100 and one time with PBS and boiled in SDS–PAGE sample buffer. The samples were separated by SDS–PAGE gel followed by CBB staining.
Immunofluorescence microscopy
MDA-MB-231 cells grown on coverslips were fixed using PBS buffer supplemented with 3.7% paraformaldehyde. After blocking with PBS with 0.05% Tween-20 (PBST) buffer containing 1% bovine serum albumin (Sigma) for 45 min at room temperature, the fixed cells were incubated with primary antibodies in a humidified chamber for 1 h at room temperature, followed by secondary antibodies for 1 h. The DNA was stained with 4′,6-diamidino-2-phenylindole from Sigma. Images were captured by DeltaVision softWoRx software (Applied Precision) and processed by deconvolution and z-stack projection.
Actin co-sedimentation assay
Nonmuscle actin proteins (APHL99, Cytoskeleton, Inc.) were thawed and centrifuged at 100000 g for 40 min (TLA 100.3 rotor, Beckman Instruments) to remove polymerized or aggregated actin. Aliquots of monomeric actin were incubated with ezrin wild-type or mutant protein for 2 h at 23°C in reaction buffer (5 mM Tris, pH 7.5, 0.5 mM ATP, 2 mM MgCl2, 100 mM KCl, and 0.2 mM DTT) to promote actin polymerization. Pellets and supernatants were solubilized in sample buffer and subjected to electrophoresis to visualize and quantify the actin binding and the affinity difference.
PIP strip assay
PIP strip assay was performed according to the user’s manual (Song et al., 2018). His-ezrin WT and mutants were purified as described above. Proteins in TBS with 0.1% Tween-20 (TBST) buffer (3 mg in 3 ml) were incubated with each PIP Strip™ membrane (P-6001, Echelon). After 1 h membrane–protein incubation, the membranes were incubated with ezrin antibody.
Circular dichroism spectroscopy
Circular dichroism (CD) spectra were recorded on a model J-810 spectropolarimeter (JASCO) using the Spectra Manager control software package (JASCO). CD spectra were recorded in NaCl 137 mM, KCl 2.7 mM, Na2HPO4 10 mM, KH2PO4 1.8 mM (pH 7.4) in 1-mm quartz glass precision cells at room temperature in a wavelength range of 260–200 nm with 1.0 nm bandwidth, using the continuous-mode setting, with 1.0 sec response and a scan speed of 50 nm/min. Three spectra were averaged; spectra were background-corrected against pure buffer. For normalization, the final protein concentrations were determined by UV/Vis-spectroscopy using pure buffer for background correction. Deconvolution of the data was performed using the Jasco Secondary Structure Estimation software by the reference CD (Yang-Us).
Atom force microscopy
Purified protein samples were diluted to 10 nM in PBS and placed on ice. Then 10 μl of protein sample was added onto a mica surface that had been pretreated with 10 mM spermidine. After 10 min, the mica surface was gently washed with water and dried by nitrogen gas. The AFM imaging was performed on Nanoscope IIIa (Digital Instruments) with a type E scanner under the Tapping Mode TM in air at room temperature. The AFM probes made of single silicon crystals with a cantilever length of 129 μm and a spring constant 33–62 Newtons/m (Olympus) were used. Images were collected in height mode and stored in the 512 × 512-pixel format. The images obtained were then plane fitted and analyzed by the computer program accompanying the imaging module.
For the statistical analysis, 1 × 1 μm images were captured using the height mode in a 512 × 512 formats. Thus, the AFM imaging provides images with sub-nanometer resolution. By measuring a contour length between two peaks of heads, we were able to measure the length between two heads of a single molecule. For quantitative analysis, fitted Gaussian curves were overlaid on the histograms. The peaks of the distributions are indicated (mean ± SD). In general, >280 molecules per group were analyzed from three independent preparations.
Cell migration assay
MDA-MB-231 cells grown on glass-bottom culture dishes (MatTek) were starved overnight and then stimulated with 20% serum. Images were taken with a DeltaVision microscopy system (Applied Precision Inc.) at a rate of 1 frame/10 min. Cell movement was tracked by Image-Pro Plus 6.0. Total migration velocity (υT) is total track distance divided by total time, and directional migration velocity (υD) is the direct distance from starting point to ending point divided by total time.
Sequence alignment and structure modeling
Protein sequence alignment were analyzed by MultAlin website and the Ezrin modeling (PDB ID: 2I1J and 1GC6) was analyzed and presented by Pymol software.
Data analyses
GraphPad Software was used to perform, evaluate, and determine significant differences between mean and unpaired Student’s t-test assuming unequal variance. Statistical analysis was considered significant when the two-sided P-value was <0.05.
Supplementary Material
Acknowledgements
We thank our lab members for stimulating discussion and Dr Jingjun Hong for his comments on this manuscript.
