

Abstract
The complexity of male reproductive impairment has hampered characterization of the underlying genetic causes of male infertility. However, in the last 20 years, more powerful and affordable tools to interrogate the genetic and epigenetic determinants of male infertility have accelerated the number of new discoveries in the characterization of male infertility. With this explosion of new data, integration in a systems-based approach—including complete phenotypic information—to male infertility is imperative. We briefly review the current understanding of genetic and epigenetic causes of male infertility and how findings may be translated into a practical component for the diagnosis and treatment of male infertility.
Keywords: Big data, epigenomics, genetic, genomics, male infertility, polymorphism
Male infertility is a common condition that impacts 4% to 12% of men worldwide (1). Although most research has focused on environmental causes of male infertility, there has been great interest in better understanding the genetics of male factor infertility. Currently, nearly 30% of male factor infertility is associated with known genetic causes. These causes include Yq chromosome micro-deletions, Klinefelter syndrome, cystic fibrosis transmembrane receptor (CFTR) gene mutations in men with bilateral absence of the vas deferens, and other cytogenetic and chromosomal abnormalities (2–5). Although these causes impact a minority of men with infertility, they do have considerable prognostic value. For example, in men with Yq microdeletions, presence of certain azoospermia factor deletions predict the chance of successful surgical sperm retrieval and the fertility of future offspring conceived through assisted reproductive technologies (ART). A genetic etiology is implicated in >50% of male infertility cases currently classified as idiopathic. The identification of additional genetic causes of male infertility would be helpful for patient counseling regarding diagnosis, potential treatments, outcomes of sperm retrieval, and ART.
In the last decade, genetic studies have focused on genomewide association studies (GWAS) and copy number variant (CNV). With GWAS, microar-rays compare the incidence of variant single-nucleotide polymorphisms (SNPs) among cases and controls. Several GWAS have suggested susceptibility loci for male infertility (6). However, GWAS are limited to variants with a small effect size and may only explain rare heritable causes of infertility traits that lack broad relevance to the majority of men with infertility. More recently, there has been a shift toward studies of CNV using microarray analyses. These CNVs characterize structural variations that occur in one’s genome where large regions are deleted or duplicated (6). Prior studies have suggested an impact of CNV burden on male infertility, with an increased frequency of rare CNVs in infertile compared with normal men (6).
More recently, epigenetic modifications to gene expression have been implicated in spermatogenesis and male factor infertility. Epigenetics is the study of the processes that can alter gene expression. Epigenetic regulation includes DNA methylation, histone modifications, and spermatozoal RNA transcripts that may serve as potential arbiters of the heritability of male infertility (7). Previously, several epigenetic signatures have been associated with sperm abnormalities (8–10).
The sperm genome and epigenome are unique. The association of genetic variants and epigenetic signatures with phenotypic traits does not imply causality of specific genes or epigenetic changes (11–13). A clear causal relationship between genetic variants and epigenetic signatures has remained largely elusive. A true systems-based approach to the study of male factor infertility requires integration of genomic, epigenomic, and environmental factors implicated in these processes to better understand fertility and spermato-genesis. This approach must also incorporate overlaps with other comorbid conditions associated with male subfertility such as certain malignancies (14–16). In the era of “big data,” increasing collaboration among researchers and sharing of genetic and epigenetic data sets in the public domain has accelerated research in each of these respective fields toward a better understanding of male infertility. Additionally, the declining costs of techniques to interrogate the sperm genome and epigenome have primed research in this area for explosive growth in the near future. However, a large comprehensive genomic and epigenomic resource from carefully phenotyped infertile men remains to be created.
Our current understanding of genetic and epigenetic causes of male infertility combined with integration of these findings through a systems-based approach may be used to create tools for the diagnosis and treatment of male infertility (Fig. 1). We review the genetic and epigenetic understanding of male factor infertility, functional validation of these associations, and cutting edge phenotyping of large cohorts of infertile males, and we describe what integration of these areas using a systems-based approach would entail.
FIGURE 1.
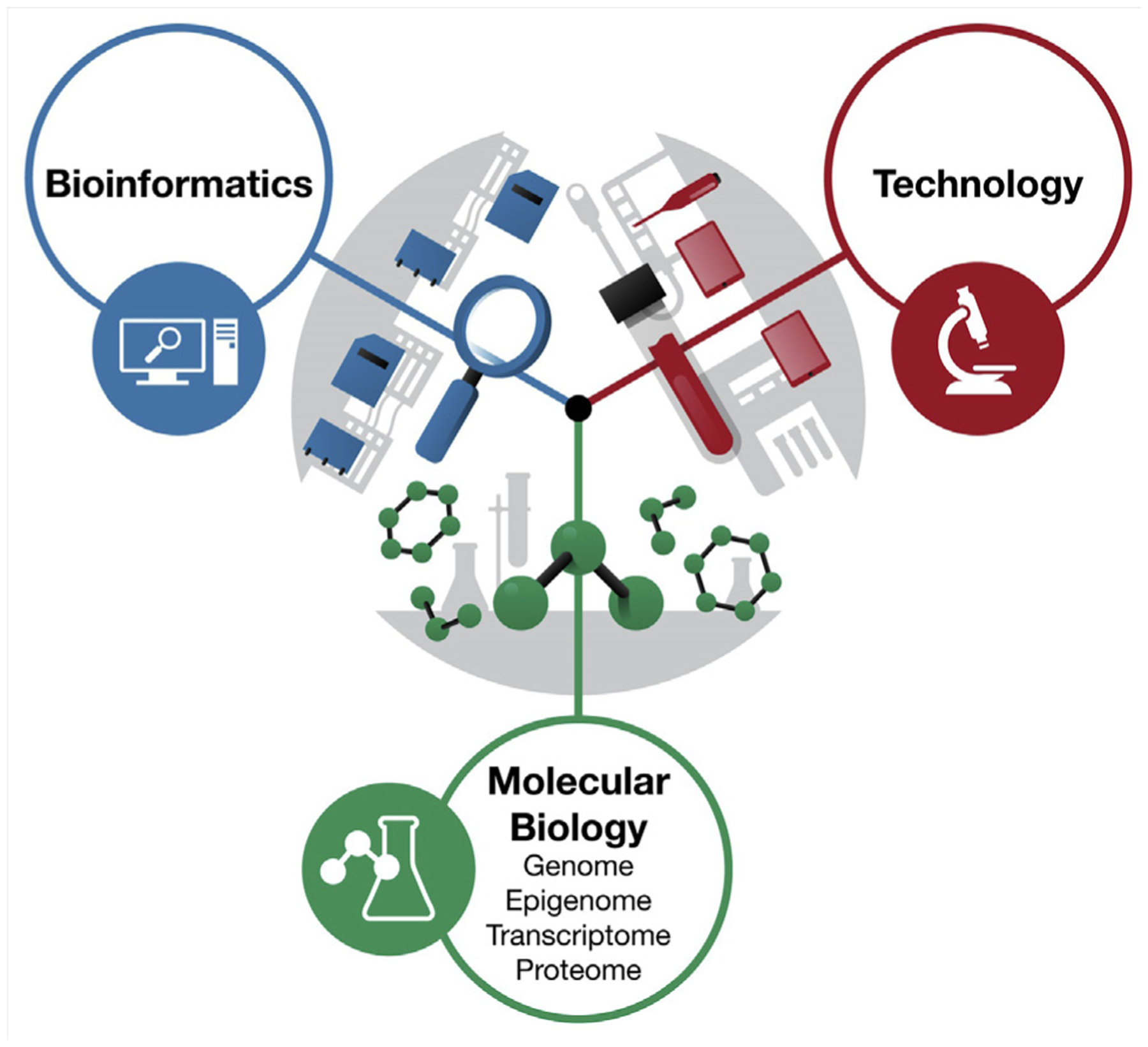
A systems-biology approach to the study of male reproductive health.
GENETICS OF MALE FACTOR INFERTILITY
The discovery of novel genetic variants associated with male infertility has accelerated rapidly over the past decade, driven by the application of tools for genomewide discovery. Although early GWAS used array-based approaches, next-generation sequencing (NGS) has more recently become the predominant tool for genetic discovery. The shift toward genomewide approaches has been driven by several factors: [1] a recognition that targeted sequencing approaches were largely ineffective, [2] growing appreciation for the vast number of genes required for spermatogenesis, and [3] increased accessibility and affordability of genomewide assessment tools. As a result, a tremendous amount of genetic data are now available.
The roles of Klinefelter syndrome and Yq chromosome microdeletions in spermatogenic failure are well recognized. These genetic anomalies were first identified through karyo-type analysis (4, 17). Subsequent development of Sanger sequencing methods more than 40 years ago enabled the democratization of DNA sequencing. This spurred a rapid rise in the number of studies targeting a gene sequence relevant to a specific phenotype in cases and controls, with the hope of identifying variants that occur more frequently in one group than another. Over the years, hundreds of spermatogenesis genes were sequenced in small cohorts ranging from tens to several hundred men. Several small effect risk variants were identified through targeted sequencing, but the yield with this approach was extremely low, and numerous spurious associations were reported, which remain to be replicated (6). Notwithstanding the shortcomings of targeted sequencing, early studies gave insight into the genetic architecture of spermatogenic impairment and paved the way for more powerful GWAS using NGS.
Microarrays permitting genomewide assessment were a tremendous advancement for the identification of genetic variants with potential roles in male infertility. Microarrays enable the interrogation of thousands to millions of variants across the genome in an efficient and robust manner. Additionally, genomewide approaches offer the significant advantage of enabling hypothesis-free variant discovery. Genomic microarrays include SNP genotyping arrays and comparative genomic hybridization arrays. The SNP arrays contain large numbers of oligonucleotide probes designed to target specific genomic loci and hybridize based on nucleotide complementarity. This enables the genotyping of common SNPs across the genome. In addition, evaluation of hybridization intensity across multiple contiguous probes allows the discovery of CNVs across the genome. Array CGH works on the principle of complementarity between two cohybridized genomes. Although this approach does not allow the identification of single-base differences, it is effective at identifying CNVs including duplications, deletions, and inversions.
Array-based approaches have been used widely in the study of male infertility, with modest success (18). Principal among the successes realized through microarray studies is insight into the genetic architecture of male infertility. Through genomewide association studies, it is becoming increasingly clear that, as expected, common variants (population incidence >1%) do not contribute appreciably to spermatogenic impairment. It was also noted in a number of studies evaluating CNVs that infertile men harbor an increased CNV content compared with fertile controls, suggesting a role for genomic instability in that population, a potential explanation for the increased risk of comorbidities observed in infertile men identified through epidemiologic studies (19).
In recent years, NGS approaches, primarily whole-exome sequencing (WES), have been used increasingly in the study of the genetics of male infertility and have largely supplanted even array-based approaches. The ~1% of the genome that codes for proteins is specifically targeted by WES, which offers significant advantages over whole-genome sequencing (WGS). First, sequencing costs are greatly reduced owing to the elimination of ~99% of the genome through the process of exome capture during library preparation. Second, because protein coding regions of the genome are relatively well characterized, and an abundance of tools exist to predict the functional consequences of variants identified within protein-coding regions, analysis and variant prioritization are greatly simplified for WES data over those from WGS.
A limitation of WES is the challenge associated with CNV calling, due to uneven and disrupted sequence coverage across the genome that is inherent in exome enrichment. However, methods exist for calling CNVs from these data. In addition, WES has the potential to miss disease-causing variants in regulatory regions of the genome; 85% of known disease-causing variants are contained within exonic regions (20). Many male infertility variants have been discovered through WES efforts over the past few years, particularly in cases of rare disorders such as globozoospermia (21, 22) and multiple morphological abnormalities of the sperm flagellum (23–26), but also in spermatogenic failure (27–30). The rate of variant discovery will undoubtedly continue to accelerate as NGS tools are increasingly deployed in clinical research and case and control data sets continue to expand.
Although WGS and WES approaches offer great promise in improving our understanding of the genetic basis for male infertility, the efficacy with which the approaches are used depends largely on experimental design and execution. Additionally, examining rare genetic perturbations in isolation is unlikely to yield broad insights into male infertility. It is only by understanding how the genetic drivers of male infertility influence other drivers of male reproductive health, including somatic health, familial health, and one’s epigenetic landscape, that we can fully begin to derive broad insights into the pathogenesis of this condition. Metabolomics may also be relevant, but this is beyond the scope of this review, and we have chosen to focus on genetics, epigenetics, and functional data derived from single-cell RNA sequencing data, which also give broad insight into gene expression data.
EPIGENETICS OF MALE INFERTILITY
Although historically there has been a greater focus on the genetics of male infertility, the sperm epigenome also plays an important role. Epigenetic signatures come in many forms, depending on the definition being used. By the most inclusive definition, an epigenetic mark is one that can be altered over an organism’s life span, can impact gene expression, and may be passed on to a developing embryo and even beyond. The molecular signatures that meet these criteria include DNA methylation (the covalent bonding of a methyl group to the 5-carbon of cytosine residues typically in the context of CpGs), small RNAs (micro-RNAs [miRNA], Piwi-interacting RNAs, etc.), and nuclear proteins and associated modifications (histones and protamines in the context of sperm). Epigenetic signatures may be indicative of pivotal events in spermatogenesis and predictive of the future potential of the cell. Further, many studies have shown that sperm epigenetic alterations may impact subtle phenotypic alterations in the offspring and even grand-offspring.