Funding
This work was supported in part by grants from the National Natural Science Foundation of China (81630080, 31430054, 91854203, 31301105, 31320103904, 31621002, 31671405, 91853115, 21922706, 81572283, 31271518, 31471275, and 31870759), National Key Research and Development Program of China (2017YFA0503600 and 2016YFA0100500), Ministry of Education (IRT_17R102 and 20113402130010), the Strategic Priority Research Program of Chinese Academy of Sciences (XDB19000000), and Central University Grants WK2340000066.
Conflict of interest: none declared.
References
- Adams G. Jr, Zhou J., Wang W., et al. (2016). The microtubule plus end tracking protein TIP150 interacts with cortactin to steer directional cell migration. J. Biol. Chem. 291, 20692–20706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akram S., Yang F., Li J., et al. (2018). LRIF1 interacts with HP1α to coordinate accurate chromosome segregation during mitosis. J. Mol. Cell Biol. 10, 527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A., Edwards K., and Fehon R.G. (2002). ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3, 586–599. [DOI] [PubMed] [Google Scholar]
- Cao D., Su Z., Wang W., et al. (2015). Signaling scaffold protein IQGAP1 interacts with microtubule plus-end tracking protein SKAP and links dynamic microtubule plus-end to steer cell migration. J. Biol. Chem. 290, 23766–23780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X., Ding X., Guo Z., et al. (2005). PALS1 specifies the localization of ezrin to the apical membrane of gastric parietal cells. J. Biol. Chem. 280, 13584–13592. [DOI] [PubMed] [Google Scholar]
- Chen J., Yao Y., Gong C., et al. (2011a). CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 19, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Wang D., Guo Z., et al. (2011b). Rho kinase phosphorylation promotes ezrin-mediated metastasis in hepatocellular carcinoma. Cancer Res. 71, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C., Kumar C., Gnad F., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- Choudhary C., Weinert B.T., Nishida Y., et al. (2014). The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550. [DOI] [PubMed] [Google Scholar]
- Clucas J., and Valderrama F. (2014). ERM proteins in cancer progression. J. Cell Sci. 127, 267–275. [DOI] [PubMed] [Google Scholar]
- Ding X., Deng H., Wang D., et al. (2010). Phospho-regulated ACAP4–Ezrin interaction is essential for histamine-stimulated parietal cell secretion. J. Biol. Chem. 285, 18769–18780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z., Miao Y., Ding X., et al. (2006). Proteomic identification and functional characterization of a novel ARF6 GTPase-activating protein, ACAP4. Mol. Cell. Proteomics 5, 1437–1449. [DOI] [PubMed] [Google Scholar]
- Fehon R.G., McClatchey A.I., and Bretscher A. (2010). Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronroos E., Hellman U., Heldin C.H., et al. (2002). Control of Smad7 stability by competition between acetylation and ubiquitination. Mol. Cell 10, 483–493. [DOI] [PubMed] [Google Scholar]
- Hamada K., Shimizu T., Matsui T., et al. (2000). Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 19, 4449–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiska L., Alfthan K., Gronholm M., et al. (1998). Association of ezrin with intercellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2). Regulation by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 273, 21893–21900. [DOI] [PubMed] [Google Scholar]
- Hunter K.W. (2004). Ezrin, a key component in tumor metastasis. Trends Mol. Med. 10, 201–204. [DOI] [PubMed] [Google Scholar]
- Inagaki N., and Katsuno H. (2017). Actin waves: origin of cell polarization and migration? Trends Cell Biol. 27, 515–526. [DOI] [PubMed] [Google Scholar]
- Jeong J., Choi J., Kim W., et al. (2019). Inhibition of ezrin causes PKCα-mediated internalization of erbb2/HER2 tyrosine kinase in breast cancer cells. J. Biol. Chem. 294, 887–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Wang W., Zhang Y., et al. (2015). Cell polarity kinase MST4 cooperates with cAMP-dependent kinase to orchestrate histamine-stimulated acid secretion in gastric parietal cells. J. Biol. Chem. 290, 28272–28285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.C., Sprung R., Chen Y., et al. (2006). Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618. [DOI] [PubMed] [Google Scholar]
- Lee J.S., Smith E., and Shilatifard A. (2010). The language of histone crosstalk. Cell 142, 682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legg J.W., Lewis C.A., Parsons M., et al. (2002). A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility. Nat. Cell Biol. 4, 399–407. [DOI] [PubMed] [Google Scholar]
- Li Q., Nance M.R., Kulikauskas R., et al. (2007). Self-masking in an intact ERM-merlin protein: an active role for the central α-helical domain. J. Mol. Biol. 365, 1446–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D., Ge L., Wang F., et al. (2007). Single-molecule detection of phosphorylation-induced plasticity changes during ezrin activation. FEBS Lett. 581, 3563–3571. [DOI] [PubMed] [Google Scholar]