There is a long-standing link between male infertility phenotypes and epigenetic perturbations in the germ line. Historically, dogma held that sperm deliver the paternal DNA blueprint to the egg. It was believed that the oocyte was solely responsible for embryonic development and that the sperm had minimal contribution. The oocyte plays a tremendous role in the process, but studies assessing the impact of sperm epigenetic perturbations have demonstrated that an aberrant sperm epigenetic landscape will result in infertility in various forms, from altered sperm motility to embryo lethality (13).
Multiple studies have assessed the impact of sperm nuclear proteins on their functional capacity as well as their relationship to infertility phenotypes. The unique nature of the sperm requires a distinctive nuclear protein landscape, the hallmark of which is the replacement of histone proteins with protamines in the mature sperm. These proteins come in two forms, protamine 1 (P1) and protamine 2 (P2), expressed in a 1:1 ratio that results in a highly compacted chromatin structure. Because histones carry with them epigenetic marks capable of altering gene transcription in the form of chemical modifications to histone tails, the removal and replacement of these marks with protamines has long been used to discount the capacity of the sperm to impact embryo-genesis and beyond. However, the removal of histones throughout spermatogenesis is incomplete. In fact, studies have shown that the small portion of the genome that remains histone bound in sperm is important in early embryonic development, suggesting that, in contrast to what was previously believed, sperm nuclear proteins are quite important to embryonic development (31). This assumption was confirmed when assessing known infertile patients (some of whom produced poor embryos during in vitro fertilization) (32). Many of these patients had altered histone-retention patterns, which confirms that normal protamine replacement and histone retention in sperm is essential for fertility and normal embryogenesis. Further, simple studies have shown that the alterations to protamine expression directly, if not produced in a 1:1 ratio, result in infertility phenotypes (32–34).
Studying sperm RNAs can be challenging for many reasons, but many investigators have produced solid data that have further brought to light the importance of a normal sperm epigenetic landscape (9, 35–37). Multiple studies have highlighted specific miRNA species that are present in sperm and that appear to play an important role in fertility and embryogenesis. Although still controversial, some sperm miRNA species have been frequently identified in multiple studies and implicated in various forms of male infertility. Foremost among the single miRNAs has been miR-34C (9, 38). This miRNA has been implicated in both spermatogenesis as well as early embryogenesis, though the precise nature of its involvement is controversial.
Larger scale analysis of sperm-borne miRNAs has suggested that many more RNA species may be implicated and that each may vary in terms of its importance to fertility. Some of the most important recent work in this regard has been performed by Krawetz’s laboratory (36, 39). Their data suggest that a specific repertoire of “RNA elements” is required for normal sperm function, fertility, and embryogenesis and that alterations to these RNA elements in sperm result in infertility phenotypes (39). The assessment of sperm RNA elements was so successful that they have suggested that the analysis of these elements may be able to predict the success rates of different fertility treatments, which may optimize clinical treatment selection.
Sperm DNA methylation has also been studied extensively and has yielded some interesting insights into infertility phenotypes. Large-scale analyses of sperm DNA methylation signatures have identified associations with many forms of infertility. One study identified DNA methylation alterations associated with various semen parameters, including motility and count (40). Another showed that there are links between sperm DNA methylation signatures and time to pregnancy (8). Remarkably, one study even demonstrated that sperm DNA methylation signatures could be used to discriminate between patients who attended an in vitro fertilization clinic for reproductive care and fertile individuals (41). Further studies have shown that sperm DNA methylation signatures are sensitive to various lifestyle choices and toxins (42, 43). These studies demonstrate the potential for clinical utility with DNA methylation signatures in the sperm.
Of the epigenetic causes, DNA methylation lends itself to helpful clinical applications. In 2013, Horvath (44) used using machine-learning approaches to generate the first epigenetic age calculator using DNA methylation signatures from somatic cells in humans. This calculation is powerfully predictive of an individual’s age and can be altered based on lifestyle decisions (45). However, this model was unable to predict age with sperm DNA methylation because as these signatures are highly unique compared with somatic cells. In 2018, our group used similar machine-learning approaches to generate an age calculator for sperm DNA methylation signatures that can predict an individual’s age with ~94% accuracy on average (46). The power of the computational tools available today is remarkable and has made these types of predictive models possible.
Epigenetics, to some extent, has validated Lamarckian evolution whereby the life course experiences of one generation are transmitted to their offspring’s epigenetic landscape to influence their developmental traits. Although we are just beginning to fuse the genetic and epigenetic determinants of male infertility, this work holds great promise to derive broad insight into a large portion of male infertility currently classified as idiopathic. However, functional validation is needed to prove the mechanism of these associations and, hence, derive causality.
FUNCTIONAL VALIDATION OF GENETIC AND EPIGENETIC FINDINGS WITHIN SPERMATOGONIAL STEM CELL CULTURES
The inability to culture the spermatogonial stem cell (SSC), the only germline stem cell within the testis, limits the functional validation of the genetic and epigenetic findings in male infertility. Without functional validation within SSC, the clinical importance of many current and future genetic and epigenetic findings is uncertain. However, our current understanding of human SSC biology is limited.
Most of what is known about SSC biology is derived from studies of rodents, which have provided a wealth of genetic, molecular, and physiological information (47). However, many differences exist between humans and mice regarding SSC development (48). For example, mouse SSC development involves multiple rounds of amplification from Asingle → Apair → Aaligned, which will then give rise to the type B spermatogonia, while human SSCs only have two rounds of amplification and consist of Adark (Ad) and Apale (Ap) (49). In addition, mouse SSCs initiate differentiation within weeks of birth whereas human SSCs experience a long-term quiescence after birth and do not differentiate until puberty, which is approximately 10 years after birth (50). These developmental differences may help explain the fact that although mouse SSCs were reliably cultured decades ago, the in vitro culture of human SSCs has been elusive, which makes functional validation of the genetic and epigenetic findings highly challenging (51–53). Therefore, we need to gain a better understanding of human SSCs to establish an efficient and authentic culture system.
Recently, work from our group and others using genomic profiling, especially single-cell transcriptome profiling, has provided multiple insights into the developmental potential and molecular features of human SSCs (54–58). For example, by applying single-cell RNA-seq (scRNA-seq) to study human SSCs and testes we identified five distinct cellular states (termed states 0–4) accompanying human spermatogonia development, with states 0–1 as the undifferentiated and reserved stem cells, and states 2–4 as the differentiating and proliferative spermatogonia, which are committed to meiosis (54, 55). Remarkably, further comparison revealed that state 0 human SSCs (from adult testes) were very similar to infant germ cells, suggesting state 0 SSCs are the most naïve and reserved stem cells in the adult human testes.
Of note, the cellular states we identified (via transcriptional features) do not correspond with previously documented Ad and Ap spermatogonia, which was described based on morphological and histological examination (59). In agreement with our observations, Jan et al. (60) profiled transcriptomes from human Ad and Ap spermatogonia via laser capture microdissection and were not able to identify transcriptional differences between Ad and Ap, suggesting that the differences exist beyond the transcriptional level.
Overall, the advancement of biotechnology, especially genomic profiling, has revolutionized the way researchers study the human male germline and has resulted in a much deeper understanding of human SSCs. However, the last frontier of being able to create a viable human SSC cell line capable of producing sperm in vitro remains.
Human SSC culture is vital to deriving mechanistic insights into how genetic and epigenetic factors work in unison to determine male reproductive potential. Until we can manipulate human SSCs, we will be unable to use techniques such as CRISPR or RNA silencing to alter the genetic or epigenetic profile of SSCs and examine the impact on spermato-genesis. Further, without culture systems that replicate the germ cell niche we will not be able to fully assess the role of Sertoli and Leydig cells in spermatogenesis. With our current data sets, we are already capable of fusing genetic, epigenetic, and transcriptomic data to yield broad insight into spermatogenesis, but culture work to determine mechanistic insights remains elusive. We are left to rely on animal models that do not adequately replicate human spermatogenesis.
TRANSGENERATIONAL IMPACT OF MALE INFERTILITY
The genetic and epigenetic heritability of male infertility has led to questions regarding the impact of one’s individual infertility on familial somatic health. Historically, the impact of male infertility on familial somatic health among the offspring and family of subfertile males was poorly understood, given the lack of comprehensive resources to begin to answer these questions. However, large population-based pedigree analyses have helped shed light on familial health among men with infertility.
Recently, analyses from the Subfertility Health and Assisted Reproduction (SHARE) study using the Utah Population Database have suggested an impact of male infertility on familial mortality and cancer risk. An increased risk of testicular cancer and thyroid cancer has been suggested in first- and second-degree relatives of subfertile men compared with fertile controls (61). Additionally, an increased risk of any childhood cancer and acute lymphoblastic leukemia in sibling and cousins of subfertile men has also been identified (62). We also found that first-degree relatives of azoospermic men had more than a twofold increased risk in congenital malformation-related death (63). These data suggest that male infertility, which is a biomarker of individual health, may also be a marker of familial health.
Similar large population-based pedigree analyses will generate a tremendous expanse of hypothesis generating data about male infertility and familial health. However, integration of these data with emerging genetic and epigenetic data is imperative. Additionally, complex phenotyping of large cohorts of infertile males and their families is critical to a comprehensive view of male infertility that leverages the abundance of genetic, epigenetic, and transcriptomic data toward clinical applications.
Sasani et al. (64) recently described the de novo mutation dynamics from large, three-generation families from Utah that were initially collected as part of the Centre d’Etude du Polymorphisme Humain consortium. This group identified a high-confidence set of germline de novo mutations that were transmitted to the following generation. There was considerable variability for parental age effects on the number of de novo mutations among different family pedigrees. Specifically, there was a nearly threefold difference between families with high and low de novo mutation rates. This is one of the first studies to suggest transmission of de novo mutational load over generations. Remarkably, this group also found that families with higher de novo mutation rates had grandparents who, on average, died 10 years younger than in families who had lower de novo mutation rates (65). This suggests a mechanistic link between higher de novo mutational load and individual and familial somatic health due to genetic or epigenetically heritable factors, which increase the de novo mutation rate and worsen fertility and overall health. This possible mechanism may help identify novel developmental, genetic, epigenetic, and environmental factors that impact familial differences in susceptibility for male reproductive impairment.
THE ROLE OF COMPLETE PHENOTYPING
The causes of male reproductive impairments are multifactorial, and each genetic, epigenetic, and environmental factor affects the male infertility phenotype. The characterization of the male infertility phenotype has historically relied on abnormalities observed on semen analyses.
Over the last 20 years, molecular genetics and epigenetic tools have changed this landscape, especially with Y-chromosome microdeletions. However, the results of more recent GWAS and CNV studies are unlikely to have a major role on the diagnosis and treatment of male infertility because they are rare. The integration of genetic and epigenetic variants into a diagnostic panel for quantitative spermatogenetic disturbances that correlates with a specific male infertility phenotype will be helpful for future personalized treatment. Complete characterization of the male infertility phenotype involving thousands of patients is required.
Table 1 presents the many components that would be included in complete phenotyping of infertile males. Genomic and epigenomic studies will be most effective when cohorts are thoroughly phenotyped and matched (66). Clinical and basic science collaborations such as the Genetics of Male Infertility Initiative (GEMINI; https://gemini.conradlab.org) and the International Male Infertility Genomics Consortium (IMIGC; http://www.imigc.org) gather large, well-characterized cohorts to increase the power of genomic and epigenomic studies.
TABLE 1.
Proposed phenotype data points for infertile males.
| Demographics |
| Age (y) |
| Partner’s age (y) |
| Race/ethnicity |
| Body mass index (kg/m2) |
| Clinical |
| Months of infertility |
| Complete medical history |
| Spermatotoxic medications and exposures |
| Complete phenotyping of female infertility factors |
| Comprehensive familial health history |
| Anatomical |
| Testis longitudinal axis (cm) |
| Presence of varicocele and associated grade |
| Scrotal ultrasound findings |
| Hormones |
| Follicle-stimulating hormone |
| Luteinizing hormone |
| Total, bioavailable, and free testosterone |
| Sex hormone-binding globulin |
| Estradiol |
| Inhibin B |
| Thyroid-stimulating hormone |
| Prolactin |
| Karyotype |
| Y chromosome microdeletion |
| Semen analysisa |
| Volume (mL) |
| Sperm count |
| Sperm concentration (M/mL) |
| % Motility |
| % Progressive motility |
| Total motile count |
| Morphology (% normal) |
| % Head defects |
| % Neck/midpiece defects |
| % Cytoplasmic defects |
| % Tail defects |
At least two semen analyses, preferably three.
SYSTEMS-BASED APPROACH
Male infertility is a complex phenotype, and traditional reductionist methods aimed at isolating a single-cause to single-phenotype relationship are unable to capture the multifactorial processes involved. A systems-based approach embraces the complexity of causation and allows for the simultaneous consideration of the genetic, epigenetic, and functional processes leading to an infertility phenotype (67). This approach uses computational modeling of multiple types of data to enable the investigation of the intricacy of causation within a multilevel and dynamic framework and simulate system behavior. For example, Zhu et al. (68) combined different types of data to construct probabilistic causal networks that simultaneously considered all data elements, leading to novel insights into segregating yeast populations. Similar approaches could provide insight into male infertility.