- Liu T.L., Upadhyayula S., Milkie D.E., et al. (2018). Observing the cell in its native state: imaging subcellular dynamics in multicellular organisms. Science 360, eaaq1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Xu L., Li J., et al. (2019). Mitotic motor CENP-E cooperates with PRC1 in temporal control of central spindle assembly. J. Mol. Cell Biol. doi: 10.1093/jmcb/mjz051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey A.I. (2003). Merlin and ERM proteins: unappreciated roles in cancer development? Nat. Rev. Cancer 3, 877–883. [DOI] [PubMed] [Google Scholar]
- Mo F., Zhuang X., Liu X., et al. (2016). Acetylation of Aurora B by TIP60 ensures accurate chromosomal segregation. Nat. Chem. Biol. 12, 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T., Parsons M., Hughes W.E., et al. (2001). Ezrin is a downstream effector of trafficking PKC–integrin complexes involved in the control of cell motility. EMBO J. 20, 2723–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie R.J., Doyle A.D., and Yamada K.M. (2009). Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 10, 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczek D., Berryman M., and Bretscher A. (1997). Identification of EBP50: a PDZ-containing phosphoprotein that associates with members of the ezrin–radixin–moesin family. J. Cell Biol. 139, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios Garcia M., Steinbauer B., Srivastava K., et al. (2017). Acetyl-CoA carboxylase 1-dependent protein acetylation controls breast cancer metastasis and recurrence. Cell Metab. 26, 842–855.e5. [DOI] [PubMed] [Google Scholar]
- Sachs N., de Ligt J., Kopper O., et al. (2018). A living biobank of breast cancer organoids captures disease heterogeneity. Cell 172, 373–386.e310. [DOI] [PubMed] [Google Scholar]
- Sarrio D., Rodriguez-Pinilla S.M., Dotor A., et al. (2006). Abnormal ezrin localization is associated with clinicopathological features in invasive breast carcinomas. Breast Cancer Res. Treat. 98, 71–79. [DOI] [PubMed] [Google Scholar]
- Sato T., Vries R.G., Snippert H.J., et al. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265. [DOI] [PubMed] [Google Scholar]
- Schlecht N.F., Brandwein-Gensler M., Smith R.V., et al. (2012). Cytoplasmic ezrin and moesin correlate with poor survival in head and neck squamous cell carcinoma. Head Neck Pathol. 6, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X., Liu W., Yuan X., et al. (2018). Acetylation of ACAP4 regulates CCL18-elicited breast cancer cell migration and invasion. J. Mol. Cell Biol. 10, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuelten C.H., Parent C.A., and Montell D.J. (2018). Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat. Rev. Cancer 18, 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Klooster J.P., Jansen M., Yuan J., et al. (2009). Mst4 and Ezrin induce brush borders downstream of the Lkb1/Strad/Mo25 polarization complex. Dev. Cell 16, 551–562. [DOI] [PubMed] [Google Scholar]
- Xia P., Wang Z., Liu X., et al. (2012). EB1 acetylation by P300/CBP-associated factor (PCAF) ensures accurate kinetochore–microtubule interactions in mitosis. Proc. Natl Acad. Sci. USA 109, 16564–16569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X., Chaponnier C., Gabbiani G., et al. (1995). Polarized distribution of actin isoforms in gastric parietal cells. Mol. Biol. Cell 6, 541–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X., Cheng L., and Forte J.G. (1996). Biochemical characterization of ezrin–actin interaction. J. Biol. Chem. 271, 7224–7229. [DOI] [PubMed] [Google Scholar]
- Yao X., and Forte J.G. (2003). Cell biology of acid secretion by the parietal cell. Annu. Rev. Physiol. 65, 103–131. [DOI] [PubMed] [Google Scholar]
- Yao X., and Smolka A.J. (2019). Gastric parietal cell physiology and helicobacter pylori-induced disease. Gastroenterology 156, 2158–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X., Thibodeau A., and Forte J.G. (1993). Ezrin–calpain I interactions in gastric parietal cells. Am. J. Phys. 265, C36–C46. [DOI] [PubMed] [Google Scholar]
- Yu H., Zhou J., Takahashi H., et al. (2014). Spatial control of proton pump H,K-ATPase docking at the apical membrane by phosphorylation-coupled ezrin–syntaxin 3 interaction. J. Biol. Chem. 289, 33333–33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X., Yao P.Y., Jiang J.Y., et al. (2017). MST4 kinase phosphorylates ACAP4 protein to orchestrate apical membrane remodeling during gastric acid secretion. J. Biol. Chem. 292, 16174–16187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Shao H., Zhu T., et al. (2013). DDA3 associates with microtubule plus ends and orchestrates microtubule dynamics and directional cell migration. Sci. Rep. 3, 1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G., Cheng Y., Gui P., et al. (2019). Dynamic acetylation of the kinetochore-associated protein HEC1 ensures accurate microtubule–kinetochore attachment. J. Biol. Chem. 294, 576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Xu W., Jiang W., et al. (2010). Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R., Cao X., Watson C., et al. (2003). Characterization of protein kinase A-mediated phosphorylation of ezrin in gastric parietal cell activation. J. Biol. Chem. 278, 35651–35659. [DOI] [PubMed] [Google Scholar]
- Zhou R., Zhu L., Kodani A., et al. (2005). Phosphorylation of ezrin on threonine 567 produces a change in secretory phenotype and repolarizes the gastric parietal cell. J. Cell Sci. 118, 4381–4391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.