Although adoption of an approach that allows for the incorporation of biological interconnections has great potential, the methods necessary for teasing out the complex relationships in infertility risk are still largely undeveloped and have several key limitations. Methods for synthesizing omic data from multiple sources are necessary for reproducibility and successful translation into clinical practice. These data sets often have different preprocessing pipelines and levels of measurement; lack of well-defined standards for integrating these data can lead to inconsistent and unreliable findings. A priori information is also critical to these models, so a strong collaborative effort between infertility experts, computer scientists, statisticians, and biologists is necessary. Application of systems- and network-based approaches in the medical field is in its infancy, but it is critical to enable a more complete view of the male infertility phenotype.
CONCLUSION
Male reproductive health is a complex process that likely relies on many gene interactions. A systems-based approach to integrate different areas of current research, including genomic and epigenetic studies, would provide deeper insight into male infertility. One of the challenges with integration analyses is the lack of truly interactive data sets and data analysts with the necessary skills to systematically analyze these data. A summary of the key studies in each of the respective areas is presented in Table 2.
TABLE 2.
Key studies for genetic, epigenetic, and transgenerational of male reproductive impairment.
| Study | Year | Study cohort | Findings | Key findings for future studies | Future integration |
|---|---|---|---|---|---|
| Genetics Aston et al. (73) | 2009 | 52 oligozoospermic men and 40 azoospermic men. | Identification of 21 SNPs associated with oligozoospermia and azoospermia. | Pilot GWAS for future studies linking SNPs with male infertility. | Identification of unique SNPs associated with male infertility. |
| Lopes et al. (74) Epigenetics | 2013 | Sample 1: 323 Caucasian men with spermatogenic impairment + 1,100 controls. Sample 2: 979 Han Chinese men with azoospermia + 6,253 controls. |
Rare autosomal deletions, rare X-linked CNVs, and rare Y-linked duplications increase an individual’s risk of spermatogenic impairment by 10%, 29%, and 88%, respectively. DMRT1 loss of function mutations are rare risk factors for spermatogenic failure. | Hypothesis generating data to direct future studies linking CNVs to azoospermia. | Identification of unique CNVs associated with male infertility. |
| Hammoud et al. (31) | 2009 | Semen samples from four men with known fertility. | Enrichment of modified nucleosomes among genes for embryonic development in sperm may provide instruction for regulation of developmental gene, noncoding RNA, and imprinted loci. | Highlight sperm epigenetic markings and links to developmental regulation. | Determination of specific epigenetic signatures associated with male infertility. |
| Aston et al. (41) | 2015 | 127 men with male factor infertility undergoing IVF and 54 normospermic controls. | Significant difference in sperm DNA methylation between men with male factor infertility undergoing IVF compared with fertile men, which was predictive of poor embryo quality. | Understand what impacts sperm DNA methylation signatures. | Use of specific epigenetic signatures to help guide treatment with ART. |
| Jenkins et al. (46) Transgenerational/familial fertility and somatic health assessments | 2018 | 329 semen samples from fertile and infertile men. | Predictive model incorporating sperm DNA methylation signatures can predict an individual’s age with >94% accuracy. | Drive investigation on the potential impacts of epigenetic signatures on aging, fertility, and somatic health. | Use of sperm DNA methylation signatures to understand environmental effects. |
| Guo et al. (54) | 2018 | Single-cell RNA sequencing of 6,500 testicular cells. | Description of key transcription and epigenetic signatures in the normal adult human testis. Suggested developmental plasticity between five transcriptional/ developmental state (including unique state 0, a novel early hSSC state). | Highlight new areas into germ cell development transitions and plasticity. | Contribute to development of hSSC for diagnostic and therapeutic uses. |
Note: ART = artificial reproductive technologies; CI = confidence interval; CNV = copy number variants; GWAS = genomewide association study; HR = hazard ratio; hSSC = human spermatogonial stem cell; IVF = in vitro fertilization; SNP = single-nucleotide polymorphism.
In other diseases such as cancer, data integration has allowed discoveries across different data sets. For example, renal cell carcinoma is a model pathology that has benefited from integration analyses (69). In 2005, only two drugs were approved for metastatic renal cell carcinoma (70). Over the next decade, 10 additional drugs were approved (70). Analyses such as cluster of cluster analysis (COCA) have shed light on shared cancer pathways that extends beyond histo-logic classifications. Chen et al. (69) performed a multiplatform analyses of 900 renal cell carcinoma cases across five data platforms including DNA methylation, DNA copy alteration, mRNA expression, miRNA expression, and protein expression. Integration analyses across various histologic subtypes of renal cell carcinoma demonstrated differences in patient survival with alterations of vascular endothelial growth factor (VEGF), nuclear erythroid 2-related factor 2 (NFF2), and mammalian target of rapamycin (mTOR). This has inspired studies of pan-urologic cancer genomic subtypes including bladder, kidney, prostate, and testes and characterization of different molecular platforms to identify drivers of cell behavior across different cancer types (71).
Currently, the evaluation of male factor infertility relies on semen analysis. However, semen analysis is a poor predictor of fertility potential and a poor prognostic test to guide approaches for assisted reproduction. There is considerable interest in personalized medicine as a diagnostic and therapeutic tool for male infertility. There are subtle differences in an individual’s fertility potential, and the goal of diagnosis and treatment should be to individualize these approaches to optimize the chance of achieving a live birth (Fig. 2).
FIGURE 2.
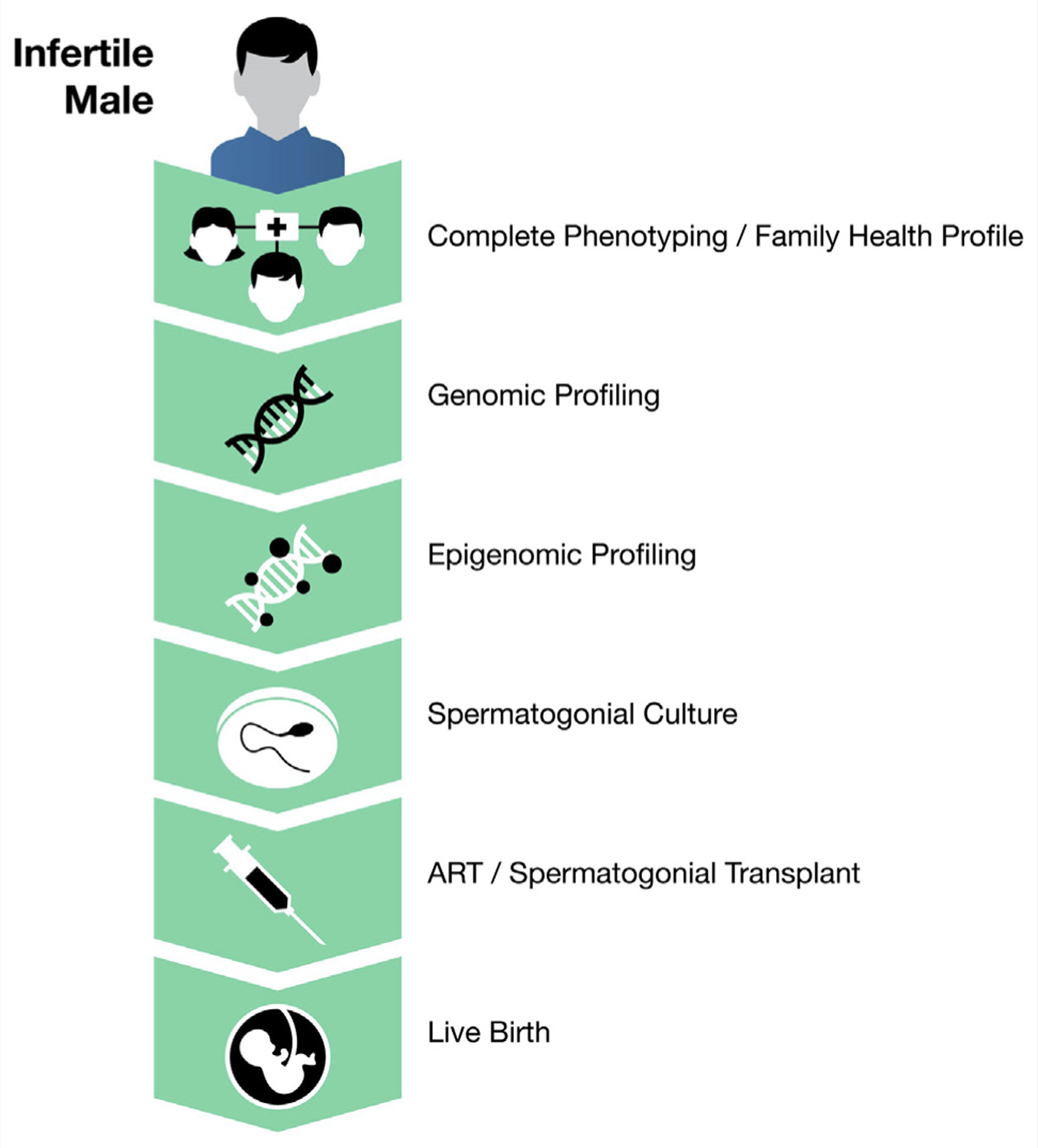
Personalized medicine for male factor infertility.
For example, consider patients with nonobstructive azoospermia. We envision an opportunity to use our understanding of the genetic and epigenetic underpinnings of sperm pathophysiology to leverage improved diagnostics. Once aberrations in the individual’s sperm are identified, SSC may be used to create functional sperm, which may undergo in vitro maturation for intracytoplasmic sperm injection or be transplanted back to the individual for natural conception. We are in the infancy of using these techniques for male factor infertility, and a great deal of research is required to shift from experimental to clinical applications.
One of the biggest frontiers in this area is the ability to create SSC culture, which holds tremendous diagnostic and therapeutic potential. In addition to the therapeutic potential already discussed, SSC culture also could unlock the mechanism linking infertility with poor individual and familial somatic health. Novel techniques such as single-sperm sequencing may be leveraged to further explore these relationships (72). Sequencing of individual sperm, in combination with the techniques we described earlier, may help us understand how a single SSC makes a population of sperm and, more importantly, how perturbations within SSC impair spermatogenesis and how these are transmitted to offspring. Understanding the pathogenic mechanisms would lead to potential diagnostics and therapeutics, which are simply lacking in male infertility.
Integration of the genetic and epigenetic platforms for male reproductive health are in their early stages. Considerable work is required to establish a truly integrated systems-based approach to the study of normal and pathological male reproductive function. Not only will this require new software to analyze the generated data, but also skilled researchers who can facilitate integration. Male infertility research is primed for a revolution from harnessing complete phenotyping of infertile males with a systems biology approach.
Acknowledgments
This work is supported by National Institute of Health (5R01HD078641-05, awarded to KIA) and the Howard Hughes Medical Institute (P30CA042014) from the National Cancer Institute to Huntsman Cancer Institute for core facilities.
Footnotes
D.P.P has nothing to disclose. T.G.J. has nothing to disclose. K.I.A. has nothing to disclose. J.G. has nothing to disclose. A.W.P. has nothing to disclose. H.A.H. has nothing to disclose. J.M.H. has nothing to disclose.
REFERENCES
- 1.Agarwal A, Mulgund A, Hamada A, Chyatte MR. A unique view on male infertility around the globe. Reprod Biol Endocrinol 2015;13:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hofherr SE, Wiktor AE, Kipp BR, Dawson DB, Van Dyke DL. Clinical diagnostic testing for the cytogenetic and molecular causes of male infertility: the Mayo Clinic experience. J Assist Reprod Genet 2011;28:1091–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter’s syndrome. Lancet 2004;364:273–83. [DOI] [PubMed] [Google Scholar]
- 4.Tiepolo L, Zuffardi O. Localization of factors controlling spermatogenesis in the nonfluorescent portion of the human Y chromosome long arm. Hum Genet 1976;34:119–24. [DOI] [PubMed] [Google Scholar]
- 5.Yatsenko AN, Yatsenko SA, Weedin JW, Lawrence AE, Patel A, Peacock S, et al. Comprehensive 5-year study of cytogenetic aberrations in 668 infertile men. J Urol 2010;183:1636–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aston KI. Genetic susceptibility to male infertility: news from genome-wide association studies. Andrology 2014;2:315–21. [DOI] [PubMed] [Google Scholar]
- 7.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol 2010;28:1057–68. [DOI] [PubMed] [Google Scholar]
- 8.Jenkins TG, Aston KI, Meyer TD, Hotaling JM, Shamsi MB, Johnstone EB, et al. Decreased fecundity and sperm DNA methylation patterns. Fertil Steril 2016;105:51–7.e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu WM, Pang RT, Chiu PC, Wong BP, Lao K, Lee KF, et al. Sperm-borne microRNA-34c is required for the first cleavage division in mouse. Proc Natl Acad Sci USA 2012;109:490–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aoki VW, Emery BR, Liu L, Carrell DT. Protamine levels vary between individual sperm cells of infertile human males and correlate with viability and DNA integrity. J Androl 2006;27:890–8. [DOI] [PubMed] [Google Scholar]
- 11.Nowakowska B Clinical interpretation of copy number variants in the human genome. J Appl Genet 2017;58:449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tam V, Patel N, Turcotte M, Bosse Y, Pare G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet 2019;20:467–84. [DOI] [PubMed] [Google Scholar]
- 13.Jenkins TG, Aston KI, James ER, Carrell DT. Sperm epigenetics in the study of male fertility, offspring health, and potential clinical applications. Syst Biol Reprod Med 2017;63:69–76. [DOI] [PubMed] [Google Scholar]
- 14.Hanson BM, Eisenberg ML, Hotaling JM. Male infertility: a biomarker of individual and familial cancer risk. Fertil Steril 2018;109:6–19. [DOI] [PubMed] [Google Scholar]
- 15.James E, Jenkins TG. Epigenetics, infertility, and cancer: future directions. Fertil Steril 2018;109:27–32. [DOI] [PubMed] [Google Scholar]
- 16.Nagirnaja L, Aston KI, Conrad DF. Genetic intersection of male infertility and cancer. Fertil Steril 2018;109:20–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klinefelter H, Reifenstein E, Albright F. Syndrome characterized by gynecomastia, aspermatogenesis without a-Leydigism, and increased excretion of follicle-stimulating hormone. J Clin Endocrinol 1942;2:615–27. [Google Scholar]
- 18.Aston KI, Conrad DF. A review of genome-wide approaches to study the genetic basis for spermatogenic defects. Methods Mol Biol 2013;927:397– 410. [DOI] [PubMed] [Google Scholar]
- 19.Aston KI, Carrell DT. Emerging evidence for the role of genomic instability in male factor infertility. Syst Biol Reprod Med 2012;58:71–80. [DOI] [PubMed] [Google Scholar]
- 20.Scacheri CA, Scacheri PC. Mutations in the noncoding genome. Curr Opin Pediatr 2015;27:659–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dam AH, Koscinski I, Kremer JA, Moutou C, Jaeger AS, Oudakker AR, et al. Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. Am J Hum Genet 2007;81:813–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koscinski I, Elinati E, Fossard C, Redin C, Muller J, Velez de la Calle J, et al. DPY19L2 deletion as a major cause of globozoospermia. Am J Hum Genet 2011;88:344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amiri-Yekta A, Coutton C, Kherraf ZE, Karaouzene T, Le Tanno P, Sanati MH, et al. Whole-exome sequencing of familial cases of multiple morphological abnormalities of the sperm flagella (MMAF) reveals new DNAH1 mutations. Hum Reprod 2016;31:2872–80. [DOI] [PubMed] [Google Scholar]
- 24.Sha YW, Wang X, Xu X, Su ZY, Cui Y, Mei LB, et al. Novel mutations in CFAP44 and CFAP43 cause multiple morphological abnormalities of the sperm flagella (MMAF). Reprod Sci 2017;26:26–34. [DOI] [PubMed] [Google Scholar]
- 25.Sha YW, Xu X, Mei LB, Li P, Su ZY, He XQ, et al. A homozygous CEP135 mutation is associated with multiple morphological abnormalities of the sperm flagella (MMAF). Gene 2017;633:48–53. [DOI] [PubMed] [Google Scholar]
- 26.Liu W, Sha Y, Li Y, Mei L, Lin S, Huang X, et al. Loss-of-function mutations in SPEF2 cause multiple morphological abnormalities of the sperm flagella (MMAF). J Med Genet 2019;56:678–84. [DOI] [PubMed] [Google Scholar]
- 27.Kasak L, Punab M, Nagirnaja L, Grigorova M, Minajeva A, Lopes AM, et al. Bi-allelic recessive loss-of-function variants in FANCM cause non-obstructive azoospermia. Am J Hum Genet 2018;103:200–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okutman O, Muller J, Baert Y, Serdarogullari M, Gultomruk M, Piton A, et al. Exome sequencing reveals a nonsense mutation in TEX15 causing spermatogenic failure in a Turkish family. Hum Mol Genet 2015;24: 5581–8. [DOI] [PubMed] [Google Scholar]
- 29.Ramasamy R, Bakircioglu ME, Cengiz C, Karaca E, Scovell J, Jhangiani SN, et al. Whole-exome sequencing identifies novel homozygous mutation in NPAS2 in family with nonobstructive azoospermia. Fertil Steril 2015;104:286–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yatsenko AN, Georgiadis AP, Ropke A, Berman AJ, Jaffe T, Olszewska M, et al. X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N Engl J Med 2015;372:2097–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR. Distinctive chromatin in human sperm packages genes for embryo development. Nature 2009;460:473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammoud SS, Nix DA, Hammoud AO, Gibson M, Cairns BR, Carrell DT. Genome-wide analysis identifies changes in histone retention and epigenetic modifications at developmental and imprinted gene loci in the sperm of infertile men. Hum Reprod 2011;26:2558–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carrell DT, Liu L. Altered protamine 2 expression is uncommon in donors of known fertility, but common among men with poor fertilizing capacity, and may reflect other abnormalities of spermiogenesis. J Androl 2001;22:604–10. [PubMed] [Google Scholar]
- 34.Cho C, Jung-Ha H, Willis WD, Goulding EH, Stein P, Xu Z, et al. Protamine 2 deficiency leads to sperm DNA damage and embryo death in mice. Biol Re-prod 2003;69:211–7. [DOI] [PubMed] [Google Scholar]
- 35.Dunn GA, Morgan CP, Bale TL. Sex-specificity in transgenerational epigenetic programming. Horm Behav 2011;59:290–5. [DOI] [PubMed] [Google Scholar]
- 36.Jodar M, Selvaraju S, Sendler E, Diamond MP, Krawetz SA. The presence, role and clinical use of spermatozoal RNAs. Hum Reprod Update 2013;19: 604–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luk AC, Chan WY, Rennert OM, Lee TL. Long noncoding RNAs in spermato-genesis: insights from recent high-throughput transcriptome studies. Reproduction 2014;147:R131–41. [DOI] [PubMed] [Google Scholar]
- 38.Yuan S, Tang C, Zhang Y, Wu J, Bao J, Zheng H, et al. mir-34b/c and mir-449a/b/c are required for spermatogenesis, but not for the first cleavage division in mice. Biol Open 2015;4:212–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jodar M, Sendler E, Moskovtsev SI, Librach CL, Goodrich R, Swanson S, et al. Absence of sperm RNA elements correlates with idiopathic male infertility. Sci Transl Med 2015;7:295re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jenkins TG, Aston KI, Hotaling JM, Shamsi MB, Simon L, Carrell DT. Teratozoospermia and asthenozoospermia are associated with specific epigenetic signatures. Andrology 2016;4:843–9. [DOI] [PubMed] [Google Scholar]
- 41.Aston KI, Uren PJ, Jenkins TG, Horsager A, Cairns BR, Smith AD, et al. Aberrant sperm DNA methylation predicts male fertility status and embryo quality. Fertil Steril 2015;104:1388–97.e1–5. [DOI] [PubMed] [Google Scholar]
- 42.Craig JR, Jenkins TG, Carrell DT, Hotaling JM. Obesity, male infertility, and the sperm epigenome. Fertil Steril 2017;107:848–59. [DOI] [PubMed] [Google Scholar]
- 43.Jenkins TG, James ER, Alonso DF, Hoidal JR, Murphy PJ, Hotaling JM, et al. Cigarette smoking significantly alters sperm DNA methylation patterns. Andrology 2017;5:1089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horvath S DNA methylation age of human tissues and cell types. Genome Biol 2013;14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosen AD, Robertson KD, Hlady RA, Muench C, Lee J, Philibert R, et al. DNA methylation age is accelerated in alcohol dependence. Transl Psychiatry 2018;8:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jenkins TG, Aston KI, Cairns B, Smith A, Carrell DT. Paternal germ line aging: DNA methylation age prediction from human sperm. BMC Genomics 2018; 19:763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanatsu-Shinohara M, Shinohara T. Spermatogonial stem cell self-renewal and development. Annu Rev Cell Dev Biol 2013;29:163–87. [DOI] [PubMed] [Google Scholar]
- 48.Boitani C, Di Persio S, Esposito V, Vicini E. Spermatogonial cells: mouse, monkey and man comparison. Semin Cell Dev Biol 2016;59:79–88. [DOI] [PubMed] [Google Scholar]
- 49.Ehmcke J, Wistuba J, Schlatt S. Spermatogonial stem cells: questions, models and perspectives. Hum Reprod Update 2006;12:275–82. [DOI] [PubMed] [Google Scholar]
- 50.Plant TM. Neuroendocrine control of the onset of puberty. Front Neuroendocrinol 2015;38:73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanatsu-Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, Toyokuni S, et al. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod 2003;69:612–6. [DOI] [PubMed] [Google Scholar]
- 52.Zheng Y, Thomas A, Schmidt CM, Dann CT. Quantitative detection of human spermatogonia for optimization of spermatogonial stem cell culture. Hum Reprod 2014;29:2497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Medrano JV, Rombaut C, Simon C, Pellicer A, Goossens E. Human spermatogonial stem cells display limited proliferation in vitro under mouse spermatogonial stem cell culture conditions. Fertil Steril 2016;106:1539–49.e8. [DOI] [PubMed] [Google Scholar]
- 54.Guo J, Grow EJ, Mlcochova H, Maher GJ, Lindskog C, Nie X, et al. The adult human testis transcriptional cell atlas. Cell Res 2018;28:1141–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo J, Grow EJ, Yi C, Mlcochova H, Maher GJ, Lindskog C, et al. Chromatin and single-cell RNA-Seq profiling reveal dynamic signaling and metabolic transitions during human spermatogonial stem cell development. Cell Stem Cell 2017;21:533–46.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hermann BP, Cheng K, Singh A, Roa-De La Cruz L, Mutoji KN, Chen IC, et al. The mammalian spermatogenesis single-cell transcriptome, from spermatogonial stem cells to spermatids. Cell Rep 2018;25:1650–67.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sohni A, Tan K, Song HW, Burow D, de Rooij DG, Laurent L, et al. The neonatal and adult human testis defined at the single-cell level. Cell Rep 2019;26:1501–17.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang M, Liu X, Chang G, Chen Y, An G, Yan L, et al. Single-Cell RNA sequencing analysis reveals sequential cell fate transition during human spermatogenesis. Cell Stem Cell 2018;23:599–614.e4. [DOI] [PubMed] [Google Scholar]
- 59.Clermont Y Spermatogenesis in man: a study of the spermatogonial population. Fertil Steril 1966;17:705–21. [PubMed] [Google Scholar]
- 60.Jan SZ, Vormer TL, Jongejan A, Roling MD, Silber SJ, de Rooij DG, et al. Unraveling transcriptome dynamics in human spermatogenesis. Development 2017;144:3659–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anderson RE, Hanson HA, Patel DP, Johnstone E, Aston KI, Carrell DT, et al. Cancer risk in first- and second-degree relatives of men with poor semen quality. Fertil Steril 2016;106:731–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anderson RE, Hanson HA, Lowrance WT, Redshaw J, Oottamasathien S, Schaeffer A, et al. Childhood cancer risk in the siblings and cousins of men with poor semen quality. J Urol 2017;197:898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hanson HA, Mayer EN, Anderson RE, Aston KI, Carrell DT, Berger J, et al. Risk of childhood mortality in family members of men with poor semen quality. Hum Reprod 2017;32:239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sasani TA, Pedersen BS, Gao Z, Baird L, Przeworski M, Jorde LB, et al. Large, three-generation human families reveal post-zygotic mosaicism and variability in germline mutation accumulation. eLife 2019;8: e46922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cawthon RM, Meeks HD, Sasani TA, Smith KR, Kerber RA, O’Brien E, et al. Lower germline mutation rates in young adults predict longer lives and longer reproductive lifespans. medRxiv 2019:19004184 Available at: 10.1101/19004184. [DOI] [Google Scholar]
- 66.Wilfert AB, Chao KR, Kaushal M, Jain S, Zollner S, Adams DR, et al. Genome-wide significance testing of variation from single case exomes. Nat Genet 2016;48:1455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carrell DT, Aston KI, Oliva R, Emery BR, De Jonge CJ. The “omics” of human male infertility: integrating big data in a systems biology approach. Cell Tissue Res 2016;363:295–312. [DOI] [PubMed] [Google Scholar]
- 68.Zhu J, Sova P, Xu Q, Dombek KM, Xu EY, Vu H, et al. Stitching together multiple data dimensions reveals interacting metabolomic and transcriptomic networks that modulate cell regulation. PLoS Biol 2012;10: e1001301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen F, Zhang Y, Senbabaoglu Y, Ciriello G, Yang L, Reznik E, et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep 2016;14: 2476–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giles RH, Choueiri TK, Heng DY, Albiges L, Hsieh JJ, Linehan WM, et al. Recommendations for the management of rare kidney cancers. Eur Urol 2017; 72:974–83. [DOI] [PubMed] [Google Scholar]
- 71.Chen F, Zhang Y, Bosse D, Lalani AA, Hakimi AA, Hsieh JJ, et al. Pan-urologic cancer genomic subtypes that transcend tissue of origin. Nat Commun 2017;8:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hinch AG, Zhang G, Becker PW, Moralli D, Hinch R, Davies B, et al. Factors influencing meiotic recombination revealed by whole-genome sequencing of single sperm. Science 2019;363:eaau8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aston KI, Carrell DT. Genome-wide study of single-nucleotide polymorphisms associated with azoospermia and severe oligozoospermia. J Androl 2009;30:711–25. [DOI] [PubMed] [Google Scholar]
- 74.Lopes AM, Aston KI, Thompson E, Carvalho F, Goncalves J, Huang N, et al. Human spermatogenic failure purges deleterious mutation load from the autosomes and both sex chromosomes, including the gene DMRT1. PLoS Genet 2013;9:e1003349. [DOI] [PMC free article] [PubMed] [Google Scholar]